

Abstract
Background and Purpose
Palmitoylethanolamide (PEA) is an endogenous fatty acid amide displaying anti-inflammatory and analgesic actions. To investigate the molecular mechanism responsible for these effects, the ability of PEA and of pain-inducing stimuli such as capsaicin (CAP) or bradykinin (BK) to influence intracellular calcium concentrations ([Ca2+]i) in peripheral sensory neurons, has been assessed in the present study. The potential involvement of the transcription factor PPARα and of TRPV1 channels in PEA-induced effects was also studied.
Experimental Approach
[Ca2+]i was evaluated by single-cell microfluorimetry in differentiated F11 cells. Activation of TRPV1 channels was assessed by imaging and patch-clamp techniques in CHO cells transiently-transfected with rat TRPV1 cDNA.
Key Results
In F11 cells, PEA (1–30 μM) dose-dependently increased [Ca2+]i. The TRPV1 antagonists capsazepine (1 μM) and SB-366791 (1 μM), as well as the PPARα antagonist GW-6471 (10 μM), inhibited PEA-induced [Ca2+]i increase; blockers of cannabinoid receptors were ineffective. PEA activated TRPV1 channels heterologously expressed in CHO cells; this effect appeared to be mediated at least in part by PPARα. When compared with CAP, PEA showed similar potency and lower efficacy, and caused stronger TRPV1 currents desensitization. Sub-effective PEA concentrations, closer to those found in vivo, counteracted CAP- and BK-induced [Ca2+]i transients, as well as CAP-induced TRPV1 activation.
Conclusions and Implications
Activation of PPARα and TRPV1 channels, rather than of cannabinoid receptors, largely mediate PEA-induced [Ca2+]i transients in sensory neurons. Differential TRPV1 activation and desensitization by CAP and PEA might contribute to their distinct pharmacological profile, possibly translating into potentially relevant clinical differences.
Keywords: palmitoylethanolamide, transient receptor potential vanilloid type-1 channels, PPAR transcription factor, bradykinin, capsaicin, intracellular calcium concentrations, pain
Introduction
Palmitoylethanolamide (PEA) is a fatty acid amide provided of anti-inflammatory and analgesic actions (Lambert et al., 2002). Exogenous administration of PEA exerts potent anti-inflammatory actions, down-regulating the release of inflammatory mediators from various cell types such as mast cells, monocytes and macrophages (Di Marzo et al., 2000). Furthermore, a broad-spectrum analgesia induced by PEA has been documented in a variety of pain models, both of inflammatory (Calignano et al., 1998; Jaggar et al., 1998; Conti et al., 2002) and neuropathic (Helyes et al., 2003) origin, leading to several small-scale studies investigating the potential clinical efficacy of PEA in inflammatory and pain conditions in humans. Endogenous PEA levels are enhanced in many inflammation and pain models in vivo (Darmani et al., 2005), leading to the suggestion that PEA represents a naturally occurring component of an homeostatic system controlling the basal threshold of inflammation and pain (Levi-Montalcini et al., 1996).
Despite the potential clinical significance of PEA, the molecular pathways responsible for its pleiotropic actions are still a matter of debate. The structural and functional similarity between the endocannabinoid anandamide (AEA) and PEA suggested that these two lipid mediators might have cannabinoid receptors (CBRs) as a common molecular target; however, although in vivo evidence indicated the involvement of CB2R or CB2-like receptors (Jaggar et al., 1998; Conti et al., 2002), at least part of the neuroprotective or anti-inflammatory effects of PEA are insensitive to CBR antagonists (Lambert et al., 2002). Moreover, PEA, like oleoyl-ethanolamide (OEA) and stearoyl-ethanolamide, exhibits very little, if any, affinity for CB1 and CB2 receptors (Sheskin et al., 1997; Lambert et al., 1999).
PEA has been also proposed to enhance the activity and/or inhibit the degradation of endogenous agonists of CBRs, the so-called ‘entourage’ effect’ (Mechoulam et al., 1998). This would lead to a reinforcement of their actions at the level of several possible targets, which include the heat-activated transient receptor potential vanilloid type 1 (TRPV1) channels, the receptors for capsaicin (CAP) (Caterina et al., 1997). In fact, in TRPV1-transfected cells, PEA potentiates [Ca2+]i responses triggered by AEA, an effect attributed to its ability to increase TRPV1 affinity for AEA (De Petrocellis et al., 2001), rather than to the inhibition of its hydrolysis mediated by the fatty acid amide hydrolase (FAAH) (Smart et al., 2000; Vandevoorde et al., 2003).
In addition, LoVerme and coll. (2005) showed that PEA activates the PPARα transcription factor, and that the anti-inflammatory actions of PEA are abolished in mice deficient in PPARα, thereby suggesting PPARα as a relevant molecular target for at least some of PEA's actions. More recently, Ryberg et al., (2007) reported that acylethanolamides such as PEA and OEA activated the orphan GPCR GPR55, producing a potent stimulation of GTPγS binding, indicative of the activation of most heterotrimeric G-proteins. However, PEA does not increase [Ca2+]i in GPR55-expressing cells (Lauckner et al., 2008); therefore, the role of this receptor in the transduction of PEA's biological actions remains doubtful.
Given the complexity of the described scenario, in order to unveil the molecular mechanisms involved in PEA-induced analgesic effects, the aim of the present work has been to investigate the concentration-dependent effects of PEA on [Ca2+]i in peripheral sensory neurons and to study the functional interactions between PEA and pain-inducing stimuli such as CAP and bradykinin (BK). Moreover, the potential involvement of the transcription factor PPARα and of TRPV1 channels in PEA-induced effects was also studied; finally, a comparative investigation of the mechanism of action of PEA and CAP at the level of TRPV1 channels was also performed.
Methods
Cell cultures
F11 and CHO cells were grown in DMEM medium supplemented with 10% FBS, 100 U·mL−1 penicillin/streptomycin and 2 mM l-glutamine. The cells were kept in a humidified atmosphere at 37°C with 5% CO2 in 100 mm plastic Petri dishes. Differentiation of F11 cells was achieved by exposure for at least 72 h to a medium containing a lower FBS concentration (2%) and 10 μM retinoic acid (Raymon et al., 1999). For Ca2+ imaging, immunocytochemistry and electrophysiological experiments, F11 and CHO cells were plated on glass coverslips (Carolina Biological Supply Co., Burlington, NC) coated with poly-L-lysine (Sigma, Milan, Italy).
2.2. Transient transfections of CHO cells
Twenty-four hours after plating, CHO cells were transiently transfected with the rat TRPV1 receptor cDNA using Lipofectamine 2000 (Invitrogen, Milan, Italy), a cationic transfection reagent able to complex negatively charged nucleic acids and to form liposomes that fuse once in contact with the cell membrane, according to the manufacturer protocols. A plasmid encoding for the Enhanced Green Fluorescent Protein (Clontech, Palo Alto, CA) was used as a transfection marker; total cDNA in the transfection mixture was kept constant at 4 μg.
RNA extraction and semiquantitative PCR
Total RNA was isolated from undifferentiated or differentiated F11 cells using TRI-Reagent (Sigma-Aldrich, Milan, Italy), reacted with 0.1 U·μL−1 DNAse-I (Sigma-Aldrich) for 15 min at room temperature (RT; 20–22°C), followed by spectrophotometric quantification. Final preparation of RNA was considered DNA- and protein-free if the OD values ratio at 260/280 nm was >1.7. cDNA was synthesized by reverse transcription using 1–5 μg of isolated RNA as a template, 2.5 U·μL−1 of MuLV high-capacity reverse transcriptase (Applied Biosystem, Monza, Italy) in a buffer containing 4 mM dNTP mix, 2.5 μM Random Primers, 1 U·μL−1 RNAse Inhibitor at 37°C for 120 min. After MuLV reverse transcriptase inactivation (10 min incubation at 95°C), the cDNA obtained was amplified in PCR gold buffer, containing 1.5 mM MgCl2, 0.8 mM dNTP mix, 0.5–1 μM forward and reverse primers and 0.1–0.25 U·μL−1 Amplitaq Gold (Applied Biosystem, Monza, Italy). The PCR amplification protocol was: denaturation at 95°C for 1 min, annealing at 52°C for 1 min, elongation at 72°C for 1 min (30–35 cycles).
Immunocytochemistry
Differentiated F11 cells were serum-starved overnight, washed with PBS and incubated with PEA, WIN55,212 or vehicle (DMSO) for 10 min at 37°C. Cells were then fixed with 4% paraformaldehyde for 30 min, blocked for 1 hr in PBS containing 5% FBS and 0.1% Tween20 at RT, and incubated overnight at 4°C with a mouse monoclonal p-ERK1/2 antibody (1:100; Santa Cruz Biotechnology Inc., Santa Cruz, CA, USA; SC-7383). Immunoreactivity was detected with an anti-mouse CY3 IgG secondary antibody (1:100; Jackson ImmunoResearch Laboratories Inc., UK). The coverslips were dried, mounted in Vectashield (Vector Laboratories, Milan, Italy) and images acquired on an inverted Leica DM IRB fluorescence microscope (Leica, Milan, Italy).
[Ca2+]i measurements
F11 or CHO cells were plated on glass coverslips and loaded with 3 μM Fura-2 acetoxymethyl ester (Fura-2 AM) for 1 h at RT in darkness in a standard solution containing (in mM): NaCl 160, KCl 5.5, CaCl2 1.5, MgSO4 1.2, HEPES 10, glucose 10, pH 7.4 adjusted with NaOH. Thereafter, the coverslips were washed twice with PBS to remove extracellular dye and placed in a perfusion chamber onto the stage of an inverted Leica DM IRB fluorescence microscope equipped with a 40× oil objective lens. Cell were perfused throughout the experiments with a medium of the above-mentioned composition; when necessary, CaCl2 was omitted, plus the addition of 1 mM EDTA (Ca2+-free solution). Fluorescence images were acquired using a digital imaging system, composed of a coolSNAP ES camera (Roper Scientific, Ottobrunn, Germany), DeltaRAM X™Microscope Illuminator (Photon Technology International, Birmingham, NJ) and MetaFluor Imaging System software (Molecular Device, Sunnyvale, CA). Cells were alternatively illuminated at wavelengths of 340 nm and 380 nm by a 100 W Xenon lamp; the emitted light was passed through a 512-nm barrier filter. Fura-2 fluorescence was recorded every 3–4 sec, and fluorescence intensity values converted in Ca2+ concentrations assuming a Kd of 224 nM (Grynkiewicz et al., 1985). In each experiment, background fluorescence was recorded in a field devoid of cells and subtracted from the measured emission of each channel. Only cells with basal [Ca2+]i in the range of 90–120 nM were analysed.
Whole-cell electrophysiology
Currents from transiently transfected CHO (1 day post transfection) or from differentiated F11 cells were recorded at RT by whole-cell patch-clamp using an Axopatch 200A amplifier (Molecular Devices, Union City, CA) with glass micropipettes of 3–5 MΩ resistance. The extracellular solution contained (in mM): 138 NaCl, 2 CaCl2, 5.4 KCl, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4 with NaOH. When necessary, an extracellular solution without Ca2+ and containing 1 mM EDTA was used. The pipette (intracellular) solution contained (mM): 140 KCl, 2 MgCl2, 10 EGTA, 10 HEPES, 5 Mg-ATP, pH 7.4 with KOH. The pCLAMP software (version 10.2; Molecular Devices) was used for data acquisition and analysis. Linear cell capacitance (C) was determined by integrating the area under the whole-cell capacity transient, evoked by short (5–10 ms) pulses from −80 to −75 mV with the whole-cell capacitance compensation circuit of the Axopatch 200A turned off. Current densities (expressed in pA/pF) were calculated as peak of the agonist-evoked inward currents at −60 mV divided by C. Ligand-gated currents were recorded both by continuous monitoring at the holding membrane voltage (−60 mV), or by using voltage ramps (from −80 to +80 mV in 100 ms).
Drugs
PEA, GW-7647, WIN 55 212-2, SR141716A and SR144528 were a kind gift of Prof A Calignano (Faculty of Pharmacy, University of Naples, Naples, Italy). Capsaicin, capsazepine, clofibrate, nimodipine and bradykinin were from SIGMA; GW-6471 and SB-366791 were from Tocris Bioscience (Bristol, UK); ω-conotoxin and ω-agatoxin were from Alomone Labs (Jerusalem, Israel). Toxins and bradykinin were prepared in distilled water; capsaicin was prepared in absolute EtOH (final concentration ≤ 0.05%); all other drugs were dissolved in DMSO (final concentration ≤ 0.6%). In each experiment, the same volume of solvent used for tested drugs was added to the control solution.
Nomenclature used in this paper follows the 5th Edition of the British Journal of Pharmacology Guide to Receptors and Channels (Alexander et al., 2011).
Statistics
Data are expressed as the mean ± SEM. Statistically significant differences between the data (P < 0.05) were evaluated with the Student's t-test or by the anova, when multiple groups were compared.
Results
Depolarization-evoked [Ca2+]i responses in F11 cells
F11 cells, obtained by the fusion of embryonic rat dorsal root ganglion (DRG) neurons with mouse neuroblastoma cells (Platika et al., 1985), express neuronal markers (β-tubulin III, NF-160 or NeuN) and nociceptive markers (delta-opioid, prostaglandin and BK receptors) (Fan et al., 1992). F11 cells display the strongest responses to BK among neuronal cell lines (Vetter and Lewis, 2010), and synthesize and release a substance P-like compound (Francel et al., 1987; Raymon et al., 1999). Differentiated F11 cells exhibited a mature neuronal morphology, with many long neurites and an increased expression of BK receptors and voltage-gated calcium channels (VGCCs) (Francel et al., 1987). Depolarization with 20 mM extracellular K+ for 30 s failed to increase [Ca2+]i in undifferentiated cells (in 20/20 cells in three experimental sessions, [Ca2+]i was 90 ± 4 nM and 95 ± 5 nM, respectively, before and after 20 mM K+e exposure; P > 0.05), whereas it caused a fivefold increase in [Ca2+]i in 33.7% of differentiated F11 cells (28/83 cells) ([Ca2+]i was 130 ± 7 nM vs. 575 ± 57 nM, respectively, before and after 20 mM K+e exposure; P < 0.05). A lower response frequency (10.4% of cells; 30/288) and a smaller [Ca2+]i increase ([Ca2+]i was 92 ± 3 nM versus 371 ± 44 nM, respectively, before and after K+e exposure; P < 0.05) were observed when undifferentiated F11 cells were exposed to 60 mM [K+]e.
PEA evokes concentration-dependent increases in [Ca2+]i in F11 cells
In 36% of differentiated F11 cells (213/584 cells), a 30 s exposure to PEA (0.5–30 μM) elicited a concentration-dependent increase in [Ca2+]i (Figure 1A), which peaked at about 450% over basal levels at the highest concentration (30 μM) (460 ± 55 nM versus 96 ± 2 nM; n = 33; P < 0.05). PEA-induced [Ca2+]i enhancement showed an EC50 of 3.0 ± 0.1 μM (Figure 1B) and was fully reversible upon drug washout. The lowest effective concentration of PEA was 1 μM, whereas a concentration of 0.5 μM was ineffective. By contrast, in undifferentiated F11 cells, PEA (10 μM) evoked a smaller [Ca2+]i increase (160 ± 29 nM vs. 74 ± 4; n = 12; P < 0.05), in only 5% (12/256 cells) of cells.
Figure 1.
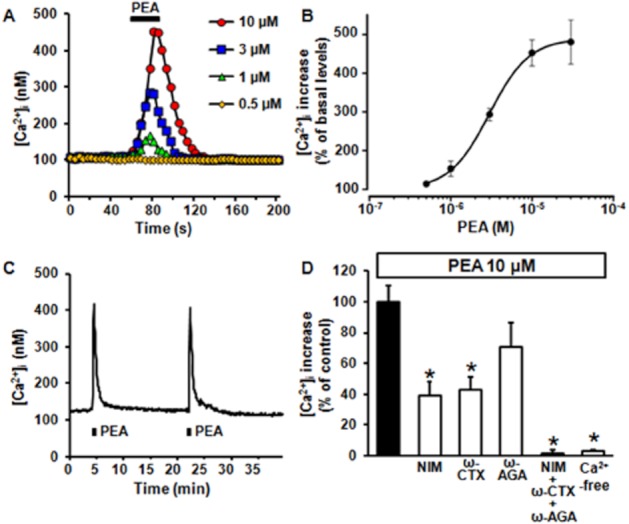
Concentration-dependent increase in [Ca2+]i elicited by PEA in differentiated F11 cells. (A) Representative traces showing the effect of PEA (0.5–10 μM) on [Ca2+]i in differentiated F11 cells. Each trace is from a single cell, representative of the entire population (n = 12–133 cells in at least three separate experimental sessions). In this and following panels and figures, the bar on the top or at the bottom of each trace corresponds to the duration of drug exposure. (B) Concentration-dependent effect of PEA. Peak [Ca2+]i values recorded in different cells after exposure to 0.5–30 μM PEA were expressed as percent of increase versus basal levels; the solid line represents the fit of the normalized data to a standard binding equation of the following form: y = max/(1 + x/EC50)n, where x is the drug concentration and n the Hill coefficient. Fitted values for n were 1.8 ± 0.1. (C) A representative trace showing the effect on [Ca2+]i of two successive 30 s exposures to PEA (10 μM), separated by a 15 min interval. (D) Quantification of the effects of a Ca2+-free extracellular solution, of the exposure to a pharmacological cocktail containing 10 μM NIM, 1 μM ω-CTX and 150 nM ω-AGA, or of 10 μM NIM, 1 μM ω-CTX, or 150 nM ω-AGA, applied separately on PEA-induced [Ca2+]i. Data are expressed as percent of [Ca2+]i increase induced by 10 μM PEA in standard Ca2+-containing solution. Each point is the mean±SEM of 20–32 separate determinations performed in at least three experimental sessions. In this and following figures, asterisks denote values statistically different from controls (P < 0.05).
In differentiated F11 cells, two 30 s exposures to 10 μM PEA separated by >15 min evoked identical [Ca2+]i increases ([Ca2+]i was 430 ± 58 nM and 412 ± 49 nM during the first and the second PEA exposure, respectively; n = 18; P > 0.05) (Figure 1C); therefore, to compare different pharmacological manipulations on PEA-induced [Ca2+]i responses, drugs were applied for 10 min before and during the second exposure to PEA, and removed subsequently. Perfusion of differentiated F11 cells with a Ca2+-free solution (plus 1 mM EDTA) or with a pharmacological cocktail containing 10 μM nimodipine (NIM), 1 μM ω-conotoxin GVIA (ω-CTX) and 150 nM ω-agatoxin IVA (ω-AGA), selective blockers of L-, N- and P/Q high voltage-activated (HVA) Ca2+ channel subtypes, respectively (Doering and Zamponi, 2003), completely prevented 10 μM PEA-induced [Ca2+]i increase. When each of the HVA Ca2+ channel blocker was applied individually, 10 μM NIM or 1 μM ω-CTX were found to inhibit 61 ± 9% (P < 0.05) or 57 ± 8% (P < 0.05), respectively, of PEA-evoked [Ca2+]i responses; by contrast, 150 M ω-agatoxin was ineffective (Figure 1D).
PEA-induced [Ca2+]i increase does not involve the activation of CBRs
In differentiated F11 cells, a 30 s exposure to the non-selective CBR agonist WIN 55 212-2 (WIN, 500 nM) (Showalter et al., 1996) increased [Ca2+]i (314 ± 33 nM vs. 100 ± 3 nM, n = 35; P < 0.05); a second exposure to the same WIN concentration after about 15 min elicited a comparable [Ca2+]i increase (310 ± 24 nM, n = 10; P > 0.05). The CB1R-selective antagonist SR141716A (SR1, 500 nM), but not the CB2R-selective antagonist SR144528 (SR2, 500 nM) or the removal of Ca2+e (plus 1 mM EDTA), abolished WIN-induced [Ca2+]i rise. By contrast, neither SR1 (500 nM) nor SR2 (500 nM) affected PEA-induced [Ca2+]i increase (Figure 2A ); in fact, PEA (10 μM)-induced [Ca2+]i peaks were 478 ± 66 nM and 423 ± 47 nM (n = 19; P > 0.05) in the absence and in the presence of SR1, respectively, and 405 ± 53 nM and 411 ± 50 nM (n = 19; P > 0.05), in the absence and in the presence of SR2 respectively. The percent of differentiated F11 cells responding to WIN (18%; 54/299) was lower than that responding to PEA; moreover, out of 74 cells recorded in four separate experimental sessions, 23 cells only responded to PEA, 11 responded only to WIN and only one cell responded to successive exposures to PEA and WIN, suggesting that the two drugs largely activated different F11 cell subpopulations.
Figure 2.
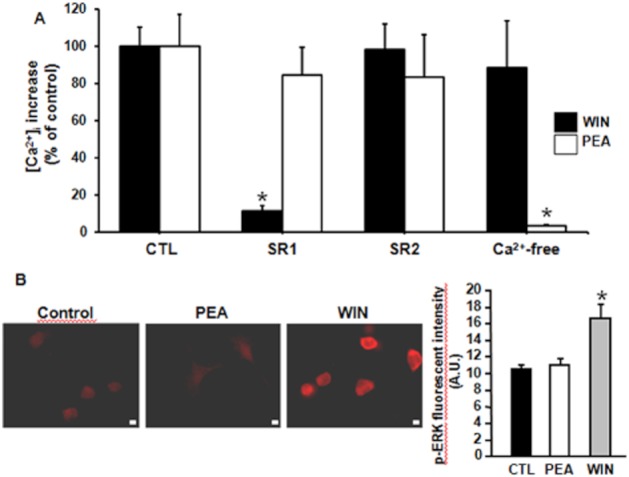
Effect of CB1R and CB2R antagonists on WIN- and PEA-induced [Ca2+]i increases in differentiated F11 cells. (A) Quantification of the effect of the CB1R-antagonist SR1 (500 nM), or of the CB2R-antagonist SR2 (500 nM), as well as of a Ca2+-free extracellular solution, on [Ca2+]i changes induced by the non-selective CBR agonist WIN 55 212-2 (WIN, 500 nM), or by PEA (10 μM). Data are expressed as percent of [Ca2+]i increase relative to respective controls (500 nM WIN or 10 μM PEA). Each point is the mean±SEM of 10–19 separate determinations performed in at least three experimental sessions. (B) pERK1/2 immunofluorescence in F11 cells in control conditions (left panel), or exposed for 10 min to PEA (10 μM; middle panel) or to WIN55,212 (500 nM; right panel). The scale bar is 10 μm. The rightmost panel shows the quantification of the data, expressed as arbitrary units (A. U.) of fluorescence intensity. Each bar is the mean of at least 4 fields from cells analysed in at least three different experimental sessions.
Finally, as with other CBR agonists (Daigle et al., 2008), a 10 min treatment of F11 cells with WIN (500 nM), but not with PEA (10 μM), increased ERK phosphorylation (Figure 2B).
Role of TRPV1 in PEA-induced [Ca2+]i increase
In differentiated F11 cells, RT-PCR experiments revealed the expression of transcripts encoding TRPV1, although high cycle numbers (35), primer concentration (1 μM), and Taq concentration (0.25 U·μL−1) were required (data not shown). In these cells, CAP (0.1–50 μM) induced a concentration-dependent increase in [Ca2+]i, which reached 563 ± 88 nM after a 30 s exposure to the highest concentration (50 μM) of the natural TRPV1 agonist (n = 30; P < 0.05 vs. basal values) (Figure 3A); the EC50 for CAP-induced [Ca2+]i increase was 1.6 ± 0.7 μM (Figure 3B). The percentage of differentiated F11 cells responding to CAP (40%; 105/260) was similar to that of cells responding to PEA (36%), and higher than that responding to WIN (18%). Moreover, 74% (14/19) of PEA-responding cells also responded to a subsequent CAP application.
Figure 3.
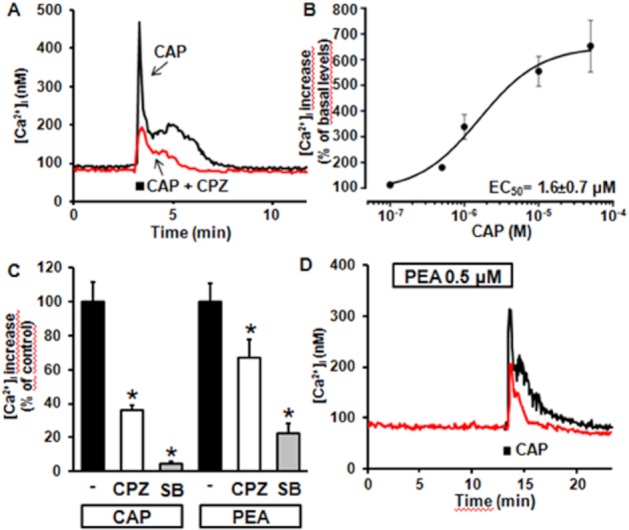
Effect of CAP on [Ca2+]i in differentiated F11 cells. (A) Superimposed representative traces showing the effect on [Ca2+]i of a single exposure to 50 μM CAP or to 50 μM CAP + 1 μM CPZ. (B) Concentration-dependent effect of CAP. Peak [Ca2+]i values recorded after exposure to 0.1–50 μM CAP were expressed as percent of increase versus basal levels; the solid line represents the fit of the normalized data to a standard binding equation of the following form: y = max/(1 + (x/EC50)n, where x is the drug concentration and n the Hill coefficient. Fitted values for n were 1.1 ± 0.4. Each point is the mean±SEM of 7–42 separate determinations performed in at least three experimental sessions. (C) Quantification of the effects of TRPV1 antagonists (CPZ and SB-366791; each at 1 μM) on CAP- or PEA-induced [Ca2+]i responses; data are expressed as percent of [Ca2+]i increase relative to respective controls (50 μM CAP or 10 μM PEA). Each point is the mean±SEM of 19–21 separate determinations performed in at least 3 experimental sessions. (D) Superimposed representative traces showing the effect on [Ca2+]i of a single exposure to 50 μM CAP in control conditions, or after 10 min pre-incubation with 0.5 μM PEA. Each trace is from a single cell, representative of the entire population (n = 35 for each experimental condition).
The kinetics of [Ca2+]i changes induced by CAP were markedly different from those elicited by high K+-induced depolarization and by PEA; in fact, although all three stimuli elicited [Ca2+]i responses, which rose rapidly and reached similar peak values, [Ca2+]i decay after stimulus removal was markedly slower for CAP, causing [Ca2+]i to remain higher than basal for several minutes (Figure 3A); similar differences in [Ca2+]i kinetics after CAP- or K+-induced depolarization also occur in primary DRG neurons (Dedov and Roufogalis, 2000).
CAP-induced [Ca2+]i responses were largely reduced by exposure to the TRPV1 antagonist capsazepine (CPZ) (Figure 3A and C), when used at a concentration (1 μM) and application times (<1 min) that do not affect VGCCs in DRG neurones (Docherty et al., 1997). SB-366791 (1 μM), a more potent and selective TRPV1 antagonist when compared with CPZ (Gunthorpe et al., 2004), largely inhibited CAP-induced [Ca2+]i increase in F11 cells (Figure 3C). Noteworthy, CPZ and SB-366791 also reduced PEA (10 μM)-induced [Ca2+]i increase (Figure 3C). Finally, exposure of differentiated F11 cells to a concentration of PEA (0.5 μM) unable to elicit [Ca2+]i responses per se, significantly blunted the peak and duration of [Ca2+]i increases triggered by CAP (50 μM) (Figure 3D) (peak [Ca2+]i was 196 ± 14 nM for CAP plus PEA vs. 343 ± 45 nM for CAP alone, n = 35; P < 0.05).
Regulation of [Ca2+]i by PPARα ligands
Exposure of differentiated F11 cells to the PPARα agonist clofibrate (CLO; 0.1–1 mM) caused a dose-dependent increase in [Ca2+]i; peak [Ca2+]i were 145 ± 27 nM and 417 ± 39 nM in the presence of 0.1 mM and 1 mM CLO respectively (n = 22–32; P < 0.05 for both conditions against respective basal values). The time-to-peak of [Ca2+]i enhancement by 1 mM CLO was 19 ± 1 s (n = 28); this value did not differ from that of PEA-induced responses (21 ± 1 s; n = 28). 1 mM CLO-induced [Ca2+]i increase was largely abolished upon perfusion with the PPARα antagonist GW-6471 (10 μM) (Xu et al., 2002), or with a Ca2+-free solution (plus 1 mM EDTA) (Figure 4A and C). GW-6471-induced blockade of CLO-induced [Ca2+]i increase was fully reversible upon drug washout; in fact, a third exposure to the same CLO concentration elicited a comparable increase in [Ca2+]i (peak [Ca2+]i were 454 ± 39 nM and 427 ± 39 nM in the first and third 1 mM CLO exposures; P > 0.05; n = 16) (Figure 4A). By contrast, GW-6471 (10 μM) failed to interfere with [Ca2+]i changes elicited by the TRPV1 agonist CAP (50 μM) (Figure 4C), suggesting that this PPARα antagonist did not act as a TRPV1 blocker in these experiments (see also below). GW-7647 (10 μM), a PPARα agonist more potent and selective than CLO (Brown et al., 2001), also triggered significant increases in [Ca2+]i in F11 cells; as with CLO, GW-6471 (10 μM) largely prevented GW-7647-induced [Ca2+]i rise (Figure 4C). Noteworthy, when used at 10 μM, the same concentration of GW-7647, CLO was ineffective (peak [Ca2+]i was 69 ± 4 nM; P > 0.05 vs. basal values; n = 15).
Figure 4.
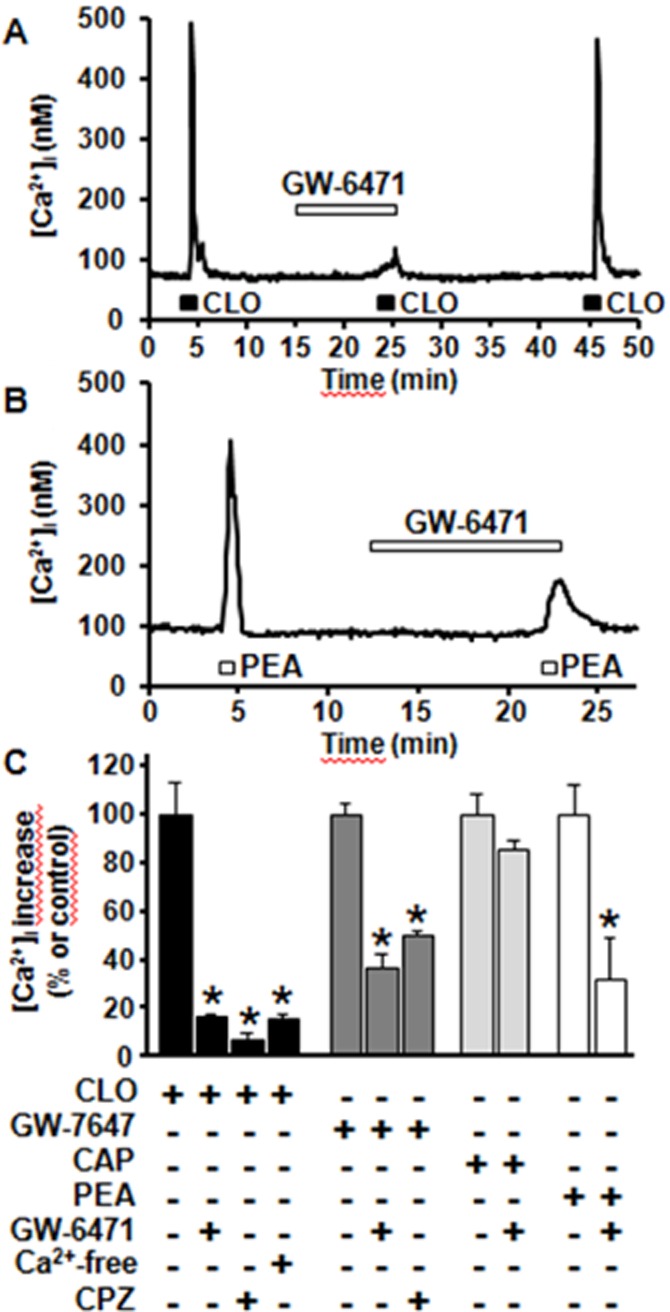
Regulation of [Ca2+]i by PPARα ligands. Representative traces showing the effect of 10 μM GW-6417 on CLO (1 mM)- (A) and PEA (10 μM)-induced (B) [Ca2+]i changes in differentiated F11 cells. The duration of GW-6417 exposure is indicated by the bar on top of the traces. (C) Quantification of the effects of 10 μM GW-6471, of a Ca2+-free extracellular solution, and of 1 μM CPZ on CLO-, GW-7647, CAP, and PEA-induced [Ca2+]i increase. Data are expressed as percent of [Ca2+]i increase relative to their respective controls (1 mM CLO; 10 μM GW-7647; 50 μM CAP; 10 μM PEA;). Each point is the mean±S.E.M. of 10–34 separate determinations performed in at least 3 experimental sessions.
PPARα-dependent modulation of [Ca2+]i in F11 cells appeared to involve the activation of TRPV1 channels; in fact, CPZ (1 μM) antagonized both CLO- and GW-7647-induced increases in [Ca2+]i (Figure 4C). Finally, the PPARα-selective antagonist GW-6471 (10 μM) reduced [Ca2+]i increase induced by PEA by 64% (Figure 4B and C), suggesting that the ability of this fatty acid amide to regulate [Ca2+]i largely depends on its PPARα agonistic properties.
Effect of PEA on BK-induced [Ca2+]i responses
Exposure of differentiated F11 cells to three subsequent BK (250 nM) exposures, each lasting 30 s and separated by >15 min, induced transient increases in [Ca2+]i having identical peak amplitudes ([Ca2+]i was 396 ± 73 nM, 381 ± 38 nM, and 382 ± 65 nM for the first, second and third stimulus, respectively; n = 20; P > 0.05) (Figure 5A). When the second exposure to BK was performed after the removal of extracellular Ca2+ (plus 1 mM EDTA; Figure 5C), [Ca2+]i peak was largely reduced ([Ca2+]i was 162 ± 10 nM in the presence of BK in Ca2+-free medium, and 427 ± 39 nM with BK in standard medium, n = 33; P < 0.05); noteworthy, BK-induced responses in the third pulse fully recovered after the removal of the Ca2+-free solution (Figure 5C). Pretreatment of differentiated F11 cells with the membrane-permeable Ca2+-chelator BAPTA-AM (50 μM) completely abolished BK-induced [Ca2+]i rise; in fact, peak [Ca2+]i during BK exposure were 430 ± 40 nM (n = 28) and 68 ± 1 nM (n = 67) in control- and BAPTA-AM-perfused cells, respectively (P < 0.05). Finally, BK-induced [Ca2+]i responses were reversibly reduced by upon co-application of CPZ (1 μM); peak [Ca2+]i was 198 ± 12 nM in the presence of BK plus CPZ versus 404 ± 39 nM with BK alone (P < 0.05, n = 32; Figure 5C). Pre-incubation for 10 min with a sub-effective concentration of PEA (0.5 μM) reversibly reduced BK-induced [Ca2+]i rise by about 25% ([Ca2+]i were 278 ± 19 nM in the presence of BK plus PEA, versus 353 ± 17 nM with BK alone; n = 34; P < 0.05) (Figure 5B and C), highlighting a functionally relevant antagonism between these two mediators.
Figure 5.
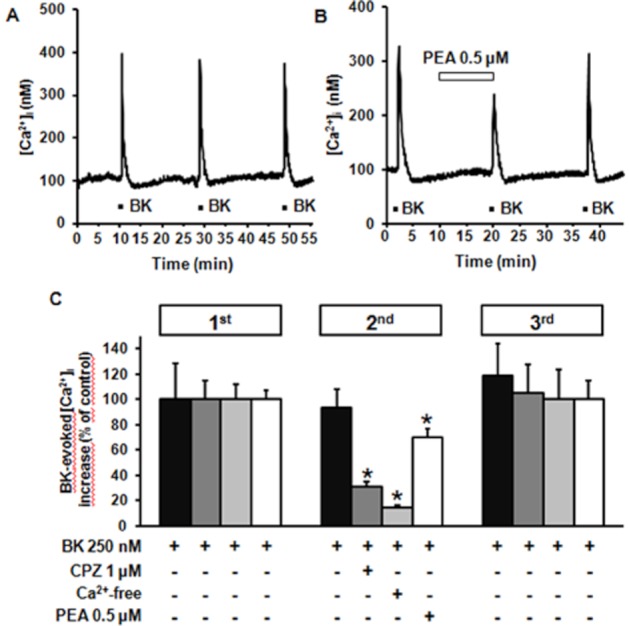
Effect of BK on [Ca2+]i in differentiated F11 cells. Representative traces showing the effect of three subsequent exposures to BK (250 nM) on [Ca2+]i in differentiated F11 cells obtained in control conditions (A) or after exposure to 0.5 μM PEA 10 min before and during the second BK exposure (B). (C) Quantification of the effects of 1 μM CPZ, of a Ca2+-free extracellular solution, and of 0.5 μM PEA on BK-induced [Ca2+]i increase (second BK exposure), and of drug washout during the third BK exposure. Data are expressed as percent of [Ca2+]i increase relative to controls (250 BK in the first pulse). Each point is the mean±SEM of 20–34 separate determinations performed in at least three experimental sessions.
Activation of TRPV1 channels by PEA
The data obtained provide pharmacological evidence suggesting that TRPV1 channels are involved in PEA-evoked [Ca2+]i responses in F11 cells. Thus, we attempted to measure CAP-induced currents in differentiated F11 cells by conventional patch-clamp recordings; however, very small (<100 pA) currents induced by 50 μM CAP were recorded in a minor fraction (<15%) of cells; among 20 cells tested, none responded to 10 μM PEA (data not shown). Thus, CHO cells transiently transfected with rat TRPV1 cDNA were used in subsequent imaging and electrophysiological experiments.
In TRPV1-transfected CHO cells, both PEA (10 μM) and CAP (5 μM) enhanced [Ca2+]i levels, an effect only observed in EGFP-positive, TRPV1-transfected cells and not in un-transfected, EGFP-negative cells (Figure 6A). In TRPV1-transfected cells, but not in non-transfected cells (data not shown), increasing concentrations of CAP (0.01–5 μM) dose-dependently activated large inward currents at −60 mV (typically of several nanoamperes at the highest CAP concentration) (Figure 6B). CAP-induced TRPV1 channel activation showed an EC50 of 0.36 ± 0.09 μM and an EMAX of 323 ± 54 pA/pF (Figure 5D). The TRPV1 antagonist CPZ (3 μM) largely abolished CAP-evoked currents (the current density was 218 ± 57 pA/pF in the presence of 0.5 μM CAP and 23 ± 10 pA/pF in the presence of 0.5 μM CAP plus 3 μM CPZ; n = 5; P < 0.05).
Figure 6.
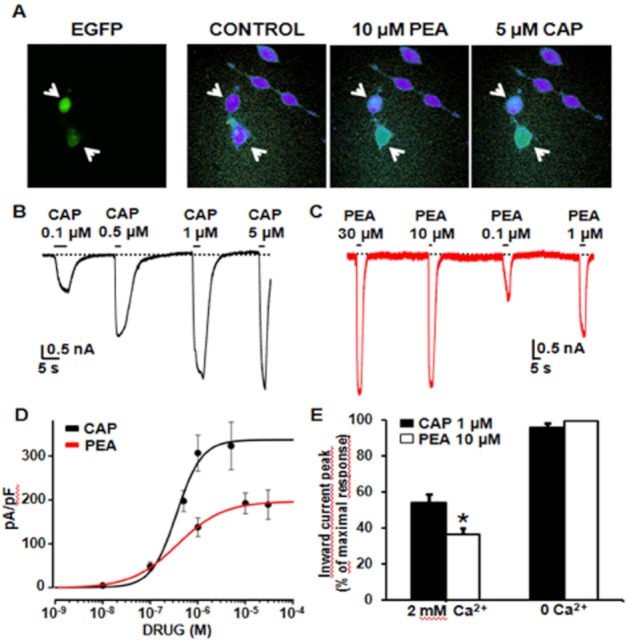
Activation of TRPV1 channels by PEA in TRPV1-transfected CHO cells. (A) Representative images showing PEA- (10 μM) and CAP- (5 μM) induced [Ca2+]i changes in CHO cells transiently-transfected with TRPV1 cDNA. To identify transfected cells, EGFP fluorescence was also monitored in the same microscopic field. Note that significant drug-induced [Ca2+]i changes only occurred in transfected (EGFP-positive) and not in non-transfeced (EGFP-negative) CHO cells. (B) Representative whole-cell inward currents from a TRPV1-transfected cell held at −60 mV and exposed to 0.1–5 μM CAP, as indicated. (C) Representative whole-cell inward currents from a TRPV1-transfected cell held at −60 mV and exposed to 0.1–30 μM PEA, as indicated. (D) Quantitative comparison of TRPV1 activation by CAP and PEA. Peak current data were expressed as pA/pF (to facilitate comparison among cells of different sizes), and expressed as a function of agonist concentrations. The solid lines represent fits of the experimental data to the following binding isotherm: y = max/(1 + x/EC50)n, where x is the drug concentration and n the Hill coefficient. The fitted values for n were 1.6 ± 0.6 for CAP, and 0.9 ± 0.1 for PEA. Each point is the mean±SEM. of 6–26 (for CAP) or 9–23 (for PEA) determinations. (E) Effect of the removal of Ca2+e on 1 μM CAP- or 10 μM PEA-induced peak inward current at −60 mV. Each point is the mean±SEM of 12–15 determinations.
In TRPV1-transfected cells, but not in un-transfected cells, PEA (0.01–30 μM) also induced the appearance of large inward currents (Figure 6C), which were sensitive to the TRPV1 antagonist CPZ (3 μM) (the current density was 227 ± 28 pA/pF in the presence of 10 μM PEA, and 1 ± 1 pA/pF in the presence of 10 μM PEA plus 3 μM CPZ; n = 6; P < 0.05). When compared with CAP, PEA-evoked TRPV1 currents showed a similar EC50 (0.37 ± 0.05 μM), but a smaller maximal amplitude (Figure 6D). Since TRPV1 channel gating (Koplas et al., 1997) and permeation (Samways and Egan, 2011) are highly dependent on [Ca2+]e, we studied whether the observed differences between CAP- and PEA-induced TRPV1 current activation were influenced by [Ca2+]e. Ca2+e removal increased by about 50% the maximal density of 1 μM CAP-induced TRPV1 currents; this effect was even more pronounced with PEA (10 μM). As a result, in the absence of Ca2+e, CAP and PEA triggered inward currents of identical amplitudes (Figure 6E).
PPARα-mediated activation of TRPV1 channels
As previously described, the PPARα agonists CLO and GW-7647 increased [Ca2+]i in F11 cells, an effect antagonized not only by the by PPARα antagonist GW-6471, but also by CPZ, a result indicating that PPARα activation might facilitate TRPV1 opening. To validate this hypothesis and to investigate whether PEA-induced TRPV1 activation involved PPARα, the effects of PPARα ligands on TRPV1 currents were studied in TRPV1-transfected cells. As shown in Figure 6A and D, CAP-induced TRPV1 currents were unaffected by GW-6471 (10 μM), a result confirming that this latter compound did not act as a TRPV1 antagonist (see also Figure 4C). On the other hand, CLO (100 μM) evoked significant TRPV1-mediated currents, an effect almost completely blocked not only by CPZ (3 μM), but also by the PPARα antagonist GW-6471 (10 μM) (Figure 7B and D). Finally, GW-6471 (10 μM) also inhibited about 50% of PEA-induced TRPV1 currents (Figure 7C and D), suggesting that the activation of PPARα mediated a significant part of TRPV1 opening triggered by this endogenous fatty acid amide.
Figure 7.
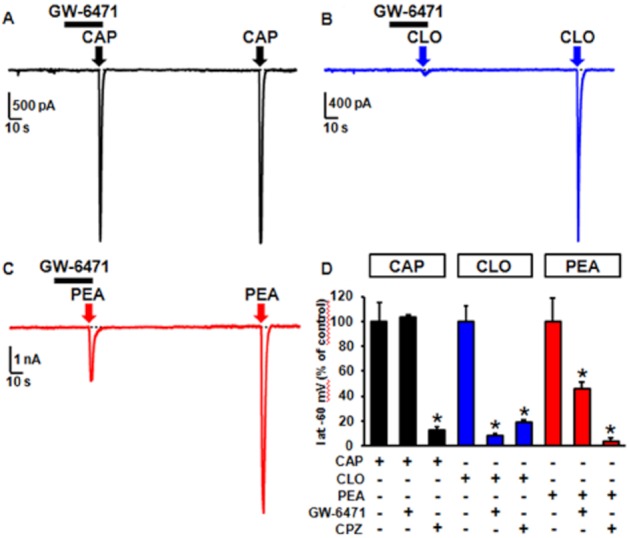
PPARα-mediated activation of TRPV1 channels by CLO and PEA, but not by CAP. Representative whole-cell current traces evoked in the same TRPV1-expressing CHO cell showing the effect of the PPARα antagonist GW-6471 (10 μM) on TRPV1 currents elicited by CAP (5 μM) (A), CLO (100 μM) (B), and PEA (10 μM) (C). CHO cells were held at −60 mV; the bars on top of each panel represent the duration of GW-6471 exposure; in each panel, the arrow indicates the addition of the agonist (exposure time: about 3 s). (D) Quantification of the effects of GW-6471 (10 μM) and of CPZ (3 μM) on CAP-, CLO-, and PEA-evoked peak TRPV1 currents. Each point is the mean±S.E.M. of 8–16 determinations for each agonist.
TRPV1 channel desensitization by CAP and PEA: implications for functional antagonism
To study the biophysical and pharmacological properties of CAP- and PEA-induced TRPV1 currents, TRPV1-transfected CHO cells were exposed to agonists during the application of voltage ramps (from −80 to +80 mV; 1.6 mV/ms) delivered at a frequency of 0.5 Hz. 1 μM CAP-induced currents showed outward rectification (+80 mV/−80 mV peak ratio = 1.5 ± 0.2) and reversed at −1.1 ± 1.6 mV (n = 8). In the continuous presence of CAP, TRPV1 currents were reduced by about 55 ± 4% (n = 9) of their peak value after 30 s of agonist exposure, indicative of agonist-dependent TRPV1 desensitization. TRPV1-mediated currents returned to basal values when CAP was removed from the extracellular solution (Figure 8A). In the same cells, PEA (10 μM) induced the appearance of currents having similar rectification properties (+80 mV/−80 mV peak ratio = 1.9 ± 0.3; n = 5; P > 0.05) and reversal potential (−1.4 ± 2.1 mV; n = 8; P > 0.05) when compared with those elicited by CAP, although their maximal amplitude was about 50% smaller (Figure 8B). PEA-induced TRPV1 currents showed an higher extent of desensitization when compared with CAP-induced currents, decreasing by 79 ± 3% (n = 21; P < 0.05 vs. CAP) after 30 s of continuous PEA exposure (Figure 8B). As expected, the same differences between CAP and PEA were also observed in experiments in which agonist-induced currents were continuously monitored at −60 mV (Figure 8C).
Figure 8.
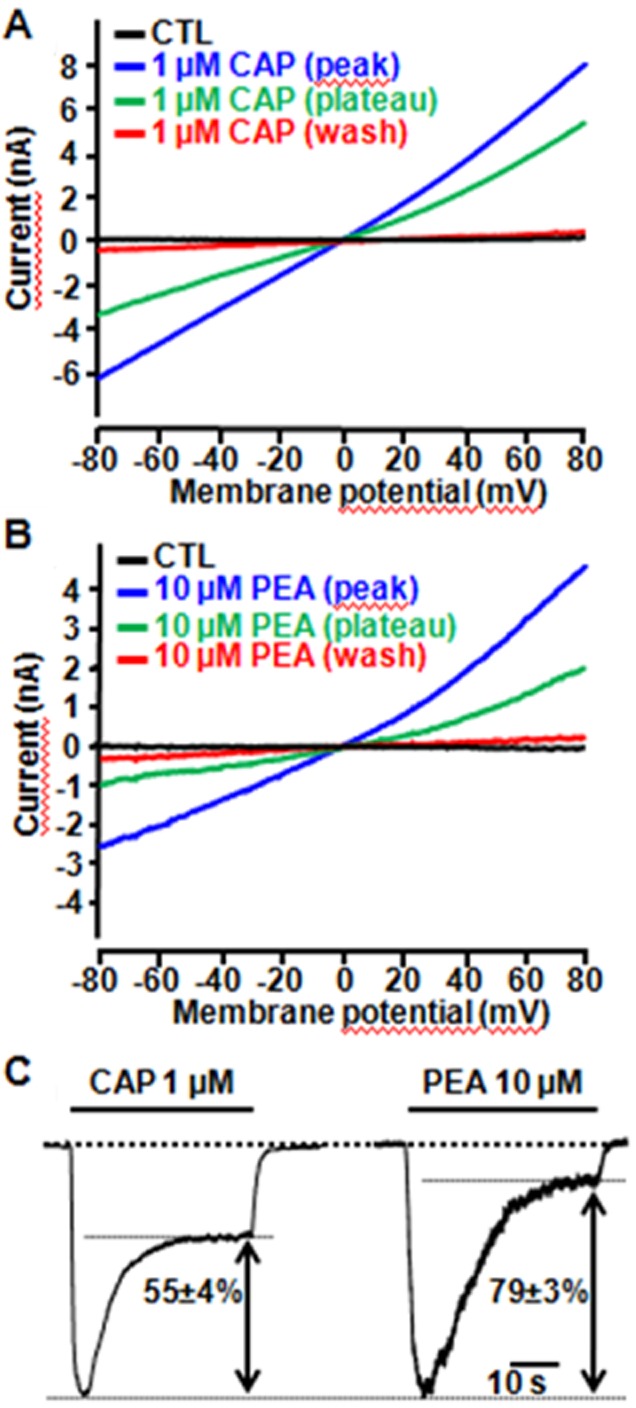
Desensitization of TRPV1-mediated currents by CAP and PEA. (A, B) Representative whole-cell current traces evoked in the same TRPV1-expressing CHO cell upon 30 s exposure to 1 μM CAP (A) or to 10 μM PEA (B), followed by drug washout; currents were recorded using a voltage ramp protocol (from −80 to +80 mV; 1.6 mV/ms). Plotted traces are: control (no agonist), peak of agonist-evoked currents, plateau of agonist-evoked currents (after 30 s of agonists exposure) and washout, as indicated. (C) Representative whole-cell inward currents evoked in TRPV1-expressing CHO cells held at −60 mV during 30 s exposures to 1 μM CAP or to 10 μM PEA, as indicated. The percentage of the current desensitized during exposure to each agonist is indicated. The bars on the top of each trace correspond to the duration of agonist exposure.
Two consecutive 0.5 μM CAP applications, each lasting only 3 s to avoid significant desensitization of TRPV1-mediated currents, separated by a 1 min interval, triggered TRPV1 currents of identical magnitudes (first: −3.3 ± 0.4 nA; second: −3.4 ± 0.4 nA; n = 4; P > 0.05) (Figure 9A); by contrast, when the second CAP exposure was preceded by a 60 s exposure to 0.01 μM PEA, a concentration which was unable to trigger measurable TRPV1-mediated currents per se, TRPV1 currents evoked by the second CAP application were reduced by 19 ± 3% (n = 12; P < 0.05) (Figure 9B).
Figure 9.
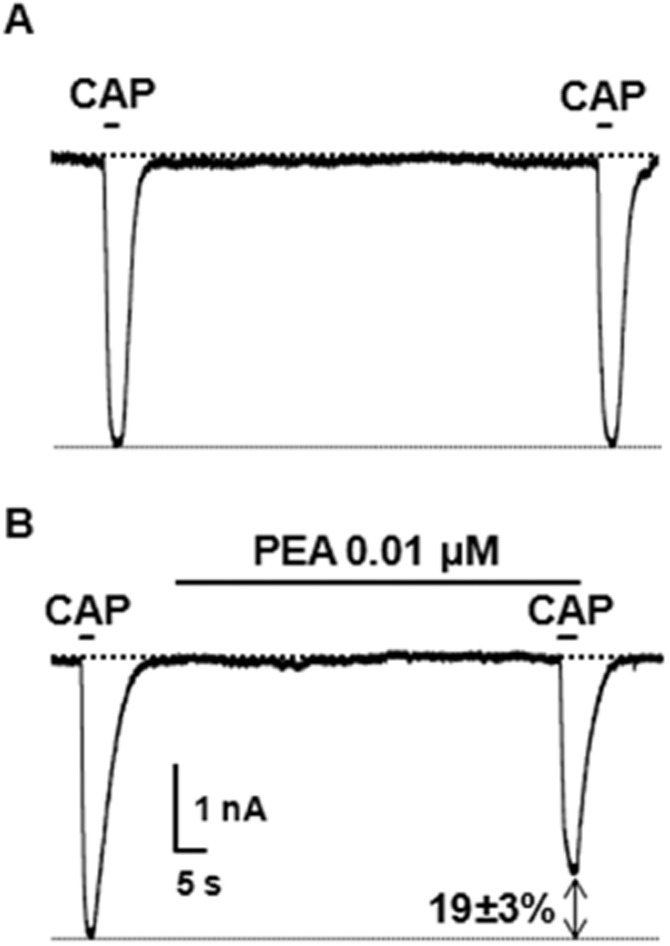
Exposure to sub-effective PEA concentrations reduce CAP-induced TRPV1 currents. Representative whole-cell current traces recorded in TRPV1-expressing CHO cells held at −60 mV in response to two subsequent 3 s exposures to 0.5 μM CAP, separated by a 1 min interval. The second CAP exposure was performed in control condition (n = 4; A), or upon pre-incubation with 0.01 μM PEA for 1 min (n = 12; B). The bars on the top of each trace correspond to the duration of drug exposures. Each trace is from a single cell, representative of the entire population. The percentage of current decrease induced by PEA exposure is indicated.
Discussion
The present results reveal that the endogenous analgesic and anti-inflammatory ethanolamide PEA dose-dependently (EC50 ∼3 μM) increased [Ca2+]i in F11 cells, a widely used cellular model for nociceptive molecular mechanisms. Although PEA-induced changes in [Ca2+]i have been previously observed in sperm cells (Ambrosini et al., 2005) and in TRPV1-transfected cells (Smart et al., 2002), the ability of PEA to regulate [Ca2+]i in neuronal cells has not been thoroughly investigated. CB1R and CB2R are both expressed in F11 cells (Ross et al., 2001); however, their activation does not seem to contribute to PEA-induced changes in [Ca2+]i in F11 cells. Furthermore, the fact that the percentage of PEA-responding cells is higher than that responding to WIN, and that the largest fraction of PEA-responding cells was not activated by the CB1R agonist, also argues against PEA acting via CB1R. Moreover, as previously reported (Liu et al., 2009), CB1R activation triggered [Ca2+]i transients in F11 cells, which were insensitive to the omission of Ca2+e, but rather depended on the release of Ca2+ from intracellular stores. By contrast, PEA-induced [Ca2+]i transients were completely abolished in the absence of Ca2+e, suggesting the activation of Ca2+e entry pathways; these seem to be mostly provided by L- and N-subtypes of VGCCs, which underlie most of the sustained high-voltage activated current in F11 cells (Boland and Dingledine, 1990). PEA does not interact directly with VGCCs when these are activated directly by membrane depolarization (Oz et al., 2005; Yoshihara et al., 2006). By contrast, in F11 cells, VGCC opening appears consequent to PEA-induced membrane depolarization triggered by TRPV1 activation. In fact, the TRPV1-antagonists CPZ and SB-366791 (Gunthorpe et al., 2004), largely reduced PEA-induced [Ca2+]i increase. Moreover, the percentage of F11 cells responding to PEA was similar to that responding to the TRPV1 agonist CAP, and most cells showing robust [Ca2+]i increases after PEA exposure also responded to CAP, strongly suggesting the TRPV1 channels participated in [Ca2+]i regulation by CAP and PEA.
These results delineate a potential molecular mechanism for PEA-induced [Ca2+]i increases, in which the activation of TRPV1 channels by this ethanolamide triggers membrane depolarization leading to a substantial enhancement of [Ca2+]i. The fact that VGCC antagonists fully abolished PEA-induced [Ca2+]i increase suggests that VGCC opening amplifies the TRPV1-initiated [Ca2+]i signal, and that, in F11 cells, TRPV1 expression is too low to generate significant Ca2+ fluxes directly contributing to PEA-induced [Ca2+]i enhancement. Although this result appears in contrast with the high Ca2+ permeability of TRPV1 channels (Caterina et al., 1997), Ca2+ ions only contribute to around 4% of total TRPV1 currents at negative (resting) membrane potentials under physiological Na+ and Ca2+ concentrations (Zeilhofer et al., 1997).
The idea that TRPV1 channels, which appear as primary mediators of pain and associated responses (Moran et al., 2011), act as transducers of some PEA effects is not novel. De Petrocellis and coll. (2001) showed that PEA reinforced [Ca2+]i increases prompted by AEA in TRPV1-transfected cells, an effect interpreted as a consequence of a PEA-induced enhancement of AEA binding to TRPV1. Later experiments also revealed that, in TRPV1-transfected cells, PEA was able to enhance [Ca2+]i also in the absence of AEA, leading to the concept that PEA acted as a partial agonist at TRPV1 receptors (Smart et al., 2002). Noteworthy, in these experiments, none of the other unsaturated ethanolamides as well as OEA, a monosaturated compound, shared PEA's ability to increase [Ca2+]i per se; moreover, although all these compounds effectively increased the cellular levels of AEA through the inhibition of the metabolizing enzyme FAAH (the so-called ‘entourage effect’; Mechoulam et al., 1998), no obvious correlation was found between FAAH inhibition and TRPV1 receptor activation (Smart et al., 2002; Vandevoorde et al., 2003). On the other hand, TRPV1 channel activation also mediates PEA-induced potentiation of the vasorelaxant responses triggered by AEA in rat mesenteric arteries (Ho et al., 2008), and calcitonin gene-related peptide release from spinal cord slices (Tognetto et al., 2001).
Recent evidence suggest that PEA acts as a PPARα agonist, and that PPARα activation participates in the anti-inflammatory actions of this naturally occurring acylethanolamide (LoVerme et al., 2005). The present result showing that the pharmacological inhibition of PPARα prevented [Ca2+]i increases triggered by PEA suggests that this lipid-activated transcription factor significantly contributes to PEA-induced Ca2+ signalling in F11 cells. An effective coupling of PPARα activation to [Ca2+]i signalling in F11 cells is also suggested by the ability of the PPARα agonists CLO and GW-7647 to enhance [Ca2+]i in these cells, an effect blocked by the PPARα antagonist GW-6471. Similar effects have also been shown in pancreatic β-cells, where PPARα agonists regulated [Ca2+]i signals via a rapid, non-genomic mechanism; in these cells, the same Authors reported that a significant fraction of the PPARα protein is located at the plasmamembrane (Ropero et al., 2009). More recently, changes in the endogenous levels of PEA have been shown to alter the acute [Ca2+]i responses evoked by depolarizing conditions in sensory neurons (Khasabova et al., 2012); although, also in this study, [Ca2+]i regulation by PEA is largely mediated by PPARα, the underlying signalling pathway appears independent on gene transcription.
The ability of CPZ to counteract [Ca2+]i changes induced by PEA as well as by CLO and GW-7647 provides pharmacological evidence for TRPV1 channels acting as crucial mediators of PPARα-dependent [Ca2+]i signalling in sensory neurons. To demonstrate that PEA-induced [Ca2+]i changes involved the activation of TRPV1 channels, and to provide direct evidence for PEA-induced TRPV1 activation, imaging and electrophysiological experiments were performed in TRPV1 transiently-transfected CHO cells (which express significant levels of endogenous PPARα; Gearing et al., 1993), to circumvent the limitation imposed by the low expression of TRPV1 channels in F11 cells (Goswami et al., 2010). In transfected cells, PEA, similarly to CAP, caused a significant increase in [Ca2+]i levels, indicating that TRPV1 over-expression allows to directly monitor Ca2+ fluxes mediated by heterologously expressed channels, without the need for VGCCs-induced signal amplification (as in F11 cells). Electrophysiological experiments also revealed that TRPV1-transfected cells displayed large CAP-activated outwardly rectifying currents that could be triggered also by PEA, as well as by the PPARα agonist CLO. The fact that GW-6471, a PPARα antagonist, which did not affect TRPV1 channels, significantly blocked TRPV1 currents elicited by PEA and CLO raises the intriguing hypothesis that a PPARα-dependent facilitation of TRPV1 channel opening mediates a large part of [Ca2+]i regulation by these compounds in sensory neurons.
By contrast, previous results obtained by Ca2+-imaging experiments in human TRPV1 stably (Appendino et al., 2009) or transiently transfected (Movahed et al., 2005) HEK-293 cells failed to reveal a significant activation of TRPV1 channels by PEA. However, it seems likely that differences among various heterologous expression systems or TRPV1 species, which are known to influence TRPV1 channel expression levels (Sándor et al., 2005), biophysical properties (Lázár et al., 2003), regulation by cytoplasmic mediators (Vellani et al., 2001) and pharmacology (Papakosta et al., 2011), might be responsible for this discrepancy. Moreover, the fact that, also in electrophysiological experiments, PEA was unable to activate rat TRPV1 heterologously expressed in HEK-293 cells (Zygmunt et al., 1999), in addition to the above-mentioned variables, could also be explained by the fact that, in these experiments, Cs+ was used as the main charge carrier cation, and that TRPV1 activation by different agonists exhibits a distinct ionic selectivity (Chung et al., 2008).
When compared with CAP, PEA activated TRPV1 channel with a similar potency (EC50 ∼0.4 μM), but a lower maximal efficacy, in line with its partial agonist activity. Although several mechanisms may lie behind this phenomenon (Pliska, 1999), agonist-dependent differences in TRPV1 channel desensitization appear to provide a plausible explanation. In fact, TRPV1 channels undergo significant ligand-dependent desensitization, both during continuous (acute desensitization) and after repeated stimulation (tachyphylaxis) (Vyklický et al., 2008). TRPV1 desensitization is highly dependent on both agonist type (Xu et al., 2005) and Ca2+e ions availability (Koplas et al., 1997), being largely abolished by the removal of the divalent cation from the extracellular medium. In keeping with this hypothesis, our results revealed that PEA and CAP displayed similar TRPV1-activating efficacy in the absence of Ca2+e. Moreover, in the presence of physiological [Ca2+]e, PEA-induced TRPV1 currents showed more tendency to desensitize when compared to CAP-induced currents.
Desensitization of TRPV1 channels appears independent on agonist concentrations, being only poorly coupled to the activation process; as a matter of fact, Koplas et al. (1997) showed that CAP desensitized a substantial fraction of TRPV1-mediated currents even at concentrations well below those causing significant TRPV1 channel opening. Our results show that PEA, when used at concentrations lower than those able to open TRPV1 channels per se, significantly reduced TRPV1 currents induced by CAP. This result might also explain the antagonistic effect exerted by low PEA concentrations on [Ca2+]i transients evoked in F11 cells by noxious stimuli, such as CAP or BK. Whether differences in hydrophobicity between CAP and PEA (xlogP3 is 3.6 for CAP and 6.2 for PEA; PubChem database) (Cheng et al., 2007) underlie this differential tendency to induce TRPV1 channel desensitization is yet unknown. Moreover, the potential role of a PPARα-dependent pathway, which would mediate at least part of TRPV1 activation by PEA and might contribute to channel desensitization, also deserves further investigation.
Altogether, these results highlight PEA-induced TRPV1 desensitization as a potential molecular mechanism underlying the analgesic actions of this acylethanolamide. In fact, low concentrations of this compound, presumably closer to those naturally found in the plasma (around 10–20 pmol·mL−1) (Zolese et al., 2005; Jean-Gilles et al., 2009), might prompt significant desensitization of TRPV1 channels in sensory neurons, thereby impeding their activation by endogenous algogenic compounds such as BK, whose nociceptive effects are known to be largely mediated by TRPV1 (Shin et al., 2002; Ferreira et al., 2004), as well as by plant vanilloid CAP. Noteworthy, this mechanism might also participate in PPARα-dependent regulation of tumor-evoked pain by endogenous levels of PEA (Khasabova et al., 2012). Moreover, an increased Ca2+-dependent synthesis of endogenous AEA has been shown to contribute to TRPV1 activation by Gq/11-coupled receptors (van der Stelt et al., 2005) such as BK receptors; the fact that chelation of Ca2+i with BAPTA completely abolishes BK-induced [Ca2+]i transient seems in line with such a mechanism.
Differences in TRPV1 desensitization between PEA and CAP might translate into potentially relevant clinical differences. In fact, topical treatment with CAP for chronic pain is associated with significant adverse events, mainly represented by sensory (burning, stinging) or autonomic (erythema) disturbances, leading to treatment discontinuation (Mason et al., 2004). It seems reasonable to hypothesize that treatment with PEA, by prompting a higher degree of TRPV1 desensitization when compared with CAP, might be associated with a lower incidence of such adverse events. A similar mechanism has been postulated to explain the potential antinociceptive and analgesic actions of camphor (Xu et al., 2005), and of the non-pungent analogue of capsaicin N-palmitoyl-vanillamide (palvanil) (De Petrocellis et al., 2011).
Acknowledgments
We acknowledge the contribution of Dr Davide Viggiano (Department of Health Sciences, University of Molise, Campobasso, Italy) and of Dr Giovanna Cacciola (Institute of Biology, Second University of Naples, Naples, Italy) for help with immunofluorescence experiments, and of Prof Antonio Calignano (Dept. of Experimental Pharmacology, Faculty of Pharmacy, University of Naples Federico II, Naples, Italy) for the generous gift of compounds used in this study, and for helpful comments throughout the study. TRPV1 cDNA was a kind gift of Prof B. Attali, University of Tel Aviv (IL).
This study was supported by grants from: the Fondazione San Paolo – IMI (Project Neuroscience), Regione Molise (Convenzione AIFA/Regione Molise), and the Italian Ministry for University (PRIN 2007 and 2009).
Glossary
Abbreviations
- ω-AGA
ω-agatoxin IVA
- ω-CTX
ω-conotoxin GVIA
- AEA
anandamide
- BK
bradykinin
- Ca2+
calcium
- CAP
capsaicin
- CBRs
cannabinoid receptors
- CLO
clofibrate
- CPZ
capsazepine
- DM
differentiation medium
- DRG
dorsal root ganglia
- FAAH
fatty acid amide hydrolase
- GM
growth medium
- K+
potassium
- NIM
nimodipine
- OD
optical density
- PEA
palmitoylethanolamide
- PPARα
peroxisome proliferator-activated receptor α
- RT
room temperature
- RT-PCR
reverse-transcription PCR
- TRPV1
transient receptor potential vanilloid type-1
- VGCCs
voltage-gated calcium channels
Conflict of interest
The authors declare no conflict of interest.
References
- Alexander SPH, Mathie A, Peters JA. Guide to receptors and channels (GRAC), 5th edn. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambrosini A, Zolese G, Ambrosi S, Bertoli E, Mantero F, Boscaro M, et al. Idiopathic infertility: effect of palmitoylethanolamide (a homologue of anandamide) on hyperactivated sperm cell motility and Ca2+ influx. J Androl. 2005;26:429–436. doi: 10.2164/jandrol.04141. [DOI] [PubMed] [Google Scholar]
- Appendino G, Ligresti A, Minassi A, Cascio MG, Allarà M, Taglialatela-Scafati O, et al. Conformationally constrained fatty acid ethanolamides as cannabinoid and vanilloid receptor probes. J Med Chem. 2009;52:3001–3009. doi: 10.1021/jm900130m. [DOI] [PubMed] [Google Scholar]
- Boland LM, Dingledine R. Multiple components of both transient and sustained barium currents in a rat dorsal root ganglion cell line. J Physiol. 1990;420:223–245. doi: 10.1113/jphysiol.1990.sp017909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown PJ, Stuart LW, Hurley KP, Lewis MC, Winegar DA, Wilson JG, et al. Identification of a subtype selective human PPARalpha agonist through parallel-array synthesis. Bioorg Med Chem Lett. 2001;11:1225–1227. doi: 10.1016/s0960-894x(01)00188-3. [DOI] [PubMed] [Google Scholar]
- Calignano A, La Rana G, Giuffrida A, Piomelli D. Control of pain initiation by endogenous cannabinoids. Nature. 1998;394:277–281. doi: 10.1038/28393. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Cheng T, Zhao Y, Li X, Lin F, Xu Y, Zhang X, et al. Computation of octanol-water partition coefficients by guiding an additive model with knowledge. J Chem Inf Model. 2007;47:2140–2148. doi: 10.1021/ci700257y. [DOI] [PubMed] [Google Scholar]
- Chung MK, Güler AD, Caterina MJ. TRPV1 shows dynamic ionic selectivity during agonist stimulation. Nat Neurosci. 2008;11:555–564. doi: 10.1038/nn.2102. [DOI] [PubMed] [Google Scholar]
- Conti S, Costa B, Colleoni M, Parolaro D, Giagnoni G. Antiinflammatory action of endocannabinoid palmitoylethanolamide and the synthetic cannabinoid nabilone in a model of acute inflammation in the rat. Br J Pharmacol. 2002;135:181–187. doi: 10.1038/sj.bjp.0704466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle TL, Kearn CS, Mackie K. Rapid CB1 cannabinoid receptor desensitization defines the time course of ERK1/2 MAP kinase signaling. Neuropharmacology. 2008;54:36–44. doi: 10.1016/j.neuropharm.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmani NA, Izzo AA, Degenhardt B, Valenti M, Scaglione G, Capasso R, et al. Involvement of the cannabimimetic compound, N-palmitoyl-ethanolamine, in inflammatory and neuropathic conditions: review of the available pre-clinical data, and first human studies. Neuropharmacology. 2005;48:1154–1163. doi: 10.1016/j.neuropharm.2005.01.001. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Davis JB, Di Marzo V. Palmitoylethanolamide enhances anandamide stimulation of human vanilloid VR1 receptors. FEBS Lett. 2001;506:253–256. doi: 10.1016/s0014-5793(01)02934-9. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Guida F, Moriello AS, De Chiaro M, Piscitelli F, de Novellis V, et al. N-palmitoyl-vanillamide (palvanil) is a non-pungent analogue of capsaicin with stronger desensitizing capability against the TRPV1 receptor and anti-hyperalgesic activity. Pharmacol Res. 2011;63:294–299. doi: 10.1016/j.phrs.2010.12.019. [DOI] [PubMed] [Google Scholar]
- Dedov VN, Roufogalis BD. Mitochondrial calcium accumulation following activation of vanilloid (VR1) receptors by capsaicin in dorsal root ganglion neurons. Neuroscience. 2000;95:183–188. doi: 10.1016/s0306-4522(99)00423-6. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, De Petrocellis L, Bisogno T. Cannabimimetic fatty acid derivatives in cancer and inflammation. Prostaglandins Other Lipid Mediat. 2000;61:43–61. doi: 10.1016/s0090-6980(00)00054-x. [DOI] [PubMed] [Google Scholar]
- Docherty RJ, Yeats JC, Piper AS. Capsazepine block of voltage-activated calcium channels in adult rat dorsal ganglion neurons in culture. Br J Pharmacol. 1997;121:1461–1467. doi: 10.1038/sj.bjp.0701272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering CJ, Zamponi GW. Molecular pharmacology of high voltage-activated calcium channels. J Bioenerg Biomembr. 2003;35:491–505. doi: 10.1023/b:jobb.0000008022.50702.1a. [DOI] [PubMed] [Google Scholar]
- Fan SF, Shen KF, Scheideler MA, Crain SM. F11 neuroblastoma x DRG neuron hybrid cells express inhibitory mu- and delta-opioid receptors which increase voltage-dependent K+ currents upon activation. Brain Res. 1992;590:329–333. doi: 10.1016/0006-8993(92)91116-v. [DOI] [PubMed] [Google Scholar]
- Ferreira J, da Silva GL, Calixto JB. Contribution of vanilloid receptors to the overt nociception induced by B2 kinin receptor activation in mice. Br J Pharmacol. 2004;141:787–794. doi: 10.1038/sj.bjp.0705546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francel PC, Harris K, Smith M, Fishman MC, Dawson G, Miller RJ. Neurochemical characteristics of a novel dorsal root ganglion X neuroblastoma hybrid cell line, F-11. J Neurochem. 1987;48:1624–1631. doi: 10.1111/j.1471-4159.1987.tb05711.x. [DOI] [PubMed] [Google Scholar]
- Gearing KL, Göttlicher M, Teboul M, Widmark E, Gustafsson JA. Interaction of the peroxisome-proliferator-activated receptor and retinoid X receptor. Proc Natl Acad Sci U S A. 1993;90:1440–1444. doi: 10.1073/pnas.90.4.1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goswami C, Rademacher N, Smalla KH, Kalscheuer V, Ropers HH, Gundelfinger ED, et al. TRPV1 acts as a synaptic protein and regulates vesicle recycling. J Cell Sci. 2010;123:2045–2057. doi: 10.1242/jcs.065144. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gunthorpe MJ, Rami HK, Jerman JC, Smart D, Gill CH, Soffin EM, et al. Identification and characterisation of SB-366791, a potent and selective vanilloid receptor (VR1/TRPV1) antagonist. Neuropharmacology. 2004;46:133–149. doi: 10.1016/s0028-3908(03)00305-8. [DOI] [PubMed] [Google Scholar]
- Helyes Z, Nemeth J, Than M, Bolcskei K, Pinter E, Szolcsanyi J. Inhibitory effect of anandamide on resiniferatoxin-induced sensory neuropeptide release in vivo and neuropathic hyperalgesia in the rat. Life Sci. 2003;73:2345–2353. doi: 10.1016/s0024-3205(03)00651-9. [DOI] [PubMed] [Google Scholar]
- Ho WS, Barrett DA, Randall MD. ‘Entourage’ effects of N-palmitoylethanolamide and N-oleoylethanolamide on vasorelaxation to anandamide occur through TRPV1 receptors. Br J Pharmacol. 2008;155:837–846. doi: 10.1038/bjp.2008.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaggar SI, Hasnie FS, Sellaturay S, Rice AS. The anti-hyperalgesic actions of the cannabinoid anandamide and the putative CB2 receptor agonist palmitoylethanolamide in visceral and somatic inflammatory pain. Pain. 1998;76:189–199. doi: 10.1016/s0304-3959(98)00041-4. [DOI] [PubMed] [Google Scholar]
- Jean-Gilles L, Feng S, Tench CR, Chapman V, Kendall DA, Barrett DA, et al. Plasma endocannabinoid levels in multiple sclerosis. J Neurol Sci. 2009;287:212–215. doi: 10.1016/j.jns.2009.07.021. [DOI] [PubMed] [Google Scholar]
- Khasabova IA, Xiong Y, Coicou LG, Piomelli D, Seybold V. Peroxisome proliferator-activated receptor α mediates acute effects of palmitoylethanolamide on sensory neurons. J Neurosci. 2012;32:12735–12743. doi: 10.1523/JNEUROSCI.0130-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koplas PA, Rosenberg RL, Oxford GS. The role of calcium in the desensitization of capsaicin responses in rat dorsal root ganglion neurons. J Neurosci. 1997;17:3525–3537. doi: 10.1523/JNEUROSCI.17-10-03525.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert DM, DiPaolo FG, Sonveaux P, Kanyonyo M, Govaerts SJ, Hermans E, et al. Analogues and homologues of N-palmitoylethanolamide, a putative endogenous CB(2) cannabinoid, as potential ligands for the cannabinoid receptors. Biochim Biophys Acta. 1999;1440:266–274. doi: 10.1016/s1388-1981(99)00132-8. [DOI] [PubMed] [Google Scholar]
- Lambert DM, Vandevoorde S, Jonsson KO, Fowler CJ. The palmitoylethanolamide family: a new class of anti-inflammatory agents? Curr Med Chem. 2002;9:663–674. doi: 10.2174/0929867023370707. [DOI] [PubMed] [Google Scholar]
- Lauckner JE, Jensen JB, Chen HY, Lu HC, Hille B, Mackie K. GPR55 is a cannabinoid receptor that increases intracellular calcium and inhibits M current. Proc Natl Acad Sci U S A. 2008;105:2699–2704. doi: 10.1073/pnas.0711278105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázár J, Szabó T, Kovács L, Blumberg PM, Bíró T. Distinct features of recombinant rat vanilloid receptor-1 expressed in various expression systems. Cell Mol Life Sci. 2003;60:2228–2240. doi: 10.1007/s00018-003-3230-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini R, Skaper SD, Dal Toso R, Petrelli L, Leon A. Nerve growth factor: from neurotrophin to neurokine. Trends Neurosci. 1996;19:514–520. doi: 10.1016/S0166-2236(96)10058-8. [DOI] [PubMed] [Google Scholar]
- Liu Q, Bhat M, Bowen WD, Cheng J. Signaling pathways from CB1 receptor activation to inhibition of NMDA-mediated calcium influx and neurotoxicity in dorsal root ganglion neurons. J Pharmacol Exp Ther. 2009;331:1062–1070. doi: 10.1124/jpet.109.156216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LoVerme J, Fu J, Astarita G, La Rana G, Russo R, Calignano A, et al. The nuclear receptor peroxisome proliferator-activated receptor-alpha mediates the anti-inflammatory actions of palmitoylethanolamide. Mol Pharmacol. 2005;67:15–19. doi: 10.1124/mol.104.006353. [DOI] [PubMed] [Google Scholar]
- Mason L, Moore RA, Derry S, Edwards JE, McQuay HJ. Systematic review of topical capsaicin for the treatment of chronic pain. BMJ. 2004;328:991. doi: 10.1136/bmj.38042.506748.EE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Fride E, Di Marzo V. Endocannabinoids. Eur J Pharmacol. 1998;359:1–18. doi: 10.1016/s0014-2999(98)00649-9. [DOI] [PubMed] [Google Scholar]
- Moran MM, McAlexander MA, Bíró T, Szallasi A. Transient receptor potential channels as therapeutic targets. Nat Rev Drug Discov. 2011;10:601–620. doi: 10.1038/nrd3456. [DOI] [PubMed] [Google Scholar]
- Movahed P, Jönsson BA, Birnir B, Wingstrand JA, Jørgensen TD, Ermund A, et al. Endogenous unsaturated C18 N-acylethanolamines are vanilloid receptor (TRPV1) agonists. J Biol Chem. 2005;280:38496–38504. doi: 10.1074/jbc.M507429200. [DOI] [PubMed] [Google Scholar]
- Oz M, Alptekin A, Tchugunova Y, Dinc M. Effects of saturated long-chain N-acylethanolamines on voltage-dependent Ca2+ fluxes in rabbit T-tubule membranes. Arch Biochem Biophys. 2005;434:344–351. doi: 10.1016/j.abb.2004.11.010. [DOI] [PubMed] [Google Scholar]
- Papakosta M, Dalle C, Haythornthwaite A, Cao L, Stevens EB, Burgess G, et al. The chimeric approach reveals that differences in the TRPV1 pore domain determine species-specific sensitivity to block of heat activation. J Biol Chem. 2011;286:39663–396672. doi: 10.1074/jbc.M111.273581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Platika D, Boulos MH, Baizer L, Fishman MC. Neuronal traits of clonal cell lines derived by fusion of dorsal root ganglia neurons with neuroblastoma cells. Proc Natl Acad Sci U S A. 1985;82:3499–3503. doi: 10.1073/pnas.82.10.3499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pliska V. Partial agonism: mechanisms based on ligand-receptor interactions and on stimulus-response coupling. J Recept Signal Transduct Res. 1999;19:597–629. doi: 10.3109/10799899909036675. [DOI] [PubMed] [Google Scholar]
- Raymon HK, Thode S, Zhou J, Friedman GC, Pardinas JR, Barrere C, et al. Immortalized human dorsal root ganglion cells differentiate into neurons with nociceptive properties. J Neurosci. 1999;19:5420–5428. doi: 10.1523/JNEUROSCI.19-13-05420.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropero AB, Juan-Picó P, Rafacho A, Fuentes E, Bermúdez-Silva FJ, Roche E, et al. Rapid non-genomic regulation of Ca2+ signals and insulin secretion by PPAR alpha ligands in mouse pancreatic islets of Langerhans. J Endocrinol. 2009;200:127–138. doi: 10.1677/JOE-08-0397. [DOI] [PubMed] [Google Scholar]
- Ross RA, Coutts AA, McFarlane SM, Anavi-Goffer S, Irving AJ, Pertwee RG, et al. Actions of cannabinoid receptor ligands on rat cultured sensory neurones: implications for antinociception. Neuropharmacology. 2001;40:221–232. doi: 10.1016/s0028-3908(00)00135-0. [DOI] [PubMed] [Google Scholar]
- Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–1101. doi: 10.1038/sj.bjp.0707460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samways DS, Egan TM. Calcium-dependent decrease in the single-channel conductance of TRPV1. Pflugers Arch. 2011;462:681–691. doi: 10.1007/s00424-011-1013-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sándor Z, Varga A, Horváth P, Nagy B, Szolcsányi J. Construction of a stable cell line uniformly expressing the rat TRPV1 receptor. Cell Mol Biol Lett. 2005;10:499–514. [PubMed] [Google Scholar]
- Sheskin T, Hanus L, Slager J, Vogel Z, Mechoulam R. Structural requirements for binding of anandamide-type compounds to the brain cannabinoid receptor. J Med Chem. 1997;40:659–667. doi: 10.1021/jm960752x. [DOI] [PubMed] [Google Scholar]
- Shin J, Cho H, Hwang SW, Jung J, Shin CY, Lee SY, et al. Bradykinin-12-lipoxygenase-VR1 signaling pathway for inflammatory hyperalgesia. Proc Natl Acad Sci U S A. 2002;99:10150–10155. doi: 10.1073/pnas.152002699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Showalter VM, Compton DR, Martin BR, Abood ME. Evaluation of binding in a transfected cell line expressing a peripheral cannabinoid receptor (CB2): identification of cannabinoid receptor subtype selective ligands. J Pharmacol Exp Ther. 1996;278:989–999. [PubMed] [Google Scholar]
- Smart D, Gunthorpe MJ, Jerman JC, Nasir S, Gray J, Muir AI, et al. The endogenous lipid anandamide is a full agonist at the human vanilloid receptor (hVR1) Br J Pharmacol. 2000;129:227–230. doi: 10.1038/sj.bjp.0703050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart D, Jonsson KO, Vandevoorde S, Lambert DM, Fowler CJ. ‘Entourage’ effects of N-acyl ethanolamines at human vanilloid receptors. Comparison of effects upon anandamide-induced vanilloid receptor activation and upon anandamide metabolism. Br J Pharmacol. 2002;136:452–458. doi: 10.1038/sj.bjp.0704732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Stelt M, Trevisani M, Vellani V, De Petrocellis L, Schiano Moriello A, Campi B, et al. Anandamide acts as an intracellular messenger amplifying Ca2+ influx via TRPV1 channels. EMBO J. 2005;24:3026–3037. doi: 10.1038/sj.emboj.7600784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tognetto M, Amadesi S, Harrison S, Creminon C, Trevisani M, Carreras M, et al. Anandamide excites central terminals of dorsal root ganglion neurons via vanilloid receptor-1 activation. J Neurosci. 2001;21:1104–1109. doi: 10.1523/JNEUROSCI.21-04-01104.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandevoorde S, Lambert DM, Smart D, Jonsson K-O FCJ. N-Morpholino- and N-Diethyl-analogues of Palmitoylethanolamide increase the sensitivity of transfected human vanilloid receptors to activation by anandamide without affecting fatty acid amidohydrolase activity. Bioorg Med Chem. 2003;11:817–825. doi: 10.1016/s0968-0896(02)00567-9. [DOI] [PubMed] [Google Scholar]
- Vellani V, Mapplebeck S, Moriondo A, Davis JB, McNaughton PA. Protein kinase C activation potentiates gating of the vanilloid receptor VR1 by capsaicin, protons, heat and anandamide. J Physiol. 2001;534:813–825. doi: 10.1111/j.1469-7793.2001.00813.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vetter I, Lewis RJ. Characterization of endogenous calcium responses in neuronal cell lines. Biochem Pharmacol. 2010;79:908–920. doi: 10.1016/j.bcp.2009.10.020. [DOI] [PubMed] [Google Scholar]
- Vyklický L, Nováková-Tousová K, Benedikt J, Samad A, Touska F, Vlachová V. Calcium-dependent desensitization of vanilloid receptor TRPV1: a mechanism possibly involved in analgesia induced by topical application of capsaicin. Physiol Res. 2008;57:S59–S68. doi: 10.33549/physiolres.931478. [DOI] [PubMed] [Google Scholar]
- Xu H, Blair NT, Clapham DE. Camphor activates and strongly desensitizes the transient receptor potential vanilloid subtype 1 channel in a vanilloid-independent mechanism. J Neurosci. 2005;25:8924–8937. doi: 10.1523/JNEUROSCI.2574-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, Cobb JE, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARalpha. Nature. 2002;415:813–817. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- Yoshihara S, Morimoto H, Ohori M, Yamada Y, Abe T, Arisaka O. Cannabinoid receptor agonists inhibit Ca(2+) influx to synaptosomes from rat brain. Pharmacology. 2006;76:157–162. doi: 10.1159/000091228. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU, Kress M, Swandulla D. Fractional Ca2+ currents through capsaicin- and proton-activated ion channels in rat dorsal root ganglion neurones. J Physiol. 1997;503:67–78. doi: 10.1111/j.1469-7793.1997.067bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zolese G, Bacchetti T, Ambrosini A, Wozniak M, Bertoli E, Ferretti G. Increased plasma concentrations of palmitoylethanolamide, an endogenous fatty acid amide, affect oxidative damage of human low-density lipoproteins: an in vitro study. Atherosclerosis. 2005;182:47–55. doi: 10.1016/j.atherosclerosis.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Zygmunt PM, Petersson J, Andersson DA, Chuang H, Sørgård M, Di Marzo V, et al. Vanilloid receptors on sensory nerves mediate the vasodilator action of anandamide. Nature. 1999;400:452–457. doi: 10.1038/22761. [DOI] [PubMed] [Google Scholar]