

Abstract
Vitamin D sufficiency is associated with protection against malignancy in a number of tissues clinically, and a strong body of evidence from animal and cell culture studies supports this protective role. Cancers in the skin differ, however, in that higher serum levels of 25OHD are associated with increased basal cell carcinomas (BCC), the most common form of epidermal malignancy. This result may be interpreted as indicating the role of UVR (spectrum 280–320) in producing vitamin D in the skin as well as causing those DNA mutations and proliferative changes that lead to epidermal malignancies. Recent animal studies have shown that mice lacking the vitamin D receptor (VDR) are predisposed to developing skin tumors either from chemical carcinogens such as 7,12 dimethylbenzanthracene (DMBA) or chronic UVR exposure. Such studies suggest that vitamin D production and subsequent signaling through the VDR in the skin may have evolved in part as a protective mechanism against UVR induced epidermal cancer formation. In this manuscript we provide evidence indicating that vitamin D signaling protects the skin from cancer formation by controlling keratinocyte proliferation and differentiation, facilitating DNA repair, and suppressing activation of the hedgehog (Hh) pathway following UVR exposure.
Keywords: vitamin D receptor; 1,25 dihydroxyvitamin D; keratinocyte; UVR; DNA damage repair; sonic hedgehog
Vitamin D and skin cancer
Skin cancer is by far the most common malignancy. Numerous studies over the past 30 years have evaluated the potential of 1,25 Dihydroxyvitamin D3 [1,25(OH)2D3] for anticancer activity [1]. Most malignant cells express VDR including basal cell (BCC) and squamous cell (SCC) carcinomas [2, [3] as well as melanomas [4]. The generally accepted potential for 1,25(OH)2D3 in the prevention and treatment of malignancy rests with its antiproliferative, prodifferentiating actions. Epidemiologic evidence linking adequate vitamin D levels to colon cancer prevention is particularly strong, although the role of vitamin D in the prevention of other cancers has also been implicated [5, [6, [7, [8, [9]. This epidemiologic evidence is lacking for skin cancers, however [10, [11, [12], with several studies suggesting a positive correlation between 25OHD levels and BCC [13]. Most likely the lack of epidemiologic evidence for a positive role for vitamin D in preventing skin cancer is due to the dual effect of UVB radiation (UVR) in promoting vitamin D3 and 1,25(OH)2D3 synthesis in the skin and in increasing DNA damage leading to skin cancer negating the benefit of UVB induced vitamin D production. However, a threshold of UVR exposure may exist that would meet the nutritional requirements for vitamin D production without increasing the risk for epidermal tumor formation.
Animal studies suggest that vitamin D plays a protective role in the skin with respect to carcinogenesis. Zinser et al. [14] treated VDR null mice orally with the carcinogen 7,12 dimethylbenzanthracene (DMBA), attempting to induce breast cancers, but to their initial surprise 85% of the VDR null mice developed skin tumors. The wildtype controls did not develop tumors. Other groups have confirmed these results with the topical administration of DMBA/TPA [15]. Ellison et al. [16] extended these results by demonstrating that the VDR null mice all developed tumors in response to UVR, with few tumors appearing in the wildtype controls. We have confirmed these results with our own studies [17] (figure 1). Surprisingly mice lacking the ability to produce 1,25(OH)2D (CYP27B1 null) did not show increased susceptibility to tumor formation following either DMBA [16] or UVR [17].
Figure 1. Tumors induced by UVR in VDR null mice.
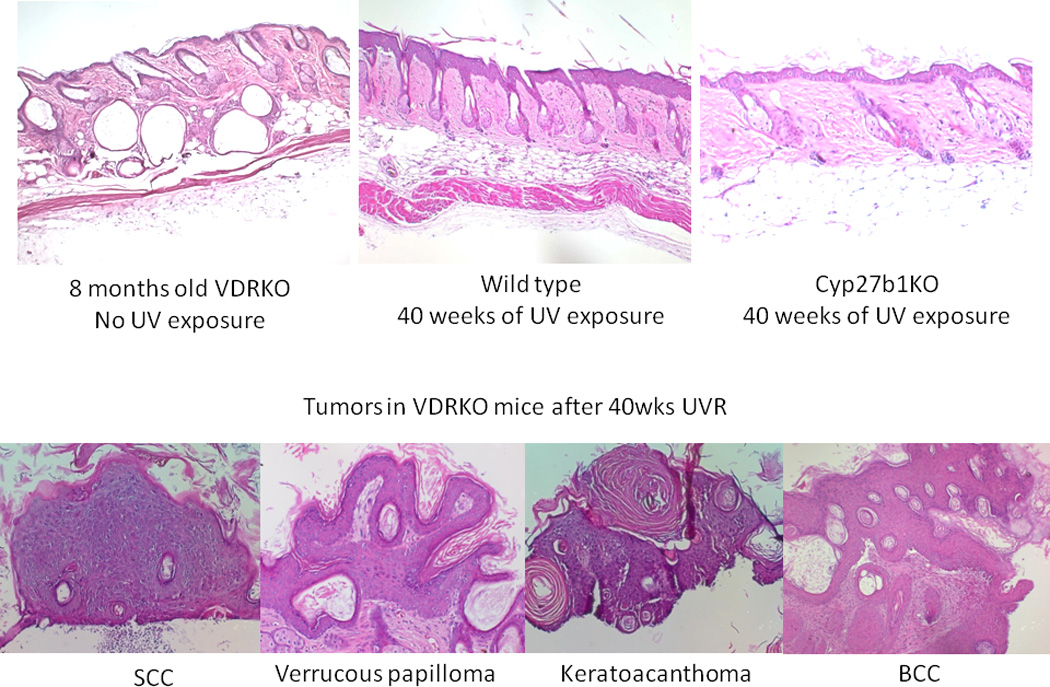
A. Representative sections of skin from 8 months old VDR null mice without UVB exposure, wild-type mice after 40 weeks of UVB exposure and CYP27B1 null mice after 40 weeks of UVB exposure. B. Tumors from VDR null mice exposed to 40 weeks of UVB irradiation were collected and classified into papillomas, squamous cell carcinomas (SCC), keratoacanthomas and basal cell carcinomas (BCC). Adapted from Teichert et al. J Invest Dermatol 131:2289-2297, 2011 with permission.
We considered three mechanisms, not necessarily independent, underlying the protective role of vitamin D in tumor formation. First, vitamin D signaling has a well established role in inhibiting proliferation and promoting differentiation of keratinocytes [18]. Thus lack of vitamin D signaling could lead to unchecked proliferation of poorly differentiated cells especially after a proliferative stimulus such as UVR. Secondly, UVR induces characteristic alterations in DNA (cyclobutane pyrimidine dimers [CPD] and 6,4 photoproducts [6,4 PP]) that if not repaired lead to mutations with the potential for initiating cancer. Studies from several research groups including our own [19, 20] have shown that vitamin D signaling enhances DNA repair. Finally, activation of hedgehog signaling is associated with essentially all BCC [21] and many SCC [22]. We [17] have found that mice lacking the VDR have constitutively active hedgehog signaling, and that 1,25(OH)2D can suppress this pathway in normal skin. Recent data from our laboratory supporting the role of these three mechanisms form the basis of this report.
Vitamin D regulation of proliferation and differentiation
The VDR null mouse shows increased proliferation and marked abnormalities in differentiation especially in the latter stages of catagen [23]. These observations in vivo were confirmed in vitro by knocking down the expression of VDR and the coactivator, DRIP205, most associated with the action of VDR in the proliferating keratinocyte [24]. The results are shown in figure 2 and demonstrate that lack of VDR and its coactivator DRIP205 increase proliferation, decrease apoptosis, and alter the morphology of the keratinocyte from the normal cuboidal form with tight intercellular junctions to a loosely aggregated collection of spindly shaped cells suggesting a change to a more primitive, less differentiated cell. The loss of differentiation assessed morphologically was confirmed by decreased expression of various differentiation dependent markers and functions [23, [24, [25]. When VDR null mice were exposed to one dose of UVR they showed a greater stimulation of proliferation than did their wildtype littermates (figure 3A), and proliferation continued to increase for at least 48h, whereas that of the wildtype littermates reached a plateau at 24h. This resulted in an almost 3 fold increase in epidermal thickness in the VDR null mice compared to wildtype littermates by 48h (figure 3B).
Figure 2. Regulation of keratinocyte proliferation and differentiation by VDR and its coactivators.
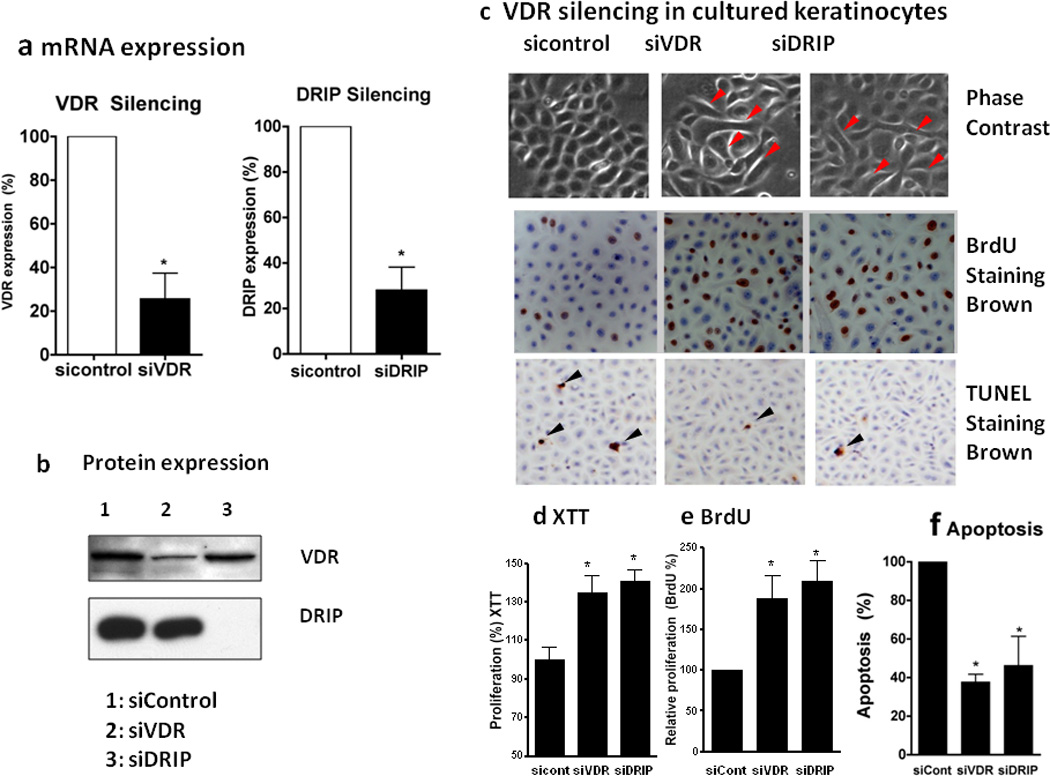
Epidermal keratinocytes were transfected with non-targeted siRNA for control (sicontrol), VDR (siVDR), and DRIP205 (siDRIP). VDR and DRIP expression was reduced as shown by qRTPCR (a) and western analysis (b). Cells were maintained in low calcium (0.03mM) to keep them proliferative. Cell proliferation was assessed by BrdU incorporation (c, e BrdU) and XTT assay (d XTT). The BrdU incorporated cells (brown) were counted using Bioquant and expressed as % total cells (blue counter staining) (c, e). Keratinocyte apoptosis was evaluated by measuring DNA fragmentation using Apoptaq In situ apoptosis peroxidase detection kit (Chemicon) (c,f TUNEL staining). The brown DNA fragmented cell nuclei (black arrows) per total cells (blue counter staining) were counted. Over 5000 cells were counted in three batches of keratinocytes to make these calculations (f, apoptosis). The hyperproliferation and decreased apoptosis were accompanied by morphologic changes from normal cuboidal epithelial cells tightly aggregated to loosely aggregated spindle shaped cells (red arrows).
Figure 3. Hyperproliferative response to UVR in VDR null epidermis.
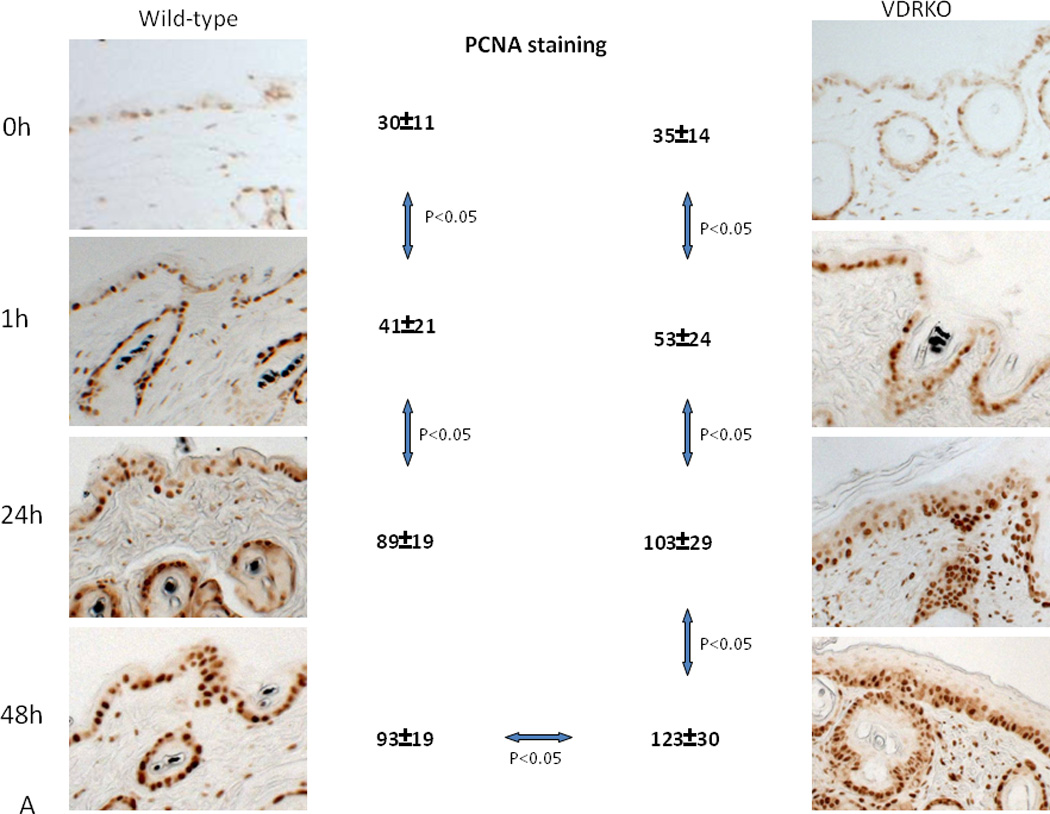
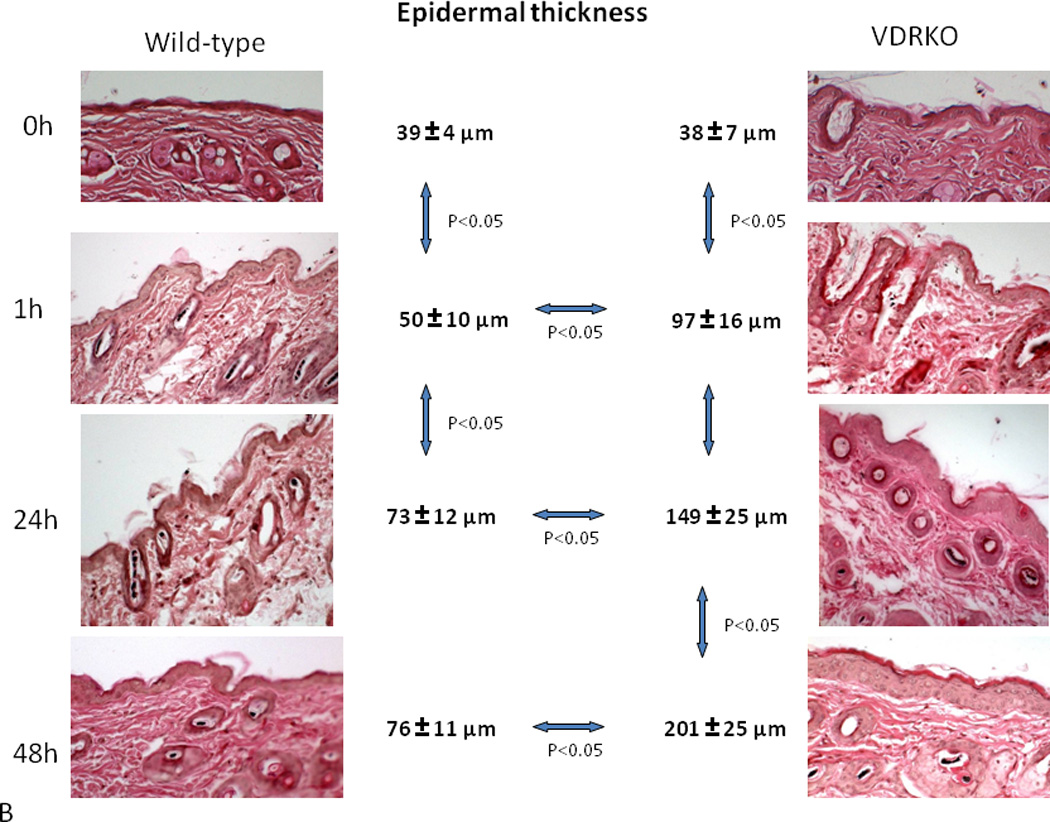
Wild-type mice exposed to 1 dose of UVB (477 mJ/cm2) showed increased proliferation (A, PCNA staining) and epidermal hyperplasia (B, H&E staining) up to 24h after treatment with no further increase at 48h. VDR null mice exposed to the same dose of UVB showed significantly more pronounced proliferation (A) and epidermal hyperplasia (B) that continued to increase at 48h. Adapted from Teichert et al. J Invest Dermatol 131:2289-2297, 2011 with permission.
Vitamin D regulation of DNA damage repair
The ozone layer protects us from UV wavelengths shorter than 280nm (UVC). UV wavelengths longer than 320nm (UVA) have been thought to have limited ability to induce the characteristic mutations in DNA seen in epidermal cancers, although recent studies indicate that UVA can cause oxidative DNA damage that is potentially mutagenic [26]. However, the major cause of skin cancer is attributed to UVB with a spectrum between 280–320nm [27]. UVR induced DNA damage includes the formation of cyclobutane pyrimidine dimers (CPD) and pyrimidine(6-4)pyrimidone photoproducts (6-4PP). If these lesions are not repaired C to T or CC to TT mutations result, the UVR “signature” lesion [28]. These mutations are often found in p53 in BCC, SCC [29, [30, [31, [32] and actinic keratoses, the precursor lesion to SCC [33]. Preventing UVR induced DNA damage from producing DNA mutations is the role of DNA damage repair (DDR) mechanisms. DDR coordinates the response of the cell cycle to DNA damage through mechanisms involving damage recognition, repair and signal transduction. These mechanisms include cell cycle checkpoints to delay the cell cycle, providing time for repair or activating senescent and apoptotic pathways. These mechanisms vary according to cell types, species, and damaging agents. The accuracy and tight control of DDR in normal primary cells keeps the spontaneous mutation rate very low [34, [35, [36]. However, with malignant transformation control of DDR is lost, and mutation rates and copy number abnormalities increase substantially [34, [35, [36, [37, [38].
Nucleotide excision repair (NER) is the principal means by which UVR damage is repaired. By removing DNA damage before DNA replication begins NER can reduce the amount of damage resulting in mutations that get incorporated into the DNA during replication [39, 40]. The two major processes [41] used by NER include transcription coupled repair (TCR) involving the repair of genes undergoing active transcription [42, 43, [44, [45, [46] and global genomic repair (GGR) for the non-transcribed regions of the genome [47]. Heritable mutations in NER genes occur in several human diseases with increased susceptibility to UVR induced epidermal malignancies such as xeroderma pigmentosum (XP) and Cockayne syndrome (CS) [47]. Identification of the genes mutated in these diseases has assisted substantially in identifying the genes and their protein products critical for DDR.
Keratinocytes in the epidermis of mice lacking a VDR show markedly retarded DDR [20]. This is demonstrated by a reduced rate of clearing CPDs and 6,4PPs following UVR whether administered in vivo (figure 4A) or in vitro in epidermal sheets from VDR null mice (figure 4B), and is associated with decreased survival after UVR exposure. The decreased clearance of CPDs in the VDR null epidermis represents a failure of global DNA repair rather than transcription coupled repair in that hydroxyurea (to block DNA synthesis) did not have a significant effect on the results in these experiments (P=0.3) (figure 4B). The Mason laboratory [48, [49] and others [50] demonstrated that 1,25(OH)2D3 topically applied protected the skin from UVR induced photodamage including increased clearance of CPDs, decreased apoptosis, increased survival, and increased expression of p53. Moll et al. [51] observed an upregulation of two genes important for DDR: XPC (xeroderma pigmentosum complementation group C) and DDB2 (damage-specific DNA binding protein 2 also known as XPE) following 1,25(OH)2D3 treatment. These actions of vitamin D signaling on DDR are likely to account for part of the reduced susceptibility of normal skin to UVR induced tumor formation.
Figure 4. Defective DNA Damage Repair in VDR null mouse epidermis following UVR.
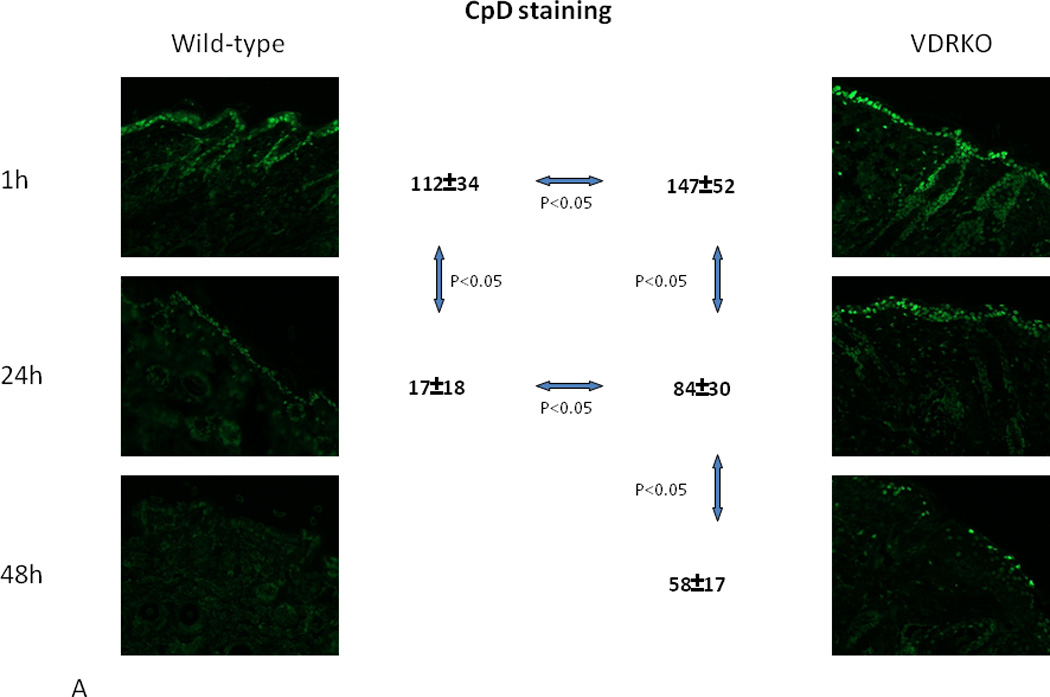
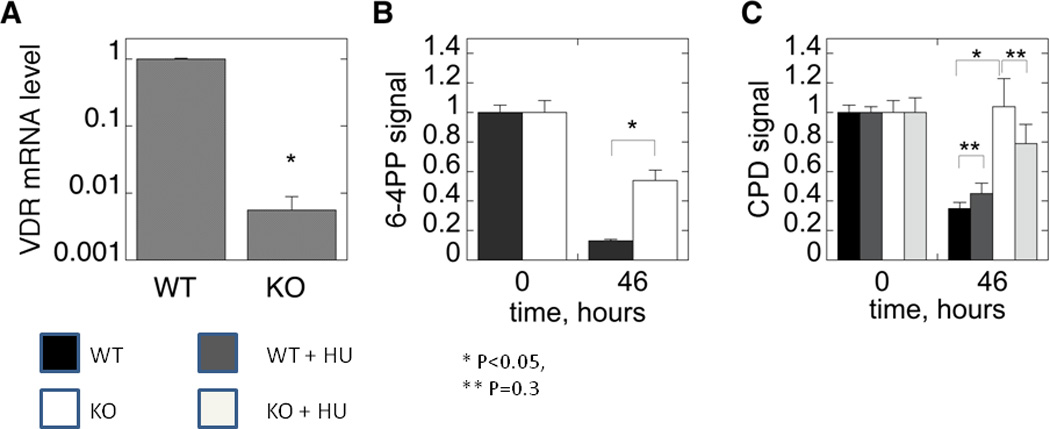
A. Wildtype and VDR null mice were exposed to 1 dose of UVB (400mJ/cm2) and the skin evaluated for the presence of CPDs over the subsequent 48hrs by immunohistochemistry (anti CPD from Cosmo Biosciences). CPDs were completely cleared by 24hr in the wildtype mouse epidermis, but persisted through 48hrs in the VDR null mouse epidermis. B. The epidermis from 2d old wildtype and VDR null mice was exposed to 35.4mJ/cm2 UVB, and CPDs and 6,4PPs (detected by immunoblots) measured immediately after irradiation and after 46hrs. In the experiment measuring CPDs, half of the epidermal explants were treated with hydroxyurea (HU) to block DNA synthesis prior to and following irradiation. Clearance of CPDs and 6,4PPs was markedly impaired in the VDR null epidermal explants consistent with the in vivo results in A. Adapted from Oh et al. J Invest Dermatol (in press, 2012).
Vitamin D regulation of hedgehog signaling
Mutations in patched 1 (Ptch1), a key component of the hedgehog signaling pathway, were discovered as the main cause of the basal cell nevus syndrome (BCNS) (Gorlin Syndrome), with its high susceptibility to the development of BCCs [21, 52]. Moreover, most sporadic BCCs have mutations in Ptch 1 or other alterations in Hh signaling [53]. The Ptch1+/− (Gorlin) mouse was then developed as the first practical model of murine BCCs [53], and they are quite susceptible to the development of BCC and SCC following UVR or ionizing radiation [53]. Ptch 1 is the membrane receptor for sonic hedgehog (Shh) which in the basal state inhibits the function of smoothened (Smoh), also in the membrane. In the presence of Shh this inhibition of Smoh is lost resulting in the activation of a family of transcription factors Gli1, Gli2, and Gli3. These Gli factors in the basal state are maintained in the cytoplasm bound to Suppressor of fused (Sufu), but with the activation of Smoh these factors are released from Sufu, enter the nucleus, and promote Hh signaling [54, [55]. Mutations in Sufu have not been found in cases of BCC but are associated with medulloblastomas, an additional feature of the Gorlin syndrome [56]. Gli1 and 2 overexpression in keratinocytes increase the expression of components of the Hh pathway, the anti apoptotic factor bcl2, cyclins D1 and D2, E2F1, cdc45 while suppressing genes associated with keratinocyte differentiation including VDR [57, [58, [59, [60, [61]. Moreover mice overexpressing Gli1, Gli2, or Shh in their basal keratinocytes or in human skin grafts [60, [61, [62, [63] develop BCC like lesions, and. BCC overexpress these Hh pathway components [64, [65, [66].
The appearance of BCC in VDR null mice following DMBA or UVR was at first surprising since UVR, ionizing radiation, or chemical carcinogens generally induce SCC not BCC [29]. However, we [17] (figure 5A) found that elements of the Hh signaling pathway are overexpressed in the epidermis and epidermal portion (utricles) of the hair follicles of adult VDR null animals. Thus we postulated that loss of 1,25(OH)2D3 and/or VDR regulation of Hh signaling is one of the causes of the increased susceptibility of the epidermis to malignant transformation. Examination of the tumors following either DMBA or UVR treatment (figure 5B) revealed overexpression of elements of the Hh signaling pathway compared to adjacent normal skin [17]. These observations raised the question whether vitamin D signaling in the skin regulates Hh signaling, and if so whether the loss of such regulation predisposes animals lacking VDR to skin cancer. Indeed, we found that in epidermal sheets and full thickness explants of skin 1,25(OH)2D3 inhibits the expression of all elements of the Hh pathway (figure 6A), and this suppression requires the VDR (figure 6B). Although our results indicate that suppression of the Hh pathway occurs via the genomic actions of 1,25(OH)2D3 acting through its receptor, other studies have shown that vitamin D itself as well as its precursor 7-dehydrocholesterol can bind to and inhibit the actions of Smoh directly [67, 68]. However, there remains uncertainty about the relative role of this mechanism vs that of the genomic suppression of the Hh pathway by 1,25(OH)2D and the VDR.
Figure 5. Overexpression of the Hh pathway in VDR null mouse skin and tumors.
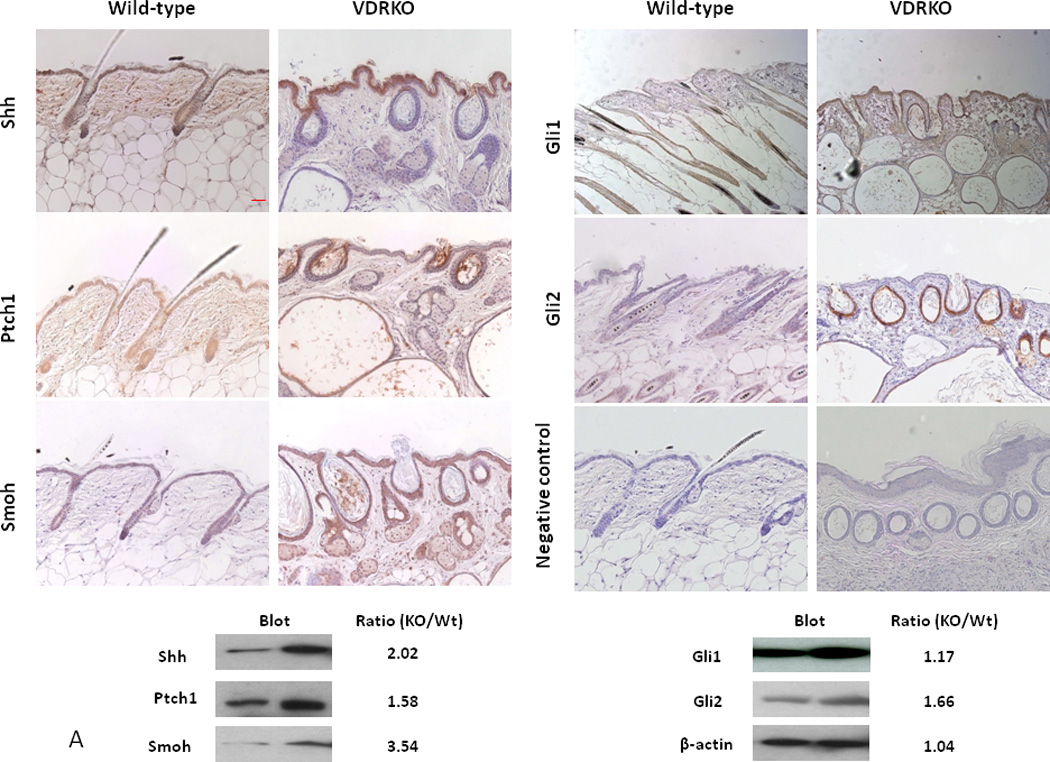
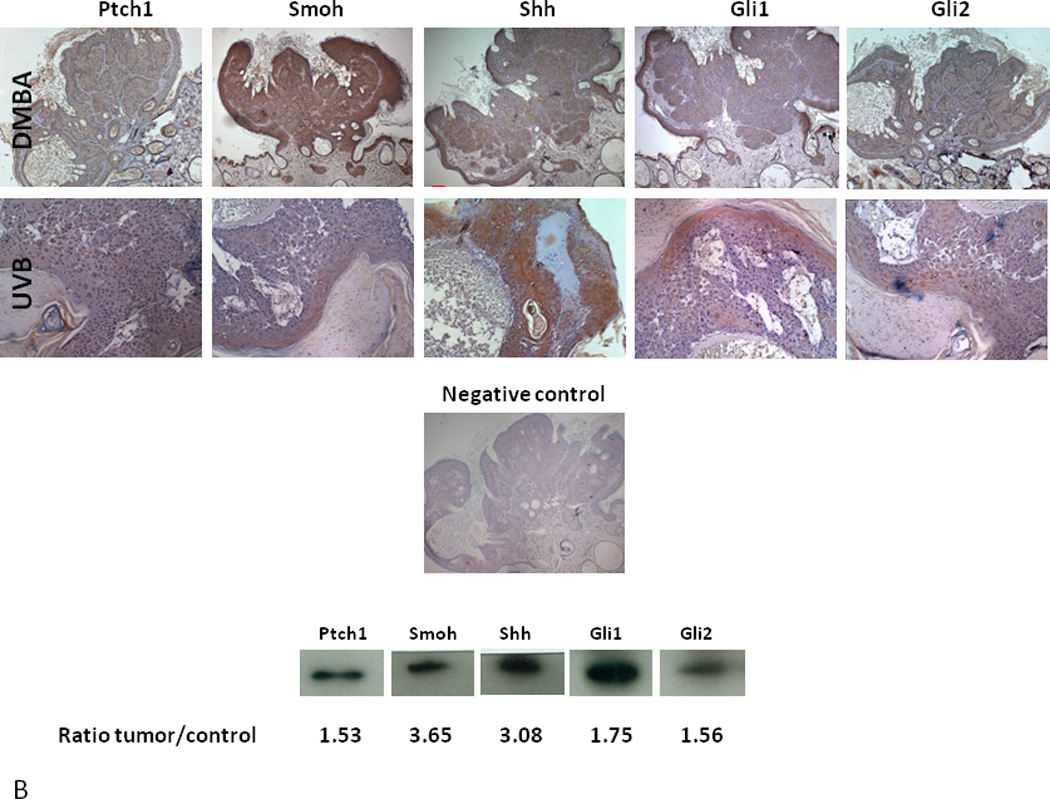
A. Shh, Ptch1, Smoh, Gli1 and Gli2 proteins as shown by the brown signal were overexpressed in the epidermis and hair follicles of VDR null mice compared to their wild-type littermates at 11 weeks after birth by immunocytochemistry. Slides were counterstained with hematoxylin (blue stain). The bar denotes 50 mm. The protein levels were quantified by western blot. The numerical value represents the average ratio of VDR null band intensity versus wild-type band intensity from three mice per group. * p<0.05. B. Shh, Ptch1, Smoh, Gli1 and Gli2 proteins as detected by immunohistochemistry in a papilloma from a VDR null mouse treated with DMBA and in a BCC from a VDR null mouse treated with UVB. Slides were counterstained with hematoxylin (blue stain). The bar denotes 50 mm. Shh, Ptch1, Smoh, Gli1 and Gli2 protein levels were also measured by western blot in skin tumors and tumor free tissue from DMBA treated VDR null mice. The numerical value represents the mean ratio of the tumor band intensity versus tumor free tissue band intensity from three mice. * p<0.05 Adapted from Teichert et al. J Invest Dermatol 131:2289-2297, 2011 with permission.
Figure 6. Suppression of Hh pathway in mouse skin by 1,25(OH)2D acting through the VDR.
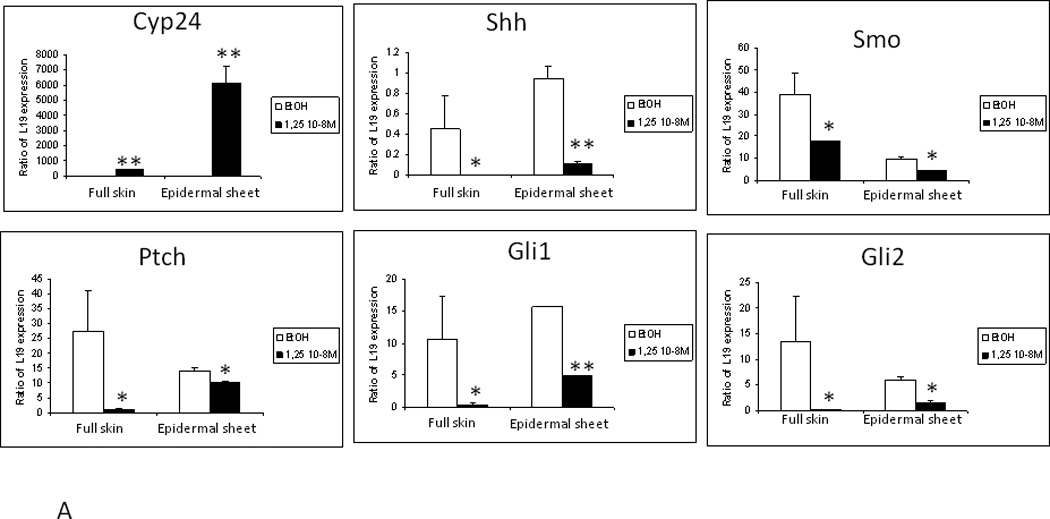
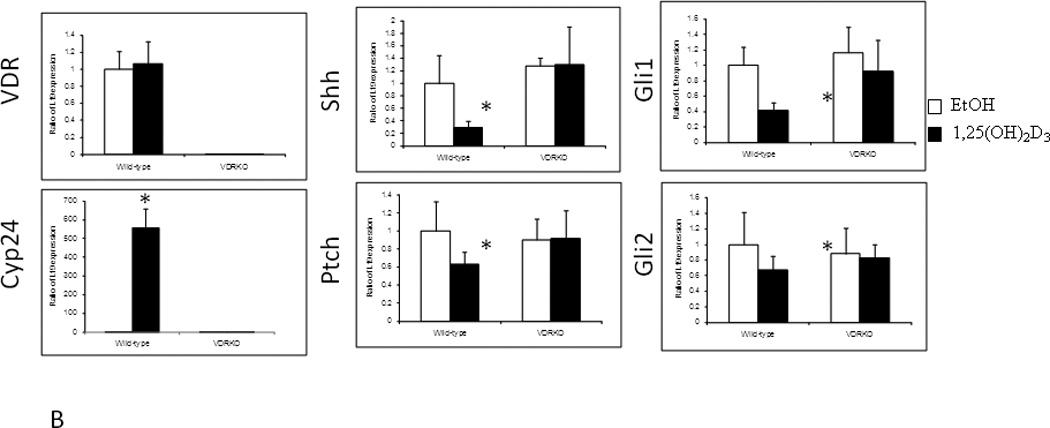
A. Treatment of epidermal preparations from wild-type mice in culture with 1,25(OH)2D3 10−8M or EtOH for 24h induced Cyp24 expression and repressed Shh, Gli1, Gli2 and Ptch1 expression. B. Epidermal preparations from wild-type and VDR null mice in culture were treated with 1,25(OH)2D3 10−8M or EtOH for 24h. Absence of VDR expression was verified in VDR null mice, and their epidermis failed to respond to 1,25(OH)2D3 induction of Cyp24 expression unlike that in wild-type mice. 1,25(OH)2D3 treatment repressed Shh, Gli1, Gli2 and Ptch1 expression only in wild-type preparations. * p<0.05. Adapted from Teichert et al. J Invest Dermatol 131:2289-2297, 2011 with permission.
Summary and Conclusions
Although clinical data regarding the protective role of vitamin D signaling in skin cancer formation remain ambiguous, the role of the VDR and its ligand in this protective function in mice is not. Lack of VDR predisposes to epidermal tumor formation. Surprisingly deletion of CYP27B1 (the 1-hydroxylase) does not by itself predispose to tumor formation with the conditions employed (DMBA, UVR in a tumor resistant mouse strain), but 1,25(OH)2D does protect against tumor formation when applied topically, and it enhances VDR regulation of proliferation, differentiation, and Hh signaling. We have explored 3 mechanisms by which vitamin D signaling might be protective against tumor formation. First we discussed the well established ability of 1,25(OH)2D and VDR to regulate keratinocyte proliferation and differentiation, noting that loss of VDR increased the proliferative response of the epidermis to UVR. Second we examined the role of vitamin D signaling in DNA damage repair (DDR), and demonstrated that lack of VDR impaired this process most likely by limiting the expression of various genes involved in DDR. Third we examined the control of Hh signaling in keratinocytes by 1,25(OH)2D and VDR. Activation of Hh signaling is essentially universal in BCC formation and in most SCCs. Lack of VDR increases Hh signaling, and 1,25(OH)2D suppresses the genes involved. Thus 3 mechanisms regulated by vitamin D signaling have been investigated, and in combination are expected to provide protection in the skin against the carcinogenic actions of UVR, enabling the beneficial actions of UVR on vitamin D production to proceed with reduced risk.
Highlights.
Skin cancer is the most common type of malignancy
UVB is the primary etiologic agent inducing skin cancer
UVB is required for vitamin D production in the skin
Vitamin D and its receptor provide protection against UVB induced skin cancer
This protection involves inhibition of proliferation, stimulation of differentiation, immune regulation, and DNA repair
Acknowledgements
The authors acknowledge the administrative support of Teresa Tong and Victoria Lee, the technical support of Sally Pennypacker, Stephanie Demetriou, and Katherine Ona-Vu, and the funding support from NIH RO1 AR050023 (DDB), DOD CA110338 (DDB), VA Merit Reviews (DDB and DHO), and the University of California Cancer Coordinating Committee grants (DHO, JEC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Eisman JA, Martin TJ, MacIntyre I, et al. 1,25-dihydroxyvitamin-D-receptor in breast cancer cells. Lancet. 1979;2(8156–8157):1335–1336. doi: 10.1016/s0140-6736(79)92816-2. [DOI] [PubMed] [Google Scholar]
- 2.Ratnam AV, Bikle DD, Su MJ, et al. Squamous carcinoma cell lines fail to respond to 1,25-Dihydroxyvitamin D despite normal levels of the vitamin D receptor. J Invest Dermatol. 1996;106(3):522–525. doi: 10.1111/1523-1747.ep12343898. [DOI] [PubMed] [Google Scholar]
- 3.Kamradt J, Rafi L, Mitschele T, et al. Analysis of the vitamin D system in cutaneous malignancies. Recent Results Cancer Res. 2003;164:259–269. doi: 10.1007/978-3-642-55580-0_19. [DOI] [PubMed] [Google Scholar]
- 4.Colston K, Colston MJ, Feldman D. 1,25-dihydroxyvitamin D3 and malignant melanoma: the presence of receptors and inhibition of cell growth in culture. Endocrinology. 1981;108(3):1083–1086. doi: 10.1210/endo-108-3-1083. [DOI] [PubMed] [Google Scholar]
- 5.Garland C, Shekelle RB, Barrett-Connor E, et al. Dietary vitamin D and calcium and risk of colorectal cancer: a 19-year prospective study in men. Lancet. 1985;1(8424):307–309. doi: 10.1016/s0140-6736(85)91082-7. [DOI] [PubMed] [Google Scholar]
- 6.Bostick RM, Potter JD, Sellers TA, et al. Relation of calcium, vitamin D, and dairy food intake to incidence of colon cancer among older women. The Iowa Women's Health Study. Am J Epidemiol. 1993;137(12):1302–1317. doi: 10.1093/oxfordjournals.aje.a116640. [DOI] [PubMed] [Google Scholar]
- 7.Kearney J, Giovannucci E, Rimm EB, et al. Calcium, vitamin D, and dairy foods and the occurrence of colon cancer in men. Am J Epidemiol. 1996;143(9):907–917. doi: 10.1093/oxfordjournals.aje.a008834. [DOI] [PubMed] [Google Scholar]
- 8.Garland FC, Garland CF, Gorham ED, et al. Geographic variation in breast cancer mortality in the United States: a hypothesis involving exposure to solar radiation. Cancer. 1990;19(6):614–622. doi: 10.1016/0091-7435(90)90058-r. [DOI] [PubMed] [Google Scholar]
- 9.Hanchette CL, Schwartz GG. Geographic patterns of prostate cancer mortality. Evidence for a protective effect of ultraviolet radiation. Cancer. 1992;70(12):2861–2869. doi: 10.1002/1097-0142(19921215)70:12<2861::aid-cncr2820701224>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 10.van Dam RM, Huang Z, Giovannucci E, et al. Diet and basal cell carcinoma of the skin in a prospective cohort of men. Am J Clin Nutr. 2000;71(1):135–141. doi: 10.1093/ajcn/71.1.135. [DOI] [PubMed] [Google Scholar]
- 11.Hunter DJ, Colditz GA, Stampfer MJ, et al. Diet and risk of basal cell carcinoma of the skin in a prospective cohort of women. Ann Epidemiol. 1992;2(3):231–239. doi: 10.1016/1047-2797(92)90055-u. [DOI] [PubMed] [Google Scholar]
- 12.Weinstock MA, Stampfer MJ, Lew RA, et al. Case-control study of melanoma and dietary vitamin D: implications for advocacy of sun protection and sunscreen use. J Invest Dermatol. 1992;98(5):809–811. doi: 10.1111/1523-1747.ep12499962. [DOI] [PubMed] [Google Scholar]
- 13.Asgari MM, Tang J, Warton ME, et al. Association of prediagnostic serum vitamin D levels with the development of basal cell carcinoma. J Invest Dermatol. 2010;130(5):1438–1443. doi: 10.1038/jid.2009.402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zinser GM, Sundberg JP, Welsh J. Vitamin D(3) receptor ablation sensitizes skin to chemically induced tumorigenesis. Carcinogenesis. 2002;23(12):2103–2109. doi: 10.1093/carcin/23.12.2103. [DOI] [PubMed] [Google Scholar]
- 15.Indra AK, Castaneda E, Antal MC, et al. Malignant transformation of DMBA/TPAinduced papillomas and nevi in the skin of mice selectively lacking retinoid-X-receptor alpha in epidermal keratinocytes. J Invest Dermatol. 2007;127(5):1250–1260. doi: 10.1038/sj.jid.5700672. [DOI] [PubMed] [Google Scholar]
- 16.Ellison TI, Smith MK, Gilliam AC, et al. Inactivation of the Vitamin D Receptor Enhances Susceptibility of Murine Skin to UV-Induced Tumorigenesis. J Invest Dermatol. 2008;128:2508–2517. doi: 10.1038/jid.2008.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teichert AE, Elalieh H, Elias PM, et al. Overexpression of hedgehog signaling is associated with epidermal tumor formation in vitamin D receptor-null mice. J Invest Dermatol. 2011;131(11):2289–2297. doi: 10.1038/jid.2011.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bikle DD. Vitamin D metabolism and function in the skin. Mol Cell Endocrinol. 2011;347(1–2):80–89. doi: 10.1016/j.mce.2011.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dixon KM, Norman AW, Sequeira VB, et al. 1alpha,25(OH)(2)-vitamin D and a nongenomic vitamin D analogue inhibit ultraviolet radiation-induced skin carcinogenesis. Cancer Prev Res (Phila) 2011;4(9):1485–1494. doi: 10.1158/1940-6207.CAPR-11-0165. [DOI] [PubMed] [Google Scholar]
- 20.Demetriou SK, Ona-Vu K, Teichert AE, et al. Vitamin D Receptor Mediates DNA Repair and Is UV Inducible in Intact Epidermis but Not in Cultured Keratinocytes. J Invest Dermatol. 2012 doi: 10.1038/jid.2012.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aszterbaum M, Rothman A, Johnson RL, et al. Identification of mutations in the human PATCHED gene in sporadic basal cell carcinomas and in patients with the basal cell nevus syndrome. J Invest Dermatol. 1998;110(6):885–888. doi: 10.1046/j.1523-1747.1998.00222.x. [DOI] [PubMed] [Google Scholar]
- 22.Ping XL, Ratner D, Zhang H, et al. PTCH mutations in squamous cell carcinoma of the skin. J Invest Dermatol. 2001;116(4):614–616. doi: 10.1046/j.1523-1747.2001.01301.x. [DOI] [PubMed] [Google Scholar]
- 23.Bikle DD, Elalieh H, Chang S, et al. Development and progression of alopecia in the vitamin D receptor null mouse. J Cell Physiol. 2006;207:340. doi: 10.1002/jcp.20578. [DOI] [PubMed] [Google Scholar]
- 24.Oda Y, Ishikawa MH, Hawker NP, et al. Differential role of two VDR coactivators, DRIP205 and SRC-3, in keratinocyte proliferation and differentiation. J Steroid Biochem Mol Biol. 2007;103(3–5):776–780. doi: 10.1016/j.jsbmb.2006.12.069. [DOI] [PubMed] [Google Scholar]
- 25.Oda Y, Uchida Y, Moradian S, et al. Vitamin D receptor and coactivators SRC 2 and 3 regulate epidermis-specific sphingolipid production and permeability barrier formation. J Invest Dermatol. 2009;129(6):1367–1378. doi: 10.1038/jid.2008.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Besaratinia A, Synold TW, Chen HH, et al. DNA lesions induced by UV A1 and B radiation in human cells: comparative analyses in the overall genome and in the p53 tumor suppressor gene. Proc Natl Acad Sci U S A. 2005;102(29):10058–10063. doi: 10.1073/pnas.0502311102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Freeman SE, Hacham H, Gange RW, et al. Wavelength dependence of pyrimidine dimer formation in DNA of human skin irradiated in situ with ultraviolet light. Proc Natl Acad Sci U S A. 1989;86(14):5605–5609. doi: 10.1073/pnas.86.14.5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hussein MR. Ultraviolet radiation and skin cancer: molecular mechanisms. J Cutan Pathol. 2005;32(3):191–205. doi: 10.1111/j.0303-6987.2005.00281.x. [DOI] [PubMed] [Google Scholar]
- 29.Daya-Grosjean L, Sarasin A. The role of UV induced lesions in skin carcinogenesis: an overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors. Mutat Res. 2005;571(1–2):43–56. doi: 10.1016/j.mrfmmm.2004.11.013. [DOI] [PubMed] [Google Scholar]
- 30.Ziegler A, Leffell DJ, Kunala S, et al. Mutation hotspots due to sunlight in the p53 gene of nonmelanoma skin cancers. Proc Natl Acad Sci U S A. 1993;90(9):4216–4220. doi: 10.1073/pnas.90.9.4216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ziegler A, Jonason AS, Leffell DJ, et al. Sunburn and p53 in the onset of skin cancer. Nature. 1994;372(6508):773–776. doi: 10.1038/372773a0. [DOI] [PubMed] [Google Scholar]
- 32.Brash DE, Rudolph JA, Simon JA, et al. A role for sunlight in skin cancer: UV-induced p53 mutations in squamous cell carcinoma. Proc Natl Acad Sci U S A. 1991;88(22):10124–10128. doi: 10.1073/pnas.88.22.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bito T, Ueda M, Ahmed NU, et al. Cyclin D and retinoblastoma gene product expression in actinic keratosis and cutaneous squamous cell carcinoma in relation to p53 expression. J Cutan Pathol. 1995;22(5):427–434. doi: 10.1111/j.1600-0560.1995.tb00758.x. [DOI] [PubMed] [Google Scholar]
- 34.Tlsty TD, Margolin BH, Lum K. Differences in the rates of gene amplification in nontumorigenic and tumorigenic cell lines as measured by Luria-Delbruck fluctuation analysis. Proc Natl Acad Sci U S A. 1989;86(23):9441–9445. doi: 10.1073/pnas.86.23.9441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tlsty TD. Normal diploid human and rodent cells lack a detectable frequency of gene amplification. Proc Natl Acad Sci U S A. 1990;87(8):3132–3136. doi: 10.1073/pnas.87.8.3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maher VM, Dorney DJ, Mendrala AL, et al. DNA excision-repair processes in human cells can eliminate the cytotoxic and mutagenic consequences of ultraviolet irradiation. Mutat Res. 1979;62(2):311–323. doi: 10.1016/0027-5107(79)90087-3. [DOI] [PubMed] [Google Scholar]
- 37.Bartkova J, Rezaei N, Liontos M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444(7119):633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 38.Bielas JH, Loeb KR, Rubin BP, et al. Human cancers express a mutator phenotype. Proc Natl Acad Sci U S A. 2006;103(48):18238–18242. doi: 10.1073/pnas.0607057103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen RH, Maher VM, McCormick JJ. Effect of excision repair by diploid human fibroblasts on the kinds and locations of mutations induced by (+/−)-7 beta,8 alpha-dihydroxy-9 alpha,10 alpha-epoxy-7,8,9,10- tetrahydrobenzo[a]pyrene in the coding region of the HPRT gene. Proc Natl Acad Sci U S A. 1990;87(21):8680–8684. doi: 10.1073/pnas.87.21.8680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wood RD. DNA damage recognition during nucleotide excision repair in mammalian cells. Biochimie. 1999;81(1–2):39–44. doi: 10.1016/s0300-9084(99)80036-4. [DOI] [PubMed] [Google Scholar]
- 41.Mellon I, Bohr VA, Smith CA, et al. Preferential DNA repair of an active gene in human cells. Proc Natl Acad Sci U S A. 1986;83(23):8878–8882. doi: 10.1073/pnas.83.23.8878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mellon I, Spivak G, Hanawalt PC. Selective removal of transcription-blocking DNA damage from the transcribed strand of the mammalian DHFR gene. Cell. 1987;51(2):241–249. doi: 10.1016/0092-8674(87)90151-6. [DOI] [PubMed] [Google Scholar]
- 43.Mellon I, Rajpal DK, Koi M, et al. Transcription-coupled repair deficiency and mutations in human mismatch repair genes. Science. 1996;272(5261):557–560. doi: 10.1126/science.272.5261.557. [DOI] [PubMed] [Google Scholar]
- 44.Bohr VA. Gene specific DNA repair. Carcinogenesis. 1991;12(11):1983–1992. doi: 10.1093/carcin/12.11.1983. [DOI] [PubMed] [Google Scholar]
- 45.Hanawalt PC. Transcription-coupled repair and human disease. Science. 1994;266(5193):1957–1958. doi: 10.1126/science.7801121. [DOI] [PubMed] [Google Scholar]
- 46.Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411(6835):366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 47.Wood RD, Mitchell M, Sgouros J, et al. Human DNA repair genes. Science. 2001;291(5507):1284–1289. doi: 10.1126/science.1056154. [DOI] [PubMed] [Google Scholar]
- 48.Dixon KM, Deo SS, Wong G, et al. Skin cancer prevention: a possible role of 1,25dihydroxyvitamin D3 and its analogs. J Steroid Biochem Mol Biol. 2005;97(1–2):137–143. doi: 10.1016/j.jsbmb.2005.06.006. [DOI] [PubMed] [Google Scholar]
- 49.Gupta R, Dixon KM, Deo SS, et al. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J Invest Dermatol. 2007;127(3):707–715. doi: 10.1038/sj.jid.5700597. [DOI] [PubMed] [Google Scholar]
- 50.De Haes P, Garmyn M, Degreef H, et al. 1,25-Dihydroxyvitamin D3 inhibits ultraviolet Binduced apoptosis, Jun kinase activation, and interleukin-6 production in primary human keratinocytes. J Cell Biochem. 2003;89(4):663–673. doi: 10.1002/jcb.10540. [DOI] [PubMed] [Google Scholar]
- 51.Moll PR, Sander V, Frischauf AM, et al. Expression profiling of vitamin D treated primary human keratinocytes. J Cell Biochem. 2007;100(3):574–592. doi: 10.1002/jcb.21061. [DOI] [PubMed] [Google Scholar]
- 52.Hahn H, Wicking C, Zaphiropoulous PG, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell. 1996;85(6):841–851. doi: 10.1016/s0092-8674(00)81268-4. [DOI] [PubMed] [Google Scholar]
- 53.Aszterbaum M, Epstein J, Oro A, et al. Ultraviolet and ionizing radiation enhance the growth of BCCs and trichoblastomas in patched heterozygous knockout mice. Nat Med. 1999;5(11):1285–1291. doi: 10.1038/15242. [DOI] [PubMed] [Google Scholar]
- 54.Barnfield PC, Zhang X, Thanabalasingham V, et al. Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple mechanisms. Differentiation. 2005;73(8):397–405. doi: 10.1111/j.1432-0436.2005.00042.x. [DOI] [PubMed] [Google Scholar]
- 55.Svard J, Heby-Henricson K, Persson-Lek M, et al. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev Cell. 2006;10(2):187–197. doi: 10.1016/j.devcel.2005.12.013. [DOI] [PubMed] [Google Scholar]
- 56.Johnson RL, Rothman AL, Xie J, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science. 1996;272(5268):1668–1671. doi: 10.1126/science.272.5268.1668. [DOI] [PubMed] [Google Scholar]
- 57.Regl G, Kasper M, Schnidar H, et al. The zinc-finger transcription factor GLI2 antagonizes contact inhibition and differentiation of human epidermal cells. Oncogene. 2004;23(6):1263–1274. doi: 10.1038/sj.onc.1207240. [DOI] [PubMed] [Google Scholar]
- 58.Regl G, Kasper M, Schnidar H, et al. Activation of the BCL2 promoter in response to Hedgehog/GLI signal transduction is predominantly mediated by GLI2. Cancer Res. 2004;64(21):7724–7731. doi: 10.1158/0008-5472.CAN-04-1085. [DOI] [PubMed] [Google Scholar]
- 59.Regl G, Neill GW, Eichberger T, et al. Human GLI2 and GLI1 are part of a positive feedback mechanism in Basal Cell Carcinoma. Oncogene. 2002;21(36):5529–5539. doi: 10.1038/sj.onc.1205748. [DOI] [PubMed] [Google Scholar]
- 60.Grachtchouk M, Mo R, Yu S, et al. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat Genet. 2000;24(3):216–217. doi: 10.1038/73417. [DOI] [PubMed] [Google Scholar]
- 61.Nilsson M, Unden AB, Krause D, et al. Induction of basal cell carcinomas and trichoepitheliomas in mice overexpressing GLI-1. Proc Natl Acad Sci U S A. 2000;97(7):3438–3443. doi: 10.1073/pnas.050467397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Oro AE, Higgins KM, Hu Z, et al. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science. 1997;276(5313):817–821. doi: 10.1126/science.276.5313.817. [DOI] [PubMed] [Google Scholar]
- 63.Fan H, Oro AE, Scott MP, et al. Induction of basal cell carcinoma features in transgenic human skin expressing Sonic Hedgehog. Nat Med. 1997;3(7):788–792. doi: 10.1038/nm0797-788. [DOI] [PubMed] [Google Scholar]
- 64.Tojo M, Mori T, Kiyosawa H, et al. Expression of sonic hedgehog signal transducers, patched and smoothened, in human basal cell carcinoma. Pathol Int. 1999;49(8):687–694. doi: 10.1046/j.1440-1827.1999.00938.x. [DOI] [PubMed] [Google Scholar]
- 65.Bonifas JM, Pennypacker S, Chuang PT, et al. Activation of expression of hedgehog target genes in basal cell carcinomas. J Invest Dermatol. 2001;116(5):739–742. doi: 10.1046/j.1523-1747.2001.01315.x. [DOI] [PubMed] [Google Scholar]
- 66.Eichberger T, Regl G, Ikram MS, et al. FOXE1, a new transcriptional target of GLI2 is expressed in human epidermis and basal cell carcinoma. J Invest Dermatol. 2004;122(5):1180–1187. doi: 10.1111/j.0022-202X.2004.22505.x. [DOI] [PubMed] [Google Scholar]
- 67.Bijlsma MF, Spek CA, Zivkovic D, et al. Repression of smoothened by patched-dependent (pro-)vitamin D3 secretion. PLoS Biol. 2006;4(8):e232. doi: 10.1371/journal.pbio.0040232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tang JY, Xiao TZ, Oda Y, et al. Vitamin D3 inhibits hedgehog signaling and proliferation in murine Basal cell carcinomas. Cancer Prev Res (Phila) 2011;4(5):744–751. doi: 10.1158/1940-6207.CAPR-10-0285. [DOI] [PMC free article] [PubMed] [Google Scholar]