

Abstract
Background and Purpose
Previous work in our laboratory showed opioid agents inhibit cytokine expression in astrocytes. Recently, Watkins and colleagues hypothesized that opioid agonists activate toll-like receptor 4 (TLR4) signalling, which leads to neuroinflammation. To test this hypothesis, we characterized LPS and opioid effects on TLR4 signalling in reporter cells.
Experimental Approach
NF-κB reporter cells expressing high levels of TLR4 were used to compare LPS and opioid effects on NF-κB activation, a pathway activated by TLR4 stimulation.
Key Results
LPS increased TLR4 signalling in a concentration-dependent manner and was antagonized by LPS antagonist (LPS-RS, from Rhodobacter sphaeroides). A concentration ratio analysis showed that LPS-RS was a competitive antagonist. The opioid agonists, morphine and fentanyl, produced minor activation of TLR4 signalling when given alone. When tested following LPS stimulation, opioid agonists inhibited NF-κB activation but this inhibition was not blocked by the general opioid antagonist, naloxone, nor by the selective μ opioid receptor antagonist, β-FNA. Indeed, both naloxone and β-FNA also inhibited NF-κB activation in reporter cells. Further examination of fentanyl and β-FNA effects revealed that both opioid agents inhibited LPS signalling in a non-competitive fashion.
Conclusions and Implications
These results show that LPS-RS is a competitive antagonist at the TLR4 complex, and that both opioid agonists and antagonists inhibit LPS signalling in a non-competitive fashion through a non-GPCR, opioid site(s) in the TLR4 signalling pathway. If confirmed, existing opioid agents or other drug molecules more selective at this novel site may provide a new therapeutic approach to the treatment of neuroinflammation.
Keywords: opioids, toll-like receptor 4 (TLR4), lipopolysaccharide (LPS), morphine, fentanyl, naltrexone, FNA, opioid-immune crosstalk
Introduction
Opioids are a large class of well-known substances that are comprised of both exogenous and endogenous substances. The established molecular targets of opioids are the three canonical GPCR opioid receptors; μ, δ and κ (Alexander et al., 2011), which are expressed throughout vertebrate phylogeny and appear to have arisen through gene duplication (Stevens, 2009). Most clinically used opioids bind to GPCR opioid receptors on neurons with high affinity in the nanomolar range, producing useful effects such as analgesia. In contrast, it is emerging from the opioid-immune literature that both classical GPCR and non-GPCR sites mediate various opioid effects on peripheral and central immune cells.
In peripheral immune cells, there are many reports of opioid immune effects mediated by classical opioid receptors (reviewed in Rogers and Peterson, 2003). Many peripheral immune cells, including macrophages, lymphocytes and T cells, express μ opioid receptors that mediate the effects of morphine on immune cell function (see Ninković and Roy, 2011). The central immune (glia) cells, microglia and astrocytes, also express μ opioid receptors (Ruzicka et al., 1995; Stiene-Martin et al., 1998; Börner et al., 2007); and studies show that μ opioid receptors mediate proinflammatory actions of morphine on glia cells (El-Hage et al., 2008; He et al., 2011).
Other studies in the opioid immune literature show that there are other actions of opioids not mediated by the canonical or GPCR opioid receptors. Using peripheral immune cells, a seminal paper by Roy et al. (1998) examined the effect of morphine on bacterial endotoxin LPS-stimulated release of cytokines from mouse peritoneal macrophages. They found that morphine differentially modulates LPS action such that a low concentration of morphine (50 nM) potentiated LPS-induced activation of NF-κB and the expression of cytokines, while a higher concentration of morphine (50 μM) had the opposite effect. They further showed that the opioid antagonist, naloxone, blocked the potentiating effect of morphine but not the inhibiting effect, and suggested a non-GPCR opioid site of action. A more recent study using the RAW264.7 macrophage cell line showed that naloxone inhibited LPS-induced NF-κB activation, and that this effect of naloxone was enhanced by morphine (Jan et al., 2011). These results point to a non-GPCR opioid site of action as both an opioid agonist and antagonist produced effects in the same direction. In triple μ, δ and κ opioid receptor knockout mice, the δ opioid receptor antagonist, naltrindole, was able to reduce graft rejection in vivo and by proxy in an in vitro assay (Gavériaux-Ruff et al., 2001). These authors concluded that naltrindole inhibited the graft rejection process by interacting with a non-GPCR opioid site yet to be discovered.
In studies examining central immune cell function, in a model using co-cultured neuron-glia cells, both (–)-naloxone and (+)-naloxone were equipotent in decreasing neurotoxicity produced by LPS, suggesting a non-GPCR opioid action (Liu et al., 2000a). Further studies by this group showed a similar lack of stereospecificity by naloxone in attenuating superoxide production and microglia activation in midbrain and cortical areas of rat brain (Liu et al., 2000b; 2002). Naloxone was also suggested to have a non-GPCR opioid site of action in blocking the effects of LPS signalling in co-cultures of astrocytes and brain endothelial cells (Hansson et al., 2008).
A possible site mediating the above non-GPCR opioid actions on central and peripheral immune cells is the toll-like receptor 4 (TLR4), which is present on immune and other cell types (O'Neill, 2008). TLR4 is one of ten closely related human toll-like receptors that is selective for recognition of Gram-negative bacterial endotoxin LPS and endogenous ligands such as heat-shock proteins and other inflammatory mediators arising from cell damage (O'Neill, 2008). Activation of TLR4 leads to the dimerization of TLR4 monomers and, ultimately, the generation of nucleus-seeking NF-κB transcription factors. Numerous pro-inflammatory cytokine and chemokine genes contain NF-κB response elements in their promoter regions, such as TNF-α and IL-6 (Kenny and O'Neill, 2008). As increasing evidence suggests that neuronal damage results in part from microglial and astroglial derived cytokines and chemokines (Deshpande et al., 2005; McCoy and Tansey, 2008; Loane and Byrnes, 2010), there is a great interest in pharmacological agents that inhibit NF-κB pathways (Sethi and Tergaonkar, 2009).
The discovery of a non-GPCR opioid site of action at TLR4 is largely due to the extensive studies performed by Watkins and colleagues (for review, see Watkins et al., 2009; Hutchinson et al., 2011). Using commercially available reporter HEK 293 cells (HEK-Blue™-hTLR4 cells from Invivogen) expressing high levels of TLR4 and its accessory proteins, their initial studies showed that both enantiomers of the opioid antagonists, naloxone and naltrexone, inhibited LPS activation of TLR4 signalling (Hutchinson et al., 2008). Additional studies identified a number of neuropharmacological agents capable of inhibiting TLR4 activation including antidepressant and antiepileptic agents (Hutchinson et al., 2010b). Further studies showed that a variety of opioid agonists produced slight but significant TLR4 activation in the absence of LPS including both enantiomers for stereoisomeric opioids such as morphine, methadone and levorphanol (Hutchinson et al., 2010a). Analysis of ligand binding sites using modelling software indicated that a potential opioid site for TLR4 interaction was located on MD-2, where it interfaces with TLR4 (Hutchinson et al., 2010c). Additional structural and functional studies confirmed a binding site for opioids on the TLR4 protein complex (Wang et al., 2012).
Recently, we found that opioids altered the TNF-α-induced expression of cytokines and NO in astrocytes by a non-GPCR opioid receptor mechanism (Davis et al., 2007; 2008). In these studies, low micromolar concentrations of the μ opioid receptor opioid agonist, morphine, and the highly selective μ opioid receptor antagonist, β-FNA, inhibited the activation of NF-κB and the expression of the chemokine CXCL10 and inducible NOS expression. To explore the possible mechanism of the non-GPCR opioid actions we observed above and to further examine opioid action on TLR4 signalling pathways linked to NF-κB, we sought to use the HEK-Blue™-hTLR4 reporter cells to assess the effect of the opioid agonists, morphine and fentanyl, and the opioid antagonists, naltrexone and β-FNA, on LPS-stimulated TLR4 signalling. We also designed an experiment with the LPS antagonist, LPS-RS (a TLR4 antagonist extracted from Rhodobacter sphaeroides), to perform a concentration ratio analysis (Schild plot) to pharmacologically characterize the nature of the antagonism.
Our results show that when cells are treated with morphine or fentanyl, select concentrations produce minor stimulation of TLR signalling. Moreover, for the first time, we note that morphine and fentanyl inhibit LPS activation of TLR4 signalling, and that this effect is not blocked by naltrexone or β-FNA. Additionally, naltrexone or β-FNA by themselves also inhibits LPS-mediated TLR4 signalling. Results of the concentration ratio analysis of LPS antagonism by increasing concentrations of LPS-RS show competitive antagonism at a single site with an apparent affinity similar to that reported by biophysical studies. However, full LPS concentration curve studies in the presence of fentanyl or β-FNA suggest that opioid antagonism is best characterized as a non-competitive mechanism of action. These findings confirm and extend previous results and add a greater pharmacological perspective to the interaction of opioid agents with putative non-GPCR opioid sites modulating glial cell function. Overall, this avenue of research may produce novel opioid-based pharmacotherapy for the treatment of neuroinflammation and related neurodegenerative diseases.
Methods
Cell culture and TLR4 signalling assay
HEK-Blue™-hTLR4 cells (Invivogen, San Diego, CA) were used in the colorimetric assays to determine TLR4 signalling. The HEK-Blue™-hTLR4 cells are HEK 293 cells stably expressing hTLR4 receptor (cloned human receptor) and the needed accessory proteins to TLR4, MD-2 and CD14. These accessory proteins interact with LPS at the TLR4 complex to induce NF-κB activation. In addition these cells also are engineered with a reporter gene, secreted embryonic alkaline phosphatase (SEAP), which is produced following NF-κB activation. The post-transcriptional level of SEAP can be quantitatively assayed using a spectrophotometer as well as visually inspected for colour change.
The growth media for the HEK-Blue™-hTLR4 cells was prepared by supplementing DMEM (Invitrogen, Carlsbad, CA, USA) with 4 mM l-glutamine, 10% FBS (Hyclone #SH30071-03, Logan UT, USA), 10 000 U·mL−1 penicillin (Invitrogen), 10 mg·mL−1 streptomycin (Invitrogen) and normocin (Invivogen). The HEK-Blue™-hTLR4 cells were maintained in the selection media. HEK-Blue Selection mix is a proprietary blend of multiple antibiotics (information not disclosed by the manufacturer). This along with normocin prevents bacterial, fungal and mycoplasmal growth. The HEK-Blue4 growth media is supplemented with 1 × HEK-Blue selection. The selection media is pre-warmed to 37°C before use and stored at 4°C. The cultures are grown in 25 cm2 flasks at 37°C and 5% CO2. Cells are subcultured when they are 60–80% confluent. The media is changed two to three times a week.
HEK-Blue Detection mix is used to detect the presence of SEAP, which correlates with TLR4 signalling though the NF-κB activation. The detection media contains nutrients for cell growth and a colour substrate, which when hydrolysed by SEAP produces a colour change. When the cells reached 60–80% confluency, they were collected, centrifuged and resuspended in the detection media. Cells (50 000) were seeded in each well of a 96-well plate (100 μl volume). The cells were then treated alone or in combinations with endotoxin free water (unstimulated; negative control), LPS (positive control), LPS-RS (known TLR4 antagonist) and varying concentrations of opioid agents (mixed in endotoxin-free water) in triplicate wells, and incubated for 24 h at 37°C in 5% CO2. The plates were read spectrophotometrically at wavelengths 620–655 nm using a BioTek Synergy 2 Microplate Reader Winooski, VT, USA. Values are reported as raw optical density absorbance units; they are abbreviated in figures as OD655.
Opioid agents and LPS ligands
The opioid agonists used in these studies were morphine sulfate and fentanyl citrate. The opioid antagonists used were naltrexone hydrochloride and β-funaltrexamine (β-FNA, (E)-4-[[(5α,6β)-17-(cyclopropylmethyl)-4,5-epoxy-3,14-dihydroxymorphinan-6-yl]amino]-4-oxo-2-butenoic acid methyl ester hydrochloride). β-FNA was a generous gift from the NIDA Drug Supply Program, and other drugs were purchased from Sigma Aldrich (St. Louis, MO). Standard, racemic formulations of all opioid agents were used. LPS (Escherichia coli K12 strain, Invivogen) was used to stimulate TLR4 signalling. The LPS antagonist, LPS-RS (a naturally occurring LPS from R. sphaeroides, Invivogen), was used as a positive control for LPS antagonism.
Statistical analysis
Prism 5.0 (GraphPad Software, La Jolla, CA) was used for plotting of the data and statistical analysis. A one-way anova with Neuman–Keuls multiple comparison post hoc tests was used to analyse differences in TLR4 activity or a Dunnett's test when one treatment group served as control. Non-linear regression was used to plot and analyse concentration–response curves and to obtain EC50 and Emax values and to produce the Schild plot. Each experiment was repeated three times with treatments performed at least in triplicate for each of the independent experiments (n = 9). SEM is represented by an error bar on each bar graph. Differences were considered significant when P < 0.05 or as evidenced by non-overlapping 95% confidence intervals.
Results
Concentration-response curves of LPS-induced TLR4 signalling
LPS produced a concentration-dependent increase in TLR4 signalling with an EC50 of 0.64 ng·mL−1 (Figure 1A, Table 1). Concurrent treatment with increasing concentrations of the LPS antagonist, LPS-RS, caused rightward, parallel shifts of the LPS curve, with LPS-RS at 10 and 100 ng·mL−1, producing significantly greater EC50 values of 3.60 and 13.58 ng·mL−1 respectively. The Emax of all concentration–response curves were not significantly different (Table 1).
Figure 1.
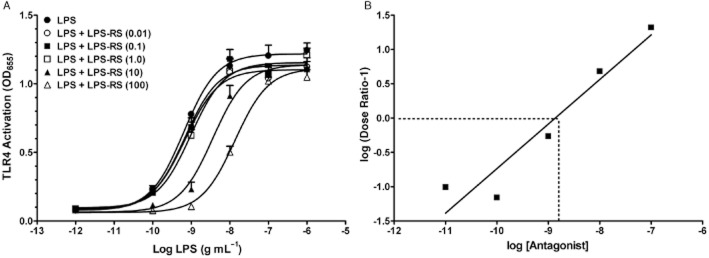
Stimulation of TLR4 signalling by LPS and inhibition by LPS-RS. (A) LPS concentration–response curve of stimulation of TLR4 activity. HEK-Blue4 cells were treated as described in Methods with LPS alone (from 10−12 to 10−6 g·mL−1) or co-treated with the LPS antagonist (RS) at increasing concentrations (in legend as ng·mL−1). EC50 and Emax values are given in Table 1. Data points are given as mean + SEM; error bars not visible are within the symbol. Experiments were carried out in triplicate with n = 9 for each treatment group. (B) Schild plot of the LPS-RS antagonism for LPS stimulation of TLR4 signalling shown in panel A. The regression line was not different than 1.0, suggesting a single site competitive antagonism and the apparent affinity (KD) of LPS-RS at the competitive site is found at the concentration where the line crosses the abscissa (−8.867 log or 1.36 ng·mL−1).
Table 1.
Pharmacological parameters of TLR4 stimulation by LPS alone and with different concentrations of LPS antagonist (RS) co-treatment
| Treatmenta | EC50b | 95% CI | Emax | 95% CI |
|---|---|---|---|---|
| LPS alone | 0.64 | 0.38–1.07 | 1.22 | 1.14–1.30 |
| LPS + RS (0.01) | 0.70 | 0.37–1.31 | 1.10 | 1.02–1.19 |
| LPS + RS (0.1) | 0.68 | 0.40–1.16 | 1.14 | 1.06–1.21 |
| LPS + RS (1.0) | 0.97 | 0.60–1.57 | 1.16 | 1.08–1.23 |
| LPS + RS (10) | 3.60* | 2.36–5.48 | 1.14 | 1.07–1.21 |
| LPS + RS (100) | 13.58* | 10.34–17.70 | 1.11 | 1.06–1.17 |
The LPS antagonist co-treatment concentrations with RS are given as ng·mL−1 in parenthesis.
Effective concentration for 50% TLR4 signalling, given in ng·mL−1.
denotes significantly different from LPS alone concentration–response curve.
A concentration ratio analysis (Schild plot) of the LPS-RS data is shown in Figure 1B. The slope of the line was 0.65 with a 95% confidence interval that included 1.0 (0.30–1.10) and was significantly different from zero at P < 0.05 by an F-test. The apparent affinity of LPS-RS was obtained by calculating the X-intercept when Y = 0 (dotted lines on graph) and was equal to a log value of −8.87 (1.36 ng·mL−1).
Effects of morphine on TLR4 signalling
Initial studies were done to assess morphine effects on TLR4 signalling (Figure 2A, left panel). Morphine at 3 and 10 μM concentrations produced slight but significant increases in TLR4 activity compared with unstimulated control cells. Co-treatment with LPS (100 ng·mL−1) and morphine (1–100 μM) resulted in significant inhibition of TLR4 signalling for morphine concentrations of 3–100 μM compared with the robust activation of TLR4 produced by LPS alone (Figure 2A, middle panel). Concurrent treatment of LPS, naltrexone (100 μM) and morphine is shown in Figure 2A, right panel. Addition of naltrexone (100 μM) to the morphine plus LPS treatment did not block morphine inhibition of LPS activation and resulted in significant inhibition at morphine concentrations of 3, 30 and 100 μM.
Figure 2.
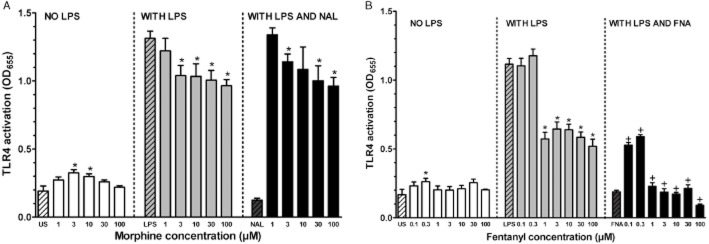
(A) Effect of morphine on TLR4 activity. Left panel: Treatment groups were unstimulated controls (US) and morphine concentrations from 1–100 μM. Asterisks denote significantly different than US control. Middle panel: Cells were co-treated with LPS (100 ng·mL−1) and morphine concentrations from 1–100 μM. Asterisks denote significantly different than LPS only-stimulated cells. Right panel: Concurrent treatment consisted of LPS (100 ng·mL−1), naltrexone (100 μM) and morphine concentrations from 1–100 μM. Asterisks denote significantly different than LPS only-stimulated cells. Data points are given as mean + SEM; error bars not visible are within the symbol. Experiments were carried out in triplicate with n = 9 for each treatment group. Hashed bars show effects of control treatments for each panel. (B) Effect of fentanyl on TLR4 activity. Left panel: Treatment groups were unstimulated controls (US) and fentanyl concentrations from 0.1–100 μM. Asterisks denote significantly different than US control. Middle panel: Cells were co-treated with LPS (30 ng·mL−1) and fentanyl concentrations from 0.1–100 μM. Asterisks denote significantly different than LPS only-stimulated cells. Right panel: Concurrent treatment consisted of LPS (30 ng·mL−1), β-FNA (30 μM) and fentanyl concentrations from 0.1–100 μM. Plus signs denote significantly different than corresponding LPS and fentanyl-treated cells in middle panel. Data points are given as mean + SEM; error bars not visible are within the symbol. Experiments were carried out in triplicate with n = 9 for each treatment group. Hashed bars show effects of control treatments for each panel.
Effects of fentanyl on TLR4 signalling
Fentanyl alone was tested in a concentration range of 0.1–100 μM. Fentanyl at 0.3 μM produced a slight but significant increase in TLR4 activation compared with unstimulated (cells only) controls (Figure 2B, left panel). Co-treatment of cells with fentanyl and LPS (30 ng·mL−1) showed that fentanyl at 01 and 0.3 μM did not alter TLR4 signalling, but fentanyl at concentrations from 1–100 μM significantly inhibited TLR4 activation compared to LPS alone-treated cells (Figure 2B, middle panel). Concurrent treatment of cells with LPS, fentanyl (0.1–100 μM) and β-FNA (30 μM), produced a significantly greater inhibition of TLR4 stimulation than LPS plus fentanyl at all concentrations of fentanyl tested (Figure 2B, right panel).
Effects of naltrexone and β-FNA on TLR4 signalling
As shown in Figure 3A (left panel), treatment of cells with naltrexone alone (3–1000 μM) did not activate TLR4 signalling. However, naltrexone plus LPS (30 ng·mL−1) significantly inhibited LPS-induced TLR4 activation at naltrexone concentrations from 30–1000 μM compared with the stimulation of TLR4 signalling of LPS alone (Figure 3A, right panel). A shown in Figure 3B (right panel), treatment of cells with β-FNA alone (3–30 μM) did not activate TLR4 signalling. However, β-FNA plus LPS (30 ng·mL−1) significantly inhibited LPS-induced TLR4 activation at β-FNA concentrations from 3–30 μM compared with the stimulation of TLR4 signalling of LPS alone (Figure 3B, right panel).
Figure 3.
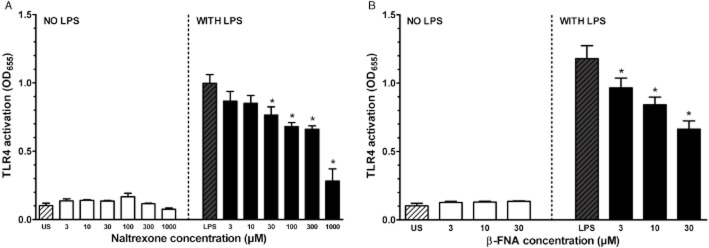
(A) Effect of naltrexone on TLR4 activity. Left panel: Treatment groups were unstimulated controls (US) and naltrexone concentrations from 3–1000 μM. Right panel: Cells were co-treated with LPS (100 ng·mL−1) and naltrexone concentrations from 3–1000 μM. Asterisks denote significantly different than LPS only-stimulated cells. Data points are given as mean + SEM; error bars not visible are within the symbol. Experiments were carried out in triplicate with n = 9 for each treatment group. (B) Effect of β-FNA on TLR4 activity. Left panel: Treatment groups were unstimulated controls (US) and β-FNA concentrations from 3–30 μM. Right panel: Cells were co-treated with LPS (30 ng·mL−1) and β-FNA concentrations from 3 to 30 μM. Asterisks denote significantly different than LPS only-stimulated cells. Data points are given as mean + SEM; error bars not visible are within the symbol. Experiments were carried out in triplicate with n = 9 for each treatment group. Hashed bars show effects of control treatments for each panel.
Concentration–response curves of LPS-induced TLR4 signalling with fentanyl and β-FNA
In a separate set of experiments, LPS produced a concentration-dependent increase in TLR4 signalling with an EC50 of 0.85 ng·mL−1 (Figure 4A, Table 2). Concurrent treatment with a single concentration (10 ng·mL−1) of the LPS antagonist, LPS-RS, caused a rightwards, parallel shift of the LPS curve, with a significantly greater EC50 value of 2.16 ng·mL−1. Consistent with the initial studies above, LPS-RS did not alter the Emax of the LPS-mediated stimulation of the TLR4 pathway. The LPS concentration–response curve in the presence of fentanyl (3 μM) shifted the curve significantly to the right (EC50 = 4.81 ng·mL−1) and downwards (Emax = 0.85). The addition of the LPS antagonist, LPS-RS, to the fentanyl plus LPS treatment did not alter the parameters of the fentanyl plus LPS curve (Table 2).
Figure 4.
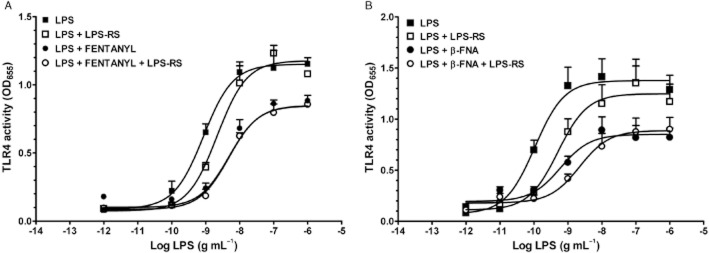
(A) Effect of fentanyl on the LPS concentration–response curve of TLR4 activity. LPS was given alone (from 10−12 to 10−6 g·mL−1), with the LPS antagonist (RS; 10 ng·mL−1), with fentanyl (FENT; 3.0 μM), or with fentanyl plus the LPS antagonist and LPS concentrations ranging from 10−12 to 10−6 g·mL−1. (B) Effect of β-FNA on the LPS concentration–response curve of TLR4 activity. LPS was given alone (concentrations ranging from 10−11 to 10−6 g·mL−1), with the LPS antagonist (RS; 10 ng·mL−1), with β-FNA (30 μM) or with β-FNA plus the LPS antagonist and LPS (concentrations ranging from 10−11 to 10−6 g·mL−1). For both panels, EC50 and Emax values are given in Table 2. Data points are given as mean ± SEM; error bars not visible are within the symbol. Experiments were carried out in triplicate with n = 9 or greater for each treatment group. Hashed bars show effects of control treatments for each panel.
Table 2.
Pharmacological parameters of TLR4 stimulation by LPS alone and with the addition of fentanyl and β-FNA
| Treatmenta | EC50b | 95% CI | Emax | 95% CI |
|---|---|---|---|---|
| Fentanyl | ||||
| LPS alone | 0.85 | 0.59–1.23 | 1.15 | 1.19–1.20 |
| LPS + RS (10 ng·mL−1) | 2.16* | 1.34–3.41 | 1.18 | 1.10–1.25 |
| LPS + fentanyl (3.0 μM) | 4.81* | 3.42–6.76 | 0.85* | 0.81–0.89 |
| LPS + fentanyl (3.0 μM) + RS (10 ng·mL−1) | 4.85* | 3.47–6.67 | 0.85* | 0.82–0.89 |
| β-FNA | ||||
| LPS alone | 0.11 | 0.04–0.24 | 1.37 | 1.24–1.52 |
| LPS + RS (10 ng·mL−1) | 0.51* | 0.26–1.59 | 1.25 | 1.08–1.42 |
| LPS + β-FNA (30 μM) | 0.60* | 0.29–1.88 | 0.85* | 0.75–0.95 |
| LPS + β-FNA (30 μM) + RS (10 ng·mL−1) | 2.20* | 0.70–6.93 | 0.89* | 0.77–1.01 |
The LPS antagonist co-treatment concentrations with RS, fentanyl, and β-FNA are given in parenthesis.
Effective concentration for 50% TLR4 signalling, given in ng·mL−1.
denotes significantly different parameter from LPS alone concentration–response curve.
In an additional set of experiments, LPS produced a concentration-dependent increase in TLR4 signalling with an EC50 of 0.11 ng·mL−1 (Figure 4B, Table 2). Concurrent treatment with a single concentration (10 ng·mL−1) of the LPS antagonist, LPS-RS, caused a rightwards, parallel shift of the LPS curve, with a significantly greater EC50 value of 0.51 ng·mL−1. Consistent with the initial studies above, LPS-RS did not alter the Emax of the LPS-mediated stimulation of the TLR4 pathway. The LPS concentration–response curve in the presence of β-FNA (30 μM) shifted the curve significantly to the right and downward. The addition of the LPS antagonist, LPS-RS, to the β-FNA plus LPS treatment did not significantly alter the parameters of the β-FNA plus LPS curve (Table 2).
Discussion and conclusion
Studies examining opioid and immune interactions in microglia and astrocytes and peripheral immune cells are complicated by the presence of GPCR opioid receptors as well as TLR4 and other immune-related receptors. For this reason, we chose to use the HEK-Blue™-hTLR4 cells to examine opioid effects targeted at TLR4, as these cells have not been reported to express GPCR opioid receptors. LPS, a potent agonist at the TLR4 complex, produced a concentration-dependent activation of the TLR4 signalling pathway, as determined by the NF-κB reporter assay in the engineered HEK-Blue™-hTLR4cell line. The EC50 values of the LPS activation of TLR4 signalling ranged from 0.11 to 0.85 ng·mL−1 in three independent experiments (Tables 2), which is consistent with the reported EC50 value of 0.21 ng·mL−1 in the same reporter cells (Hutchinson et al., 2007). Increasing concentrations of the LPS antagonist, LPS-RS, co-treated in separate groups of cells with the complete LPS concentration–response curves, produced progressive rightwards parallel shifts of the LPS concentration–response curves, with no decrease in the maximal effect. The use of multiple concentrations of LPS-RS allowed for a concentration ratio analysis, a classic pharmacological technique first described more than 50 years ago (Schild, 1947). The results of the Schild plot (Figure 1B) yielded an apparent affinity (KD) of the LPS-RS binding site of 1.36 ng·mL−1 and confirmed the competitive, single-site antagonism by LPS-RS of LPS-induced TLR4 signalling. To our knowledge, this is the first time that LPS-RS antagonism of LPS was characterized by a pharmacological (Schild) analysis.
Morphine, and the more potent opioid and μ opioid receptor-selective agonist, fentanyl, produced minor but significant stimulation of TLR signalling at a few doses in the absence of LPS (Figure 2A,B). However, stimulation of TLR4 signalling was not observed with the general opioid antagonist, naltrexone, or the highly μ opioid receptor-selective antagonist, β-FNA (Figure 3). These data confirm previous results using the same reporter cells showing that single concentrations (10 μM) of morphine, fentanyl and a number of other opioid agonists non-stereoselectively stimulate TLR4 signalling (Hutchinson et al., 2010a). The magnitude of TLR4 activation in these studies, while significant when compared to the control cells, did not reach the level of TLR4 stimulation produced by a very low concentration of LPS (0.05 ng·mL−1) included for comparison. This LPS concentration falls far to the left of their LPS concentration–response curve, which had an EC50 of 0.21 ng·mL−1 (Hutchinson et al., 2007; 2008).
In contrast, morphine and fentanyl both produced a potent inhibition of LPS-induced TLR4 activation across a wide concentration range (Figure 2). Comparing opioid agonist effects on TLR4 signalling when cells were treated alone with either morphine or fentanyl (previous studies) to their antagonist effect on TLR4 signalling when cells were co-treated with LPS and morphine or fentanyl (present studies), the agonist effects on TLR4 signalling yields about a 10% stimulation, whereas the antagonist effects were in the range of 50–80% inhibition. These data, and the previous studies cited above, suggest that opioid agonists may have both a minor pro-inflammatory effect and a more major anti-inflammatory effect mediated through TLR4 signalling pathways. For all opioid agents tested, there was not a clear concentration-dependent inhibition of LPS-induced signalling, which suggests either a low capacity site that is easily saturated or a type of non-competitive antagonism. To our knowledge, these are the first pharmacological data examining the effects of opioid agonists in combination with LPS in a TLR4 signalling cell system.
Co-treatment of cells with naltrexone added to the morphine plus LPS treatment did not block the inhibition of LPS produced by the morphine plus LPS treatment (Figure 2A). Co-treatment of cells with β-FNA added to the fentanyl plus LPS treatment did not block the inhibition of LPS produced by the fentanyl plus LPS treatment, but rather produced an even greater inhibition of LPS-induced TLR4 signalling (Figure 2B). As observed for the opioid agonists, co-treatment of a single concentration of LPS with various concentrations of the general opioid antagonist, naltrexone, or the selective μ opioid receptor antagonist, β-FNA, blocked the stimulation of TLR4 signalling at most concentrations used (Figure 3). This is consistent with similar studies testing naloxone and naltrexone in the same reporter cells (Hutchinson et al., 2008), although these authors also showed that the GPCR opioid receptor-inactive enantiomers of naloxone and naltrexone were equipotent as the GPCR active drugs used here. These prior results, as well as the present data, support the hypothesis of a non-classical, non-GPCR site of opioid action. The same site that LPS binds to on the MD-2 accessory protein of the TLR4 receptor was recently characterized as an opioid pro-inflammatory site of action (Wang et al., 2012); however, the present data showing that LPS-RS does not alter the inhibition of LPS-induced TLR4 signalling suggests an alternative site of action.
As noted above, both opioid agonists and antagonists were not concentration-dependent in their inhibition of single concentrations of LPS stimulation. This suggests an opioid effect that is non-competitive in nature. This hypothesis is supported by the action of opioid agents on the full LPS concentration–response curve shown in Figure 4. For both fentanyl, an opioid agonist, and β-FNA, an opioid antagonist, opioid-mediated decreases in the maximal effect and non-parallel concentration–response curve shifts support a non-competitive antagonist mode of action. Furthermore, the addition of the competitive LPS antagonist, LPS-RS, to these opioid treatments did not alter the nature of the opioid-mediated antagonism. In contrast, in previous studies, the stimulation of TLR4 signalling observed after treatment of morphine 3-glucoronide alone, a major morphine metabolite, was shown to be blocked by the LPS antagonist, LPS-RS, in a competitive fashion (Lewis et al., 2010). Further structure–activity studies, with complete concentration–response curves of opioid agonists and antagonists will help to clarify the nature of the opioid interaction.
Opioid immune effects are not limited to the TLR4 pathway but were observed in functional studies of TLR2 and TLR9 pathways (He et al., 2011; Zhang et al., 2011). TLR2 knock-out mice show a reduced effect of microglia activation by morphine and reduced signs of withdrawal when made tolerant to morphine (Zhang et al., 2011). In microglia, morphine treatment increases the expression of TLR9, which is dependent on the expression of μ opioid receptors (He et al., 2011). Further pharmacological studies are needed to determine if the opioid-immune interaction described here extends to other toll-like receptors and if classical opioid receptors and/or non-GPCR sites are involved. Future studies should also examine modulation of TLR4 signalling produced by endogenous ligands of the TLR4 complex, of which there are many, and appear to be released during cell stress, apoptosis and neuroinflammation (Erridge, 2010). Additionally, opioids may be exerting their effects at intracellular signalling molecules beyond the TLR4 receptor complex and target key molecules in NF-κB pathway. For instance, morphine was shown to decrease NF-κB activity by increasing the expression of inhibitor of NF-κB (IκB) in neuronal cell lines (Börner et al., 2012). Finally, in cells containing both μ opioid receptors and TLR4, there is evidence of crosstalk between these signalling pathways. For example, morphine treatment decreases the mRNA and protein of TLR4 in mouse macrophages, an effect reversed by the opioid antagonist, naltrexone (Franchi et al., 2012).
In conclusion, the present results show that both opioid agonists and antagonists inhibit LPS-induced TLR4 signalling in a reporter cell line at a non-GPCR opioid site. The nature of the opioid interaction is different than the competitive antagonism observed with the LPS antagonist, LPS-RS, and appears to be non-competitive. Given the opioid stimulation of TLR4 signalling observed sporadically here and in previous studies, and the inhibition of LPS-induced TLR4 signalling shown here, the most parsimonious characterization of opioids in the present model system may be as weak partial agonists. These results suggest that previous and forthcoming studies using cells that express both GPCR opioid receptors and toll-like receptors need to be carefully interpreted as commonly used opioids, such as morphine and naltrexone, may be exerting their effects at multiple sites of action. Ultimately, therapeutic manipulation of the novel opioid site(s) characterized here, at the TLR4 complex or elsewhere in the signalling pathway, may provide a new avenue for the treatment of neuroinflammatory disorders.
Acknowledgments
This work was supported in part by NIH grants NS 062664 (RLD) and an Oklahoma Center for the Advancement of Science and Technology (OCAST) contract HR10-031 (CWS). We are grateful to Kelly McCracken and Daniel Buck for their technical assistance.
Glossary
Abbreviations
- β-FNA
β -funaltrexamine
- LPS-RS
LPS antagonist
- TLR4
toll-like receptor 4
Conflicts of interest
The authors declare that they have no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Börner C, Stumm R, Höllt V, Kraus J. Comparative analysis of mu-opioid receptor expression in immune and neuronal cells. J Neuroimmunol. 2007;188:56–63. doi: 10.1016/j.jneuroim.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Börner C, Höllt V, Kraus J. Mechanisms of the inhibition of nuclear factor-κB by morphine in neuronal cells. Mol Pharmacol. 2012;81:587–597. doi: 10.1124/mol.111.076620. [DOI] [PubMed] [Google Scholar]
- Davis RL, Buck DJ, Saffarian N, Stevens CW. The opioid antagonist, beta-funaltrexamine, inhibits chemokine expression in human astroglial cells. J Neuroimmunol. 2007;186:141–149. doi: 10.1016/j.jneuroim.2007.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RL, Buck DJ, Saffarian N, Mohan S, DeSilva U, Fernando SC, et al. Beta-funaltrexamine inhibits inducible nitric-oxide synthase expression in human astroglial cells. J Neuroimmune Pharmacol. 2008;3:150–153. doi: 10.1007/s11481-008-9102-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deshpande M, Zheng J, Borgmann K, Persidsky R, Wu L, Schellpeper C, et al. Role of activated astrocytes in neuronal damage: potential links to HIV-1-associated dementia. Neurotox Res. 2005;7:183–192. doi: 10.1007/BF03036448. [DOI] [PubMed] [Google Scholar]
- El-Hage N, Bruce-Keller AJ, Yakovleva T, Bazov I, Bakalkin G, Knapp PE, et al. Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca(2+)](i), NF-κB trafficking and transcription. PLoS ONE. 2008;3:e4093. doi: 10.1371/journal.pone.0004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erridge C. Endogenous ligands of TLR2 and TLR4: agonists or assistants? J Leukoc Biol. 2010;87:989–999. doi: 10.1189/jlb.1209775. [DOI] [PubMed] [Google Scholar]
- Franchi S, Moretti S, Castelli M, Lattuada D, Scavullo C, Panerai AE, et al. Mu opioid receptor activation modulates Toll like receptor 4 in murine macrophages. Brain Behav Immun. 2012;26:480–488. doi: 10.1016/j.bbi.2011.12.010. [DOI] [PubMed] [Google Scholar]
- Gavériaux-Ruff C, Filliol D, Simonin F, Matthes HW, Kieffer BL. Immunosuppression by delta-opioid antagonist naltrindole: delta- and triple mudeltakappa-opioid receptor knockout mice reveal a nonopioid activity. J Pharmacol Exp Ther. 2001;298:1193–1198. [PubMed] [Google Scholar]
- Hansson E, Westerlund A, Bjorklund U, Olsson T. Mu-opioid agonists inhibit the enhanced intracellular Ca(2+) responses in inflammatory activated astrocytes co-cultured with brain endothelial cells. Neuroscience. 2008;155:1237–1249. doi: 10.1016/j.neuroscience.2008.04.027. [DOI] [PubMed] [Google Scholar]
- He L, Li H, Chen L, Miao J, Jiang Y, Zhang Y, et al. Toll-like receptor 9 is required for opioid-induced microglia apoptosis. PLoS ONE. 2011;6:e18190. doi: 10.1371/journal.pone.0018190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Bland ST, Johnson KW, Rice KC, Maier SF, Watkins LR. Opioid-induced glial activation: mechanisms of activation and implications for opioid analgesia, dependence, and reward. Sci World J. 2007;7:98–111. doi: 10.1100/tsw.2007.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Brown K, Coats BD, Shridhar M, Sholar PW, et al. Non-stereoselective reversal of neuropathic pain by naloxone and naltrexone: involvement of toll-like receptor 4 (TLR4) Eur J Neurosci. 2008;28:20–29. doi: 10.1111/j.1460-9568.2008.06321.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Zhang Y, Shridhar M, Evans JH, Buchanan MM, Zhao TX, et al. Evidence that opioids may have toll-like receptor 4 and MD-2 effects. Brain Behav Immun. 2010a;24:83–95. doi: 10.1016/j.bbi.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Loram LC, Zhang Y, Shridhar M, Rezvani N, Berkelhammer D, et al. Evidence that tricyclic small molecules may possess toll-like receptor and myeloid differentiation protein 2 activity. Neuroscience. 2010b;168:551–563. doi: 10.1016/j.neuroscience.2010.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Lewis SS, Coats BD, Rezvani N, Zhang Y, Wieseler JL, et al. Possible involvement of toll-like receptor 4/myeloid differentiation factor-2 activity of opioid inactive isomers causes spinal proinflammation and related behavioral consequences. Neuroscience. 2010c;167:880–893. doi: 10.1016/j.neuroscience.2010.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson MR, Shavit Y, Grace PM, Rice KC, Maier SF, Watkins LR. Exploring the neuroimmunopharmacology of opioids: an integrative review of mechanisms of central immune signaling and their implications for opioid analgesia. Pharmacol Rev. 2011;63:772–810. doi: 10.1124/pr.110.004135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jan WC, Chen CH, Hsu K, Tsai PS, Huang CJ. L-type calcium channels and mu-opioid receptors are involved in mediating the anti-inflammatory effects of naloxone. J Surg Res. 2011;167:263–272. doi: 10.1016/j.jss.2010.03.039. [DOI] [PubMed] [Google Scholar]
- Kenny EF, O'Neill LA. Signaling adaptors used by Toll-like receptors: an update. Cytokine. 2008;43:342–349. doi: 10.1016/j.cyto.2008.07.010. [DOI] [PubMed] [Google Scholar]
- Lewis SS, Hutchinson MR, Rezvani N, Loram LC, Zhang Y, Maier SF, et al. Evidence that intrathecal morphine-3-glucuronide may cause pain enhancement via toll-like receptor 4/MD-2 and interleukin-1beta. Neuroscience. 2010;165:569–583. doi: 10.1016/j.neuroscience.2009.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Du L, Kong LY, Hudson PM, Wilson BC, Chang RC, et al. Reduction by naloxone of lipopolysaccharide-induced neurotoxicity in mouse cortical neuron-glia co-cultures. Neuroscience. 2000a;97:749–756. doi: 10.1016/s0306-4522(00)00057-9. [DOI] [PubMed] [Google Scholar]
- Liu B, Du L, Hong JS. Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther. 2000b;293:607–617. [PubMed] [Google Scholar]
- Liu Y, Qin L, Wilson BC, An L, Hong JS, Liu B. Inhibition by naloxone stereoisomers of beta-amyloid peptide (1-42)-induced superoxide production in microglia and degeneration of cortical and mesencephalic neurons. J Pharmacol Exp Ther. 2002;302:1212–1219. doi: 10.1124/jpet.102.035956. [DOI] [PubMed] [Google Scholar]
- Loane DJ, Byrnes KR. Role of microglia in neurotrauma. Neurother. 2010;7:366–377. doi: 10.1016/j.nurt.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy MK, Tansey MG. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation. 2008;5:45–58. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninković J, Roy S. Role of the mu-opioid receptor in opioid modulation of immune function. Amino Acids. 2011 doi: 10.1007/s00726-011-1163-0. doi: 10.1007/s00726-011-1163-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill LA. The interleukin-1 receptor/Toll-like receptor superfamily: 10 years of progress. Immunol Rev. 2008;226:10–18. doi: 10.1111/j.1600-065X.2008.00701.x. [DOI] [PubMed] [Google Scholar]
- Rogers TJ, Peterson PK. Opioid G protein-coupled receptors: signals at the crossroads of inflammation. Trends Immunol. 2003;24:116–121. doi: 10.1016/s1471-4906(03)00003-6. [DOI] [PubMed] [Google Scholar]
- Roy S, Cain KJ, Chapin RB, Charboneau RG, Barke RA. Morphine modulates NF-κB activation in macrophages. Biochem Biophys Res Commun. 1998;245:392–396. doi: 10.1006/bbrc.1998.8415. [DOI] [PubMed] [Google Scholar]
- Ruzicka BB, Fox CA, Thompson RC, Meng F, Watson SJ, Akil H. Primary astroglial cultures derived from several rat brain regions differentially express mudelta and kappa opioid receptor mRNA. Brain Res Mol Brain Res. 1995;34:209–220. doi: 10.1016/0169-328x(95)00165-o. [DOI] [PubMed] [Google Scholar]
- Schild HO. The use of drug antagonists for the identification and classification of drugs. Br J Pharmacol. 1947;2:251–258. doi: 10.1111/j.1476-5381.1947.tb00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi G, Tergaonkar V. Potential pharmacological control of the NF-κB pathway. Trends Pharmacol Sci. 2009;30:313–321. doi: 10.1016/j.tips.2009.03.004. [DOI] [PubMed] [Google Scholar]
- Stevens CW. The evolution of vertebrate opioid receptors. Front Biosci. 2009;14:1247–1269. doi: 10.2741/3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stiene-Martin A, Zhou R, Hauser KF. Regional, developmental, and cell cycle-dependent differences in mudelta, and kappa-opioid receptor expression among cultured mouse astrocytes. Glia. 1998;22:249–259. [PMC free article] [PubMed] [Google Scholar]
- Wang X, Loram LC, Ramos K, de Jesus AJ, Thomas J, Cheng K, et al. Morphine activates neuroinflammation in a manner parallel to endotoxin. Proc Natl Acad Sci U S A. 2012;17:6325–6330. doi: 10.1073/pnas.1200130109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins LR, Hutchinson MR, Rice KC, Maier SF. The ‘toll’ of opioid-induced glial activation: improving the clinical efficacy of opioids by targeting glia. Trends Pharmacol Sci. 2009;30:581–591. doi: 10.1016/j.tips.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Li H, Li Y, Sun X, Zhu M, Hanley G, et al. Essential role of toll-like receptor 2 in morphine-induced microglia activation in mice. Neurosci Lett. 2011;489:43–47. doi: 10.1016/j.neulet.2010.11.063. [DOI] [PMC free article] [PubMed] [Google Scholar]