

Abstract
Antiapoptotic proteins are commonly overexpressed in gliomas, contributing to therapeutic resistance. We recently reported that clinically achievable concentrations of the Bcl-2/Bcl-xL inhibitor ABT-737 failed to induce apoptosis in glioma cells, with persistent expression of survivin and Mcl-1. To address the role of these mediators in glioma apoptosis resistance, we analyzed the effects of YM-155, a survivin suppressant, on survival on a panel of glioma cell lines. YM-155 inhibited growth, and downregulated survivin and Mcl-1 in a dose- and cell line-dependent manner. Whereas U373, LN18, LNZ428, T98G, LN229, and LNZ308 cells exhibited an IC50 of 10–75 nM, A172 cells were resistant (IC50 ~ 250 nM). No correlation was found between sensitivity to YM-155 and baseline expression of survivin or cIAP-1/cIAP-2/XIAP. However, strong correlation was observed between EGFR activation levels and YM-155 response, which was confirmed using EGFR-transduced versus wild-type cells. Because we postulated that decreasing Mcl-1 expression may enhance glioma sensitivity to ABT-737, we examined whether cotreatment with YM-155 promoted ABT-737 efficacy. YM-155 synergistically enhanced ABT-737-induced cytotoxicity and caspase-dependent apoptosis. Down-regulation of Mcl-1 using shRNA also enhanced ABT-737-inducing killing, confirming an important role for Mcl-1 in mediating synergism between ABT-737 and YM-155. As with YM-155 alone, sensitivity to YM-155 and ABT-737 inversely correlated with EGFR activation status. However, sensitivity could be restored in highly resistant U87-EGFRvIII cells by inhibition of EGFR or its downstream pathways, highlighting the impact of EGFR signaling on Mcl-1 expression and the relevance of combined targeted therapies to overcome the multiple resistance mechanisms of these aggressive tumors.
Keywords: YM-155, ABT-737, EGFR activation, glioma, synergy, apoptosis
Introduction
Glioblastoma (GBM) is the most common primary malignant brain tumor and one of the most aggressive forms of cancer. GBM exhibits a high resistance to standard chemotherapy and radiotherapy. Resistance of tumor cells to the induction of apoptosis is an important reason for the failure of anticancer treatments in patients with gliomas, and several factors working in concert have been implicated as sources of this treatment resistance (1). We, among others, have indicated that dysregulation of the NF-κB, Bcl-2, and Akt pathways may be integrally involved in mediating glioma resistance to apoptotic signaling (1–9). Bcl-2 is a prime target for novel therapeutics because it is overexpressed in many forms of cancer and contributes to cancer progression and therapy resistance based on its ability to inhibit apoptosis. New anticancer therapeutics, such as ABT-737, are being developed to specifically target the prosurvival members of the Bcl-2 family by using small-molecule mimetics of BH3-only proteins (10). As a single agent, ABT-737 triggers apoptosis in various types of human cancers including multiple myeloma, leukemia, and lymphoma (11–15). We, among others have shown only weak activity against various solid tumor cell lines, such as gliomas (6, 16, 17). In our studies, ABT-737 in combination with bortezomib induced apoptosis (6); however, inactivation of PTEN limited the therapeutic efficacy of ABT-737 in glioma. Others have shown that sensitivity to ABT-737 depends on the expression levels of Mcl-1, Bcl-2, Bcl-xL, Bim and Noxa (12–14, 18–20), and that Mcl-1 down-regulation dramatically increases ABT-737 lethality in diverse malignant cell types (14, 16, 21, 22). Moreover, Bak and Bax activation is essential in potentiation of ABT-737 lethality by agents that down-regulate Mcl-1 (12, 14, 22).
High expression of survivin has also been associated with the increasing malignancy grade among gliomas (23–25). Survivin is a member of the Inhibitor of Apoptosis Protein (IAP) family and has gained interest as a potential molecular target in the treatment of cancer (26). Recent reports suggest that survivin protein levels are upregulated by co-expression of human epidermal growth factor receptor 2 (HER2) and epidermal growth factor receptor (EGFR) through the PI3K/Akt signaling pathway in breast cancer cells (27). In previous studies, we demonstrated that overexpression of survivin is found in more than 70% of pediatric and adult malignant gliomas (28).
YM-155 is a novel survivin suppressant that is currently in clinical trials (29, 30). YM-155 has been shown to suppress survivin expression, with little effect on expression levels of other IAP family members, and to inhibit growth and viability of diverse human cancer cell lines (31, 32). Because YM-155-induced apoptosis is associated with down-regulation of Mcl-1 (33), which is a significant mediator of resistance to ABT-737, we hypothesized that combining YM-155 and ABT-737 would potentiate apoptosis induction in gliomas, either by direct inhibitory effects on survivin levels or by associated effects on Mcl-1. Our results validated this hypothesis; moreover, we observed a strong association between treatment response and EGFR expression status, which was enhanced by targeted inhibition of this pathway.
Materials and Methods
Cell lines
U87, T98G, U373, A172 and LN229 were obtained from the American Type Culture Collection (Manassas, VA). LN18, LNZ308, and LNZ428 were provided by Dr. Nicolas de Tribolet. The establishment of the parental human glioblastoma cell line, U87 and its derivatives, which overexpress exogenous wild-type EGFR (U87-EGFR), or constitutively active EGFR with a genomic deletion of exons 2–7 (U87-EGFRvIII) has been described elsewhere (34). The cell lines were kindly provided by Dr. W.K. Cavenee (Ludwig Institute for Cancer Research, San Diego, CA, USA). Human astrocytes (HA) and growth media were obtained from ScienCell Research Laboratories (Carlsbad, CA). Cell lines used in this study were not authenticated. Cell culture conditions of these cell lines were as previously described (3, 35).
Reagents and antibodies
ABT-737, YM-155, BEZ-235, gefitinib, and PI-103 were purchased from Chemie Tek (Indianapolis, IN). N-Acetyl-L-cysteine (NAC) and LY294002, U0126 were purchased from Cell Signaling Technology (Beverly, MA). Pan-caspase inhibitor, Z-VAD-fmk was purchased from R & D Systems (Minneapolis, MN). The following antibodies were used: Bcl-2, Bcl-xL, Mcl-1, Bim, Bak, Bax, cleaved PARP, cleaved caspase 3, cleaved caspase 7, survivin, cIAP-1, cIAP-2, XIAP, Mcl-1, Bax, Bak, pERK, total ERK, pAkt, total Akt, pS6K (Ser 235/236) and s-Actin were from Cell Signaling Technology (Beverly, MA). Monoclonal anti-Bax (6A7) was from Sigma. Phospho EGFR antibodies were from Invitrogen. All primary antibodies were used at 1: 1000 dilutions. Secondary antibodies conjugated to horseradish peroxidase, used at 1:2000 dilutions, were purchased from Santa Cruz Biotechnology, CA, and visualized by enhanced chemiluminescence (Cell Signaling Technology).
Cell proliferation assay
Cells (5 × 103/well) were plated in 96-well microtiter plates (Costar, Cambridge, MA) in 100 μl of growth medium, and after overnight attachment, exposed for 3 days to inhibitors or vehicle (DMSO). After the treatment interval, cells were washed in medium, and the number of viable cells was determined using a colorimetric cell proliferation assay (CellTiter96 Aqueous NonRadioactive Cell Proliferation Assay; Promega, Madison, WI) as described previously (3).
Annexin V apoptosis assay
Apoptosis was evaluated using fluorescein isothiocyanate conjugated annexin V/propidium iodide assay kit (Molecular Probes, Invitrogen) based on annexin-V binding to phosphatidylserine exposed on the outer leaflet of the plasma membrane lipid bilayer of cells entering the apoptotic pathway as described previously (3, 6). Briefly, cells were treated with or without inhibitors for the indicated duration, collected by trypsin-EDTA, pelleted by centrifugation (1,000 rpm for 5 min), washed in ice-cold PBS, and resuspended in the annexin V-FITC and 1 μg/ml propidium iodide reagent in the dark for 15 min before flow cytometric analysis. Labeling was analyzed by flow cytometry with a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA). A minimum of 20,000 cells per sample was collected.
DiOC6 labeling and detection of mitochondrial membrane depolarization
Mitochondrial membrane depolarization was measured as described previously (6). In brief, floating cells were collected, and attached cells were trypsinized and resuspended in PBS. Cells were loaded with 50 nmol/L 3′,3′-dihexyloxacarbo-cyanine iodide (DiOC6, Invitrogen) at 37°C for 15 min. The positively charged DiOC6 accumulates in intact mitochondria, whereas mitochondria with depolarized membranes accumulate less DiOC6. Cells were spun at 3,000 × g, rinsed with PBS and resuspended in 1 ml of PBS. Fluorescence intensity was detected by flow cytometry and analyzed with CellQuest (Becton Dickinson) and FlowJo (Tree Star, Inc., Ashland, OR) analysis software.
Immunoprecipitation and Western blotting analysis
Cells were washed in cold PBS and lysed in buffer containing 30 mM HEPES, 10% glycerol, 1% Triton X-100, 100 mmol/L NaCl, 10 mmol/L MgCl2, 5 mM EDTA, 2mM Na3VO4, 2 mM β-glycerophosphate, 1 mM phenylmethylsulfonyl fluoride, 1 mmol/L 4-(2-aminoethyl) benzenesulfonyl fluoride, 0.8 μmol/L aprotinin, 50 μmol/L bestatin, 15 μmol/L E-64, 20 μmol/L leupeptin, and 10 μmol/L pepstatin A for 15 min on ice. Samples were centrifuged at 12,000g for 15 min, supernatants were isolated, and protein was quantified using Protein Assay Reagent (Pierce Chemical, Rockford, IL). Equal amounts of protein were separated by SDS polyacrylamide gel electrophoresis (PAGE) and electrotransferred onto a nylon membrane (Invitrogen). Nonspecific antibody binding was blocked by incubation of the membranes with 4% bovine serum albumin in Tris-buffered saline (TBS)/Tween 20 (0.1%). The membranes were then probed with appropriate dilutions of primary antibody overnight at 4°C. The antibody-labeled blots were washed three times in TBS/Tween 20 and incubated with a 1:2000 dilution of horseradish peroxidase-conjugated secondary antibody in TBS/Tween 20 at room temperature for 1 h. Proteins were visualized by Western Blot Chemiluminescence Reagent (Cell Signaling). Where indicated, the membranes were reprobed with antibodies against β-actin to ensure equal loading and transfer of proteins.
For immunoprecipitation, cell extracts were prepared by lysing 5 × 106 cells on ice for 30 min in CHAPS lysis buffer (10 mmol/L HEPES (pH 7.4), 150 mmol/L NaCl, 1% CHAPS, protease, phosphatase inhibitors). Lysates were clarified by centrifugation at 15,000 × g for 10 min at 4 °C, and the protein concentrations in the supernatants were determined. Equal amounts of protein extracts were incubated overnight with primary antibody. Afterward, Dynabeads Protein G (Invitrogen) was added for 2 hours, followed by magnetic separation of the immunoprecipitated fraction; Western blot analysis was carried out as described above. Scanning densitometry was performed using acquisition into Adobe Photoshop (Adobe Systems, Inc) followed by image analysis (UN-SCAN-IT gel, version 6.1; Silk Scientific).
Transient transfection
Optimal 29mer-pRS-shRNA constructs were obtained from Origene (Rockville, MD). Sequences specific for human Mcl-1 (ACC TAG AAG GTG GCA TCA GGA ATG TGC TG) and control sequences (GCA CTA CCA GAG CTA ACT CAG ATA GTA CT) (non-target shRNA) were used for this study. Glioma cells were seeded in six-well plates and allowed to reach 70% confluence. Transfection of targeting or control shRNA was performed by using FuGene 6 according to the manufacturer’s recommendations (Roche Applied Science, Indianapolis, IN). One μg of Mcl-1 or non-targeting shRNA in 100 μL Opti-MEM medium was mixed with 2 μL of FuGene 6. After the mixture was incubated at room temperature for 20 min, complete medium was added to make the total volume up to 2 mL. After 48 h, media was changed and cells were incubated with inhibitors for 24 h. Cell viability (annexin V binding) or Western blot analysis was carried out as described above.
Statistical analysis
Unless otherwise stated, data are expressed as mean ± S.D. The significance of differences between experimental conditions was determined using a two-tailed Student’s t test. Differences were considered significant at p values <0.05.
Results
YM-155 sensitizes glioma cells to ABT-737 but not non-neoplastic astrocytes
Glioma cells were treated with ABT-737 or YM-155 or both (Fig. 1A) and apoptotic cell death was examined by Annexin V/PI staining. As shown in Fig. 1B, YM-155 significantly increased the sensitivity of LN18, U373, LNZ428, LN229, T98G, and LNZ308 cells to ABT-737 treatment compared with cells treated with ABT-737 alone. Simultaneous treatment with ABT-737 and YM-155 resulted in a significant increase in the appearance of cleaved fragments of caspase-7, caspase-3 and PARP (Fig. 1C). This apoptotic response was circumvented by the broad-specificity caspase inhibitor z-VAD-fmk (Fig. 1D). In contrast to the above cell lines, a more modest effect was seen in A172 cells (Fig. 1E). As shown in Supplementary Fig. S1A, ABT-737 had no effect on normal human astrocytes (HA) even at high concentrations (72 h; IC50, > 25 μmol/L). Simultaneous treatment of ABT-737 plus YM-155 also had little or no effect on cell proliferation (Supplementary Fig. S1A), caspase or PARP activation (Supplementary Fig. S1B), suggesting selectivity against glioma cells versus non-neoplastic astrocytes.
Figure 1. YM-155 sensitizes glioma cells to ABT-737 toxicity.

A, Chemical structure of YM-155, LY294002, PI-103, BEZ-235, ABT-737 and U0126. B, Glioma cells were treated with the indicated concentrations of ABT-737 or YM-155, or both for 24 h. Apoptosis was assessed by Annexin V-FITC and propidium iodide (PI) staining and FACS analysis. Data are representative of three independent experiments. In parallel, cell extracts were prepared, and equal amounts of protein were separated by SDS-PAGE and subjected to Western blotting analysis with the indicated antibodies (C). D, U373 and LNZ308 cells were treated with ABT-737 (2.5 μmol/L), YM-155 (25 nmol/L) or the combination (A + Y). In parallel, cells were pretreated with 25 μM Z-VAD-FMK (pan caspase inhibitor) for 2 h followed by the combination of ABT-737 plus YM-155 for 24 h. Control cells received an equivalent amount of DMSO. Apoptosis (upper panel, representative annexin V binding histogram; lower panel, bar chart representing three independent experiments) was analyzed by flow cytometry. The percentages of cells in each quadrant are indicated. E, A172 cells were treated with the indicated concentrations of ABT-737 or YM-155 (25 nmol/L) or the combination for 24 h. Apoptosis was analyzed as in D.
Sensitivity of glioma cell lines to YM-155
To investigate the independent activity of YM-155 in glioma cells, we examined the cytotoxic efficacy of YM-155 in a panel of glioma cell lines under normal culture conditions. After a 24, 48 and 72-hour treatment, inhibition of cell proliferation and viability was assessed by MTS assay. All glioma cell lines responded in a dose (Fig. 2A) and time-dependent (data not shown) manner to YM-155 with reduction of cell numbers. U373, LN18, LNZ428, T98G, LN229, and LNZ308 cells exhibited an IC50 of ~ 10–75 nmol/L, whereas, A172 cells were resistant to YM-155 with IC50 ~ 250 nmol/L. To investigate the mechanisms underlying the differential sensitivity to YM-155, a series of Western blot experiments were done to compare the expression of various IAP family proteins. No correlation was found between the sensitivity of cells to YM-155 and the expression of survivin, cIAP-1, cIAP-2 and XIAP (Fig. 2B). We then examined the ability of YM-155 to inhibit survivin expression. In accordance with previous reports (32, 33), survivin was downregulated in a concentration-dependent manner (Fig. 2C). However, YM155 had no effect on XIAP, cIAP-1 and cIAP-2 (data not shown). Because survivin expression is regulated by EGFR via the PI3K/Akt signaling pathway (27), we investigated the levels of EGFR activation (phosphorylation of tyrosine residues at 845, 1068, 1086, 1148, and 1173) by Western immunoblotting analysis. EGFR phosphorylation levels varied substantially among the cell lines tested, and strong correlation was observed between EGFR activation levels and response to YM-155 (Fig. 2D).
Figure 2. YM-155 inhibits cell proliferation in malignant human glioma cell lines.
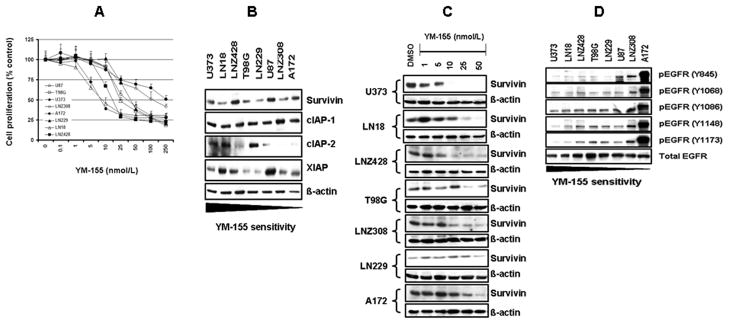
A, Glioma cells were incubated with varying concentrations of YM-155 for 72 hours; proliferation inhibition was assessed semiquantitatively by spectrophotometric measurement of MTS bioreduction. Points represent the mean of three measurements ± S.D. B, Glioma cells were seeded at 60% confluence, and allowed to attach overnight. Cell extracts were subjected to Western blotting analysis with the indicated antibodies. C, Glioma cells were treated with the indicated concentrations of YM-155 for 24 h. Cell extracts were subjected to Western blotting analysis with anti-survivin antibody. D, Glioma cells were grown as in B, and subjected to Western blotting analysis with the indicated antibodies. The blots were subsequently stripped and reprobed with total EGFR.
YM-155 downregulates Mcl-1 and sensitizes glioma cells to ABT-737 by enhancing Bax conformational changes and disrupting Bak/Bax interactions with Mcl-1
Because downregulation of Mcl-1 has been observed to confer sensitivity to ABT-737 in other tumor types (14, 22), we questioned whether the potentiating effect of YM-155 on ABT-737 response was due to an effect on Mcl-1 expression. YM-155 caused down-regulation of Mcl-1 in a dose-dependent manner (Fig. 3A) but did not affect other Bcl-2 family members (data not shown). As shown in Fig. 3B, Mcl-1 shRNA also caused significant enhancement of apoptosis induction (Fig. 3B). Because Mcl-1 blocks the progression of apoptosis by binding and sequestering the pro-apoptotic protein Bak, we examined the association between Bak and Mcl-1. Glioma cells were treated with ABT-737 or YM-155 or both and immunoprecipitated using Bak antibody followed by Western blotting using Mcl-1 antibody. As shown in Fig. 3C, the association between Mcl-1 and Bak, was not changed by ABT-737 treatment, whereas, this effect was largely abrogated by the combination of ABT-737 and YM-155, suggesting that cotreatment with ABT-737 plus YM-155 was able to disrupt the Bak-Mcl-1 interaction.
Figure 3. YM-155 downregulates Mcl-1 and sensitizes glioma cells to ABT-737 by enhancing Bax conformational changes and disrupting Bak/Bax interactions with Mcl-1.

A, Logarithmically growing glioma cells were treated with the indicated concentrations of YM-155 for 24 h and subjected to Western blotting analysis with anti-Mcl-1 antibody. B, LN18, U373, and LNZ308 cells were transfected with non-target (NT) or Mcl-1 shRNA. Forty-eight hours post-transfection, cells were treated with the indicated concentrations of ABT-737 for 24 h, and viability was assessed by Annexin V/PI apoptosis assay. In parallel, cell lysates were collected and protein was subjected to Western blot analysis using anti-Mcl-1 antibody. C, LN18, U373, LN229, and LNZ308 cells were treated with inhibitors for 24 h. An equal amount of protein (400 μg) was immunoprecipitated (IP) with Bak antibody. Immunoprecipitates were subjected to Western blot analysis using anti-Mcl-1 antibody. D, Lysates were subjected to immunoprecipitation using anti-Bax 6A7 (Sigma-Aldrich) that recognizes only Bax protein that has undergone conformational change. Immunoprecipitates were subjected to immunoblotting using anti-Bax polyclonal antibodies (CST).
To examine the effect of YM-155 and ABT-737 on Bax activation in glioma cells, we analyzed activity-related conformational changes of Bax (12) by immunoprecipitation followed by Western blot analysis with antibodies recognizing N-terminal epitopes of Bax (monoclonal anti-bax, 6A7, Sigma). When cells were separately exposed to ABT-737 (2.5μM) or YM-155 (25 nM), a minimal Bax conformational change was observed. In contrast, cells co-exposed to YM-155 and ABT-737 displayed a significant increase in Bax conformational change compared with cells treated with ABT-737 alone (Fig. 3D), suggesting that down-regulation of Mcl-1 and activation of Bax may be responsible for the marked induction of apoptosis in cells co-exposed to YM-155 and ABT-737.
Combination of YM-155 and ABT-737 induces loss of mitochondrial membrane potential
Because Bcl-2 family proteins are key regulators of the mitochondrial apoptotic pathway, and changes in mitochondrial membrane potential (Δψm) are thought to represent an early event in the induction of apoptosis, and likely capture the effects of agents on various aspects of Bcl-2 family member homeostasis, we evaluated the effect ABT-737 with or without YM-155 on Δψm. The integrity of the mitochondrial membranes of the cells was examined by DiOC6 staining and flow cytometry; the decrease in fluorescence intensity reflected the loss of Δψm. As shown in Fig. 4A, exposure of U373 and LN18 (YM-155 sensitive) glioma cells to 25 nmol/L of YM-155 for 24 h caused a marked loss of Δψm compared with the control group. However, no significant change in Δψm was seen in A172 (YM-155 resistant cell line). Cotreatment with ABT-737 and YM-155 produced an even more dramatic loss of Δψm (Fig. 4B) in U373 and LN18 but only a modest change in A172 cell lines. The use of NAC, a scavenger of reactive oxygen species (ROS), almost completely rescued apoptosis (Fig. 4C) and reversed the disruption in Δψm (Fig. 4D) induced by YM-155 + ABT-737.
Figure 4. Combination of YM-155 and ABT-737 induces loss of mitochondrial membrane potential.

A, U373, LN18 and A172 cells were treated with the indicated concentrations of YM-155 for 24 h. The integrity of the mitochondrial membranes of the cells was examined by DiOC6 staining and flow cytometry. Decrease in fluorescence intensity reflected loss of Δψm. Histogram (upper panel) and bar chart (lower panel) represent the mean number of Δψm cells acquired from three independent experiments. B, U373, LN18 and A172 cells were treated with the indicated concentrations of YM-155 with ABT-737 (2.5 μmol/L) for 24 h. Loss of mitochondrial membrane potential was determined. C and D, U373 and LN18 cells were treated with ABT-737 (2.5 μmol/L) or YM-155 (25 nmol/L) or the combination of both (A + Y). In parallel, cells were pretreated with NAC (5 mmol/L, ROS scavenger) for 2 h followed by the combination of ABT-737 plus YM-155 for 24 h. Apoptosis (C) or loss of mitochondrial membrane potential (D) was analyzed by flow cytometry.
Overactivation of the EGFR pathway confers resistance to YM-155 and ABT-737-induced cytotoxicity
In view of our observation the glioma cells with high levels of EGFR activation, such as A172 (Figure 2D), appeared to be less sensitive to both YM-155 and the combination of YM-155 and ABT-737 (Figure 1E), we questioned whether EGFR activation status was a key contributor to treatment response. We therefore compared dose-response relationships to YM-155 in native U87 cells with those transduced to highly overexpress EGFR-WT and EGFRvIII, a truncated constitutively active EGFR variant. As shown in Fig. 5A., cell proliferation in vitro was inhibited (assessed after 3 days of exposure to YM-155 by MTS assay) by YM-155 to a substantially greater degree in native versus EGFR-transduced cell lines, with IC50 of ~60, 270 and 280 nmol/L for U87, U87-EGFR (WT), and U87-EGFRvIII, respectively (Fig. 5B). YM-155 had a significantly lower inhibitory effect on survivin and Mcl-1 expression in U87-EGFR and U87-EGFRvIII than U87 cells (Fig. 5C) and a correspondingly reduced induction of apoptosis in the EGFR-transduced cells (Fig. 5D). To examine the relation between EGFR status and treatment response to YM-155 and ABT-737, cells (U87 and U87-EGFRvIII) were treated with ABT-737 or YM-155 or both. Annexin V assay demonstrated that U87 cells were strikingly more sensitive to the combination of ABT-737 and YM-155 than U87-EGFRvIII cells (Fig. 5E). Western blot analysis further validated this differential efficacy. Co-treatment with YM-155 and ABT-737 induced substantially less caspase-7, 3 and PARP cleavage in U87-EGFR and U87-EGFRvIII cell lines than in parental U87 cells (Fig. 5F). Differential effects were also observed in terms of Bax activation (Fig. 5G).
Figure 5. Overactivation of the EGFR pathway confers resistance to YM-155 and ABT-737-induced cytotoxicity.

A, Logarithmically growing U87, U87-EGFR or U87-EGFRvIII glioma cells were incubated with varying concentrations of YM-155. After a 72-h treatment, cell proliferation inhibition was assessed by MTS assay. B,C, Cells were seeded at 60% confluence, allowed to attach overnight. Cell extracts were subjected to Western blotting analysis with the indicated antibodies.. D and E, Logarithmically growing glioma cells were treated with the indicated concentrations of YM-155, ABT-737 or both for 24 h. Apoptosis was determined by Annexin V/PI staining. F, Cell extracts were subjected to Western blotting analysis with the indicated antibodies. G, U87 and U87-EGFRvIII cells were treated with inhibitors for 24 h. An equal amount of protein (400 μg) was immunoprecipitated (IP) with monoclonal anti-Bax (6A7, Sigma) antibody and then immunoblotted (WB) with polyclonal anti-Bax antibody (CST).
EGFR provides major survival signals through both ERK and Akt in glioma cells
Because survivin is involved in multiple signaling mechanisms initiated by EGFR activation (26), we characterized whether certain components of downstream EGFR signaling might correlate with the observed resistance in EGFR-overactivated glioma cells. Cells were incubated with U0126 (MEK1 inhibitor), LY294002 (PI3K/Akt inhibitor), BEZ-235 or PI-103 (PI3K/mTOR dual kinase inhibitors) or gefitinib (EGFR inhibitor) for 24 h, and levels of respective phosphoproteins and survivin were analyzed by Western blotting. The survivin levels were significantly decreased by U0126, LY294002, BEZ-235, PI-103 and gefitinib (Fig. 6A). To test whether inhibition of ERK or PI3K/Akt/mTOR pathways could sensitize U87-EGFRvIII cells to YM-155 + ABT-737-induced toxicity, cells were pretreated with either U0126, LY294002, BEZ235 or PI-103 and the effect of ABT-737 and YM-155 on viability was assessed. The viability of EGFRvIII cells was not affected by the combination of YM-155 and ABT-737. In contrast, pretreated cells demonstrated significantly increased apoptotic sensitivity to YM-155 + ABT-737 (Fig. 6B) and increased Bax activation (Fig. 6C). As shown in Fig. 6D, exposure of U87 and U87-EGFRvIII cells to YM-155 for 24 h caused minimal or no loss of Δψm compared with the control group. Cotreatment with ABT-737 and YM-155 produced a significant loss of Δψm in U87 but not in U87-EGFRvIII cell lines (Fig. 6E). However, pretreatment of U87-EGFRvIII cells with LY294002, BEZ235 or PI-103 (Fig. 6F) led to a significant enhancement of Δψm loss.
Figure 6. EGFR provides major survival signals through both ERK and Akt in glioma cells.

A, U87 and U87-EGFRvIII cells were incubated with the indicated concentrations of inhibitors for 24 h. Control cells received an equivalent amount of DMSO. Cell extracts were subjected to Western blotting analysis with the indicated antibodies.. B, U87-EGFRvIII cells were incubated with U0126, LY294002 (LY), BEZ-235 (BEZ), or PI-103 (PI) for 2 h followed by the combination of ABT-737 (2.5 μmol/L) plus YM-155 (25 nmol/L) for 24 h (YA). Apoptosis was analyzed by annexin V/PI assay. C, U87-EGFRvIII cells were treated with ABT-737 (2.5 μmol/L) plus YM-155 (25 nmol/L) (ABT + YM) with the indicated inhibitors for 24 h. An equal amount of protein (400 μg) was immunoprecipitated (IP) with monoclonal anti-Bax (6A7, Sigma) antibody and then immunoblotted (WB) with polyclonal anti-Bax antibody (CST). D, E, U87 and U87-EGFRvIII cells were treated with the indicated concentrations of YM-155 (D), with or without ABT or YM-155 (2.5 μmol/L) for 24 h. Loss of mitochondrial membrane potential was determined by DiOC6 staining and flow cytometry. F, U87-EGFRvIII cells were pretreated with LY294002 (LY), BEZ-235 (BEZ), or PI-103 (PI) for 2 h followed by the combination of ABT-737 (2.5 μmol/L) plus YM-155 (25 nmol/L) [A + Y] for 24 h. Loss of mitochondrial membrane potential was determined as in 6D (**, p<0.005).
Discussion
Previous studies have demonstrated that the cell killing effect of ABT-737 varies widely among cancer types or cell lines from the same type of cancers (36, 37). Our recent study (6) showed that, unlike hematologic malignancies (12, 15, 38) glioma cells were resistant to apoptosis induction in response to ABT-737 alone, which may reflect activation of apoptosis-inhibitory molecules, such as Mcl-1 that are not affected by ABT-737, and survival signaling pathways that counteract apoptosis induction. In this regard, there has been much recent interest in the role of survivin as a potential apoptosis-modulating target in the treatment of cancer (26). In this study, we found that a survivin inhibitor, YM-155, potently inhibited growth of a subset of malignant human glioma cell lines. This agent significantly reduced Mcl-1 expression in sensitive cell lines, consistent with recent observations of Tang et al. (33), while having no effect on the expression levels of c-IAP-1, c-IAP-2, XIAP, Bcl-2, or Bcl-xL.
Because ABT-737 has been observed to be most effective against cancers in which Mcl-1, an antiapoptotic Bcl-2 family member, is down-regulated or absent (12, 22), we speculated that approaches directed at inhibiting Mcl-1 could sensitize glioma cells to ABT-737. Our results showed that YM-155 in combination with ABT-737 induced cell death, mediated by mitochondrial membrane depolarization and caspase activation. Cotreatment of glioma cells with low concentrations of ABT-737 and YM-155 resulted in apoptotic cell death as verified by annexin V staining, associated with the activation of caspases at the initiative and executive stages. The role of Mcl-1 in this process was further validated by a genetic (shRNA) approach to reduce MCL-1 levels, which independently increased the ABT-737 response.
In addition to delineating the important role of Mcl-1 in ABT-737 mediated apoptosis, our results demonstrated that sensitivity to YM-155 and the combination of YM-155 and ABT-737 varied substantially between different glioma cell lines, correlating strongly with levels of EGFR activation. EGFR overexpression, often resulting from gene amplification and/or mutation, has been associated with cancer progression in glioma patients. The most common mutant found is EGFRvIII, a ligand-independent, constitutively active receptor that has been observed to correlate with poor prognosis (39, 40). In our experiments, YM-155 enhanced ABT-737-induced cytotoxicity to a substantially different extent depending on the cell lines used, with significant synergistic enhancement of tumor cell killing in glioma cells without high levels of EGFR activation. Conversely, we observed that overactivation of the EGFR signaling pathway, which is noted in approximately half of malignant gliomas, was associated with resistance to the apoptosis induced by YM-155 and ABT-737.
Studies using various cell types imply that multiple pathways involved in EGFR signaling, including PI3K, JAK/STAT3, and MEK/ERK, are involved in the regulation of Mcl-1 transcription (41). We demonstrate in this study that the downstream effectors of aberrantly activated EGFR signaling pathways are involved in promoting Mcl-1 expression, and resultant ABT-737 and YM-155 resistance, and that EGFR or MAPK or Akt inhibitors significantly reduced Mcl-1 protein expression levels and promoted the apoptotic effect of YM-155/ABT-737 co-treatment. Previously, our group has demonstrated that the activation of Bax, including conformational changes and oligomerization, seems to play a crucial role in the initiation of apoptosis after other signaling-targeted therapies in gliomas (6, 42). Moreover, proapoptotic Bak is sequestered by the antiapoptotic Mcl-1 and Bcl-XL, its displacement by BH3-only proteins being required for cell death (43). The results of the present study indicate that cotreatment with YM-155 and ABT-737 indeed causes conformational change of Bax and this event allows Bak displacement from its antiapoptotic counterparts to occur.
In summary, here we demonstrated that the combination of YM-155 and ABT-737 potently triggered apoptosis in a subset of glioma cells, associated with reduced Mcl-1 protein expression, loss of mitochondrial transmembrane potential, activation of Bak and Bax, and stimulation of caspase-3 activity. EGFR signaling, which has been known to activate multiple survival signaling pathways, including Ras/MAPK and PI3K/Akt signaling, was a critical modulator of this apoptotic response. Our study demonstrated that the sensitivity to YM-155 and ABT-737 was inversely correlated with EGFR activation status, and was partially restored in highly resistant U87-EGFRvIII cells by combining ABT-737 and YM-155 with either gefitinib, U0126, LY294002, BEZ-235, or PI-103 to inhibit various aspects of EGFR signaling. Since more than one acquired resistance mechanism may simultaneously exist in malignant gliomas, targeting a series of rationally defined survival signaling pathways in concert might provide a useful strategy to overcome the multiple nonoverlapping resistance mechanisms that characterize these highly aggressive tumors.
Supplementary Material
Acknowledgments
Grant support: This work was supported by National Institutes of Health Grant P01NS40923 (I.F. Pollack) and by the Walter L. Copeland fund of The Pittsburgh Foundation (D.R. Premkumar); NIH Grants CA130966, CA158911 (S.Y. Cheng. and B. Hu.), a Research Award for Researching Brain Cancer from James S. McDonnell Foundation (B. Hu.) a grant with the Pennsylvania Department of Health, and Innovative Research Scholar Awards of the Hillman Foundation (S.Y. Cheng., B. Hu.) and a Zeller Scholar Award from the Zeller Family Foundation (S.Y. Cheng).
The authors thank Robert Lacomy and Alexis Styche for FACS analysis.
Footnotes
Conflict of Interest: None declared
Authors contributed equally to this study: Esther P. Jane, Daniel R. Premkumar, and Joseph D. DiDomenico
Authorship contributions:
Participated in research design: Daniel R. Premkumar and Ian F. Pollack
Conducted experiments: Esther P. Jane, Daniel R. Premkumar, and Joe D. DiDomenico
Performed data analysis: Esther P. Jane, Daniel R. Premkumar, Joe D. DiDomenico and Ian F. Pollack
Reagent contribution and discussions regarding experiments: Bo Hu and Shiyuan Cheng
Wrote or contributed to the writing of the manuscript: Daniel R. Premkumar, Shiyuan Cheng and Ian F. Pollack
References
- 1.de Groot JF, Gilbert MR. New molecular targets in malignant gliomas. Curr Opin Neurol. 2007;20:712–718. doi: 10.1097/WCO.0b013e3282f15650. [DOI] [PubMed] [Google Scholar]
- 2.Bredel M, Scholtens DM, Yadav AK, Alvarez AA, Renfrow JJ, Chandler JP, et al. NFKBIA deletion in glioblastomas. N Engl J Med. 2011;364:627–637. doi: 10.1056/NEJMoa1006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jane EP, Premkumar DR, Pollack IF. Bortezomib sensitizes malignant human glioma cells to TRAIL, mediated by inhibition of the NF-{kappa}B signaling pathway. Mol Cancer Ther. 2011;10:198–208. doi: 10.1158/1535-7163.MCT-10-0725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kitchens CA, McDonald PR, Shun TY, Pollack IF, Lazo JS. Identification of chemosensitivity nodes for vinblastine through small interfering RNA high-throughput screens. J Pharmacol Exp Ther. 2011;339:851–858. doi: 10.1124/jpet.111.184879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knobbe CB, Merlo A, Reifenberger G. Pten signaling in gliomas. Neuro Oncol. 2002;4:196–211. [PMC free article] [PubMed] [Google Scholar]
- 6.Premkumar DR, Jane EP, DiDomenico JD, Vukmer NA, Agostino NR, Pollack IF. ABT-737 synergizes with bortezomib to induce apoptosis, mediated by Bid cleavage, Bax activation, and mitochondrial dysfunction in an Akt-dependent context in malignant human glioma cell lines. J Pharmacol Exp Ther. 2012;341:859–872. doi: 10.1124/jpet.112.191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thaker NG, McDonald PR, Zhang F, Kitchens CA, Shun TY, Pollack IF, et al. Designing, optimizing, and implementing high-throughput siRNA genomic screening with glioma cells for the discovery of survival genes and novel drug targets. J Neurosci Methods. 2010;185:204–212. doi: 10.1016/j.jneumeth.2009.09.023. [DOI] [PubMed] [Google Scholar]
- 8.Thaker NG, Zhang F, McDonald PR, Shun TY, Lazo JS, Pollack IF. Functional genomic analysis of glioblastoma multiforme through short interfering RNA screening: a paradigm for therapeutic development. Neurosurg Focus. 2010;28:E4. doi: 10.3171/2009.10.FOCUS09210. [DOI] [PubMed] [Google Scholar]
- 9.Thaker NG, Zhang F, McDonald PR, Shun TY, Lewen MD, Pollack IF, et al. Identification of survival genes in human glioblastoma cells by small interfering RNA screening. Mol Pharmacol. 2009;76:1246–1255. doi: 10.1124/mol.109.058024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, et al. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 11.Hann CL, Daniel VC, Sugar EA, Dobromilskaya I, Murphy SC, Cope L, et al. Therapeutic efficacy of ABT-737, a selective inhibitor of BCL-2, in small cell lung cancer. Cancer Res. 2008;68:2321–2328. doi: 10.1158/0008-5472.CAN-07-5031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, et al. Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell. 2006;10:375–388. doi: 10.1016/j.ccr.2006.10.006. [DOI] [PubMed] [Google Scholar]
- 13.Tahir SK, Yang X, Anderson MG, Morgan-Lappe SE, Sarthy AV, Chen J, et al. Influence of Bcl-2 family members on the cellular response of small-cell lung cancer cell lines to ABT-737. Cancer Res. 2007;67:1176–1183. doi: 10.1158/0008-5472.CAN-06-2203. [DOI] [PubMed] [Google Scholar]
- 14.van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, et al. The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralized. Cancer Cell. 2006;10:389–399. doi: 10.1016/j.ccr.2006.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bodet L, Gomez-Bougie P, Touzeau C, Dousset C, Descamps G, Maiga S, et al. ABT-737 is highly effective against molecular subgroups of multiple myeloma. Blood. 2011;118:3901–3910. doi: 10.1182/blood-2010-11-317438. [DOI] [PubMed] [Google Scholar]
- 16.Lestini BJ, Goldsmith KC, Fluchel MN, Liu X, Chen NL, Goyal B, et al. Mcl1 downregulation sensitizes neuroblastoma to cytotoxic chemotherapy and small molecule Bcl2-family antagonists. Cancer Biol Ther. 2009;8:1587–1595. doi: 10.4161/cbt.8.16.8964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li R, Zang Y, Li C, Patel NS, Grandis JR, Johnson DE. ABT-737 synergizes with chemotherapy to kill head and neck squamous cell carcinoma cells via a Noxa-mediated pathway. Mol Pharmacol. 2009;75:1231–1239. doi: 10.1124/mol.108.052969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hauck P, Chao BH, Litz J, Krystal GW. Alterations in the Noxa/Mcl-1 axis determine sensitivity of small cell lung cancer to the BH3 mimetic ABT-737. Mol Cancer Ther. 2009;8:883–892. doi: 10.1158/1535-7163.MCT-08-1118. [DOI] [PubMed] [Google Scholar]
- 19.Olejniczak ET, Van Sant C, Anderson MG, Wang G, Tahir SK, Sauter G, et al. Integrative genomic analysis of small-cell lung carcinoma reveals correlates of sensitivity to bcl-2 antagonists and uncovers novel chromosomal gains. Mol Cancer Res. 2007;5:331–339. doi: 10.1158/1541-7786.MCR-06-0367. [DOI] [PubMed] [Google Scholar]
- 20.Deng J, Carlson N, Takeyama K, Dal Cin P, Shipp M, Letai A. BH3 profiling identifies three distinct classes of apoptotic blocks to predict response to ABT-737 and conventional chemotherapeutic agents. Cancer Cell. 2007;12:171–185. doi: 10.1016/j.ccr.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 21.Olberding KE, Wang X, Zhu Y, Pan J, Rai SN, Li C. Actinomycin D synergistically enhances the efficacy of the BH3 mimetic ABT-737 by downregulating Mcl-1 expression. Cancer Biol Ther. 2010;10:918–929. doi: 10.4161/cbt.10.9.13274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen S, Dai Y, Harada H, Dent P, Grant S. Mcl-1 down-regulation potentiates ABT-737 lethality by cooperatively inducing Bak activation and Bax translocation. Cancer Res. 2007;67:782–791. doi: 10.1158/0008-5472.CAN-06-3964. [DOI] [PubMed] [Google Scholar]
- 23.Chakravarti A, Delaney MA, Noll E, Black PM, Loeffler JS, Muzikansky A, et al. Prognostic and pathologic significance of quantitative protein expression profiling in human gliomas. Clin Cancer Res. 2001;7:2387–2395. [PubMed] [Google Scholar]
- 24.Chakravarti A, Noll E, Black PM, Finkelstein DF, Finkelstein DM, Dyson NJ, et al. Quantitatively determined survivin expression levels are of prognostic value in human gliomas. J Clin Oncol. 2002;20:1063–1068. doi: 10.1200/JCO.2002.20.4.1063. [DOI] [PubMed] [Google Scholar]
- 25.Zhen HN, Zhang X, Hu PZ, Yang TT, Fei Z, Zhang JN, et al. Survivin expression and its relation with proliferation, apoptosis, and angiogenesis in brain gliomas. Cancer. 2005;104:2775–2783. doi: 10.1002/cncr.21490. [DOI] [PubMed] [Google Scholar]
- 26.Kanwar JR, Kamalapuram SK, Kanwar RK. Targeting survivin in cancer: the cell-signalling perspective. Drug Discov Today. 2011;16:485–494. doi: 10.1016/j.drudis.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Asanuma H, Torigoe T, Kamiguchi K, Hirohashi Y, Ohmura T, Hirata K, et al. Survivin expression is regulated by coexpression of human epidermal growth factor receptor 2 and epidermal growth factor receptor via phosphatidylinositol 3-kinase/AKT signaling pathway in breast cancer cells. Cancer Res. 2005;65:11018–11025. doi: 10.1158/0008-5472.CAN-05-0491. [DOI] [PubMed] [Google Scholar]
- 28.Okada H, Low KL, Kohanbash G, McDonald HA, Hamilton RL, Pollack IF. Expression of glioma-associated antigens in pediatric brain stem and non-brain stem gliomas. Journal of Neuro-Oncology. 2008;88:245–250. doi: 10.1007/s11060-008-9566-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Giaccone G, Zatloukal P, Roubec J, Floor K, Musil J, Kuta M, et al. Multicenter phase II trial of YM155, a small-molecule suppressor of survivin, in patients with advanced, refractory, non-small-cell lung cancer. J Clin Oncol. 2009;27:4481–4486. doi: 10.1200/JCO.2008.21.1862. [DOI] [PubMed] [Google Scholar]
- 30.Satoh T, Okamoto I, Miyazaki M, Morinaga R, Tsuya A, Hasegawa Y, et al. Phase I study of YM155, a novel survivin suppressant, in patients with advanced solid tumors. Clin Cancer Res. 2009;15:3872–3880. doi: 10.1158/1078-0432.CCR-08-1946. [DOI] [PubMed] [Google Scholar]
- 31.Nakahara T, Kita A, Yamanaka K, Mori M, Amino N, Takeuchi M, et al. Broad spectrum and potent antitumor activities of YM155, a novel small-molecule survivin suppressant, in a wide variety of human cancer cell lines and xenograft models. Cancer Sci. 2011;102:614–621. doi: 10.1111/j.1349-7006.2010.01834.x. [DOI] [PubMed] [Google Scholar]
- 32.Nakahara T, Takeuchi M, Kinoyama I, Minematsu T, Shirasuna K, Matsuhisa A, et al. YM155, a novel small-molecule survivin suppressant, induces regression of established human hormone-refractory prostate tumor xenografts. Cancer Res. 2007;67:8014–8021. doi: 10.1158/0008-5472.CAN-07-1343. [DOI] [PubMed] [Google Scholar]
- 33.Tang H, Shao H, Yu C, Hou J. Mcl-1 downregulation by YM155 contributes to its synergistic anti-tumor activities with ABT-263. Biochem Pharmacol. 2011;82:1066–1072. doi: 10.1016/j.bcp.2011.07.064. [DOI] [PubMed] [Google Scholar]
- 34.Huang HS, Nagane M, Klingbeil CK, Lin H, Nishikawa R, Ji XD, et al. The enhanced tumorigenic activity of a mutant epidermal growth factor receptor common in human cancers is mediated by threshold levels of constitutive tyrosine phosphorylation and unattenuated signaling. J Biol Chem. 1997;272:2927–2935. doi: 10.1074/jbc.272.5.2927. [DOI] [PubMed] [Google Scholar]
- 35.Nagane M, Coufal F, Lin H, Bogler O, Cavenee WK, Huang HJ. A common mutant epidermal growth factor receptor confers enhanced tumorigenicity on human glioblastoma cells by increasing proliferation and reducing apoptosis. Cancer Res. 1996;56:5079–5086. [PubMed] [Google Scholar]
- 36.Kang MH, Reynolds CP. Bcl-2 inhibitors: targeting mitochondrial apoptotic pathways in cancer therapy. Clin Cancer Res. 2009;15:1126–1132. doi: 10.1158/1078-0432.CCR-08-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Labi V, Grespi F, Baumgartner F, Villunger A. Targeting the Bcl-2-regulated apoptosis pathway by BH3 mimetics: a breakthrough in anticancer therapy? Cell Death Differ. 2008;15:977–987. doi: 10.1038/cdd.2008.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.High LM, Szymanska B, Wilczynska-Kalak U, Barber N, O’Brien R, Khaw SL, et al. The Bcl-2 homology domain 3 mimetic ABT-737 targets the apoptotic machinery in acute lymphoblastic leukemia resulting in synergistic in vitro and in vivo interactions with established drugs. Mol Pharmacol. 2010;77:483–494. doi: 10.1124/mol.109.060780. [DOI] [PubMed] [Google Scholar]
- 39.Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proc Natl Acad Sci U S A. 1990;87:8602–8606. doi: 10.1073/pnas.87.21.8602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shinojima N, Tada K, Shiraishi S, Kamiryo T, Kochi M, Nakamura H, et al. Prognostic value of epidermal growth factor receptor in patients with glioblastoma multiforme. Cancer Res. 2003;63:6962–6970. [PubMed] [Google Scholar]
- 41.Craig RW. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia. 2002;16:444–454. doi: 10.1038/sj.leu.2402416. [DOI] [PubMed] [Google Scholar]
- 42.Premkumar DR, Jane EP, Pollack IF. Co-administration of NVP-AEW541 and dasatinib induces mitochondrial-mediated apoptosis through Bax activation in malignant human glioma cell lines. Int J Oncol. 2010;37:633–643. doi: 10.3892/ijo_00000712. [DOI] [PubMed] [Google Scholar]
- 43.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, et al. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.