

Abstract
Background and Purpose
The present study assessed the effects of cilostazol on LPS-stimulated TLR4 signal pathways in synovial macrophages from patients with rheumatoid arthritis (RA). These effects were confirmed in collagen-induced arthritis (CIA) in mice.
Experimental Approach
Expression of TLR4, PU.1, NF-κB p65 and IκBα on synovial fluid macrophages from RA patients was determined by Western blotting, and cytokines were measured by elisa. Anti-arthritic effects were evaluated in CIA mice.
Key Results
Intracellular cAMP was concentration-dependently raised by cilostazol (1–100 μM). Cilostazol significantly suppressed LPS-stimulated increase of TLR4 expression by blocking PU.1 transcriptional activity in RA macrophages. In addition, cilostazol decreased LPS-induced myeloid differentiation factor 88 (MyD88) expression, but not that of TNF receptor-associated factor 6 (TRAF6). Cilostazol also suppressed IkBα degradation and NF-κB p65 nuclear translocation. Moreover, LPS-induced increase of cytokine production (TNF-α, IL-1β) was inhibited by cilostazol, an effect which was accompanied by suppression of IκBα degradation, and NF-κB p65 nuclear translocation. However, expression of anti-inflammatory IL-10 was elevated by cilostazol and forskolin/IBMX. In mice with CIA, post-treatment with cilostazol (30 mg kg−1 day−1) decreased expression of TLR4 in knee joints in association with decreased recruitment of macrophages. Consequently, synovial inflammation, proteoglycan depletion and bone erosion were significantly inhibited by cilostazol treatment.
Conclusions and Implications
Cilostazol down-regulated LPS-stimulated PU.1-linked TLR4 expression and TLR4/MyD88/NF-κB signal pathways, and then suppressed inflammatory cytokine production in synovial macrophages from RA patients. Also cilostazol markedly inhibited the severity of CIA in mice.
Keywords: TLR4, PU.1, MyD88, Cilostazol, Cytokine, RA Macrophages, CIA
Introduction
Rheumatoid arthritis (RA) is a chronic inflammatory and destructive joint disease, characterized by synovial hyperplasia and subintimal infiltration of inflammatory cells into the joints. Activated monocytes/macrophages are found in abundance in synovial fluid of patients with RA (Firestein and Svaifler, 1987), contributing to chronic inflammation and joint destruction through release of pro-inflammatory cytokines and chemokines (Feldmann et al., 1996).
Toll-like receptors (TLRs), a family of conserved pattern-recognition receptors, have an important role in innate and adaptive immunity, and are mainly expressed on cells, such as macrophages and dendritic cells, where these cells act as primary sensors recognizing endogenous and exogenous stimuli (Kawai and Akira, 2007). Increased expression of TLR4 has been demonstrated in peripheral blood monocytes and synovial tissue of RA patients (Kyburz et al., 2003; receptor nomenclature follows Alexander et al., 2011). TLR4 recognizes LPS, an integral component of the outer membranes of Gram-negative bacteria (Akira and Takeda, 2004) and TLR4-induced intracellular signalling recruits the adaptor proteins MyD88 and TRAF6 which, in turn,leads to the activation of the transcription factor, NF-κB, resulting in the production of inflammatory cytokines, chemokines and tissue destructive enzymes (Janeway and Medzhitov, 2002; Gohda et al., 2004). On the other hand, macrophages, as key effector cells in innate immunity, abundantly express TLR4. In macrophages, another transcription factor, PU.1, has been shown to increase transcriptional expression of various inflammatory genes, including that for TLR4 (Rehli et al., 2000; Lichtinger et al., 2007).
Cilostazol, an inhibitor of PDE type III, exhibits potent anti-inflammatory effects, such as suppression of NAD(P)H oxidase–dependent superoxide formation and cytokine production in human umbilical vein endothelial cells (Shin et al., 2004). Cilostazol was previously demonstrated to suppress expression of adhesion molecules and chemokine release in association with reduced monocyte adhesion to HUVECs (Park et al., 2005). Furthermore, the anti-inflammatory effects of cilostazol were ascribed to cAMP-dependent protein kinase activation–coupled suppression of NF-κB gene transcription (Park et al., 2006).
TLR4 is activated in macrophages in RA synovium and PU.1 regulates the expression of inflammatory genes through expression of mRNA for TLR4 and the anti-inflammatory effect of cilostazol has been ascribed to the suppression of NF-κB gene transcription. Taken together these findings led us to hypothesize that inhibition by cilostazol of PU.1-linked TLR4 signal pathways may provide an effective therapeutic strategy for the suppression of inflammation. Therefore, we examined the effects of cilostazol on LPS-stimulated TLR4 signal pathways, focussing on its effects on the PU.1-linked TLR4 and MyD88 expression. In addition we examined the effects of cilostazol on NF-κB activity and inflammatory cytokine production in macrophages obtained from synovial fluid of RA patients.
Methods
Patients and cell preparation
Peripheral blood was obtained from five normal volunteer donors, and synovial fluid samples were obtained from the knee joints of 23 patients with RA who fulfilled the 1987 revised criteria of the American College of Rheumatology, at the time of therapeutic arthrocentesis. All patients who attended the Rheumatology Clinic at Dong-A University Hospital and Pusan University Hospital (Busan, Korea) provided informed consent, and approval was obtained from the ethics committee of University Hospitals. Monocytes from peripheral blood and synovial fluid were isolated by Ficoll–Paque (GE Healthcare, Uppsala, Sweden) density-gradient centrifugation and were cultured in RPMI 1640 containing 10% FBS. Adherent cells from peripheral blood were allowed to differentiate into normal control macrophages, as described by Liu et al. (2003). Briefly, blood was diluted in PBS (pH 7.4) and 30 mL of diluted blood was layered over 10 mL of Ficoll–Paque (GE Healthcare), and centrifuged at 800× g for 30 min. Peripheral blood monocytes were collected and then monocytes were obtained by adhesion for 24 h in RPMI-1640 containing 10% FBS. After 24 h, non-adherent cells were removed, and the adherent cells were cultured in the culture media with 25 ng ml−1 M-CSF (Peprotech Inc., Rocky Hill, NJ). After 5 days, non-adherent or dead cells were removed, and the remaining cells were used for experiments. Synovial fluid was diluted 1:1 with PBS and 30 mL of diluted synovial fluid was layered over 10 mL of Ficoll–Paque, and centrifuged at 800× g for 30 min. Synovial fluid mononuclear cells were collected, and then monocytes were obtained by adhesion for 24 h in RPMI-1640 containing 10% FBS. After 5 days, non-adherent or dead cells were removed and the remaining cells were used for experiments. Flow cytometric analyses showed that adherent peripheral blood macrophages were 81 ± 7% CD14+ cells (n = 5), and adherent RA synovial fluid macrophages were 89 ± 3% CD14+ cells (n = 5).
Enyme linked immunosorbent assay
Levels of TNF-α, IL-1β and IL-10 in culture supernatants (Assay Design Inc., Ann Arbor, MI) and intracellular cAMP levels (R&D systems, Minneapolis, MN) were measured using an elisa kits according to the manufacturer's protocols.
Western blot analysis
Each sample (30 μg protein) was loaded into a 10% SDS-PAGE and transferred to a nitrocellulose membrane (Amersham Biosciences, Inc., Piscataway, NJ). The blocked membrane was then incubated with antibodies for NF-κB p65, IκB-α (Santa Cruz Biotechnology, Inc, CA), PU.1 (Cell signaling Technology, Danvers, MA) and TLR4 (Abcam, Cambridge, MA). Immunoreactive bands were visualized with a chemiluminescent reagent of the Supersignal West Dura Extended Duration Substrate Kit (Pierce Chemical, Rockford, IL). Signals of the bands were quantified using the GS-710 calibrated imaging densitometer (Bio-Rad, Hercules, CA). Results are expressed as a relative density.
Quantitative real-time reverse transcription
Total RNA was extracted from cells using TRIzol reagent (Invitrogen, San Diego, CA). TLR4 gene expression was measured by real-time PCR using a LightCycler (Roche Molecular Biochemicals, Mannheim, Germany) instrument with the LightCycler DNA Master SYBR Green I (Roche Molecular Biochemicals) starting with 1 ng of reverse-transcribed total RNA. PCR was performed under the following conditions: 95°C × 10 min, 50 × (95°C × 10 s, 62°C × 30 s, 72°C × 1 s). The following oligonucleotide primers specific for human TLR4 and GAPDH were used: TLR4, 5′-GAC CAG AAC TGC TAC AAC AGAT-3′ (sense) and 5′-GCA ACA CCT TCA GAT AAG GAG-3′ (antisense) and GAPDH, 5′-TGT AGT TGA GGT CAA TGA AGGG′ (sense) and 5′-ACA TCG CTC AGA CAC CATG-3′ (antisense). The quantification data were analysed using Rotor-Gene Real-Time Analysis Software 6.0 (Corbett Research, Mortlake, Australia).
Immunocytochemistry
Cells were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and incubated with antibodies to PU.1 (Cell signalling Technology), MyD88 and TRAF6 (Santa Cruz Biotechnology, Inc), for 1 h and then incubated with FITC-, cy3-conjugated secondary antibody for 30 min. Fluorescent images were obtained using a confocal microscope (Nikon).
Small interfering RNA (siRNA) preparation and transfection
PU.1 siRNA oligonucleotide (GenBank accession no.NM_003120.1) was synthesized by Bioneer (Daejeon, Korea). The siRNA negative control duplex was used as a control oligonucleotide. siRNA molecules were transfected into cells using Lipofectamine 2000 according to Invitrogen's protocols. The final concentration of 100 nM PU.1 siRNA was determined as a concentration to maximally suppress PU.1 expression. siRNA was dissolved to the medium 48 h before the treatment of compounds.
NF-κB transcription factor assay
For measuring NF-κB transcription factor in nuclear extracts, cell lysates were extracted using a nuclear extraction kit (Chemicon International, Temecula, CA) according to the manufacturer's protocol. DNA binding activity of NF-κB p65 was measured using a colorimetric NF-κB p65 transcription factor assay kit (Rockland Immunochemicals, Gilbertsville, PA) according to the manufacturer's protocol.
Collagen-induced arthritis (CIA)
All experimental procedures were conducted in accordance with the Animal Care Guidelines of the Animal Experimental Committee of the College of Medicine, Pusan National University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). A total of 30 animals were used in the experiments described here. Male DBA/1J mice (ages 8–12 weeks) were purchased from Japan SLC, Inc. (Shizuoka, Japan). All animals were housed in the facility with a 12 h light–dark cycle. For a CIA model, 100 μg of bovine type II collagen (Chondrex, Redmond, WA) dissolved in 0.1 M acetic acid was emulsified with an equal volume of Freund's complete adjuvant (Sigma, St. Louis, MO) and administered i.d. at the base of the tail into DBA/1J mice. On day 18, a booster emulsion prepared with type II collagen and Freund's incomplete adjuvant was injected i.d. near the primary injection. The mice were divided into 3 experimental groups: (1) Sham animals were maintained up to day 38. (2) vehicle-treated CIA mice received 25% DMSO solution 0.3 ml i.p.. (3) Cilostazol-treated CIA mice received 30 mg kg−1 day−1 cilostazol i.p. for 20 days after the booster injection. Mice were killed at day 38 after primary injection and knee joints were isolated.
Tissue preparation and histological analysis
Whole knee joints from mice were dissected and fixed for one day in 4% paraformaldehyde, decalcified in 10% EDTA, dehydrated and embedded in paraffin blocks. Blocks of 6 μm thickness were cut and stained with haematoxylin and eosin (H&E) for assessment of synovial inflammation and bone erosion; stained with safranin-O for proteoglycan in articular cartilage; and with tartrate-resistant acid phosphatase (TRAP) for detection of osteoclasts. TRAP staining was performed using a leukocyte acid phosphatase staining kit (Sigma). The histological severity of arthritis was scored on a scale of 0–3, where 0 = normal; 1 = minimal synovial inflammation, with cartilage and bone erosion limited to discrete foci; 2 = synovial inflammation and moderate erosion, with normal joint architecture intact; and 3 = severe inflammation and severe erosion, with joint architecture disrupted, as described previously (Williams et al., 1992). Progressive loss of articular cartilage was scored on a scale of 0–3, ranging from fully stained cartilage (no cartilage loss) to de-stained cartilage or complete loss of articular cartilage (Lubberts et al., 2004). Bone erosion was graded on a scale of 0–5, ranging from no damage to complete loss of bone structure. Scoring was performed by two observers without knowledge of the experimental group.
Immunohistochemistry and immunofluorescence
Serial sections (6 μm thickness) were cut on a paraffin block and endogenous peroxidase activity was quenched by incubation with 0.3% H2O2 and non-specific antibody binding was blocked with CAS Block (Zymed Laboratories, Inc., San Francisco, CA) for 10 min. The preparations were incubated with a rabbit anti-TLR4 antibody (Santa Cruz Biotechnology, Inc.) at 4°C overnight. These were subsequently incubated with a biotinylated anti-rabbit antibody (Vector Laboratories, Inc., Burlingame, CA) for 1 h and then with an avidin–biotin peroxidase complex solution (Vectastain Elite ABC kit, Vector Laboratories, Inc.) for 1 h. The sections were rinsed after each reaction. Finally, the immunoreaction products were visualized with a solution of 0.02% 3,3′-diaminobenzidine tetrahydrochloride (diaminobenzidine substrate kit, Vector laboratories, Inc.). In the double immunofluorescence experiments, the preparations were incubated for 1 h at room temperature with a goat anti-CD68 and a rabbit anti-TLR4 antibody. After washing, the sections were incubated with a secondary antibody solution for 30 min at room temperature, and mounted using Vectashield mounting medium (Vector Laboratories, Inc.). Fluorescence was detected using a fluorescence microscope (Axiovert 200, Carl Zeiss, Oberkochem, Germany).
Data analysis
Results are expressed as the mean ± SEM. The concentration-dependent responses were analysed using one-way analysis of variance followed by Tukey's multiple comparison tests. Student's t-test was used to determine the significance of the difference between treated and untreated groups. P-values less than 0.05 were considered significant.
Materials
Cilostazol (OPC-13013), [6-[4-(1-cyclohexyl-1H-tetrazol-5-yl)butoxy]-3,4-dihydro-2-(1H)-quinolinone] was donated by Otsuka Pharmaceutical Co. Ltd. (Tokushima, Japan) and was dissolved in dimethyl sulfoxide as a 10 mM stock solution. Forskolin, IBMX and LPS were obtained from Sigma-Aldrich. Rp-cAMPS was obtained from Alexis (San Diego, CA). Anti-TLR4 monoclonal antibody was obtained from eBioscience (San Diego, CA).
Results
Inhibition of TLR4 expression by cilostazol
In order to assess a possible involvement of cAMP in the intracellular signalling pathway related to the inhibitory effect of cilostazol, we measured intracellular cAMP levels in macrophages obtained from RA patients. Application of cilostazol (1–100 μM) to the culture media for 1 h caused a significant increase in intracellular cAMP levels in a concentration-dependent manner, as did application of forskolin (10 μM) + IBMX (100 μM) (Figure 1A). We compared the expression of TLR4 in peripheral blood macrophages from normal volunteers and synovial fluid macrophages from patients with RA. With or without LPS (1 μg mL−1), TLR4 protein was more prominently expressed in the RA synovial fluid macrophages than in the peripheral blood macrophages (Figure 1B).
Figure 1.

Inhibitory effects of cilostazol and forskolin/IBMX on LPS-induced TLR4 mRNA and protein expression in RA macrophages. (A) Intracellular cAMP level in RA macrophages after treatment with cilostazol (1–100 μM) or forskolin (10 μM) + IBMX (100 μM) for 1 h was measured by elisa. Results are expressed as the mean ± SEM (n = 4). *P < 0.05, **P < 0.01, ***P < 0.001 versus no treatment. (B) Comparison of TLR4 protein expression in the absence and the presence of LPS for 24 h between synovial fluid (SF) macrophages and peripheral blood (PB) macrophages from each five donors. Results are expressed as the mean ± SEM (n = 5). $$P < 0.01, $$$P < 0.001 versus no treatment. (C and D) Cilostazol and forskolin/IBMX induced decrease in TLR4 mRNA and protein, as revealed by real-time PCR and Western blot. RA macrophages were treated with cilostazol (1, 10, 30 μM) and forskolin (10 μM) + IBMX (100 μM) for 4 h, followed by exposure to LPS for 12 h (real-time PCR) and 24 h (Western blot). (E) Reversal by Rp-cAMPS of decreased TLR4 protein expression by cilostazol and forskolin + IBMX. Rp-cAMPS (1 μM) was applied at 30 min prior to treatment with cilostazol. Results are expressed as the mean ± SEM (n = 4–5). ##P < 0.01, ###P < 0.001 versus no treatment; *P < 0.05, **P < 0.01, ***P < 0.001 versus LPS alone; †P < 0.05, ††P < 0.01 significant effect of Rp-cAMPS.
To determine whether cilostazol modulated LPS-induced TLR4 transcriptional and translational expression in RA macrophages, we performed real-time PCR and Western blot analysis. The LPS-stimulated expression of mRNA for TLR4 in RA macrophages, measured by real-time PCR, was suppressed by cilostazol (1–30 μM) and by forskolin + IBMX (Figure 1C). TLR4 protein levels measured by Western blotting showed that LPS increased TLR4 protein expression, which was concentration-dependently inhibited by cilostazol (1–30 μM) or by forskolin + IBMX (Figure 1D). The suppression of TLR4 expression by cilostazol (10 μM) or forskolin + IBMX was reversed by pretreatment with Rp-cAMPS (1 μM), an inhibitor of cAMP-dependent protein kinase (Figure 1E).
Inhibition by cilostazol of LPS-induced nuclear PU.1 expression
PU.1, an Ets family transcription factor, regulates TLR4 expression (Rehli et al., 2000; Lichtinger et al., 2007). Therefore, we determined the suppression of PU.1 activity by cilostazol in RA macrophages. Accumulation of PU.1 protein in the nucleus was enhanced after exposure to LPS and cilostazol (1–30 μM) concentration-dependently inhibited nuclear PU.1 protein expression in LPS-stimulated cells (Figure 2A). In the confocal study, translocation of PU.1 protein into the nucleus was markedly stimulated by LPS, an effect was largely inhibited by cilostazol (Figure 2B). The involvement of PU.1 in TLR4 expression was confirmed in RA macrophages transfected with PU.1 siRNA, which showed significantly reduced PU.1 mRNA and protein expression up to 10–20% of the negative control. Neither LPS nor cilostazol showed any effects on TLR4 expression in these PU.1-knockdown cells, in contrast to the findings in negative control cells. These observations suggested that nuclear PU.1 was required to induce expression of TLR4 in response to LPS, and that cilostazol suppressed TLR4 expression via inhibition of PU.1 activation (Figure 2C).
Figure 2.

Effects of LPS and cilostazol on expression of PU.1 transcription factor in the RA macrophages. (A) Time-dependent increase in PU.1 expression by LPS (left) and concentration-dependent decrease in PU.1 expression by cilostazol (right) in the nuclear fraction of RA macrophages. (B) Confocal microscopic images detecting nuclear localization of PU.1 (red) and DAPI (blue) in the presence of LPS for 1 h under treatment with and without cilostazol (10 μM, for 4 h) in RA macrophages. (C) Analysis of PU.1-knockdown cells: PU.1 mRNA and protein expression were reduced, in contrast to negative controls (Ca). In PU.1-knockdown cells, LPS and cilostazol showed little effect on TLR4 expression, in contrast to the findings in negative control cells (Cb). Results are expressed as the mean ± SEM of four experiments. ##P < 0.01; ###P < 0.001 versus no treatment; *P < 0.05 versus LPS alone.
Inhibition by cilostazol of MyD88 expression
We next investigated the effect of cilostazol on the intracellular adaptor proteins involved in signal transduction by TLR4. RA macrophages were incubated with cilostazol (10 μM) for 4 h, followed by exposure to LPS for 24 h. Rp-cAMPS (1 μM) was applied 30 min prior to treatment with cilostazol. LPS increased MyD88, but not TRAP6, expression in RA macrophages and cilostazol inhibited the increased expression of MyD88. Rp-cAMPS (1 μM) reversed the inhibitory effect of cilostazol on LPS-induced MyD88 expression (Figure 3A). However, TRAF6 showed constitutively high expression and cilostazol did not affect expression of TRAF6, when assessed by Western blotting (Figure 3B). These results were confirmed by immunocytochemistry (Figure 3C), suggesting that cilostazol inhibits MyD88 expression, but not that of TRAF6.
Figure 3.
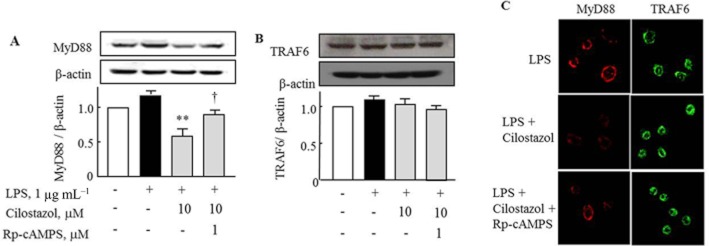
Effect of cilostazol on the expression of the adaptor proteins, MyD88 and TRAF6 in RA macrophages. (A) Inhibitory effect of cilostazol on LPS-induced increased expression of MyD88, and its reversal by Rp-cAMPS (1 μM). (B) Expression of TRAF6 after treatment with cilostazol in the absence and presence of Rp-cAMPS. (C) Confocal microscopic images indicating expression of MyD88 and TRAF6 under treatment with cilostazol in the absence and presence of Rp-cAMPS. RA macrophages were incubated with cilostazol (10 μM) for 4 h, followed by exposure to LPS for 24 h. Rp-cAMPS (1 μM) was applied at 30 min before treatment with cilostazol. Results are expressed as the mean and SEM of four experiments. ** = P < 0.01 versus LPS alone; † = P < 0.05 significant effect of Rp-cAMPS.
Inhibition of NF-κB p65 activation by cilostazol
We assessed suppression by cilostazol of NF-κB p65 activation in LPS-stimulated RA macrophages. Stimulation of macrophages with LPS (1 μg mL−1) for 30 min decreased IκBα expression in the cytosol, whereas NF-κB p65 expression was significantly increased in the nucleus. Pretreatment with cilostazol (1–30 μM) for 4 h resulted in increased IκBα expression in the cytosol and decreased NF-κB p65 expression in the nucleus in a concentration-dependent manner. Similar findings were obtained with forskolin + IBMX (Figure 4A). The effects of both cilostazol and forskolin + IBMX were reversed by Rp-cAMPS (Figure 4B). DNA binding activity of NF-κB p65 was determined using a NF-κB p65 transcription factor assay kit and showed that cilostazol or forskolin + IBMX inhibited the LPS-induced increase of nuclear NF-κB-dependent DNA binding activity (Figure 4C). These results suggest that cilostazol down-regulated TLR4-mediated inflammatory cytokine responses through inhibition of NF-κB activity.
Figure 4.
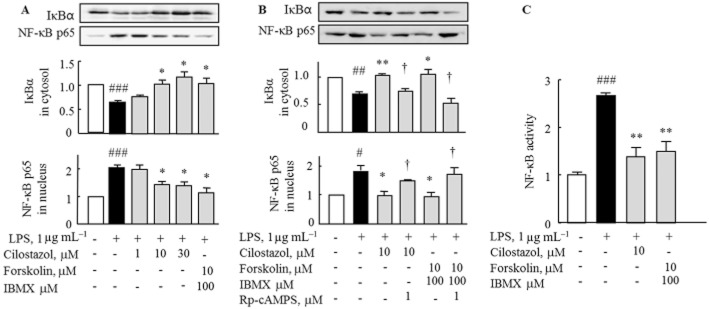
Effect of cilostazol and forskolin + IBMX on LPS-stimulated IκBα degradation in the cytoplasm and NF-κB p65 expression in the nucleus following LPS stimulation. (A) Treatment with cilostazol and forskolin + IBMX for 4 h increased IκBα expression in the cytoplasm and decreased nuclear NF-κB p65 expression in the presence of LPS for 30 min. (B) Reversal by pretreatment with Rp-cAMPS (1 μM, for 30 min) of increased IκBα in the cytoplasm and decreased NF-κB p65 expression in the nucleus, which were induced by cilostazol and forskolin + IBMX. (C) Treatment with cilostazol and forskolin + IBMX for 4 h prevented LPS-induced increase in NF-κB activity. Results are expressed as the mean ± SEM of three to four experiments. #P < 0.05, ##P < 0.01, ###P < 0.001 versus no treatment; *P < 0.05, **P < 0.01 versus LPS alone; †P < 0.05 significant effect of Rp-cAMPS.
Inhibition of TLR4-induced cytokine production in RA macrophages by cilostazol
In order to determine the functional correlation of TLR4 expression with pro-inflammatory cytokine levels and inhibition by cilostazol, we examined changes in pro-inflammatory cytokine levels in response to LPS stimulation, using elisa. As shown in Figure 5A and B, stimulation of RA macrophages by LPS (1 μg mL−1) for 48 h increased production of TNF-α and IL-1β in the culture medium. Levels of both cytokines were significantly suppressed by 4h pretreatment with cilostazol (10 μM) or forskolin + IBMX. In contrast, IL-10, an anti-inflammatory cytokine was elevated by cilostazol (Figure 5C). We next assessed whether TLR4 was involved in these inhibitory effects of cilostazol on cytokine production. When RA macrophages were treated with anti-TLR4 antibody (10 μg ml−1) for 4 h before LPS stimulation, LPS-induced TNF-α production was specifically suppressed, comparable to the effects of cilostazol (Figure 5D).
Figure 5.
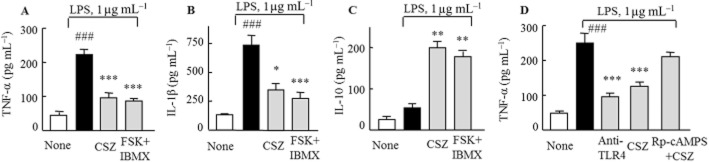
Effects of cilostazol (CSZ) (10 μM) and forskolin (10 μM) +IBMX (100 μM) on TNF-α (A), IL-1β (B) and IL-10 (C) levels that were released into the incubation media of RA macrophages following LPS treatment. (D) Inhibition by anti-TLR4 antibody (10 μg mL−1) and cilostazol (10 μM) of the LPS-induced TNF-α production. Reversal by Rp-cAMPS (1 μM) of the decreased TNF-α production induced by cilostazol. Results are expressed as the mean ± SEM of four experiments. ###P < 0.001 versus no treatment *P < 0.05; **P < 0.01, ***P < 0.001 versus LPS alone.
Decreased expression of TLR4 and CD68 by cilostazol in a CIA mouse model
The concurrent expression of TLR4 and CD68, as a marker of the recruitment of macrophages, was measured in the knee joints of mice. Numbers of cells with immunoreactive TLR4 were markedly increased in joints from mice with CIA, compared with those from untreated mice. After treatment with cilostazol (30 mg kg−1 day−1) in CIA mice, the TLR4-positive cells were decreased (Figure 6A and B). Immunofluorescent double staining for CD68 (a marker for macrophages) and TLR4 showed increased expression of both CD68 and TLR4 in sections of knee joints from vehicle-treated CIA mice, and the majority of cells expressing TLR4 were co-localized with synovial macrophages. Increased expression of both CD68 and TLR4 was markedly reduced in cilostazol-treated CIA mice (Figure 6C), indicating a close association of increased expression of TLR4 with increased numbers of macrophages in the joint, both of which were suppressed by cilostazol.
Figure 6.
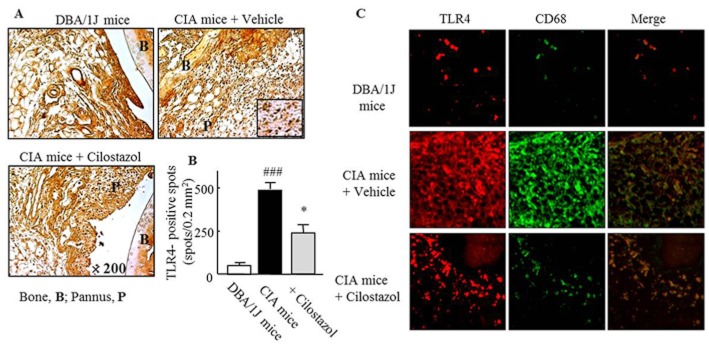
Inhibition of TLR4 expression by cilostazol in a mouse model of CIA. (A) Representative photomicrographs of TLR4-positive cells in knee joint sections on day 38 from mice treated with vehicle or cilostazol (30 mg kg−1 day−1). (B) Quantitative analysis: TLR4-positive cells are expressed as the mean ± SEM of six experiments. ###P < 0.001 versus control mice; *P < 0.05 versus mice with CIA treated with vehicle. (C) Double-immunofluorescence images showing TLR4 and CD68 (a marker for macrophages). Co-localization of CD68/TLR4 was markedly reduced by cilostazol treatment.
Suppression of cartilage depletion and bone erosion with reduced arthritis scores by cilostazol
We next evaluated the effects of cilostazol on cartilage depletion and bone erosion in knee joints from CIA mice. On day 38, knee joints obtained from mice subjected to CIA showed structural alterations, including thickened synovium with depleted proteoglycan and severe bone erosion, when compared with normal DBA/1J mice. Arthritis scores (histological severity of arthritis) of vehicle-treated CIA mice were reduced in cilostazol-treated CIA mice (Figure 7A and D). In addition, CIA mice showed markedly reduced proteoglycan content when assessed by safranin-O staining. Treatment with cilostazol preserved the proteoglycan content of knee joint cartilage, as shown in Figure 7B and E. In addition, treatment with cilostazol effectively reduced the occurrence of bone erosion when evaluated by TRAP staining, and erosion scores were reduced by cilostazol (Figure 7C and F). These results indicate that cilostazol effectively protected joints from synovial inflammation, cartilage depletion and bone erosion during the arthritic reaction.
Figure 7.

Inhibition of disease progression and bone erosion by cilostazol in a mouse model of CIA. Histological sections of knee joints (on day 38) from DBA/1J and CIA mice treated with vehicle or 30 mg kg−1 day−1 of cilostazol were stained with haematoxylin–eosin (A), safranin-O (B) and TRAP (C). Representative histological sections stained with TRAP (C) show osteoclast features with bone erosion. Quantitative analyses represent scores for arthritis (D), proteoglycan deletion (E) and bone erosion (F) in the knee joints. Results are expressed as the mean ± SEM of six experiments. *P < 0.05; **P < 0.01 versus CIA mice treated with vehicle.
Discussion
Data from the present study demonstrated that, in synovial macrophages from RA patients, cilostazol suppressed LPS-induced increased nuclear translocation of PU.1 and then down-regulated PU.1-linked TLR4 and MyD88 expression via activation of a cAMP-dependent protein kinase. Cilostazol inhibited the LPS-stimulated increase in IκBα degradation and NF-κB p65 nuclear translocation, thereby suppressing the production of inflammatory cytokines. In addition, cilostazol strongly protected knee joints from synovial inflammation, cartilage depletion and bone destruction in CIA mice, an effect which was accompanied by a significantly decreased synovial TLR4 expression and macrophage recruitment.
According to the report of Simmonds and Foxwell (2008), synovium from patients with RA displays a large number of macrophages with T cells and synovial fibroblasts. Macrophages, an important component of the innate immune system, are the principal source of TNFα, IL-1β and other cytokines that are pivotal in promoting inflammation and joint destruction in RA (Firestein, 2005)and there is a high level of endogenous TLR4 ligands in the joints of patients with RA (Radstake et al., 2004). Our results revealed that LPS-induced TLR4 expression was inhibited by cilostazol as well as by forskolin + IBMX, and that these effects were reversed by pretreatment with Rp-cAMPS, implicating cAMP-dependent protein kinase activation in the suppression of TLR4 expression.
The transcription factor, PU.1, is a central regulator of TLR4. In the nucleus of macrophages, PU.1, after binding to the TLR4 promoter, regulates transcriptional expression of TLR4 (Rehli et al., 2000; Akira and Takeda, 2004; Lichtinger et al., 2007). In this study, we have demonstrated that LPS-induced accumulation of PU.1 protein in the nucleus of synovial macrophages from RA patients, was markedly attenuated in a concentration-dependent manner, by cilostazol, suggesting that the suppressive effect of cilostazol on TLR4 expression was dependent on suppression of the nuclear expression of PU.1. This hypothesis was further supported by the observation that when RA macrophages were transfected with PU.1 siRNA, LPS did not elicit TLR4 expression. These results indicated that PU.1 was involved in the expression of TLR4, and that cilostazol reduced TLR4 expression via inhibition of PU.1 expression.
The transcription factor NF-κB is the regulator of TLR signalling, which, in turn, controls the expression of many genes involved in the inflammatory response (Akira et al., 2001; Janeway and Medzhitov, 2002). As reported by Handel et al. (1995), our results showed that LPS increased nuclear NF-κB p65 expression, in association with enhanced IκBα degradation, in synovial macrophages from RA patients. This increase in NF-κB p65 activation was accompanied by a significant increase in the production of TNF-α and IL-1β, which, in turn, was significantly attenuated by treatment with cilostazol or forskolin + IBMX, effects consistent with earlier reports (Park et al., 2005; 2006).
MyD88 and Mal/TIRAP are essential for LPS-induced IκBα phosphorylation, NF-κB p65 activation and IL-6 production in fibroblasts (Andreakos et al., 2004) and a high level of TLR4 is expressed at the cell surface of macrophages and neutralization of TLR4 blocks LPS signalling (Akira and Takeda, 2004; Andreakos et al., 2004). We determined whether cilostazol could suppress adaptor proteins, such as MyD88 and TRAF6, in macrophages. Indeed, expression of MyD88 protein was up-regulated by exposure to LPS in RA macrophages, and cilostazol significantly decreased its expression, and this decrease was reversed by Rp-cAMPS. In addition, it has been suggested that TRAF6 is essential for MyD88-dependent signalling (Gohda et al., 2004). However, although we found constitutively high expression of TRAF6 in RA macrophages, its expression was not altered by cilostazol, as assessed by Western blot and immunocytochemistry. In view of these results, it is likely that cilostazol inhibited pro-inflammatory cytokine production through suppression of TLR4-linked MyD88.
Inhibition of TLR4 suppressed the severity of experimental arthritis and resulted in lower IL-1 expression in arthritic joints (Abdollahi-Roodsaz et al., 2007) and we found that LPS-induced increases in cytokine secretion, including TNF-α and IL-1β, from synovial macrophages were suppressed by cilostazol and forskolin + IBMX, and that LPS-induced TNF-α production was specifically suppressed by treatment with anti-TLR4 antibody, as well as by cilostazol. However, IL-10 was significantly elevated by cilostazol in the incubation media. IL-10 has been found in RA synovium (Isomaki et al., 1996), and both IL-4 and IL-10 can inhibit the secretion of TNF-α, IL-1β, and IL-6 in macrophages (Gordon, 2003). However, the the mechanism by which cilostazol caused an increase in IL-10 expression remains unclear.
In contrast to its effects in macrophages from RA patients, cilostazol attenuated LPS-induced NF-κB activation with subsequent decrease in cytokine expression in RAW 264.7 cells, via MAPK inhibition (Park et al. 2010b). Furthermore this effect involved a cAMP-independent pathway, because cilostazol did not elevate intracellular cAMP in the RAW 264.7 cells. Thus, further studies are required in order to assess whether cilostazol can suppress LPS-stimulated MAPK phosphorylation via a cAMP-dependent pathway in RA macrophages.
The present study measured the effects of cilostazol in a murine CIA model, in terms of synovial inflammation, cartilage depletion and bone erosion. Although the exact pathogenesis of RA has not been fully defined, joint inflammation with infiltration of monocytes/macrophages and participation of locally produced cytokines, as well as loss of cartilage and bone destruction, are critical components (Redlich et al., 2002; Schett et al., 2008). Our present study showed, with immunofluorescent double staining, abundant TLR4-expressing cells co-localized with CD68-positive macrophage-like cells in the knee joint synovium of CIA mice, and both cell types were markedly reduced by treatment with cilostazol. In addition, treatment with cilostazol significantly preserved the proteoglycan content of knee joint cartilage which was markedly reduced in vehicle-treated CIA mice. Many reports have suggested that the pathogenic mechanisms of bone destruction in a murine CIA model are largely dependent on osteoclast formation and their activity in RANKL generation (Redlich et al., 2002; Zwerina et al., 2004). Recently, Park et al. (2010a) demonstrated that cilostazol suppressed proliferation of synovial fibroblasts from RA patients by enhancing apoptosis with increased cytochrome c release and apoptosis-inducing factor translocation, along with increased caspase 3 activation via increased HO-1 expression, linked to Nrf and activation of cAMP-dependent protein kinase. However, in the present study, we did not determine the role of HO-1 expression in the action of cilostazol in the suppression of LPS-induced increased nuclear translocation of PU.1 and subsequent down-regulation of TLR4 expression in synovial macrophages from RA patients. Therefore, more work is required to determine whether cilostazol enhances the apoptosis of synovial macrophages via an LPS-induced increased PU-1-linked TLR signal pathway.
Varani et al. (2011) emphasized a central role of adenosine receptors (A2A and A3) in inflammatory and clinical responses to RA by inhibiting the NF-κB pathway and diminishing cytokines such as TNF-α, IL-1β and IL-6. Intriguingly, cilostazol reduced infarct size in a rabbit model of myocardial infarction by increasing myocardial adenosine during ischaemia (Bai et al., 2011). In the present experiments, we did not assess a possible involvement of adenosine in the intracellular signalling pathways related to the inhibitory effects of cilostazol. However, Kim et al. (2012) have observed that concurrent administration of cilostazol and methotrexate suppressed production of cytokines, including TNF-α, IL-1β, IL-6 and CCL2 in an additive manner, with prevention of joint damage in RA via the interactive action of adenosine A2A receptors and cAMP–protein kinase.
Taken together, the present results demonstrated that cilostazol suppressed the increased nuclear translocation of PU.1 stimulated by LPS and down-regulated PU.1-linked TLR4 expression. Cilostazol also suppressed MyD88 expression, IκBα degradation and NF-κB activation via cAMP-dependent protein kinase activation in synovial macrophages from RA patients. We also confirmed decreased synovial inflammation, proteoglycan depletion and bone erosion by cilostazol in mice with CIA.
Acknowledgments
This work was supported by the MRC program of the NRF (2005–0049477), a National Research Foundation of Korea (NRF) grant funded by the Korea government (MEST) (2011–0009020). We are most grateful to Dr Dai Hyon Yu (Otsuka Pharmaceutical Co., Ltd, Otsuka International Asia Arab Division, Korea) for his helpful suggestions and generous comments.
Glossary
Abbreviations
- CIA
collagen-induced arthritis
- MyD88
myeloid differentiation factor 88
- RA
rheumatoid arthritis
- TLR
Toll-like receptors
- TRAP
tartrate-resistant acid phosphatase
References
- Abdollahi-Roodsaz S, Joosten LA, Roelofs MF, Radstake TR, Matera G, Popa C, et al. Inhibition of toll-like receptor 4 breaks the inflammatory loop in autoimmune destructive arthritis. Arthritis Rheum. 2007;56:2957–2967. doi: 10.1002/art.22848. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th edn. Br. J. Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreakos E, Sacre SM, Smith C, Lundberg A, Kiriakidis S, Stonehouse T, et al. Distinct pathways of LPS-induced NF-κB activation and cytokine production in human myeloid and nonmyeloid cells defined by selective utilization of MyD88 and Mal/TIRAP. Blood. 2004;103:2229–2237. doi: 10.1182/blood-2003-04-1356. [DOI] [PubMed] [Google Scholar]
- Bai Y, Murakami H, Iwasa M, Sumi S, Yamada Y, Ushikoshi H, et al. Cilostazol protects the heart against ischaemia reperfusion injury in a rabbit model of myocardial infarction: focus on adenosine, nitric oxide and mitochondrial ATP-sensitive potassium channels. Clin Exp Pharmacol Physiol. 2011;38:658–665. doi: 10.1111/j.1440-1681.2011.05550.x. [DOI] [PubMed] [Google Scholar]
- Feldmann M, Brennan FM, Maini RN. Role of cytokines in rheumatoid arthritis. Annu Rev Immunol. 1996;14:397–440. doi: 10.1146/annurev.immunol.14.1.397. [DOI] [PubMed] [Google Scholar]
- Firestein GS. Immunologic mechanisms in the pathogenesis of rheumatoid arthritis. J Clin Rheumatol. 2005;11(Suppl):S39–S44. doi: 10.1097/01.rhu.0000166673.34461.33. [DOI] [PubMed] [Google Scholar]
- Firestein GS, Svaifler NJ. Peripheral blood and synovial fluid monocyte activation in inflammatory arthritis. II. Low levels of synovial fluid and synovial tissue interferon suggest that gamma-interferon is not the primary macrophage activating factor. Arthritis Rheum. 1987;30:864–871. doi: 10.1002/art.1780300804. [DOI] [PubMed] [Google Scholar]
- Gohda J, Matsumura T, Inoue J. Cutting edge: TNFR-associated factor (TRAF) 6 is essential for MyD88-dependent pathway but not toll/IL-1 receptor domain-containing adaptor-inducing IFN-beta (TRIF)-dependent pathway in TLR signalling. J Immunol. 2004;173:2913–2917. doi: 10.4049/jimmunol.173.5.2913. [DOI] [PubMed] [Google Scholar]
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- Handel ML, McMorrow LB, Gravallese EM. Nuclear factor–kB in rheumatoid synovium: localization of p50 and p65. Arthritis Rheum. 1995;38:1762–1770. doi: 10.1002/art.1780381209. [DOI] [PubMed] [Google Scholar]
- Isomaki P, Luukkainen R, Saario R, Toivanen P, Punnonen J. Interleukin-10 functions as an antiinflammatory cytokine in rheumatoid synovium. Arthritis Rheum. 1996;39:386–395. doi: 10.1002/art.1780390306. [DOI] [PubMed] [Google Scholar]
- Janeway CA, Jr, Medzhitov R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Signalling to NF-kB by Toll-like receptors. Trends Mol Med. 2007;13:460–469. doi: 10.1016/j.molmed.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Kim HY, Lee SW, Park SY, Baek SH, Lee CW, Hong KW, Kim CD. Efficacy of concurrent administration of cilostazol and methotrexate in rheumatoid arthritis: pharmacologic and clinical significance. Life Sci. 2012;91:250–257. doi: 10.1016/j.lfs.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Kyburz D, Rethage J, Seibl R, Lauener R, Gay RE, Carson DA, et al. Bacterial peptidoglycans but not CpG oligodeoxynucleotides activate synovial fibroblasts by toll-like receptor signalling. Arthritis Rheum. 2003;48:642–650. doi: 10.1002/art.10848. [DOI] [PubMed] [Google Scholar]
- Lichtinger M, Ingram R, Hornef M, Bonifer C, Rehli M. Transcription factor PU.1 controls transcription start site positioning and alternative TLR4 promoter usage. J Biol Chem. 2007;282:26874–26883. doi: 10.1074/jbc.M703856200. [DOI] [PubMed] [Google Scholar]
- Liu H, Ma Y, Cole SM, Zander C, Chen KH, Karras J, et al. Serine phosphorylation of STAT3 is essential for Mcl-1 expression and macrophage survival. Blood. 2003;102:344–352. doi: 10.1182/blood-2002-11-3396. [DOI] [PubMed] [Google Scholar]
- Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, et al. Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum. 2004;50:650–659. doi: 10.1002/art.20001. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SY, Lee JH, Kim YK, Kim CD, Rhim BY, Lee WS, et al. Cilostazol prevents remnant lipoprotein particle-induced monocyte adhesion to endothelial cells by suppression of adhesion molecules and monocyte chemoattractant protein-1 expression via lectin-like receptor for oxidized low-density lipoprotein receptor activation. J Pharmacol Exp Ther. 2005;312:1241–1248. doi: 10.1124/jpet.104.077826. [DOI] [PubMed] [Google Scholar]
- Park SY, Lee JH, Kim CD, Lee WS, Park WS, Han J, et al. Cilostazol suppresses superoxide production and expression of adhesion molecules in human endothelial cells via mediation of cAMP-dependent protein kinase-mediated maxi-K channel activation. J Pharmacol Exp Ther. 2006;317:1238–1245. doi: 10.1124/jpet.105.098509. [DOI] [PubMed] [Google Scholar]
- Park SY, Lee SW, Shin HK, Lee WS, Rhim BY, Hong KW, et al. Cilostazol inhibits proliferation and enhances apoptosis of synovial cells from patient with rheumatoid arthritis with suppression of inflammatory cytokine production via Nrf2-mediated heme oxygenase-1 induction. Arthritis Rheum. 2010a;62:732–741. doi: 10.1002/art.27291. [DOI] [PubMed] [Google Scholar]
- Park WS, Jung WK, Lee DY, Moon C, Yea SS, Park SG, et al. Cilostazol protects mice against endotoxin shock and attenuates LPS-induced cytokine expression in RAW 264.7 macrophages via MAPK inhibition and NF-kappaB inactivation: not involved in cAMP mechanisms. Int Immunopharmacol. 2010b;10:1077–1085. doi: 10.1016/j.intimp.2010.06.008. [DOI] [PubMed] [Google Scholar]
- Radstake TR, Roelofs MF, Jenniskens YM, Oppers-Walgreen B, van Riel PL, Barrera P, et al. Expression of Toll-like receptors 2 and 4 in rheumatoid synovial tissue and regulation by proinflammatory cytokines interleukin-12 and interleukin-18 via interferon-g. Arthritis Rheum. 2004;50:3856–3865. doi: 10.1002/art.20678. [DOI] [PubMed] [Google Scholar]
- Redlich K, Hayer S, Ricci R, David J-P, Tohidast-Akrad M, Kollias G, et al. Osteoclasts are essential for TNF-alpha-mediated joint destruction. J Clin Invest. 2002;110:1419–1427. doi: 10.1172/JCI15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehli M, Poltorak A, Schwarzfischer L, Krause SW, Andreesen R, Beutler B. PU.1 and interferon consensus sequence-binding protein regulate the myeloid expression of the human Toll-like receptor 4 genes. J Biol Chem. 2000;275:9773–9781. doi: 10.1074/jbc.275.13.9773. [DOI] [PubMed] [Google Scholar]
- Schett G, Stach C, Zwerina J, Voll R, Manger B. How antirheumatic drugs protect joints from damage in rheumatoid arthritis. Arthritis Rheum. 2008;58:2936–2948. doi: 10.1002/art.23951. [DOI] [PubMed] [Google Scholar]
- Shin HK, Kim YK, Kim KY, Lee JH, Hong KW. Remnant lipoprotein particles induce apoptosis in endothelial cells by NAD(P)H oxidase-mediated production of superoxide and cytokines via lectin-like oxidized low-density lipoprotein receptor-1 activation: prevention by cilostazol. Circulation. 2004;109:1022–1028. doi: 10.1161/01.CIR.0000117403.64398.53. [DOI] [PubMed] [Google Scholar]
- Simmonds RE, Foxwell BM. Signalling, inflammation and arthritis: NF-kappaB and its relevance to arthritis and inflammation. Rheumatology. 2008;47:584–590. doi: 10.1093/rheumatology/kem298. [DOI] [PubMed] [Google Scholar]
- Varani K, Padovan M, Vincenzi F, Targa M, Trotta F, Govoni M, et al. A2A and A3 adenosine receptor expression in rheumatoid arthritis: upregulation, inverse correlation with disease activity score and suppression of inflammatory cytokine and metalloproteinase release. Arthritis Res Ther. 2011;13:R197. doi: 10.1186/ar3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams RO, Feldmann M, Maini RN. Anti-tumor necrosis factor ameliorates joint disease in murine collagen-induced arthritis. Proc Natl Acad Sci U S A. 1992;89:9784–9788. doi: 10.1073/pnas.89.20.9784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwerina J, Hayer S, Tohidast-Akrad M, Bergmeister H, Redlich K, Feige U, et al. Single and combined inhibition of tumor necrosis factor, interleukin-1, and RANKL pathways in tumor necrosis factor-induced arthritis: effects on synovial inflammation, bone erosion, and cartilage destruction. Arthritis Rheum. 2004;50:277–290. doi: 10.1002/art.11487. [DOI] [PubMed] [Google Scholar]