

Highlights
► Systems-level approaches are increasingly being used in influenza virus research. ► The timing and magnitude of the host response is a key determinant of disease outcome. ► The kinetics of immune activation must be considered for successful antiviral therapy. ► Collaborative Cross mice are providing new insights into the role of host genetics. ► New computational approaches are leading to more robust and predictive network models.
Keywords: Computational biology, Genomics, Inflammation, Influenza virus, Interferon, Systems biology
Abstract
Influenza virus research has recently undergone a shift from a virus-centric perspective to one that embraces the full spectrum of virus–host interactions and cellular signaling events that determine disease outcome. This change has been brought about by the increasing use and expanding scope of high-throughput molecular profiling and computational biology, which together fuel discovery in systems biology. In this review, we show how these approaches have revealed an uncontrolled inflammatory response as a contributor to the extreme virulence of the 1918 pandemic and avian H5N1 viruses, and how this response differs from that induced by the 2009 H1N1 viruses responsible for the most recent influenza pandemic. We also discuss how new animal models, such as the Collaborative Cross mouse systems genetics platform, are key to the necessary systematic investigation of the impact of host genetics on infection outcome, how genome-wide RNAi screens have identified hundreds of cellular factors involved in viral replication, and how systems biology approaches are making possible the rational design of new drugs and vaccines against an ever-evolving respiratory virus.
1. Introduction
Each winter brings a new flu season and with it the possibility of unpleasant surprises for the human population. Most years, influenza outbreaks are relatively predictable, causing 30,000–40,000 deaths in the United States. More serious threats come in the form of exceptionally transmissible or virulent influenza virus variants, which can give rise to worldwide pandemics and greatly increased morbidity and mortality. New viral variants arise through a combination of antigenic drift—gradual changes brought about by point mutations in viral genes—and antigenic shift—abrupt changes brought about by the reassortment of complete viral gene segments. But while the mechanisms underlying the evolution of new influenza virus variants are relatively well understood, comparably less is known about the virus–host interactions that determine disease outcome.
Traditionally, research on viral pathogenesis has focused on the contribution of viral genes and gene products, with little emphasis on the host. In the case of influenza virus, which encodes less than a dozen genes, hundreds of papers have been published on the viral NS1 protein, which has been reported to have numerous functions that can potentially impact virulence [1]. The viral hemagglutinin (HA) and neuraminidase (NA) genes have also been implicated as virulence determinants, as have components of the viral polymerase. Although knowledge of viral protein function has been instrumental in the development of anti-influenza drugs, which include inhibitors of NA (oseltamivir and zanamivir) and the viral M2 ion channel (amantadine and rimantadine), drug-resistant influenza viruses continue to emerge and there remains a need for new and effective antiviral therapies [2]. Similarly, challenges remain in developing influenza vaccines that are protective across a broad range of strains, as the most effective vaccines promote an immune response against HA, which is the most variable of the viral proteins [3].
The advent of high-throughput genome-based technologies brought the ability to expand from a virus-centric perspective to one that encompasses the entire spectrum of virus–host interactions and their impact on the global host response to infection. We were among the first to bring this approach to influenza virus research with genome-scale analyses of the host response to active or heat-inactivated influenza virus [4], to a deletion mutant lacking the NS1 gene [5], and to recombinant viruses containing the HA and NA genes from the 1918 pandemic virus [6]. Our group was also the first to use genomic approaches to analyze a macaque model of influenza virus infection [7]. These studies showed the power of genomic analyses to provide a global view of the host transcriptional response to infection [8], but only hinted at the complexities of the response that remains to be uncovered.
Despite these early advances, as late as 2007, a review of the use of genomics to study influenza virus–host interactions yielded only a handful of additional studies [9]. Over the past 5 years, however, the landscape has changed dramatically. Gene expression profiling is being augmented by proteomics and metabolomics, global RNAi screens are being used to identify host factors involved in influenza virus replication, and gene knockout and Collaborative Cross mouse models are providing new insights into the host genetic determinants that regulate disease outcome (Fig. 1 ). Moreover, new technologies, such as next-generation sequencing, are allowing us to peer ever deeper into the host response, and computational approaches are becoming increasingly sophisticated.
Fig. 1.
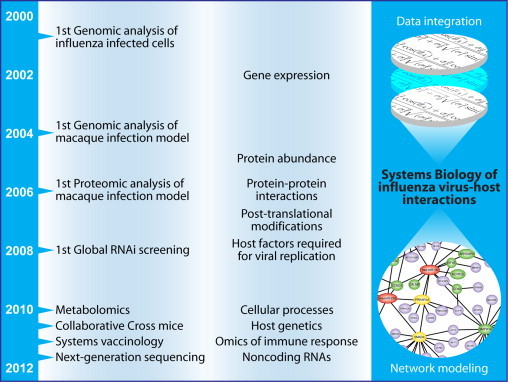
Overview of advances in the use of high-throughput approaches to study influenza virus–host interactions and viral pathogenesis. Advances in computational methods are now beginning to provide the means to integrate the diverse types of data and information obtained from these approaches to yield systems-level insights into virus–host interactions, the antiviral response, and innate and adaptive immunity.
In this review, we touch upon each of these topics as well as provide an overview of what systems biology approaches are revealing about the host response to highly pathogenic viruses, such as the reconstructed 1918 pandemic virus and avian H5N1 strains, and to newly emerging viruses, such as the H1N1 virus responsible for the 2009 pandemic. We also discuss how systems biology and predictive methodology approaches are contributing to drug and vaccine development and speculate on what the future holds for the systems biology of influenza virus–host interactions.
2. 1918 and avian H5N1 viruses: pandemics of the past and future?
Why do some influenza viruses cause severe, even fatal disease, while others cause relatively mild respiratory infections? While this is not a new question, finding the answer took on renewed urgency with the first reports of human infection with avian H5N1 viruses [10], [11]. In the initial outbreak in Hong Kong, 6 of 18 infected individuals died, and “bird flu” became an international concern. Since then, highly pathogenic avian H5N1 viruses have continued to appear throughout Southeast Asia and into Europe and Africa with hundreds more human infections and a mortality rate of up to 60% [12], [13]. The possibility that such a virus could acquire the ability to readily transmit between humans is justifiable cause for concern. Research on the mechanisms underlying highly virulent influenza virus infection gained further momentum with the reconstruction of the 1918 pandemic virus [14], a virus responsible for the greatest infectious disease outbreak in human history and over 50 million deaths worldwide [15]. Not surprisingly, the reconstructed 1918 virus and avian H5N1 strains have also become a prominent focus of genomic and systems-level analyses aimed at understanding the virus–host interactions underlying severe respiratory virus infection.
2.1. Animal models: inflammation gone awry
Genomic analysis of the host response to the reconstructed 1918 pandemic virus (r1918) began in 2006 with studies using a mouse infection model [16]. Mice infected with r1918 developed severe lung pathology, including intense infiltration of neutrophils, and died within 5 days of infection. At the genomic level, the lungs of these animals exhibited an early (day 1) increase in the expression of cytokine, chemokine, and apoptosis-related genes, including genes associated with death receptor, IL-6, type I IFN, and Toll-like receptor responses. This response was unique to the r1918-infected animals and was not observed in the lungs of mice infected with a contemporary (and less pathogenic) H1N1 strain. Increased inflammatory gene expression paralleled destruction of the respiratory epithelium and was sustained until the death of the animals, providing a first glimpse of an inflammatory signature that has since become pathognomonic with severe respiratory virus infection (Fig. 2 ).
Fig. 2.
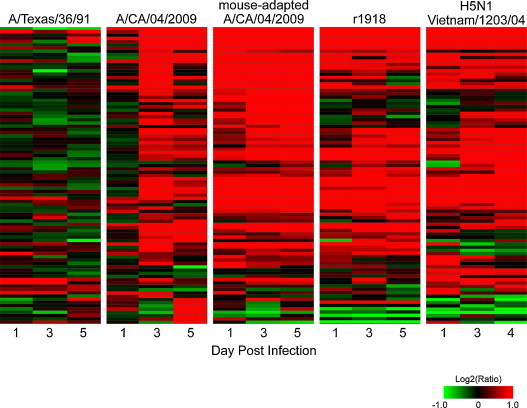
Differential induction of inflammatory gene expression by influenza viruses causing mild or severe respiratory disease. Shown are changes in inflammatory gene expression profiles over time in the lungs of mice infected with A/Texas/36/91 (a nonpathogenic seasonal isolate), A/CA/04/2009 (a mildly pathogenic 2009 H1N1 pandemic isolate), or highly pathogenic mouse-adapted 2009 H1N1, r1918, or avian H5N1 viruses. Expression values are represented as the average of the log2 ratio of infected to respective mock-infected samples for three biological replicates per condition. Red and green indicate that gene expression is increased or decreased relative to mock, respectively.
Whereas the virulence of r1918 in mice was unusual—most influenza viruses require adaptation to cause lethal infection in mice—its virulence in macaques proved to be exceptional. The first experimental infection of cynomolgus macaques with r1918 resulted in acute respiratory distress and death, with all animals euthanized by day 8 due to the severity of symptoms [17]. Genomic analysis of bronchial tissue from r1918-infected animals (chosen because of less immune cell infiltration than in the lung) also revealed an early and sustained induction of inflammatory cytokine (particularly IL-6) and chemokine genes, including CXCL1 and CXCL6, which are important for the recruitment of neutrophils. In contrast, macaques infected with a less pathogenic H1N1 strain showed increased expression of DDX58 (RIG-I), IFIH1 (MDA5), type 1 IFN, and IFN-stimulated genes (ISGs) early after infection, but this response was down-regulated at later time points as the animals recovered. Fewer ISGs were induced in r1918-infected macaques, but those that were induced retained high levels of expression until the death of the animals. Interestingly, a similar lack of attenuation of early immune activation leading to death is observed in macaques infected with simian immunodeficiency virus, indicating that uncontrolled immune responses might be a general mechanism of pathogenicity across a wide range of viruses and hosts [18].
Studies using the macaque model to examine the host response to H5N1 viruses have also reported severe lung pathology and the induction of a strong innate immune transcriptional response, although macaques infected with H5N1 viruses typically recover [19]. In a direct comparison of the host response to r1918 and H5N1 infection, both viruses were found to cause early and severe lung pathology and both induced similar ISG expression at 12-h post infection. However, by 24-h post infection, while both viruses reached similar titers, only the r1918 virus induced the expression of numerous genes related to cell death and inflammation, including the inflammasome components NLRP3 and IL-1β [20]. Although the inflammasome has been reported to be an essential component of the innate immune response to influenza A viruses [21], [22], [23], the excessive activation of this response appears to be detrimental.
Whereas r1918 has repeatedly been found to be more virulent than H5N1 viruses in macaques, the opposite is seen when using a mouse model of infection, where H5N1 viruses exhibit greater virulence. In a study by Cilloniz et al. [24], both viruses reached similar titers in mice and both elicited an early and marked increase in the expression of IFN-regulated and inflammatory response genes. However, in mice, it is the H5N1 virus that elicits strong induction of inflammasome genes. The H5N1 virus also disseminated to the brain and spleen, and a computational approach identified a correlation between tissue dissemination and the up-regulation of pro-inflammatory responses and the down-regulation of anti-inflammatory genes. Most notable was the down-regulation of Alox5, responsible for the biogenesis of lipoxins—eicosanoids with anti-inflammatory properties [25]—and Socs2, encoding a suppressor of cytokine signaling that can be induced by lipoxins to control the inflammatory response [26].
H5N1 infection of ferrets also induces the strong up-regulation of IFN response genes [27], including the expression of CXCL10, a chemoattractant for activated Th1 lymphocytes and natural killer cells. Treatment of ferrets with an antagonist of CXCR3, the receptor for CXCL10, resulted in reduced viral loads and pathology, but only delayed eventual mortality. Thus, multiple genomic profiling studies using a variety of animal models have pointed to an early, sustained, and excessive inflammatory response as a hallmark of severe influenza virus infection.
2.2. In vitro systems: transcriptional regulation of inflammation
Although animal models arguably provide the most biologically relevant system for analyzing the host response to infection, in vitro systems using lung epithelial cells [28] and primary human macrophages [29] have also provided evidence for the early up-regulation of pro-inflammatory cytokine genes in response to infection with highly pathogenic or modified H5N1 viruses. In vitro systems have further been used to examine the extent to which the transcription factor NF-κB, a major regulator of inflammatory gene expression, has on the host response to H5N1 infection [30]. Infecting primary human endothelial cells in the presence or absence of a dominant negative mutant of IκB kinase 2, which blocks the activation of NF-κB, revealed that the majority of H5N1-induced genes were NF-κB-dependent. Some genes, such as IFN-β, were strictly NF-κB-dependent, whereas others were only partially dependent upon NF-κB for their induction. In contrast, a less pathogenic influenza virus, A/WSN/33, induced a weaker overall host response that was less dependent upon NF-κB.
In an extension of these studies, promoter analyses were used to identify additional transcriptional regulators necessary for H5N1-induced gene expression in endothelial cells [31]. These analyses were performed using the gene expression profiles induced by H5N1 infection but not by infection with mildly pathogenic H7N7 avian or H1N1 viruses. Coupled with targeted siRNA knockdown, these analyses confirmed the role of IRF3 in H5N1 induction of IFN response genes and also identified a requirement for the transcription factors HMGA1 and NFATC4 in mediating the strong inflammatory response induced by H5N1 infection. The combination of NF-κB, HMGA1, and NFATC4 signaling may therefore be of particular importance in the inflammatory response induced in endothelial cells by highly pathogenic H5N1 viruses.
More recently, weighted gene correlation network analysis (WGCNA) was used to identify the host regulatory network response to H5N1 infection of human bronchial epithelial (Calu-3) cells [32]. This method identifies gene expression patterns on the basis of underlying correlation structures and groups genes into signaling networks or modules. The two most prominent networks identified included genes associated with the immune response or keratinization processes. The mechanisms by which changes in keratin gene expression contribute to the host response to H5N1 virus infection are not clear, but alterations in keratin may impact such processes as intracellular viral transport and budding. As discuss later, several quantitative proteomic studies have also shown increases in the abundance of keratin in response to influenza virus infection.
2.3. Therapeutic implications: targeting the inflammatory response
The finding that an excessive and uncontrolled inflammatory response—frequently referred to as a cytokine storm—is a common factor in the virulence of r1918 and avian H5N1 viruses suggests that therapy to limit the inflammatory response may be beneficial in treating infections caused by highly pathogenic influenza viruses. Yet as reviewed elsewhere [33], despite efforts to target the host response with a variety of anti-inflammatory drugs, this approach has been largely unsuccessful. At least part of the difficulty may rest with the timing at which anti-inflammatory or immunomodulating drugs are administered. Just as genomic profiling studies have revealed that the timing and duration of the inflammatory response is an important factor in disease outcome, so too is it likely that the timing at which elements of the host response are suppressed or enhanced through drug therapy needs to be carefully controlled. With the increasing sophistication of computational approaches, one challenge for the future will be to garner a deeper understanding of the rapidly changing dynamics of the immune response (see Section 10). This may lead to rational improvements in both the targets and timing of immunomodulatory therapy.
3. The 2009 H1N1 pandemic: emergence of a novel influenza virus
In early 2009, interest in the 1918 and H5N1 viruses took a temporary back seat with reports of unusually severe influenza outbreaks in Mexico. By April 2009, the virus appeared in the United States, causing clusters of illness in schools and communities. Although most infections were mild, some individuals had severe symptoms, and the virus was clearly capable of rapid human-to-human transmission. By June 2009, the World Health Organization announced that a global pandemic was underway. The virus was determined to carry a unique reassortment of gene segments from avian, human, and swine influenza viruses [34], [35], resulting in a virus for which few people under age 60 had any significant level of preexisting immunity [36]. Such rapid emergence of new reassortment viruses by genetic shift illuminates the challenges for the host immune system as well as for vaccination and therapeutic strategies.
3.1. Animal models: macaques, mice, and pigs too
The cynomolgus macaque model used to study r1918 and H5N1 pathogenesis has also proven useful in characterizing the host response to 2009 H1N1 pandemic viruses [37], [38]. In particular, infection of macaques with either of two genetically similar but clinically distinct human isolates resulted in clinical disease that ranged from mild to severe pneumonia, closely mimicking human infection with these viruses [38]. Gene expression profiling of lung tissue from these animals revealed the induction of numerous genes related to the inflammatory response, and in animals infected with the virus causing more severe clinical symptoms, there was also up-regulation of a network of NF-κB signaling molecules, which correlated with increased pro-inflammatory plasma cytokine levels (e.g., IL-6 and MCP-1) early after infection. However, unlike r1918 or H5N1 infection, these responses resolved and the animals recovered from infection. Comparing the host response to highly pathogenic and 2009 H1N1 viruses might therefore lead to the identification of immune activation attenuation signals that could represent potential targets for therapy against r1918 and H5N1 infection.
Most 2009 H1N1 pandemic viruses cause only mild disease in mice [37], [39]. However, serial lung passage can be used to generate mouse-adapted viruses with markedly increased virulence [40]. Genomic profiling of the host response to a mouse-adapted virus—containing a total of five mutations in HA, PB2, and NP and causing 100% lethality—provided an opportunity for comparative analyses of the host response to mild or severe 2009 H1N1 infection [41]. Perhaps not surprisingly, the mouse-adapted virus elicited an early and sustained inflammatory response, reminiscent of that induced by r1918 and H5N1 viruses (Fig. 2). This observation was confirmed computationally using nonparametric multidimensional scaling to visualize global gene expression concordance across the transcriptional profiles elicited by these viruses (Fig. 3 ). In addition, animals infected with the mouse-adapted virus exhibited perturbation in the expression of lipid metabolism genes later in infection, as was observed previously in macaques infected with H5N1 virus. Transcription factor activation state prediction identified IRF1 and NF-κB as the primary drivers of the sustained inflammatory response along with inhibition of the negative regulator TRIM24.
Fig. 3.
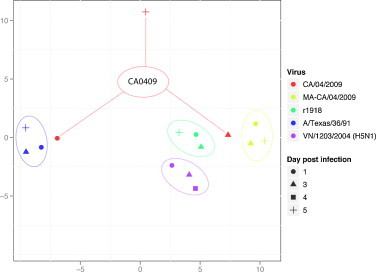
Comparison of transcriptional profiles elicited by A/Texas/36/91 (a nonpathogenic seasonal isolate), CA/04/2009 (a mildly pathogenic 2009 H1N1 pandemic isolate), or highly pathogenic mouse-adapted 2009 H1N1, r1918, or avian H5N1 viruses. Nonparametric multidimensional scaling was used to represent the Euclidian distance between samples on each day post infection. The matrix distance was calculated using the list of genes that are represented in Fig. 2.
Because the 2009 H1N1 virus derived in part from swine influenza viruses, there has also been interest in determining the extent to which these viruses are able to cause disease in pigs [42], [43]. In a study by Ma et al., pigs were infected with a human 2009 H1N1 isolate, a 2009 swine isolate, or with a 1918-like classical swine influenza virus [42]. Both of the 2009 viruses caused mild clinical symptoms, whereas the classical swine virus induced only fever. Animals infected with the 2009 viruses exhibited an increase in the transcription of inflammatory and immune response pathways that included genes associated with pattern recognition receptor signaling. By day 5 after infection, these animals also exhibited increased expression of genes associated with cell death and lipid metabolism. In contrast, these responses were largely absent in animals infected with the classical swine virus, and the expression of genes associated with lipid metabolism was actually suppressed. It is possible that the suppression of lipid metabolism genes may be due in part by the actions of the NS1 protein of the 1918-like virus. Previous transcriptional studies have shown that infection of a lung epithelial cell line with a recombinant seasonal virus containing the 1918 NS1 gene results in the down-regulation of a network of genes associated with lipid metabolism [44].
The differential response of pigs to the 2009 H1N1 viruses and the 1918-like classical swine influenza virus is particularly intriguing in light of the fact that pigs infected with r1918 exhibit only transient fever and mild respiratory disease [45], in stark contrast to the rapidly lethal infections that result in macaques and mice when infected with the r1918 virus. This makes the pig a unique model for the future generation of more detailed high-throughput and systems level analyses that may help to identify unique aspects of the host response that make pigs largely resistant to the effects of the r1918 virus. Such knowledge could be used to rationally design therapeutic approaches to suppress or augment targeted aspects of the host response in macaques or other animal models (and eventually humans) to mitigate pathogenic outcome.
3.2. Animal models: bacterial co-infection
Morbidity and mortality associated with primary influenza virus infection is often exacerbated by a secondary bacterial infection, an event that was common during the 2009 pandemic [46]. To study the relationship between viral and bacterial infection, Kash et al. used a model in which mice were infected with either a 2009 seasonal isolate or a 2009 pandemic virus followed by inoculation with Streptococcus pneumoniae [47]. All mice infected with virus alone survived the infection and exhibited similar lung transcriptional profiles characterized by moderate increases in inflammatory gene expression. Animals infected with the seasonal virus plus S. pneumonia also survived infection, whereas bacterial co-infection with the 2009 pandemic virus resulted in 100% mortality. Interestingly, lethal bacterial co-infection was not accompanied by a significant increase in the inflammatory response, but rather was characterized by increased basal epithelial cell apoptosis and a lack of cellular gene transcription associated with re-proliferation and repair (a transcriptional response that was elicited by the seasonal virus). Thus, the extent to which bacterial co-infection increases morbidity and mortality appears to be at least in part dependent upon the virus–host interactions that lead to lung injury and subsequent repair.
In general, gene expression profiling of macaques, pigs, mice, and ferrets [48] infected with 2009 H1N1 pandemic isolates has revealed an early increase in inflammatory and innate immune gene expression that correlates with mild-to-moderate lung pathology. In all cases, this increase in gene expression resolves during later stages of infection and tissue repair and recovery responses ensue. In vitro analyses using a lung epithelial cell line [49] or primary human alveolar epithelial cells [50] have also revealed a transient increase in inflammatory and IFN response genes. These observations once more point to the dynamics of the host immune response as being essential to outcome and underline the importance of good timing of therapeutic intervention. With increasing evidence of synergy between different pathogens, the infectious history of the host also emerges as a major determinant of pathology. Taken together with the evident impact of host genetic variation (see Section 5), considerable challenges lie ahead for the development of broad-spectrum vaccines and therapies.
4. Viral genetic determinants of pathogenesis
In the studies described above, high-throughput and computational approaches were used to study influenza virus pathogenesis primarily by analyzing and comparing the host response to wild-type viruses. This approach can be augmented, however, to gain additional insight into the viral and host genetic determinants of virulence. One tactic for studying viral determinants is to generate recombinant viruses in which a gene (or genes) from a highly pathogenic virus replaces the corresponding gene of an otherwise less virulent strain.
Several studies have used this approach to better understand the contribution of the NS1 gene to r1918 virulence. For example, when used to infect lung epithelial cells, an A/WSN/33 (mouse-adapted) virus containing the 1918 NS1 gene efficiently blocked the expression of IFN-regulated genes, whereas the parental A/WSN/33 virus elicited significant induction of IFN-regulated gene expression [5]. This approach was also taken a step further by swapping the NS1 genes of the 1918 and seasonal A/Texas/36/91 viruses [44]. Again, the recombinant virus containing the 1918 NS1 gene induced cytokine and chemokine gene expression, but blocked the transcription of many IFN-regulated genes and genes associated with lipid metabolism. In contrast, the opposite effect was observed in cells infected with r1918 engineered to contain the A/Texas/36/91 NS1 gene. Similarly, infection of primary human tracheobronchial epithelial cells with an A/Texas/36/91 virus containing a deletion of the C-terminal effector domain of NS1 was used to define the contributions of this domain in the suppression of IFN signaling [51]. Together, these studies have demonstrated the importance of the NS1 protein in regulating the host response to infection, particularly with regard to IFN-regulated gene expression, and point to this protein as a key contributor to the high virulence of the 1918 pandemic virus. In the same way, additional studies have focused on recombinant viruses expressing HA and NA from the 1918 virus [6] and H5N1 viruses containing single amino acid substitutions in the viral polymerase (PB2) [52] or PB1-F2 protein [53] that result in increased virulence.
5. Host genetic determinants of pathogenesis: knockout and Collaborative Cross mice
Just as alterations to the viral genome can be used to study viral determinants of pathogenesis, so too can systematic changes to the host genome be used to study host determinants of disease outcome. A common method in this regard is the use of targeted gene knockout mice. While not a new concept, the addition of genomic profiling adds an important dimension by providing the ability to learn how loss of function in one aspect of the host response affects other aspects of viral recognition or innate immune signaling. One of the first studies to profile the transcriptional response of a knockout mouse to influenza virus infection looked at the role of P58IPK, an inhibitor of the eIF2α kinases PKR and PERK, which respectively function to regulate protein synthesis during virus infection and the unfolded protein response [54]. Infection of P58IPK-knockout mice with a mouse-adapted virus, or with r1918, resulted in greatly amplified expression of inflammatory and apoptotic response genes, providing the first evidence that P58IPK functions during virus infection to inhibit the over-activation of inflammatory and cell death responses.
Whereas the use of P58IPK-knockout mice represented the first use of a mammalian infection model to demonstrate the role of P58IPK in protection against virus infection, a number of additional studies have examined the transcriptional response of mice containing knockouts in genes well known for their role in the antiviral response. To examine the impact of deficiencies in the key inflammatory mediators TNF and IL1 on virus infection, mice containing gene knockouts in the receptors for these molecules were infected with the r1918 virus [55]. Although the virus was lethal in all animals, TNF receptor knockout mice survived statistically longer than wild-type mice and exhibited delayed or decreased expression of genes associated with antiviral and innate immune signaling and negative acute-phase response. IL1 receptor knockout mice, in contrast, exhibited an increase in the expression of genes associated with dendritic and natural killer cell processes and a compensatory increase in TNF expression. Signaling through the IL1 receptor therefore appears to be protective, whereas signaling through the TNF receptor increases the severity of infection.
Similar approaches have been used to examine the role of the Mx1 protein, which is lacking in most laboratory strains of mice [56], and signaling through the IFN receptor, a primary mediator of innate immunity. Mx1+/+ mice are partially protected against r1918 infection, and when treated with IFN, fully survive infection and exhibit down-regulated expression of cytokine and chemokine genes normally induced by the r1918 virus [57]. With respect to IFN signaling, infection of mouse embryonic fibroblasts lacking the α/β IFN receptor results in decreased expression of antiviral genes (e.g., TLR3, PKR, and STAT1) and increased viral replication, but there is no effect on the induction of genes related to inflammatory and apoptotic responses, which are induced to the levels observed in the presence of the receptor [58]. This study, and the above-mentioned interplay between IL1 and TNF, also reveals a significant amount of redundant and compensatory signaling in antiviral and innate immune responses. While informative, such redundancy can also complicate and lead to misleading interpretation of data resulting from the use of gene knockout animals.
A more recent and comprehensive approach to identifying novel host genetic determinants associated with disease susceptibility and outcome is the mouse Collaborative Cross systems genetics platform [59]. The Collaborative Cross is a panel of recombinant inbred mouse lines derived from a genetically diverse set of eight inbred founder strains. The approach is designed to yield high levels of genetic variation uniformly across the genome and to capture the majority of genetic variation in this animal. This unique collection of animals can be used to identify individual and multiple genetic traits that contribute to complex immune phenotypes and the generation of protective or pathologic outcomes following influenza virus infection (Fig. 4 ).
Fig. 4.
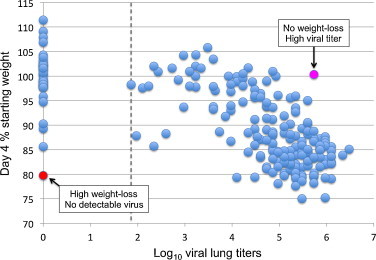
Genetic diversity across pre-Collaborative Cross mouse lines results in differential host response to influenza virus infection. Shown is the relationship between weight loss and viral titer for 209 pre-Collaborative Cross mouse lines infected with influenza virus A/PR/8. Dashed line indicates limit of detection for viral titer.
Although the Collaborative Cross resource is not yet fully developed, Bottomly et al. used 44 pre-Collaborative Cross lines, determined to have extreme high or low responses to influenza virus infection based on clinical readouts, to identify expression quantitative trait loci (eQTL)—genes that regulate the expression of mRNAs—associated with the host transcriptional response to infection [60]. This approach yielded 17 validated eQTLs, which were then used for structural equation modeling to identify additional genes with putative downstream relationships with the validated eQTLs. The genes identified belonged to a variety of functional categories, including immune response, tissue regeneration, and cellular adhesion. Thus, the approach enables the generation of networks of gene relationships that can drive the discovery of additional genes important to the host antiviral response.
A similar approach, using BXD recombinant inbred mice [61] and genome-wide linkage analysis, identified genetic elements responsible for survival following H5N1 infection [62]. This study yielded five quantitative trait loci associated with disease severity, and gene expression analysis identified candidate genes within these loci with functions related to innate immunity and inflammation. However, in comparison to the Collaborative Cross, the BXD platform does not capture the full genetic diversity present in outbred populations [63], and it is therefore less suitable for a systems genetics approach aimed at identifying multiple genetic elements, including small-effect-size and modifier genes that can significantly impact phenotypic outcomes.
6. Proteomics and metabolomics: proteins and processes
The studies described so far have relied extensively upon the use of microarray-based genomic profiling. However, additional high-throughput data types are necessary for building a true systems-level view of the events that occur in a virus-infected cell or tissue. Proteomics, with its focus on the biologically active products of genes and the post-translational modifications that regulate protein function; and metabolomics, with its focus on small molecules and cellular processes; are essential to systems biology and are becoming increasingly integrated into the study of influenza virus–host interactions.
Despite the challenges of profiling mammalian tissues, the first use of global proteomics to study influenza virus infection was directed at the nonhuman primate model, where tandem mass spectrometry identified over 3500 proteins in macaque lung tissue [64]. Quantitation by relative peptide abundance revealed that infection with the mildly pathogenic A/Texas/36/91 seasonal isolate led to increases in the abundance of a variety of proteins involved in innate immune signaling. The approach also identified changes in the abundance of proteins that were not predicted from transcriptional profiling, thereby demonstrating the utility of proteomics for both confirming and expanding upon the views provided by genomic analyses.
A subsequent and more in-depth profiling of the macaque lung proteome identified over 4200 proteins, 400 of which increased in abundance in response to infection with A/Texas/36/91, a recombinant virus containing the HA and NA genes from the 1918 virus, or a highly pathogenic avian H5N1 isolate [65]. The increased abundance of these 400 proteins was considered to represent a “core” response to influenza virus infection. Linear regression analysis further revealed that increased abundance of 96 of these proteins correlated with severe disease (caused by the H5N1 virus), including such proteins as PKR, MX1, and RIG-I (DDX58). In addition, while the abundance of proteins involved in the immune response, metabolism, and transport increased rapidly after infection with the H5N1 virus, this response was delayed in the animals infected with the less pathogenic viruses. To a large extent, these findings mirror the results of genomic analyses [19] and reinforce the hypothesis that the dynamics of immune activation are essential to disease outcome.
A range of proteomic approaches, including two-dimensional differential in-gel electrophoresis (2-D DIGE) coupled with tandem mass spectrometry [66], [67], stable isotope labeling by amino acids in cell culture (SILAC) [68], [69], [70], and subcellular proteome analysis using iTRAQ (isobaric tag for absolute and relative quantitation) [71] have also been used to analyze influenza virus-induced changes in protein abundance in a variety of cultured cell types. Collectively, these studies have identified changes in the abundance of numerous IFN-regulated proteins, components of the cytoskeleton, keratin, and other processes. Proteomic approaches have also been used to identify protein interaction partners, including cellular factors that associate with the viral ribonucleoprotein [72] and the trimeric viral RNA polymerase complex [73]. In addition, the mass spectrometric approaches common to proteomics are being extended to metabolic and lipidomic profiling, yielding new insights into the effects of influenza virus infection on intermediates of glycolysis and the tricarboxylic acid (TCA) cycle [74], fatty acid biosynthesis and cholesterol metabolism [75], and the role of palmitoylation on the activity of innate immune effector proteins, including IFITM3 [76].
Given the considerable technical and computational challenges associated with high-throughput proteomics and metabolomics, many of these studies have pushed the envelope in terms of bringing new approaches to the difficult problems of peptide, protein, and metabolite detection, identification, and quantification and the application of these approaches to complex model systems. Nevertheless, these approaches to date have provided only modest gains with respect to our knowledge of influenza virus biology. Technical and algorithmic improvements that allow the identification and quantification of ever greater numbers of proteins, protein post-translational modifications, and metabolites will certainly continue to move the field forward. However, significant advances in biological knowledge are only likely to come with computational approaches that can successfully integrate diverse types of high-throughput data into meaningful network models of signaling pathways and inter-molecular interactions.
7. Genome-wide screens: host factors involved in influenza virus replication
Like all viruses, influenza virus depends upon cellular factors and machinery to support its replication. While high-throughput “omic” technologies provide a wealth of information regarding the changes in gene transcription and protein and metabolite abundance that occur during infection, they provide comparatively little information about the identity of host factors required for influenza virus replication. Yet knowledge of such factors should help to put high-throughput data into context as well as provide new targets for therapeutic intervention.
In an attempt to fill this knowledge gap, half a dozen studies have reported the use of genome-wide screens to identify host factors involved in influenza virus replication. The majority of these screens have used RNA interference (RNAi) to direct homology-dependent gene suppression on a genome-wide scale. The first such study established an influenza virus infection system using Drosophila cells, and after screening a RNAi library against over 13,000 genes, identified 110 genes whose suppression impacted reporter gene expression from a modified influenza virus [77]. Subsequent studies using mammalian cell RNAi screens identified 133 [78], 295 [79], and 287 [80] genes encoding proteins that affected various steps in the viral life cycle ranging from entry and uncoating to assembly and budding. In a variation on this approach, Shapira et al. [81] used the results from yeast two-hybrid analysis and gene expression profiling of influenza virus-infected primary human bronchial epithelial cells to computationally predict factors and pathways that affect the viral life cycle. These predictions were then tested by suppressing the expression of these genes through RNAi, resulting in 616 genes whose products affected influenza virus replication. Together, these studies identified cellular factors involved in a variety of functions, including cellular signaling, translation initiation, and nuclear transport.
As an alternative to RNAi screening, Sui et al. used the technique of random homozygous gene perturbation (RHGP) to identify host genes that prevent influenza virus-mediated killing of the host cell [82]. This technique uses a lentiviral-based genetic element that can integrate at a single site in one allele of a gene in either a sense or anti-sense orientation. No prior knowledge or annotation of the gene is necessary, and the result can be either knock-down or over-expression of the gene. The approach generates a library of independent clones that can be screened for a desired phenotype. In this case, the isolation of clones resistant to killing by influenza virus yielded 110 human genes. Only four of these genes had been previously linked to influenza virus infection (MDN1, GRK6, AKT1, and STXBP1), demonstrating the potential of the approach to identify novel targets.
The combined results of these studies have been the subject of extensive review [83], [84]. In general, meta-analysis exposed a striking lack of overlap in the genes identified by these screens. Pair-wise comparisons revealed only 128 genes (out of a total of 1449 identified) that affected influenza virus replication in at least two screens, and the number of genes in common between pairs of screens is even smaller, ranging from zero to 32 genes [83]. This low degree of overlap could be due to a variety of factors, including differences in screening systems, cell types, and viruses and was also observed in RNAi screens used to identify host factors for HIV-1 replication, where only three genes were identified by all three screens [85]. Importantly, concordance might also be affected by the statistical prioritization approaches used and might be generally hampered by a large number of host factors falling within a narrow band of positive or negative correlation with pathogenicity, reminiscent of the challenges faced by gene signature definition [86]. Moreover, only a handful of the genes identified have been subjected to functional analyses to confirm their role in influenza virus replication. These studies, while impressive in the number of genes identified, therefore represent only the initial steps in a much longer road toward a deeper understanding of the virus–host interplay necessary for viral replication and the identification of new drug targets.
8. Vaccine development: predicting immunogenicity
Vaccines against influenza virus are strain-specific (targeted against one influenza A H1N1 strain, one influenza A H3N2 strain, and one influenza B strain), and the formulation may be changed annually on the basis of global surveillance and the emergence of new strains [87]. The effectiveness of the vaccine depends upon the similarity between the viruses in the vaccine and the viruses in circulation as well as upon individual variation among vaccine recipients. The development of influenza vaccines is therefore an ongoing process, as is the search for improved efficacy and broad-spectrum protection. Over the past few years, the application of high-throughput and computational approaches to vaccine development, variously termed systems vaccinology [88] or vaccinomics [89], has increasingly come to the forefront with the goal of rational vaccine development and the identification of useful predictors of vaccine efficacy.
Genomic approaches were first applied to influenza vaccine research in a nonhuman primate study aimed at evaluating the protective efficacy of a live influenza virus vaccine produced by truncating the NS1 gene of the human H1N1 isolate A/Texas/36/91 [90]. The live vaccine was highly immunogenic and transcriptional profiling of bronchial cells revealed the strong induction of IFN-regulated genes. After challenge, animals receiving the live vaccine had reduced viral replication and lung pathology and less activation of cytokine, chemokine, and IFN-regulated genes compared with that observed in animals vaccinated with a formalin-killed wild-type virus. The study therefore demonstrated the vaccine potential of live influenza virus attenuated through modification of the NS1 gene as well as the use of gene expression profiling to identify transcriptional signatures after both vaccination and challenge.
More recently, a combination of traditional, high-throughput, and computational approaches have been used to evaluate immune responses to vaccination against influenza virus in human vaccine studies. Genomic analysis of peripheral blood samples from 119 individuals immunized with a trivalent influenza vaccine revealed an early increase in the expression of genes associated with IFN signaling and antigen presentation, which correlated with the magnitude of the antibody response [91]. In a more comprehensive approach, Nakaya et al. evaluated innate and adaptive immune responses to vaccination against influenza virus using blood samples obtained over a 3-year period from individuals receiving a trivalent inactivated vaccine or a live attenuated vaccine [92]. Although the clinical effectiveness of these vaccines was similar, only the live attenuated vaccine induced robust expression of genes associated with an IFN response (as did only the live vaccine in the macaque study described above). In contrast, the inactivated vaccine induced higher antibody titers, and only the inactivated vaccine induced the expression of genes associated with a plasma B cell response. Using the computational approach of discriminant analysis via mixed integer programming, this gene expression signature was shown to be predictive of later antibody titer.
Together, these studies demonstrate the ability of molecular profiling and computational approaches to predict immunogenicity (as measured by antibody titers) and to provide new insights into the mechanisms of action of vaccines. Such information is clearly of value to future vaccine development and evaluation (including the rapid identification of individuals who fail to respond to vaccination), and systems-level approaches are destined to play an ever increasing role in vaccine research.
9. Digging deeper into the data: analysis across experiments and model systems
The rapidly rising numbers of studies using high-throughput approaches (both within and outside of the influenza virus field) are generating enormous amounts of data. Unfortunately, this data is often only analyzed for a single publication and is subsequently archived, perhaps never to be used again. Although gene expression databases such as the Gene Expression Omnibus (GEO) [93] have been in existence for over a decade, new database resources, such as the Influenza Research Database [94] and the Virus Pathogen Resource [95] promise to put multiple data types relevant to influenza virus research in a centralized location. This should provide increased opportunities for data integration and the analysis of data across multiple experiments or experimental systems.
As an example, Chang et al. used meta-analysis—statistical methods to combine and contrast findings from multiple independent studies—to derive gene expression signatures from 12 studies that used mouse models to measure transcriptional responses to influenza virus, respiratory syncytial virus, mengovirus, or SARS coronavirus [96]. The viruses used in these studies caused clinical outcomes that ranged from 100% survival to 100% lethality, and the goal of the analysis was to identify gene expression signatures with the capacity to segregate and predict mild or highly pathogenic infections. A series of computational techniques generated signatures that were either oppositely expressed (referred to as a “digital” relationship) or that were expressed on a continuum (referred to as an “analog” relationship) when comparing mild and highly pathogenic infections. The best predictor of a highly pathogenic infection was a 57-gene analog signature that included the induction of genes associated with the inflammatory response and chemokine signaling and decreased expression of genes associated with lung repair. The analog nature of this signature indicates that highly pathogenic viruses (including r1918, avian H5N1 viruses, and SARS coronavirus) induce or suppress the expression of many of the same genes as mildly pathogenic viruses, but to a much greater degree. The magnitude of the host response, rather than the induction or suppression of a unique set of genes, therefore appears to be a critical factor in determining disease outcome.
Whereas the above study looked at a single infection model and a range of respiratory viruses and clinical outcomes, McDermott et al. analyzed the host transcriptional response to a single avian H5N1 strain across cell culture, mouse, and macaque infection models [97]. This study used three complementary approaches: functional similarity analysis, the identification of patterns of coexpression between systems, and a network inference method to identify conserved regulatory influences. Because these approaches do not rely on matching comparable time points between experiments, they can be used to compare data from systems with different sampling times and dynamics. These combined approaches identified functions and pathways that displayed similar behavior or regulation across all three systems, most notably in IFN and inflammatory responses, indicating that significant portions of the response to influenza virus are conserved across cell culture, mouse, and macaque models. Predicted regulatory influences driving this response included GTF2B (a factor also known as TFIIB and targeted by a number of viruses to disrupt transcriptional initiation) and ATF4 (a transcription factor involved in the endoplasmic reticulum stress response). Overall, this study demonstrates the ability of computational approaches to reveal relationships between in vitro and in vivo systems and for gaining deeper insight into biological responses to infection through the use of pre-existing data sets. It also serves to bolster the much-debated relevance of cell culture systems for predicting responses in animal models. In the future, it will also be interesting to focus on host-specific differences and to gain insight into the variability of pathogenicity as a function of host genetics.
10. Necessary paradigm shifts
Just as systems biology contributed to the shift from a virus-centric view to the integrated study of virus–host interactions, a similar paradigm shift will be necessary in the upcoming, second phase of computational analysis of viral pathogenicity. Three somewhat unexpected themes of viral pathology have emerged from the initial application of systems approaches: (i) pathogenicity is to a larger-than-expected extent a function of host genetics and likely infectious history [13], (ii) low versus high pathogenicity is not so much a function of different host gene networks responding to the infection but rather similar or identical networks responding to a different degree [96], and (iii) chronic, non-attenuated activation of these networks highly correlates with outcome, and attenuation might be kinetically controlled as a function of the dynamics of the virus–host interaction [18]. All three observations can be reconciled into a single hypothesis of time-dependent encoding of host responses, making the timing of components of the host response of utmost importance for the virus–host interactions themselves and for disease outcome and treatment (Fig. 5 ).
Fig. 5.
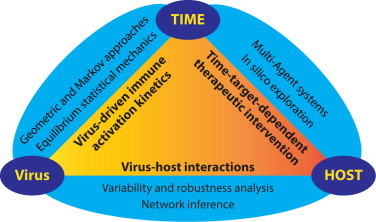
The kinetics of virus-driven immune activation is a central component of virus–host interactions and a key factor in viral pathogenesis. Mathematical approaches discussed in the text for studying highly time-resolved data, time-dependent therapeutic intervention, and virus–host interactions are indicated.
An understanding of the time-dependency of immune activation and its control is therefore likely to be key to understanding pathogenicity and our ability to alter disease outcome. Few data are currently available that follow molecular events over time, and even less so on the appropriate time scales. Immune activation is a process that happens on a single-cell level within minutes to hours rather than hours to days. It will therefore be necessary to generate omics data with high time resolution during the early and relevant transition phases of immune activation. Acquisition of such data faces technical challenges and is limited by problems of synchronization of events, cells, and tissues. However, novel computational approaches should be able to overcome these difficulties. Geometric approaches, for instance low-dimensional, non-linear representations of gene networks based on their activity [98], can be used to renormalize poorly time-resolved data to absolute references (pre-infection versus post-infection; low-pathogenicity versus high-pathogenicity; ressortant versus wild-type viruses) and therefore reinterpret activity differences as differences in observation time. Even more interesting in this context are continuous-time Markov processes defined on a path space spanned by low-resolution experiments. Here, the few available experimental data points over time are expanded through inference methodology allowing approximation and subsequent comparisons of the system's internal time dynamics across different viruses or genetic variants. In order to be successful, these methods will require, in addition to the available data sets, the generation of data from controlled experiments in cell culture systems where key components of immune activation networks are studied individually.
Future research will also have to address the local robustness of virus–host networks. Today, robustness (a measure of the system's tolerance against external and internal variability) is only studied superficially with respect to detail and quantity. Typically, survival or other global properties of the system are quantified over large time intervals as fractions of affected individuals in a given experiment with at best hundreds of individuals. In order to better understand the molecular network structure and dynamics leading to immune responses, redundancy in these networks, and the relative importance of highly connected (“hubs”) as well as sparse regions of the networks (“bottlenecks”), is required to quantify robustness at the level of individual network components. To this end, Collaborative Cross mice and human population genetic studies will provide sources of genetic variability of the host. These will have to be combined with systematic studies of reassortant viruses to explore in detail the interactions between virus and host genetics. It will also be important to focus on subtle variations rather than extreme cases in order to better appreciate the contribution of individual network components.
Furthermore, in order to better appreciate robustness, it will be necessary to quantify not only differences but also similarity or equivalency under noise [99]. In addition, computational research will have to focus on developing better approaches for the inference of network dynamics from network topology similar to what has recently been achieved for metabolic networks [100], as it is not possible to measure all relevant network sub-dynamics in sufficient detail for lack of accessible experimental systems. Finally, we will need to better establish causality in the virus–host networks. Current computational methodology largely focuses on statistical correlations between observables. In order to understand cause and effect relationships in the networks, the above mentioned time-series analyses will need to be accompanied by network inference methodology based on first principle physics [101], [102].
Finally, the generation of detailed molecular models is needed to be able to simulate parts of the virus–host networks in silico. Such modeling will be required to develop better screening of effective drugs once high-priority targets have been established through the above approaches. To this end, the multi-agent systems (MAS) paradigm shows great promise. MAS aims to model complex systems by the use of autonomous programs, named agents, which have independent behaviors and which interact among each other or with a virtual environment [103], [104]. MAS has been applied in social science, economics, and ecology to solve problems such as collective decision making, process optimization, or the analysis of emerging properties. In the context of system biology, MAS represents a system that can simulate the biological mechanisms involved between the major actors of the host immune response across different scales. On a macroscopic level, cell-to-cell and virus-cell interactions and communications can be modeled and integrated with a microscopic level that captures the dynamics of signaling and the triggering of metabolic pathways. Such modeling will help to refine the most important components involved during infection and contribute to a better understanding of emerging properties. Different MAS platforms [105], [106], [107] have already been used to model host immune responses and physical interactions at a single-cell level [108], [109], [110], [111]. Once a better understanding of network sub-structures and dynamics is achieved, better parameter sets will be available to increase the effectiveness of such models in the systematic in silico exploration of virus–host networks in view of possible drug targets and theoretical targeting strategies.
11. Conclusions
The types of computational analyses just described will need to play an increasing role in the future, as ever-expanding amounts and types of high-throughput data are generated. In addition, it will be critical to develop computational methods and visualization techniques capable of integrating diverse types of data, in a quantitative manner, and displaying them in a meaningful and understandable fashion. Computational biology has become quite adept at generating giant “hairball” networks, but such visualizations provide too little in the way of helping us understand the biology of the system or in providing targets for future experimentation.
Similarly, improved computational methods, predictive models, and more targeted experimentation are needed to find the sweet spot between broad conclusions made on the basis of functional categories (such as hypercytokinemia, inflammation, and cell death) and the identification of changes in the expression of individual genes or the abundance of individual proteins. Focusing on the former provides too high level a view, and focusing on the latter brings us back to a reductionist approach that fails to take advantage of the bigger picture provided by global analyses. Moreover, it is difficult to know whether targeting an individual component of a system—and knowing which one to pick—will impact disease outcome, as there are many examples of compensatory pathways and responses. Nevertheless, progress is being made in this regard, including the development of methods for identifying important genes or proteins (network hubs or bottlenecks) on the basis of topological analysis of protein-protein interaction or inferred networks [112], [113], and targets identified by this approach have been successfully validated through gene knockdown [114].
Computational approaches must also evolve to deal with new high-throughput technologies that are bringing different types of data into play. For example, next-generation sequencing technologies have recently revealed that influenza virus infection induces the differential expression of a variety of long (greater than 200 nucleotide) noncoding RNAs [115]. Many of these RNAs are similarly regulated by IFN treatment, suggesting that long noncoding RNAs may be involved in regulating the host response, including innate immunity. Similarly, there is increasing evidence that microRNAs play a role in the host response to numerous viruses [116], including influenza virus [117]. Next-generation sequencing has also revealed that additional forms of small noncoding RNAs, such as small nuclear RNAs (snoRNAs) and piwi-associated small RNAs (piRNA), are also differentially expressed in response to influenza virus infection [118]. Moreover, noncoding RNAs have been found to directly regulate protein function, as exemplified by the identification of the 7SK snRNA as a regulator of HMGA1 function [119]. Next-generation sequencing is therefore adding a critical new analytical component that may alter the way we think about virus–host interactions and gene regulatory mechanisms. It is likely that a detailed knowledge of noncoding RNA regulation and function will be necessary for a full understanding of influenza virus pathogenesis.
Systems biology, while not without its critics, has become well established in biomedical research, particularly in the areas of drug development and cancer biology. Although slower to come to the infectious disease field, the approach has made substantial inroads into influenza virus research, as the studies described in this review attest. An influx of new investigators, together with ongoing improvements to computational methods and technologies, should continue to propel the field forward. As an additional driver, the approach has also received support from the National Institute of Allergy and Infectious Diseases, which has funded four systems biology centers, including two focused on influenza virus [120]. Influenza virus, with its ability to evolve in surprising new ways, is certain to be a threat well into the future. Similarly, the methods used to study this virus must continue to evolve if we are to develop the methods of surveillance, vaccination, and therapy needed to protect the public health.
Acknowledgments
We thank Sean Proll and Laurence Josset for figure preparation and Mark Heise and Martin Ferris for Collaborative Cross data. Research in the author's laboratory is supported by Public Health Service grants R2400011172, R2400011157, P30DA015625, P51RR00166, and U54AI081680 and by federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under contract HHSN272200800060C.
References
- 1.Hale B.G., Randall R.E., Ortin J., Jackson D. The multifunctional NS1 protein of influenza A viruses. Journal of General Virology. 2008;89:2359–2376. doi: 10.1099/vir.0.2008/004606-0. [DOI] [PubMed] [Google Scholar]
- 2.Pizzorno A., Abed Y., Boivin G. Influenza drug resistance. Seminars in Respiratory and Critical Care Medicine. 2011;32:409–422. doi: 10.1055/s-0031-1283281. [DOI] [PubMed] [Google Scholar]
- 3.Ellebedy A.H., Webby R.J. Influenza vaccines. Vaccine. 2009;27(Suppl. 4):D65–D68. doi: 10.1016/j.vaccine.2009.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geiss G.K., An M.C., Bumgarner R.E., Hammersmark E., Cunningham D., Katze M.G. Global impact of influenza virus on cellular pathways is mediated by both replication-dependent and -independent events. Journal of Virology. 2001;75:4321–4331. doi: 10.1128/JVI.75.9.4321-4331.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Geiss G.K., Salvatore M., Tumpey T.M., Carter V.S., Wang X., Basler C.F. Cellular transcriptional profiling in influenza A virus-infected lung epithelial cells: the role of the nonstructural NS1 protein in the evasion of the host innate defense and its potential contribution to pandemic influenza. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:10736–10741. doi: 10.1073/pnas.112338099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kash J.C., Basler C.F., Garcia-Sastre A., Carter V., Billharz R., Swayne D.E. Global host immune response: pathogenesis and transcriptional profiling of type A influenza viruses expressing the hemagglutinin and neuraminidase genes from the 1918 pandemic virus. Journal of Virology. 2004;78:9499–9511. doi: 10.1128/JVI.78.17.9499-9511.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baskin C.R., Garcia-Sastre A., Tumpey T.M., Bielefeldt-Ohmann H., Carter V.S., Nistal-Villan E. Integration of clinical data, pathology, and cDNA microarrays in influenza virus-infected pigtailed macaques (Macaca nemestrina) Journal of Virology. 2004;78:10420–10432. doi: 10.1128/JVI.78.19.10420-10432.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Korth M.J., Kash J.C., Baskin C.R., Katze M.G. Insights into influenza virus–host interactions through global gene expression profiling: cell culture systems to animal models. In: Kawaoka Y., editor. Influenza virology: current topics. Horizon Scientific Press; Wymondham, UK: 2006. pp. 341–360. [Google Scholar]
- 9.Fornek J.L., Korth M.J., Katze M.G. Use of functional genomics to understand influenza–host interactions. Advances in Virus Research. 2007;70:81–100. doi: 10.1016/S0065-3527(07)70003-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Subbarao K., Klimov A., Katz J., Regnery H., Lim W., Hall H. Characterization of an avian influenza A (H5N1) virus isolated from a child with a fatal respiratory illness. Science. 1998;279:393–396. doi: 10.1126/science.279.5349.393. [DOI] [PubMed] [Google Scholar]
- 11.Claas E.C., Osterhaus A.D., van Beek R., De Jong J.C., Rimmelzwaan G.F., Senne D.A. Human influenza A H5N1 virus related to a highly pathogenic avian influenza virus. Lancet. 1998;351:472–477. doi: 10.1016/S0140-6736(97)11212-0. [DOI] [PubMed] [Google Scholar]
- 12.Influenza Yong E. Five questions on H5N1. Nature. 2012;486:456–458. doi: 10.1038/486456a. [DOI] [PubMed] [Google Scholar]
- 13.Wang T.T., Parides M.K., Palese P. Seroevidence for H5N1 influenza infections in humans: meta-analysis. Science. 2012;335:1463. doi: 10.1126/science.1218888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tumpey T.M., Basler C.F., Aguilar P.V., Zeng H., Solorzano A., Swayne D.E. Characterization of the reconstructed 1918 Spanish influenza pandemic virus. Science. 2005;310:77–80. doi: 10.1126/science.1119392. [DOI] [PubMed] [Google Scholar]
- 15.Johnson N.P., Mueller J. Updating the accounts: global mortality of the 1918–1920 Spanish influenza pandemic. Bulletin of the History of Medicine. 2002;76:105–115. doi: 10.1353/bhm.2002.0022. [DOI] [PubMed] [Google Scholar]
- 16.Kash J.C., Tumpey T.M., Proll S.C., Carter V., Perwitasari O., Thomas M.J. Genomic analysis of increased host immune and cell death responses induced by 1918 influenza virus. Nature. 2006;443:578–581. doi: 10.1038/nature05181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kobasa D., Jones S.M., Shinya K., Kash J.C., Copps J., Ebihara H. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature. 2007;445:319–323. doi: 10.1038/nature05495. [DOI] [PubMed] [Google Scholar]
- 18.Benecke A., Gale M., Jr., Katze M.G. Dynamics of innate immunity are key to chronic immune activation in AIDS. Current Opinion in HIV AIDS. 2012;7:79–85. doi: 10.1097/COH.0b013e32834dde31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baskin C.R., Bielefeldt-Ohmann H., Tumpey T.M., Sabourin P.J., Long J.P., Garcia-Sastre A. Early and sustained innate immune response defines pathology and death in nonhuman primates infected by highly pathogenic influenza virus. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:3455–3460. doi: 10.1073/pnas.0813234106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cilloniz C., Shinya K., Peng X., Korth M.J., Proll S.C., Aicher L.D. Lethal influenza virus infection in macaques is associated with early dysregulation of inflammatory related genes. PLoS Pathogens. 2009;5:e1000604. doi: 10.1371/journal.ppat.1000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thomas P.G., Dash P., Aldridge J.R., Jr., Ellebedy A.H., Reynolds C., Funk A.J. The intracellular sensor NLRP3 mediates key innate and healing responses to influenza A virus via the regulation of caspase-1. Immunity. 2009;30:566–575. doi: 10.1016/j.immuni.2009.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ichinohe T., Lee H.K., Ogura Y., Flavell R., Iwasaki A. Inflammasome recognition of influenza virus is essential for adaptive immune responses. Journal of Experimental Medicine. 2009;206:79–87. doi: 10.1084/jem.20081667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Allen I.C., Scull M.A., Moore C.B., Holl E.K., McElvania-TeKippe E., Taxman D.J. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity. 2009;30:556–565. doi: 10.1016/j.immuni.2009.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cilloniz C., Pantin-Jackwood M.J., Ni C., Goodman A.G., Peng X., Proll S.C. Lethal dissemination of H5N1 influenza virus is associated with dysregulation of inflammation and lipoxin signaling in a mouse model of infection. Journal of Virology. 2010;84:7613–7624. doi: 10.1128/JVI.00553-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Serhan C.N., Chiang N., Van Dyke T.E. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nature Reviews of Immunology. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Machado F.S., Johndrow J.E., Esper L., Dias A., Bafica A., Serhan C.N. Anti-inflammatory actions of lipoxin A4 and aspirin-triggered lipoxin are SOCS-2 dependent. Nature Medicine. 2006;12:330–334. doi: 10.1038/nm1355. [DOI] [PubMed] [Google Scholar]
- 27.Cameron C.M., Cameron M.J., Bermejo-Martin J.F., Ran L., Xu L., Turner P.V. Gene expression analysis of host innate immune responses during lethal H5N1 infection in ferrets. Journal of Virology. 2008;82:11308–11317. doi: 10.1128/JVI.00691-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chakrabarti A.K., Vipat V.C., Mukherjee S., Singh R., Pawar S.D., Mishra A.C. Host gene expression profiling in influenza A virus-infected lung epithelial (A549) cells: a comparative analysis between highly pathogenic and modified H5N1 viruses. Virology Journal. 2010;7:219. doi: 10.1186/1743-422X-7-219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S.M., Gardy J.L., Cheung C.Y., Cheung T.K., Hui K.P., Ip N.Y. Systems-level comparison of host-responses elicited by avian H5N1 and seasonal H1N1 influenza viruses in primary human macrophages. PLoS ONE. 2009;4:e8072. doi: 10.1371/journal.pone.0008072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schmolke M., Viemann D., Roth J., Ludwig S. Essential impact of NF-kappaB signaling on the H5N1 influenza A virus-induced transcriptome. Journal of Immunology. 2009;183:5180–5189. doi: 10.4049/jimmunol.0804198. [DOI] [PubMed] [Google Scholar]
- 31.Viemann D., Schmolke M., Lueken A., Boergeling Y., Friesenhagen J., Wittkowski H. H5N1 virus activates signaling pathways in human endothelial cells resulting in a specific imbalanced inflammatory response. Journal of Immunology. 2011;186:164–173. doi: 10.4049/jimmunol.0904170. [DOI] [PubMed] [Google Scholar]
- 32.Li C., Bankhead A., III, Eisfeld A.J., Hatta Y., Jeng S., Chang J.H. Host regulatory network response to infection with highly pathogenic H5N1 avian influenza virus. Journal of Virology. 2011;85:10955–10967. doi: 10.1128/JVI.05792-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tisoncik J.R., Korth M.J., Simmons C.P., Farrar J., Martin T.R., Katze M.G. Into the eye of the cytokine storm. Microbiology and Molecular Biology Reviews. 2012;76:16–32. doi: 10.1128/MMBR.05015-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Garten R.J., Davis C.T., Russell C.A., Shu B., Lindstrom S., Balish A. Antigenic and genetic characteristics of swine-origin 2009 A(H1N1) influenza viruses circulating in humans. Science. 2009;325:197–201. doi: 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith G.J., Vijaykrishna D., Bahl J., Lycett S.J., Worobey M., Pybus O.G. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature. 2009;459:1122–1125. doi: 10.1038/nature08182. [DOI] [PubMed] [Google Scholar]
- 36.Hancock K., Veguilla V., Lu X., Zhong W., Butler E.N., Sun H. Cross-reactive antibody responses to the 2009 pandemic H1N1 influenza virus. New England Journal of Medicine. 2009;361:1945–1952. doi: 10.1056/NEJMoa0906453. [DOI] [PubMed] [Google Scholar]
- 37.Itoh Y., Shinya K., Kiso M., Watanabe T., Sakoda Y., Hatta M. In vitro and in vivo characterization of new swine-origin H1N1 influenza viruses. Nature. 2009;460:1021–1025. doi: 10.1038/nature08260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Safronetz D., Rockx B., Feldmann F., Belisle S.E., Palermo R.E., Brining D. Pandemic swine-origin H1N1 influenza A isolates show heterogeneous virulence in macaques. Journal of Virology. 2011;85:1214–1223. doi: 10.1128/JVI.01848-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maines T.R., Jayaraman A., Belser J.A., Wadford D.A., Pappas C., Zeng H. Transmission and pathogenesis of swine-origin 2009 A(H1N1) influenza viruses in ferrets and mice. Science. 2009;325:484–487. doi: 10.1126/science.1177238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ilyushina N.A., Khalenkov A.M., Seiler J.P., Forrest H.L., Bovin N.V., Marjuki H. Adaptation of pandemic H1N1 influenza viruses in mice. Journal of Virology. 2010;84:8607–8616. doi: 10.1128/JVI.00159-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Josset L., Belser J.A., Pantin-Jackwood M., Chang J.H., Chang S.T., Belisle S.E. Implication of inflammatory macrophages, nuclear receptors and interferon regulatory factors in increased virulence of pandemic 2009 H1N1 influenza A virus after host adaptation. Journal of Virology. 2012;86:7192–7206. doi: 10.1128/JVI.00563-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ma W., Belisle S.E., Mosier D., Li X., Stigger-Rosser E., Liu Q. 2009 pandemic H1N1 influenza virus causes disease and upregulation of genes related to inflammatory and immune responses, cell death, and lipid metabolism in pigs. Journal of Virology. 2011;85:11626–11637. doi: 10.1128/JVI.05705-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li Y., Zhou H., Wen Z., Wu S., Huang C., Jia G. Transcription analysis on response of swine lung to H1N1 swine influenza virus. BMC Genomics. 2011;12:398. doi: 10.1186/1471-2164-12-398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Billharz R., Zeng H., Proll S.C., Korth M.J., Lederer S., Albrecht R. The NS1 protein of the 1918 pandemic influenza virus blocks host interferon and lipid metabolism pathways. Journal of Virology. 2009;83:10557–10570. doi: 10.1128/JVI.00330-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weingartl H.M., Albrecht R.A., Lager K.M., Babiuk S., Marszal P., Neufeld J. Experimental infection of pigs with the human 1918 pandemic influenza virus. Journal of Virology. 2009;83:4287–4296. doi: 10.1128/JVI.02399-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gill J.R., Sheng Z.M., Ely S.F., Guinee D.G., Beasley M.B., Suh J. Pulmonary pathologic findings of fatal 2009 pandemic influenza A/H1N1 viral infections. Archives of Pathology & Laboratory Medicine. 2010;134:235–243. doi: 10.5858/134.2.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kash J.C., Walters K.A., Davis A.S., Sandouk A., Schwartzman L.M., Jagger B.W. Lethal synergism of 2009 pandemic H1N1 influenza virus and Streptococcus pneumoniae coinfection is associated with loss of murine lung repair responses. mBio. 2011:2011. doi: 10.1128/mBio.00172-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rowe T., Leon A.J., Crevar C.J., Carter D.M., Xu L., Ran L. Modeling host responses in ferrets during A/California/07/2009 influenza infection. Virology. 2010;401:257–265. doi: 10.1016/j.virol.2010.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mukherjee S., Vipat V.C., Mishra A.C., Pawar S.D., Chakrabarti A.K. Pandemic (H1N1) 2009 influenza virus induces weaker host immune responses in vitro: a possible mechanism of high transmissibility. Virology Journal. 2011;8:140. doi: 10.1186/1743-422X-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lee S.M., Chan R.W., Gardy J.L., Lo C.K., Sihoe A.D., Kang S.S. Systems-level comparison of host responses induced by pandemic and seasonal influenza A H1N1 viruses in primary human type I-like alveolar epithelial cells in vitro. Respiratory Research. 2010;11:147. doi: 10.1186/1465-9921-11-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tisoncik J.R., Billharz R., Burmakina S., Belisle S.E., Proll S.C., Korth M.J. The NS1 protein of influenza A virus suppresses interferon-regulated activation of antigen-presentation and immune-proteasome pathways. Journal of General Virology. 2011;92:2093–2104. doi: 10.1099/vir.0.032060-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fornek J.L., Gillim-Ross L., Santos C., Carter V., Ward J.M., Cheng L.I. A single-amino-acid substitution in a polymerase protein of an H5N1 influenza virus is associated with systemic infection and impaired T-cell activation in mice. Journal of Virology. 2009;83:11102–11115. doi: 10.1128/JVI.00994-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Conenello G.M., Tisoncik J.R., Rosenzweig E., Varga Z.T., Palese P., Katze M.G. A single N66S mutation in the PB1-F2 protein of influenza A virus increases virulence by inhibiting the early interferon response in vivo. Journal of Virology. 2011;85:652–662. doi: 10.1128/JVI.01987-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goodman A.G., Fornek J.L., Medigeshi G.R., Perrone L.A., Peng X., Dyer M.D. P58(IPK): a novel CIHD member of the host innate defense response against pathogenic virus infection. PLoS Pathogens. 2009;5:e1000438. doi: 10.1371/journal.ppat.1000438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Belisle S.E., Tisoncik J.R., Korth M.J., Carter V.S., Proll S.C., Swayne D.E. Genomic profiling of tumor necrosis factor alpha (TNF-α) receptor and interleukin-1 receptor knockout mice reveals a link between TNF-α signaling and increased severity of 1918 pandemic influenza virus infection. Journal of Virology. 2010;84:12576–12588. doi: 10.1128/JVI.01310-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tumpey T.M., Szretter K.J., Van Hoeven N., Katz J.M., Kochs G., Haller O. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. Journal of Virology. 2007;81:10818–10821. doi: 10.1128/JVI.01116-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cilloniz C., Pantin-Jackwood M.J., Ni C., Carter V.S., Korth M.J., Swayne D.E. Molecular signatures associated with Mx1-mediated resistance to highly pathogenic influenza virus infection: mechanisms of survival. Journal of Virology. 2012;86:2437–2446. doi: 10.1128/JVI.06156-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goodman A.G., Zeng H., Proll S.C., Peng X., Cilloniz C., Carter V.S. The alpha/beta interferon receptor provides protection against influenza virus replication but is dispensable for inflammatory response signaling. Journal of Virology. 2010;84:2027–2037. doi: 10.1128/JVI.01595-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Collaborative-Cross-Consortium The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics. 2012;190:389–401. doi: 10.1534/genetics.111.132639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bottomly D., Ferris M.T., Aicher L.D., Rosenzweig E., Whitmore A., Aylor D.L. Expression quantitative trait Loci for extreme host response to influenza a in pre-collaborative cross mice. G3 (Bethesda) 2012;2:213–221. doi: 10.1534/g3.111.001800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Peirce J.L., Lu L., Gu J., Silver L.M., Williams R.W. A new set of BXD recombinant inbred lines from advanced intercross populations in mice. BMC Genetics. 2004;5:7. doi: 10.1186/1471-2156-5-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Boon A.C., deBeauchamp J., Hollmann A., Luke J., Kotb M., Rowe S. Host genetic variation affects resistance to infection with a highly pathogenic H5N1 influenza A virus in mice. Journal of Virology. 2009;83:10417–10426. doi: 10.1128/JVI.00514-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Roberts A., Pardo-Manuel de Villena F., Wang W., McMillan L., Threadgill D.W. The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mammalian Genome. 2007;18:473–481. doi: 10.1007/s00335-007-9045-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Baas T., Baskin C.R., Diamond D.L., Garcia-Sastre A., Bielefeldt-Ohmann H., Tumpey T.M. Integrated molecular signature of disease: analysis of influenza virus-infected macaques through functional genomics and proteomics. Journal of Virology. 2006;80:10813–10828. doi: 10.1128/JVI.00851-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brown J.N., Palermo R.E., Baskin C.R., Gritsenko M., Sabourin P.J., Long J.P. Macaque proteome response to highly pathogenic avian influenza and 1918 reassortant influenza virus infections. Journal of Virology. 2010;84:12058–12068. doi: 10.1128/JVI.01129-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vester D., Rapp E., Gade D., Genzel Y., Reichl U. Quantitative analysis of cellular proteome alterations in human influenza A virus-infected mammalian cell lines. Proteomics. 2009;9:3316–3327. doi: 10.1002/pmic.200800893. [DOI] [PubMed] [Google Scholar]
- 67.Vester D., Rapp E., Kluge S., Genzel Y., Reichl U. Virus–host cell interactions in vaccine production cell lines infected with different human influenza A virus variants: a proteomic approach. Journal of Proteomics. 2010;73:1656–1669. doi: 10.1016/j.jprot.2010.04.006. [DOI] [PubMed] [Google Scholar]
- 68.Emmott E., Wise H., Loucaides E.M., Matthews D.A., Digard P., Hiscox J.A. Quantitative proteomics using SILAC coupled to LC–MS/MS reveals changes in the nucleolar proteome in influenza A virus-infected cells. Journal of Proteome Research. 2010;9:5335–5345. doi: 10.1021/pr100593g. [DOI] [PubMed] [Google Scholar]
- 69.Coombs K.M., Berard A., Xu W., Krokhin O., Meng X., Cortens J.P. Quantitative proteomic analyses of influenza virus-infected cultured human lung cells. Journal of Virology. 2010;84:10888–10906. doi: 10.1128/JVI.00431-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dove B.K., Surtees R., Bean T.J., Munday D., Wise H.M., Digard P. A quantitative proteomic analysis of lung epithelial (A549) cells infected with 2009 pandemic influenza A virus using stable isotope labelling with amino acids in cell culture. Proteomics. 2012;12:1431–1436. doi: 10.1002/pmic.201100470. [DOI] [PubMed] [Google Scholar]
- 71.Lietzen N., Ohman T., Rintahaka J., Julkunen I., Aittokallio T., Matikainen S. Quantitative subcellular proteome and secretome profiling of influenza A virus-infected human primary macrophages. PLoS Pathogens. 2011;7:e1001340. doi: 10.1371/journal.ppat.1001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mayer D., Molawi K., Martinez-Sobrido L., Ghanem A., Thomas S., Baginsky S. Identification of cellular interaction partners of the influenza virus ribonucleoprotein complex and polymerase complex using proteomic-based approaches. Journal of Proteome Research. 2007;6:672–682. doi: 10.1021/pr060432u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bradel-Tretheway B.G., Mattiacio J.L., Krasnoselsky A., Stevenson C., Purdy D., Dewhurst S. Comprehensive proteomic analysis of influenza virus polymerase complex reveals a novel association with mitochondrial proteins and RNA polymerase accessory factors. Journal of Virology. 2011;85:8569–8581. doi: 10.1128/JVI.00496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ritter J.B., Wahl A.S., Freund S., Genzel Y., Reichl U. Metabolic effects of influenza virus infection in cultured animal cells: intra- and extracellular metabolite profiling. BMC Systems Biology. 2010;4:61. doi: 10.1186/1752-0509-4-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lin S., Liu N., Yang Z., Song W., Wang P., Chen H. GC/MS-based metabolomics reveals fatty acid biosynthesis and cholesterol metabolism in cell lines infected with influenza A virus. Talanta. 2010;83:262–268. doi: 10.1016/j.talanta.2010.09.019. [DOI] [PubMed] [Google Scholar]
- 76.Yount J.S., Moltedo B., Yang Y.Y., Charron G., Moran T.M., Lopez C.B. Palmitoylome profiling reveals S-palmitoylation-dependent antiviral activity of IFITM3. Nature Chemical Biology. 2010;6:610–614. doi: 10.1038/nchembio.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hao L., Sakurai A., Watanabe T., Sorensen E., Nidom C.A., Newton M.A. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature. 2008;454:890–893. doi: 10.1038/nature07151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brass A.L., Huang I.C., Benita Y., John S.P., Krishnan M.N., Feeley E.M. The IFITM proteins mediate cellular resistance to influenza A H1N1 virus, West Nile virus, and dengue virus. Cell. 2009;139:1243–1254. doi: 10.1016/j.cell.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Konig R., Stertz S., Zhou Y., Inoue A., Hoffmann H.H., Bhattacharyya S. Human host factors required for influenza virus replication. Nature. 2010;463:813–817. doi: 10.1038/nature08699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Karlas A., Machuy N., Shin Y., Pleissner K.P., Artarini A., Heuer D. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature. 2010;463:818–822. doi: 10.1038/nature08760. [DOI] [PubMed] [Google Scholar]
- 81.Shapira S.D., Gat-Viks I., Shum B.O., Dricot A., de Grace M.M., Wu L. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell. 2009;139:1255–1267. doi: 10.1016/j.cell.2009.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sui B., Bamba D., Weng K., Ung H., Chang S., Van Dyke J. The use of random homozygous gene perturbation to identify novel host-oriented targets for influenza. Virology. 2009;387:473–481. doi: 10.1016/j.virol.2009.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Watanabe T., Watanabe S., Kawaoka Y. Cellular networks involved in the influenza virus life cycle. Cell Host and Microbes. 2010;7:427–439. doi: 10.1016/j.chom.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Meliopoulos V.A., Andersen L.E., Birrer K.F., Simpson K.J., Lowenthal J.W., Bean A.G. Host gene targets for novel influenza therapies elucidated by high-throughput RNA interference screens. FASEB Journal. 2012;26:1372–1386. doi: 10.1096/fj.11-193466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Chan E.Y., Korth M.J., Katze M.G. Decoding the multifaceted HIV-1 virus–host interactome. Journal of Biology. 2009;8:84. doi: 10.1186/jbiol183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ein-Dor L., Kela I., Getz G., Givol D., Domany E. Outcome signature genes in breast cancer: is there a unique set. Bioinformatics. 2005;21:171–178. doi: 10.1093/bioinformatics/bth469. [DOI] [PubMed] [Google Scholar]
- 87.Fiore A.E., Uyeki T.M., Broder K., Finelli L., Euler G.L., Singleton J.A. Prevention and control of influenza with vaccines: recommendations of the Advisory Committee on Immunization Practices (ACIP), 2010. MMWR Recommendations and Reports. 2010;59:1–62. [PubMed] [Google Scholar]
- 88.Pulendran B., Li S., Nakaya H.I. Systems vaccinology. Immunity. 2010;33:516–529. doi: 10.1016/j.immuni.2010.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Poland G.A., Ovsyannikova I.G., Kennedy R.B., Haralambieva I.H., Jacobson R.M. Vaccinomics and a new paradigm for the development of preventive vaccines against viral infections. OMICS. 2011;15:625–636. doi: 10.1089/omi.2011.0032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Baskin C.R., Bielefeldt-Ohmann H., Garcia-Sastre A., Tumpey T.M., Van Hoeven N., Carter V.S. Functional genomic and serological analysis of the protective immune response resulting from vaccination of macaques with an NS1-truncated influenza virus. Journal of Virology. 2007;81:11817–11827. doi: 10.1128/JVI.00590-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bucasas K.L., Franco L.M., Shaw C.A., Bray M.S., Wells J.M., Nino D. Early patterns of gene expression correlate with the humoral immune response to influenza vaccination in humans. Journal of Infectious Diseases. 2011;203:921–929. doi: 10.1093/infdis/jiq156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Nakaya H.I., Wrammert J., Lee E.K., Racioppi L., Marie-Kunze S., Haining W.N. Systems biology of vaccination for seasonal influenza in humans. Nature Immunology. 2011;12:786–795. doi: 10.1038/ni.2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Edgar R., Domrachev M., Lash A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Research. 2002;30:207–210. doi: 10.1093/nar/30.1.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Squires R.B., Noronha J., Hunt V., Garcia-Sastre A., Macken C., Baumgarth N. Influenza Research Database: an integrated bioinformatics resource for influenza research and surveillance. Influenza and Other Respiratory Viruses. 2012 doi: 10.1111/j.1750-2659.2011.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Pickett BE, Sadat EL, Zhang Y, Noronha JM, Squires RB, Hunt V, et al. ViPR: an open bioinformatics database and analysis resource for virology research. Nucleic Acids Research, 2012;D593–8. [DOI] [PMC free article] [PubMed]
- 96.Chang S.T., Tchitchek N., Ghosh D., Benecke A., Katze M.G. A chemokine gene expression signature derived from meta-analysis predicts the pathogenicity of viral respiratory infections. BMC Systems Biology. 2011;5:202. doi: 10.1186/1752-0509-5-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McDermott J.E., Shankaran H., Eisfeld A.J., Belisle S.E., Neuman G., Li C. Conserved host response to highly pathogenic avian influenza virus infection in human cell culture, mouse and macaque model systems. BMC Systems Biology. 2011;5:190. doi: 10.1186/1752-0509-5-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Becavin C., Tchitchek N., Mintsa-Eya C., Lesne A., Benecke A. Improving the efficiency of multidimensional scaling in the analysis of high-dimensional data using singular value decomposition. Bioinformatics. 2011;27:1413–1421. doi: 10.1093/bioinformatics/btr143. [DOI] [PubMed] [Google Scholar]
- 99.Tchitchek N., Golib-Dzib J.F., Targat B., Noth S., Benecke A., Lesne A. CDS: a fold-change based statistical test for concomitant identifcation of distinctness and similarity in gene expression analysis. Genomics, Proteomics, Bioinformatics. 2012;10:127–135. doi: 10.1016/j.gpb.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sonnenschein N., Golib Dzib J.F., Lesne A., Eilebrecht S., Boulkroun S., Zennaro M.C. A network perspective on metabolic inconsistency. BMC Systems Biology. 2012;6:41. doi: 10.1186/1752-0509-6-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Berg J. Out-of-equilibrium dynamics of gene expression and the Jarzynski equality. Physical Review Letters. 2008;100:188101. doi: 10.1103/PhysRevLett.100.188101. [DOI] [PubMed] [Google Scholar]
- 102.Benecke A. Gene regulatory network inference using out of equilibrium statistical mechanics. HFSP Journal. 2008;2:183–188. doi: 10.2976/1.2957743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Wooldridge M. An introduction to multiagent systems. Computer. 2002;86:348. [Google Scholar]
- 104.Weiss G. Multiagent systems: a moder approach to distributed artificial intelligence. AI Magazine. 2000;1:619. [Google Scholar]
- 105.Bordini R.H., Hubner J.F., Wooldridge M.J. 2007. Programming multi-agent systems in AgenSpeak using Jason. [Google Scholar]
- 106.Luke S., Cioffi-Revilla C., Panait L., Sullivan K., Balan G. MASON: a multiagent simulation environment. Simulation. 2005;81:517–527. [Google Scholar]
- 107.Bellifemine F.L., Caire G., Greenwood D. John Wiley Sons Ltd.; 2007. Developing multi-agent systems with JADE. [Google Scholar]
- 108.Baldazzi V., Castiglione F., Bernaschi M. An enhanced agent based model of the immune system response. Cellular Immunology. 2006;244:77–79. doi: 10.1016/j.cellimm.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 109.Sun T., McMinn P., Holcombe M., Smallwood R., MacNeil S. Agent based modelling helps in understanding the rules by which fibroblasts support keratinocyte colony formation. PLoS ONE. 2008;3:e2129. doi: 10.1371/journal.pone.0002129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Richmond P., Walker D., Coakley S., Romano D. High performance cellular level agent-based simulation with FLAME for the GPU. Brief Bioinformatics. 2010;11:334–347. doi: 10.1093/bib/bbp073. [DOI] [PubMed] [Google Scholar]
- 111.Folcik V.A., Broderick G., Mohan S., Block B., Ekbote C., Doolittle J. Using an agent-based model to analyze the dynamic communication network of the immune response. Theoretical Biology and Medical Modelling. 2011;8:1. doi: 10.1186/1742-4682-8-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.McDermott J.E., Taylor R.C., Yoon H., Heffron F. Bottlenecks and hubs in inferred networks are important for virulence in Salmonella typhimurium. Journal of Computational Biology. 2009;16:169–180. doi: 10.1089/cmb.2008.04TT. [DOI] [PubMed] [Google Scholar]
- 113.Diamond D.L., Syder A.J., Jacobs J.M., Sorensen C.M., Walters K.A., Proll S.C. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathogens. 2010;6:e1000719. doi: 10.1371/journal.ppat.1000719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rasmussen A.L., Diamond D.L., McDermott J.E., Gao X., Metz T.O., Matzke M.M. Systems virology identifies a mitochondrial fatty acid oxidation enzyme, dodecenoyl coenzyme A delta isomerase, required for hepatitis C virus replication and likely pathogenesis. Journal of Virology. 2011;85:11646–11654. doi: 10.1128/JVI.05605-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Peng X., Gralinski L., Armour C.D., Ferris M.T., Thomas M.J., Proll S. Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. mBio. 2010;1:e00206–e00210. doi: 10.1128/mBio.00206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Skalsky R.L., Cullen B.R. Viruses, microRNAs, and host interactions. Annual Review of Microbiology. 2010;64:123–141. doi: 10.1146/annurev.micro.112408.134243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Li Y., Chan E.Y., Li J., Ni C., Peng X., Rosenzweig E. MicroRNA expression and virulence in pandemic influenza virus-infected mice. Journal of Virology. 2010;84:3023–3032. doi: 10.1128/JVI.02203-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Peng X., Gralinski L., Ferris M.T., Frieman M.B., Thomas M.J., Proll S. Integrative deep sequencing of the mouse lung transcriptome reveals differential expression of diverse classes of small RNAs in response to respiratory virus infection. mBio. 2011;2 doi: 10.1128/mBio.00198-11. e00198–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Eilebrecht S., Brysbaert G., Wegert T., Urlaub H., Benecke B.J., Benecke A. 7SK small nuclear RNA directly affects HMGA1 function in transcription regulation. Nucleic Acids Research. 2011;39:2057–2072. doi: 10.1093/nar/gkq1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Aderem A., Adkins J.N., Ansong C., Galagan J., Kaiser S., Korth M.J. A systems biology approach to infectious disease research: innovating the pathogen–host research paradigm. mBio. 2011;2 doi: 10.1128/mBio.00325-10. e00325–10. [DOI] [PMC free article] [PubMed] [Google Scholar]