

Abstract
Background and Purpose
The Sigma-1 receptor (Sig1R) impacts on calcium ion signalling and has a plethora of ligands. This study investigated Sig1R and its ligands in relation to endogenous calcium events of endothelial cells and transient receptor potential (TRP) channels.
Experimental Approach
Intracellular calcium and patch clamp measurements were made from human saphenous vein endothelial cells and HEK 293 cells expressing exogenous human TRPC5, TRPM2 or TRPM3. Sig1R ligands were applied and short interfering RNA was used to deplete Sig1R. TRP channels tagged with fluorescent proteins were used for subcellular localization studies.
Key Results
In endothelial cells, 10–100 μM of the Sig1R antagonist BD1063 inhibited sustained but not transient calcium responses evoked by histamine. The Sig1R agonist 4-IBP and related antagonist BD1047 were also inhibitory. The Sig1R agonist SKF10047 had no effect. Sustained calcium entry evoked by VEGF or hydrogen peroxide was also inhibited by BD1063, BD1047 or 4-IBP, but not SKF10047. 4-IBP, BD1047 and BD1063 inhibited TRPC5 or TRPM3, but not TRPM2. Inhibitory effects of BD1047 were rapid in onset and readily reversed on washout. SKF10047 inhibited TRPC5 but not TRPM3 or TRPM2. Depletion of Sig1R did not prevent the inhibitory actions of BD1063 or BD1047 and Sig1R did not co-localize with TRPC5 or TRPM3.
Conclusions and Implications
The data suggest that two types of Sig1R ligand (BD1047/BD1063 and 4-IBP) are inhibitors of receptor- or chemically activated calcium entry channels, acting relatively directly and independently of the Sig1R. Chemical foundations for TRP channel inhibitors are suggested.
Keywords: calcium channels, cationic channels, TRP channels, endothelial cells, Sigma-1 receptor, histamine, hydrogen peroxide, vascular endothelial growth factor
Introduction
Endothelial cells line the inner walls of blood vessels to provide physiological regulation of blood pressure (Aird, 2007). The cells also mediate elements of vascular adaptation during adverse events such as oxidative stress and inflammation, with hydrogen peroxide (H2O2) and histamine acting as modifiers of endothelial properties (Xu and Touyz, 2006; Aird, 2007). Endothelial cells also provide the basis for new vessel growth during development or in adult life (Carmeliet, 2005; Aird, 2007; Folkman, 2007). New growth from pre-existing blood vessels (angiogenesis) has pivotal roles in physiology and enables tumour expansion, driving research efforts to develop novel anticancer agents through inhibition of angiogenesis (Carmeliet, 2005; Folkman, 2007). A key extracellular signalling molecule in physiological and tumour angiogenesis is VEGF, which acts via VEGF receptors of the receptor tyrosine kinase superfamily (Olsson et al., 2006). Clinical trials have shown benefit of VEGF antagonism in cancer treatment, albeit with problems of resistance and limited efficacy (Bergers and Hanahan, 2008).
The calcium ion (Ca2+) plays critical roles in intracellular signalling and is considered to be a dominant intracellular messenger (Berridge, 2003; Clapham, 2007). The intracellular Ca2+ concentration is profoundly influenced by Ca2+ influx across the plasma membrane, which is controlled dynamically by multiple Ca2+ channels that vary in type between different cells. The Ca2+ has diverse effects spanning short and long timescales, for example, evoking or modulating neurotransmitter release, cytokine secretion, muscle contraction, migration, proliferation, apoptosis and gene expression (Berridge et al., 2003). In the endothelium, Ca2+ has roles, for example, in the regulation of nitric oxide synthase and vascular permeability (Nilius and Droogmans, 2001). Extracellular agonists that evoke such effects include histamine, ATP and acetylcholine acting via GPCRs. H2O2 also evokes Ca2+ signals in endothelial cells, acting relatively directly on redox-sensitive plasma membrane Ca2+ channels to evoke Ca2+ entry (Yoshida et al., 2006; Hecquet et al., 2008). There is a suggested relevance of Ca2+ signalling in VEGF responses through phospholipase C-γ, inositol 1,4,5-trisphosphate-evoked Ca2+ release and DAG (Faehling et al., 2002; Jho et al., 2005). There is evidence for VEGF-evoked Ca2+ entry mediated by transient receptor potential (TRP) and Orai1 CRAC channels (Faehling et al., 2002; Jho et al., 2005; Hamdollah Zadeh et al., 2008; Li et al., 2011).
In mammals, there are 28 genes encoding TRP proteins, most of which are capable of forming homo- or heteromultimeric Ca2+ and/or Na+-permeable channels (Damann et al., 2008; Venkatachalam and Montell, 2007). The channels are often stimulated by chemical and physical factors. One subtype is the canonical TRPs (TRPCs). Specific physiological stimulators are not known. Instead, there are multiple relatively non-specific stimulators that include GPCR agonists, oxidized phospholipids, redox factors, acidification and toxic metal ions (Jiang et al., 2011). Another subtype of the TRP channels is the melastatin (TRPM) type. Multiple but less diverse stimuli have been suggested to impinge on these channels, including H2O2 and pregnenolone sulphate (PregS) (Grimm et al., 2005; Wagner et al., 2008; Jiang et al., 2011). Involvement in endothelial cell functions has been suggested for TRPC and TRPM channels (Jho et al., 2005; Yoshida et al., 2006; Hecquet et al., 2008; Ge et al., 2009). There is relatively limited information on the pharmacology of the channels and subtype- or isoform-specific small-molecule inhibitors are mostly lacking. Known inhibitory modulators of TRPC or TRPM channels include 2-aminoethoxydiphenyl borate, flufenamic acid, resveratrol, diethylstilbestrol, ethyl-1-(4-(2,3,3-trichloroacrylamide)phenyl)-5-(trifluoromethyl)-1H-pyrazole-4-carboxylate and N-(p-amylcinnamoyl) anthranilic acid (Jiang et al., 2011; Naylor et al., 2011).
Sigma receptors were initially suggested to be a type of opioid receptor but are now known to be distinct, rather enigmatic, membrane proteins of two types: the Sigma-1 receptor (Sig1R) and Sigma-2 receptor (Monnet, 2005; Maurice and Su, 2009; Su et al., 2010). Most is known about Sig1R. A defining characteristic is diverse pharmacology, with a catalogue of chemicals binding the protein, including opioids (e.g. pentazocine), steroids (e.g. PregS) and antipsychotic drugs (e.g. haloperidol) (Monnet, 2005; Maurice and Su, 2009). While there has been lack of clarity about the biological function of Sig1R, it has quite consistently been associated with Ca2+ signalling, both at intracellular Ca2+ compartments and the plasma membrane through regulation of ion channels (Monnet, 2005). Intracellular function is suggested to arise at mitochondrion-associated endoplasmic reticulum membrane interfaces (Su et al., 2010). There has also been lack of clarity about which substances are agonists and which are antagonists at Sig1R because of uncertainty about Sig1R's functional roles. However, Sig1R agonists are considered to include SKF10047 and 4-IBP, and Sig1R antagonists BD1047 and BD1063 (see Figure 1 for the chemical structures). Sig1R is widely expressed in mammalian cell types, but there has been little or no attention to its relevance in endothelial cells or other cardiovascular cells. A few recent studies have, however, suggested importance in cardiovascular biology, including effects on endothelial nitric oxide synthase (Tagashira et al., 2010).
Figure 1.

Inhibition of histamine-induced Ca2+ entry in endothelial cells by Sig1R ligands. (A) Two-dimensional chemical structures of four Sig1R ligands. (B–F) Intracellular Ca2+ measurement data from SVECs. (B–C) Example of data showing effects of 10 μM histamine in the presence of 100 μM BD1047 (B) or 1–100 μM BD1063 (C) compared with vehicle controls (N = 4 for each). The ligands were applied 30 min before testing histamine and maintained throughout the experiments. (D–F) Mean data for the types of experiment illustrated in (B, C), showing analysis of the sustained (5 min) response to 10 μM histamine, 100 μg·mL−1 VEGF, or 1 mM H2O2 in the presence of 100 μM of the Sig1R ligand indicated (n/N = 3/12 for each experiment). The 4-IBP experiments have a separate vehicle control because they used DMSO rather than water as the solvent.
In this study, we investigated whether Sig1R might have importance for endothelial cell Ca2+ signalling by exploring whether there are effects of Sig1R ligands and short inferring RNA (siRNA) targeted to Sig1R expression. We observed specific effects of Sig1R ligands on Ca2+ entry and hypothesized that the ligands might be TRP channel modulators acting independently of Sig1Rs. Studies of selected TRP channels over-expressed in HEK 293 cells supported this hypothesis.
Methods
Endothelial cell culture
For preparation of human saphenous vein endothelial cells (SVECs), saphenous vein samples were obtained under aseptic condition and with informed written consent from patients undergoing coronary bypass surgery in the General Infirmary at Leeds, UK. Approval was granted by the Leeds Teaching Hospitals Local Research Ethics Committee. The cells were isolated using previously described methods (Bauer et al., 2010). Briefly, saphenous vein samples were chopped into pieces about 1 cm2, opened longitudinally and pinned onto silicone elastomer-coated 60 mm Petri dishes with the lumen exposed. The samples were then incubated in M199 medium (Sigma, Gillingham, UK) supplemented with 1 mg·mL−1 type II collagenase (Type II; Worthington, Lakewood, USA) for 10 min at 37°C. The collagenase suspension was collected along with 2 × 10 mL wash solution; minimal essential medium (Life Technologies, Paisley, UK) was supplemented with 5% fetal calf serum (FCS) and 1% penicillin/streptomycin, and then centrifuged for 6 min at 600× g. The supernatant was discarded and the pellet was resuspended in 25 mL wash solution and recentrifuged. The supernatant was removed again and the final pellet was resuspended in 4 mL of complete endothelial cell culture medium; M199 (Gibco) was supplemented with 20% FCS, 100 U·mL−1 penicillin-streptomycin, 15 μg·mL−1 endothelial cell growth supplement (Sigma), 20 mM HEPES (Sigma), 5 U·mL−1 heparin (Leo Laboratories, Risborough, UK) and 1 mM pyruvate (Sigma). The cells were plated onto T25 flasks (Sarstedt, Leicester, UK) and maintained in a humidified incubator at 37°C gassed with 95% air and 5% CO2. After 2–3 days of plating the cells, the culture medium was changed to remove any non-adherent, non-viable cells and this was considered passage 0. The culture medium was completely changed every 2–3 days. Cells were passaged at 60–70% confluence. SVECs were used for experiments at passages 3–6.
HEK 293 cell culture
Human TRPM3, TRPC5 or TRPM2 was conditionally over-expressed in HEK293 cells as described previously (McHugh et al., 2003; Zeng et al., 2004; Naylor et al., 2008; 2011). cDNA encoding the TRP channels was under the control of a tetracycline-regulated expression system, such that addition of 1 μg·mL−1 tetracycline induced expression of TRP channels. Cells were maintained in DMEM-F12 + GLUTAMAX (Gibco) supplemented with 10% FCS and 100 U·mL−1 penicillin/streptomycin (Sigma) in the presence of selection of antibiotics (10 μg·mL−1 blasticidin and 400 μg·mL−1 zeocin; Life Technologies) at 37°C in a humidified incubator gassed with 95% air and 5% CO2. The cells were treated with tetracycline 24 h prior to experiments (Tet+). Cells not treated with tetracycline (Tet–) were used as a control.
Intracellular Ca2+ measurement
Twenty-four hours before experimentation, endothelial cells were seeded on 96-well clear non-coated plates (Nunc, Kamstrup, Denmark) and HEK 293 cells on poly-D-lysine black-walled, clear-bottomed plates (Corning, Edison, NJ, USA) at a confluence of 80–90%. The cells were incubated with fura-2AM (2 μM) in the presence of 0.01% pluronic acid at 37°C for 1 h followed by a 0.5 h wash at room temperature (21 ± 2°C). Measurements were made at room temperature on a 96-well plate reader (FlexStationП384; Molecular Devices, Sunnyvale, CA, USA). The change (Δ) in intracellular calcium (Ca2+) concentration is indicated as the ratio of fura-2 emission intensities for 340 and 380 nm excitation (F ratio). Wells within columns of the 96-well plate were loaded alternately for test and control conditions. Recordings were made in standard bath solution (SBS) containing (in mM): NaCl 135, KCl 5, MgCl2 1.2, CaCl2 1.5, glucose 8 and HEPES 10; pH was titrated to 7.4 using 4 M NaOH and the osmolarity was ∼290 mOsm·kg−1. Ca2+-free extracellular solution (0 Ca2+) was prepared by excluding CaCl2 and adding 0.4 mM EGTA. Cells were pretreated with Sig1R ligands for 0.5 h (endothelial cells or TRPC5 cells) or 1 h (TRPM3/M2 cells) and maintained throughout Ca2+ recordings; a longer incubation period was used for TRPM recordings because pilot experiments using SKF10047 suggested that shorter exposures were less effective.
Whole-cell patch clamp
Voltage clamp was performed at room temperature using the whole-cell patch clamp configuration on TRPM3 or TRPC5-induced cells plated on 13 mm glass coverslips at low density (∼20–30%) and induced by tetracycline 24 h before experiments. Borosilicate glass capillaries (Harvard Apparatus, Holliston, MA, USA) were pulled to a tip diameter of approximately 1 μm using a PP-830 vertical two-stage pipette puller (Narishige, Tokyo, Japan). Pipette resistances after fire polishing and filling with pipette solution were 3–5 MΩ. Electrodes comprised silver wires coated with chloride ions. Electrical signals were amplified and recorded using an Axopatch 200B amplifier and pCLAMP 10 software (Molecular Devices, Sunnyvale, CA, USA). Data were filtered at 1 kHz and sampled digitally at 2 kHz via a Digidata 1440A analogue to digital converter. Series resistances were <10 MΩ. The voltage protocol consisted of a step from a holding potential of 0 to −100 mV followed by a 0.1 s ramp to +100 mV, before returning to 0 mV (repeated every 10 s). Analysis was performed offline using Clampfit 10.2 (Axon Instruments) and Origin 7.5 software (Origin Lab Corporation, Northampton, MA, USA). The extracellular bath solution for TRPC5 recording contained (in mM): NaCl 140, KCl 5, MgCl2 1.2, BaCl2 1.5, D-glucose 8 and HEPES 10. The extracellular bath solution for TRPM3 recording contained (in mM): NaCl 130, KCl 5, CsCl 10, MgCl2 1.2, CaCl2 1.5, glucose 8 and HEPES 10. For both solutions, the pH was titrated to 7.4 using 4 M NaOH and the osmolality was adjusted to ∼295 mOsm·kg−1 using mannitol. The patch pipette solution for TRPC5 recording contained (in mM): CsCl 135, MgCl2 2, EGTA 1, HEPES 10, Na2ATP 5 and Na2GTP 0.1. The patch pipette solution for TRPM3 recording contained (in mM): CsAspartate 80, CsCl 45, HEPES 10, BAPTA 10 and Na2ATP 4. For both solutions, the pH was titrated to 7.2 using 4 M CsOH and the osmolality was 295 mOsm·kg−1. The pipette solutions were filtered using 0.2 μM membrane filters (Minisart, Goettingen, Germany), divided into aliquots of ∼50 μL and stored at −20°C.
Short interfering RNA (siRNA)
Accell SMART pool siRNA (Dharmacon, Inc., Pittsburgh, PA, USA) targeted four sequences within the human Sig1R transcript (CUAUUAAUAAAGAUUUGUU, CGAGUAGUGCUGCUCUUC, GGGAUAUCCAUGCUUAUGU and GUUCUAGAGUUAAGGAUGG). A 100 μM stock of siRNA was prepared in RNase-free water and stored at −20°C. Endothelial cells were cultured in a 6-well plate (Sarstedt) at 70–80% confluence for 24 h and then the culture medium in each well was replaced with 1 μM of Sig1R siRNA or control non-target Accell siRNA comprising a scrambled sequence with no significant homology to human gene sequences in Accell siRNA delivery medium (Dharmacon, Inc.). Cells were incubated in a 5% CO2 incubator at 37°С for 72 h before quantification of mRNA abundance by real-time reverse-transcription PCR (RT-PCR) and gel electrophoresis. For Ca2+ measurements, SVECs were plated in a clear non-coated 96-well plate for 24 h at 80–90% confluence. The culture medium was then replaced with Accell siRNA delivery medium containing 1 μM of Sig1R siRNA or control siRNA. Cells were incubated in a 5% CO2 incubator at 37°С for 3 days before Ca2+ measurement. HEK293 (Tet–) cells at 60–70% confluence were transfected with 1 μM Sig1R siRNA or control SMART pool siRNA using an Amaxa electroporation device set to program T-16. The transfected cells were incubated in a 5% CO2 incubator at 37°С in a 6-well plate for 48 h before quantification of mRNA abundance by real-time RT-PCR and gel electrophoresis. For intracellular Ca2+ measurements, 48 h after the transfection, the cells were induced by tetracycline and plated at equal density (80–90% confluence) in a 96-well poly-D-lysine plate for another 24 h. To confirm knock-down by RT-PCR, total RNA was collected by phase separation and DNase I treated (Life Technologies). A 600 ng RNA was reverse transcribed in a 20 μL reaction containing AMV reverse transcriptase (Promega, Southampton, UK) and 500 ng Oligo dT15 (Invitrogen) primers. RT-PCR was carried out using primers for human Sig1R that span exon 3 (Forward GGTGTTCGTGAATGCG; Reverse CCGTGTACTACCGTCT). The PCR product was sequenced to confirm identity (Leeds University service). Western blot was carried out 72 h after siRNA transfection by using an affinity-purified goat polyclonal Sig1R (S-18) antibody, 1:50 (Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA) and secondary peroxidase-conjugated donkey anti-goat antibody, 1:20 000 (Jackson ImmunoResearch Lab, West Grove, PA, USA).
Detection of proteins by fluorescence
Endogenous Sig1R in HEK293 (Tet–) was detected using goat polyclonal Sig1R (S-18) antibody. The cells on glass coverslips were fixed and permeabilized, and non-specific binding was blocked with 10% donkey serum for 1 h. The cells were incubated either with Sig1R antibody (1:50) or the antibody pre-adsorbed to its antigenic blocking peptide (Santa Cruz Biotechnology, Inc.) in 2.5% donkey serum for 1 h. The detection of primary antibody was performed by Cy3-conjugated donkey anti-goat (Jackson ImmunoResearch Lab) secondary antibody (1:300) for 30 min. DAPI staining for 5 min allowed detection of cell nuclei. The coverslips were then mounted and stored at 4°C until analysis. The fluorescence of the secondary antibody was detected by Deltavision deconvolution system (Applied Precision Instruments, Seattle, WA, USA) containing an Olympus IX-70 inverted microscope fitted with a ×100 UPLAN objective (NA1.35). To investigate the co-localization of endogenous Sig1R and TRP channels, HEK293 (Tet–) cells were transfected either with TRPC5-GFP or TRPM3-YFP cDNA using FuGene HD transfection reagent according to the manufacturer's protocol (Roche, Burgess Hill, UK). Briefly, 0.5 μg of plasmid DNA and 1.5 μL FuGene HD were mixed in 300 μL serum-free DMEM culture medium for 20–30 min and then pipetted onto the cells on glass coverslips and incubated at 37°C in a tissue culture incubator for 24 h. The cells were then fixed, permeabilized, blocked and stained for endogenous Sig-1R as described above. Fluorescence was detected using Deltavision deconvolution microscopy.
Chemicals and stock solutions
Unless indicated, salts and other chemicals were from the Sigma Chemical Company. SKF10047, BD1063, BD1047 and 4-IBP (Figure 1A) were from Tocris Bioscience (Bristol, UK). Histamine (50 mM) stock was prepared in distilled water and stored at 4°C. VEGF (100 μg·mL−1) stock was prepared in distilled water and stored at −20°C. The 30% w/w H2O2 stock was stored at 4°C. Stock solutions of 100, 100 and 50 mM concentrations of SKF10047, BD1063 and BD1047, respectively, were in distilled water and stored at −20°C. 4-IBP (10 mM) and PregS (100 mM) stock solutions were prepared in 100% dimethyl sulfoxide (DMSO) and stored at 4°C. Nifedipine (50 mM) stock solutions were prepared in 100% DMSO and stored at −20°C. Gadolinium(III) chloride (100 mM) stock solution was prepared in distilled water and stored at room temperature. Sphingosine-1-phosphate (S1P) (10 mM) and lysophosphatidylcholine (LPC) (50 mM) stock solutions were prepared in methanol and stored at −20°C. Fura-2AM (acetoxymethyl) (Life Technologies) stock (1 mM) was prepared in 100% DMSO and stored at −20 °С. Pluronic acid F-127 (10%) stock was prepared in 100% DMSO and stored at room temperature.
Data analysis
Mean data are presented as mean ± SE mean. Data were produced in pairs (test and control) and compared using independent Student t-tests (for intracellular Ca2+ measurement) or paired Student's t-test (for patch clamp recording) with statistical significance indicated by * (P < 0.05) and no significant difference by NS (P > 0.05). One-way anova followed by Bonferroni post hoc test was used for multiple groups. The rate of rise of the VEGF response was calculated by measuring the slope of the initial component. All intracellular Ca2+ measurement data were presented as n/N, where n indicates the number of independent experiments and N indicates the number of tested wells in the 96-well plates. For patch clamp recordings, n is the number of cells from which recordings were made. Representative traces and mean data were obtained from at least three independent experiments. Endothelial cell experiments were performed on cells from at least eight different patients. Origin software (Origin Lab Corporation) was used for data analysis and presentation.
Results
Inhibition of endothelial cell Ca2+ entry by BD1047, BD1063 and 4-IBP, but not SKF10047
Extracellular application of histamine to SVECs elicited a rise in the intracellular Ca2+ concentration that decayed to a plateau and then to a second plateau over a 5 min period (Figure 1B). In the presence of the Sig1R antagonist BD1047 or BD1063 at 100 μM, the initial response to histamine was similar to the control but the plateau phases were strongly inhibited (Figure 1B–D; Supporting Information Fig. S1a). At 10 μM BD1063 had a similar effect but 1 μM was ineffective (Figure 1C). Similar inhibition occurred with the Sig1R agonist 4-IBP but not the archetypal Sig1R agonist SKF10047, which failed to modify any aspect of the histamine response (Figure 1; Supporting Information Fig. S1b,c). The data suggest that BD1047, BD1063 and 4-IBP are strong inhibitors of sustained histamine-evoked Ca2+ signals but that SKF10047 has no effect.
To investigate if the effects of Sig1R ligands were specific to histamine responses, we also investigated responses to VEGF and H2O2. VEGF evoked a slower and more sustained response compared with histamine (Supporting Information Fig. S1d). BD1047, BD1063 and 4-IBP inhibited the sustained but not the transient response and SKF10047 had no effect (Figure 1; Supporting Information Fig. S1e). H2O2 evoked a relatively slow and sustained response that depended largely on Ca2+ influx (Supporting Information Fig. S1f). BD1047, BD1063 and 4-IBP inhibited the response but SKF10047 had no effect (Figure 1; Supporting Information Fig. S1g). The data suggest that three of the four Sig1R ligands inhibit Ca2+ entry evoked by VEGF or H2O2 but that inhibition is not as strong as that seen when histamine is the agonist.
Sig1R ligands did not affect histamine-evoked Ca2+ release (Figure 1; Supporting Information Fig. S1a–c), suggesting lack of histamine receptor antagonism, lack of artefact in the Ca2+ measurement technique and lack of non-specific effects on cellular Ca2+ handling. However, as additional controls for non-specific effects, we measured responses to thapsigargin in the absence of extracellular Ca2+, which reflected passive Ca2+ leak from intracellular stores and the handling of this Ca2+ by storage and extrusion mechanisms (Supporting Information Fig. S2a–d). There were no effects of BD1063, BD1047, 4-IBP or SKF10047 on this Ca2+ signal (Supporting Information Fig. S2a–e). The data suggest that effects of Sig1R ligands arise through inhibition of Ca2+ entry.
Inhibition of TRPC5 expressed in HEK 293 cells by BD1047, BD1063, 4-IBP and SKF10047
The molecular identities of channels mediating Ca2+ entry evoked by histamine in SVECs are not yet known, but TRPC5 channels are implicated in endothelial cell responses to H2O2 (Yoshida et al., 2006; Chaudhuri et al., 2008; Wong et al., 2010). We therefore investigated if there are effects of Sig1R ligands on TRPC5 channels over-expressed in HEK 293 cells.
Activity of TRPC5 was first evoked by gadolinium ions (Gd3+), leading to Ca2+ influx that reached a maximum after about 4 min (Figure 2A). BD1047 (100 μM) inhibited this Ca2+ influx (Figure 2A). The effect did not depend on the agonist that first stimulated TRPC5 because Ca2+ influx evoked by LPC was also inhibited by BD1047 (Figure 2; Supporting Information Fig. S3a). There was also inhibition of TRPC5 activity evoked by S1P (Supporting Information Fig. S3b). Inhibition was confirmed in whole-cell patch clamp recordings (Figure 2C–F). In these experiments, the inhibitory effect of BD1047 against Gd3+-evoked current was seen to occur rapidly and reversed readily on washout (Figure 2C). A non-specific TRPC5 channel inhibitor, 2-APB, was applied for comparison (Figure 2C). In response to 10 μM BD1047, there was ∼59% inhibition of current at −80 mV and further inhibition when the concentration was elevated during the same recording (Figure 2D). There was also inhibition of spontaneous TRPC5 current (Figure 2E), again suggesting that blockade was independent of the mode of activation of TRPC5 (Figure 2E,F). The expected shape of the TRPC5 current–voltage relationship was observed and seen to be inhibited by BD1047 across the voltage range from −100 to +100 mV (Figure 2E). In response to 6 μM BD1047, there was ∼40% inhibition of spontaneous TRPC5-mediated current and greater inhibition at higher concentrations (Figure 2F). BD1063 and 4-IBP inhibited TRPC5 and, in contrast to endothelial Ca2+ entry, SKF10047 was also an inhibitor (Figure 2; Supporting Information Fig. S3b). None of the Sig1R ligands was an activator of TRPC5 (Supporting Information Fig. S4a). The data suggest that TRPC5 has broad susceptibility to inhibition by Sig1R ligands, which includes inhibition by SKF10047.
Figure 2.
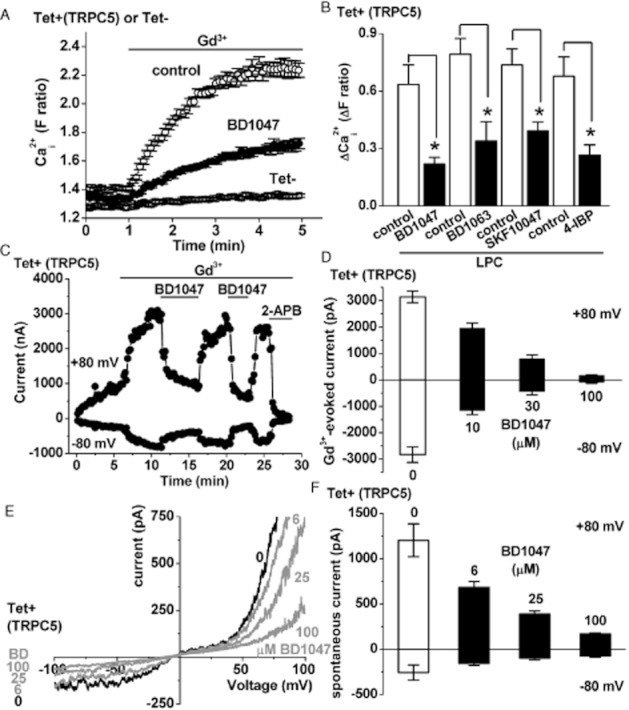
Inhibition of TRPC5 by Sig1R ligands. (A) Example of Ca2+ measurement data for TRPC5-expressing cells, showing responses to 20 μM Gd3+ in the presence of 100 μM BD1047 or the vehicle control (N = 6 for each). Also shown is the lack of effect of Gd3+ in cells without TRPC5 expression (Tet–). (B) Mean data for the type of experiment illustrated in (A) but showing analysis of the maximum response to 10 μM LPC (in place of Gd3+) and effects of 100 μM of the Sig1R ligand indicated (n/N = 3/18 for each condition). (C–F) Whole-cell voltage clamp data from TRPC5-expressing cells. (C) Time series plot of outward and inward currents stimulated by 20 μM Gd3+, showing repeated effects of 100 μM BD1047. 2-aminoethoxydiphenyl borate (2-APB, 75 μM) was applied at the end of the experiment. (D) Mean data for Gd3+-stimulated TRPC5 currents in the presence of 0, 10, 30 and 100 μM BD1047 applied cumulatively (n = 13). (E) Current–voltage relationships for spontaneous currents show the effects of 6, 25 and 100 μM BD1047 applied cumulatively. (F) Mean data for spontaneous TRPC5 currents in the presence of 0, 6, 25 and 100 μM BD1047 applied cumulatively (n = 5).
Inhibition of TRPM3 by BD1047, BD1063 and 4-IBP, but not SKF10047
TRPM2 channels are also implicated in endothelial cell responses to H2O2 (Hecquet et al., 2008). TRPM2 over-expressed in HEK 293 cells was not however sensitive to any of the four Sig1R ligands applied at 100 μM concentration (Figure 3; Supporting Information Fig. S3c). However, in parallel experiments, we tested Sig1R ligands against another TRPM channel, TRPM3, and unexpectedly observed inhibitory effects (Figure 3B–E). TRPM3 was first stimulated by a high concentration of nifedipine (Wagner et al., 2008), leading to Ca2+ influx (Figure 3B,C). BD1047, BD1063 and 4-IBP inhibited the Ca2+ influx but SKF10047 had no effect (Figure 3B,C). The inhibitory effect of BD1047 was confirmed in whole-cell patch clamp recordings in which TRPM3 was first stimulated by an alternative TRPM3 agonist PregS (Figure 3D,E). The effect of BD1047 was seen to occur rapidly and reversed readily on washout (Figure 3D). There was a tendency for the inhibitory effect to be stronger at +80 mV compared with −80 mV (Figure 3E), but the small amplitudes of currents at −80 mV prevented thorough analysis. None of the Sig1R ligands was an activator of TRPM3 (Supporting Information Fig. S4b). The data suggest that TRPM3, but not TRPM2, is susceptible to inhibition by Sig1R ligands and that the profile of inhibition is similar to that seen in endothelial cells.
Figure 3.

Inhibition of TRPM3 but not TRPM2 by Sig1R ligands. (A) Mean Ca2+ measurement data for TRPM2 cells, showing responses to 1 mM H2O2 in the presence of 100 μM of the specified inhibitor or the vehicle control (n/N = 3/4 for each). (B–E) Data from TRPM3-expressing cells. (B) Example of Ca2+ measurement data showing responses to 20 μM nifedipine in the continuous presence of 100 μM BD1047 or the vehicle control (N = 6 for each). Also shown is the lack of effect of nifedipine in cells without TRPM3 expression (Tet–). (C) Mean data for the types of experiment illustrated in (B), showing analysis of the maximum responses to nifedipine in the presence of 100 μM BD1047, BD1063, SKF10047 or 4-IBP (n/N = 3/18 for each condition). The 4-IBP experiments used DMSO rather than water as the solvent. (D, E) Whole-cell voltage clamp data from TRPM3-expressing cells. (D) Time series plot of outward and inward currents, showing stimulation by 5 μM PregS and the effect of 100 μM BD1047. Horizontal bars indicate the periods of compound application. (E) As for (D) but mean data showing the percentage of PregS-induced current remaining after application of BD1047 (n = 5).
Sig1R does not mediate the effects of Sig1R ligands
The similar effects of Sig1R antagonists and agonists, and lack of effect of SKF10047 in some experiments, were indications that the Sig1R was not mediating the effects of Sig1R ligands on Ca2+ entry. Nevertheless, to directly investigate the role of Sig1R, we knocked down its expression using Sig1R siRNA in endothelial cells or HEK 293 cells (Supporting Information Fig. S5). Histamine-evoked Ca2+ elevation was unaffected by Sig1R siRNA and the inhibitory effect of BD1063 remained intact (Figure 4; Supporting Information Fig. S6a,b). Sig1R siRNA also did not affect activation of TRPC5 by LPC and its inhibition by BD1047 (Figure 4; Supporting Information Fig. S6c), or affect stimulation of TRPM3 by PregS and its inhibition by BD1047 (Supporting Information Fig. S6d,e). The data suggest that Sig1R does not mediate the actions of Sig1R ligands on Ca2+ entry in endothelial cells or the actions on TRPC5 or TRPM3 in HEK 293 cells.
Figure 4.
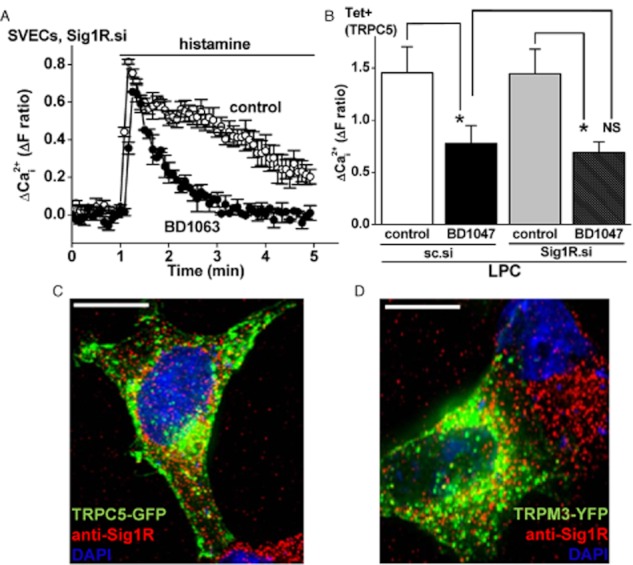
Lack of role of the Sig1R. (A) Example of Ca2+ measurement results for the effect of 50 μM BD1063 (30 min treatment) on 10 μM histamine-evoked Ca2+ signals in SVECs treated with Sig1R siRNA (Sig1R.si). (B) Mean Ca2+ measurement data for the maximum responses to 10 μM LPC in TRPC5-expressing HEK 293 cells treated with sc.si (control) or Sig1R.si and then exposed to 100 μM BD1047 or vehicle control (n/N = 3/16 for each condition). (C, D) Merged images showing typical non-induced HEK 293 cells transiently expressing TRPC5-GFP (C) or TRPM3-YFP (D). Fluorescence from the GFP or YFP is in green. Cells were stained with anti-Sig1R antibody (red) and the nuclear stain DAPI (blue). Cells were pretreated with 100 μM SKF10047 (C) or BD1047 (D). The scale bars are 10 μm.
To further explore the relevance of the Sig1R, we performed co-localization studies between Sig1R and TRP channels. Either GFP-tagged TRPC5 or YFP-tagged TRPM3 (Figure 4C,D) was expressed to observe the channels via green fluorescence. Localization of endogenous Sig1R was observed in the same cells using anti-Sig1R antibody and the red fluorescence of its secondary antibody (Figure 4C,D; Supporting Information Fig. S7a,b). Data are shown for cells in the presence of SKF10047 or BD1047, although neither ligand modified the localization of Sig1R or TRP channel (data not shown). TRPC5 and Sig1R, or TRPM3 and Sig1R, were largely independent of each other (Figure 4C,D; Supporting Information Fig. S7b). The data suggest that Sig1R does not co-localize with TRPC5 or TRPM3, consistent with the lack of functional involvement of Sig1R in the actions of Sig1R ligands on these ion channels.
Discussion and conclusions
The study shows previously unrecognized chemical sensitivities of endogenous endothelial cell Ca2+ entry and two types of Ca2+-permeable TRP channel, TRPC5 and TRPM3. The compounds BD1047, BD1063 and 4-IBP were strong inhibitors of sustained histamine-evoked Ca2+ entry in endothelial cells whereas SKF10047 had no effect. The same profile existed for VEGF- and H2O2-evoked Ca2+ entries, although the inhibition was less. The finding that Sig1R agonists and antagonists both inhibited the Ca2+ entry and had apparently lower potencies than observed against Sig1Rs argued against a role for Sig1R in mediating the effects of Sig1R ligands on Ca2+ entry. Such a conclusion was supported by results of Sig1R knockdown experiments. Similarly, Sig1R independence but sensitivity to Sig1R ligands was observed for over-expressed TRPC5 and TRPM3 channels in HEK 293 cells, although sensitivity to SKF10047 of TRPC5 (but not TRPM3) contrasted with the endothelial cell studies. We suggest, therefore, that Sig1R ligands are previously unrecognized inhibitors of TRPC5, TRPM3 and endogenous endothelial cell Ca2+ entry, acting independently of Sig1Rs.
BD1047, BD1063 and 4-IBP are suggested to be blockers of Ca2+ entry channels in endothelial cells because they inhibited components of the responses that depended on extracellular Ca2+, they acted independently of the trigger for activation of Ca2+ entry (i.e. histamine, VEGF or H2O2), and they did not affect Ca2+ release. The lack of effect on the initial (Ca2+ release) response to histamine or VEGF suggested that the compounds did not act as antagonists of histamine or VEGF receptors or affect phospholipase C-dependent signalling. Therefore, the compounds may act relatively directly as Ca2+ channel inhibitors. The effects on over-expressed TRP channels support this hypothesis, especially given the relatively rapid inhibitory and washout effects on whole-cell currents.
BD1047, BD1063 and 4-IBP are chemically quite similar (Supporting Information Fig. S8). BD1063 and 4-IBP both contain a halogenated aromatic ring attached to a saturated six-membered ring (piperazine or piperidine respectively) by a two-atom spacer (ethylene or amide respectively). In their minimal energy conformations (chair, all equatorial), they share a common three-dimensional shape and a similarly positioned basic nitrogen. BD1047 can be considered a ring-opened analogue of BD1063, and therefore one of its possible conformations would be highly similar to that of BD1063. In addition, being a tetramethylethylenediamine analogue, BD1047 might act as a metal chelator. These common structural features may provide a clue for developing more potent and selective inhibitors of the Ca2+ entry channels. We previously reported that resveratrol and diethylstilbestrol are also inhibitors of TRPC5, acting via different mechanisms (Naylor et al., 2011). Resveratrol and diethylstilbestrol, which lack basic nitrogens and saturated ring systems, are structurally clearly distinct from BD1047, BD1063 and 4-IBP. Moreover, although they share the hydroxylated stilbene scaffold, resveratrol is a flat, fully conjugated molecule, whereas the ethyl groups of diethylstilbestrol force the phenyl groups to twist by approximately 90°, thereby reducing conjugation with the olefin (Supporting Information Fig. S8). Therefore, resveratrol, diethylstilbestrol and BD1047/BD1063/4-IBP would seem to represent distinct groups of chemical modulators of TRP channels. SKF10047 may represent a fourth.
The molecular compositions of the Ca2+ entry channels activated by histamine, VEGF and H2O2 in endothelial cells remain to be determined. Previous studies suggested involvement of TRPC1 and TRPC6 in VEGF responses (Jho et al., 2005; Hamdollah Zadeh et al., 2008; Ge et al., 2009), and TRPC5 in H2O2 responses (Yoshida et al., 2006). Orai1 CRAC channels also contribute to the VEGF response (Li et al., 2011). The histamine-activated channels have not been determined, but in over-expression systems histamine receptor activation is capable of stimulating TRPC5 and TRPC6 channels (Hofmann et al., 1999; Ma et al., 2010). Histamine might be expected to activate Orai1 CRAC channels but a CRAC channel inhibitor, Synta 66, had relatively little effect on histamine-evoked Ca2+ entry in SVECs (M. Amer, unpubl. obs.). Expression of TRPM3 is low in SVECs (Naylor et al., 2010). TRPM3 in HEK 293 cells was not stimulated by H2O2 (Naylor et al., 2010) and is not known to be activated by histamine or VEGF in endothelial cells. Nevertheless, the data suggest commonality in the pharmacology of endothelial Ca2+ entry and TRP channels, and show that the inhibitory effects are relevant to endogenous channel mechanisms.
The study suggests the absence of a relationship between Sig1Rs and endothelial cell Ca2+ entry or TRPC5 and TRPM3 channels, even though Sig1Rs are expressed in these cells and have been commonly associated with Ca2+ signalling systems. The study reveals, however, new pharmacology for suppressing endothelial cell Ca2+ entry or for suppressing TRPC5 and TRPM3 channels. The pharmacology arises from the Sig1R ligands even though the Sig1R is not involved in the effects. The information has the potential to be used as the basis for developing potent and subtype-selective inhibitors of receptor- and chemically activated Ca2+ entry mechanisms, for which currently used inhibitors are inadequate. If developed, such inhibitors could have therapeutic value for the suppression of unwanted increases in vascular permeability, unwanted angiogenesis or other events related to these Ca2+ entry mechanisms.
Acknowledgments
This research was supported by research grants from the Wellcome Trust, the British Heart Foundation and the Medical Research Council. M. S. A. was supported by a PhD Scholarship from the Egyptian Ministry of Higher Education. B. H. was supported by a scholarship from the University of Leeds and the China Scholarship Council. L. A. W. was supported by a BBSRC-AstraZeneca PhD Studentship.
Glossary
Abbreviations
- 4-IBP
4-(N-benzylpiperidin-4-yl)-4-iodobenzamide
- BD1047
([2-((3,4-dichlorophenyl)ethyl][2-(dimethylamino)ethyl]methylamine dihydrobromide
- BD1063
1-[2-(3,4-dichlorophenyl)ethyl]-4-methylpiperazine dihydrobromide
- Sig1R
Sigma-1 receptor
- siRNA
short interfering RNA
- SKF10047
(+)-N-allylnormetazocine hydrochloride
- SVECs
saphenous vein endothelial cells
- TRPC
transient receptor potential canonical
- TRPM
transient receptor potential melastatin
Conflict of interest
The authors state no conflict of interest.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's web-site:
Figure S1 Supporting data for effects of Sig1R ligands on Ca2+ signals in SVECs. Data are from intracellular Ca2+ measurements. (A) Mean data to accompany those in Figure 1, showing analysis of the peak responses to histamine (n/N = 3/12 for each experiment). (B,C) Example of data showing effects of 10 μM histamine in the presence of 100 μM 4-IBP (B) or 100 μM SKF10047 (C) compared with vehicle controls (N = 4 for each). The ligands were applied 30 min before testing histamine and maintained throughout the experiments. (D) Example of data showing effects of 100 ng·mL−1 VEGF in the presence of 100 μM BD1063 compared with the vehicle control. BD1063 was applied for 30 min before testing of VEGF and was maintained throughout the experiment. (E) Mean data for the effects of Sig1R ligands (100 μM) on the rate of rise of the VEGF response (n/N = 3/12 for each), from the same experiments analysed in Figure 1. (F) Dependence of the response to H2O2 on extracellular Ca2+. Typical experiment showing 1 mM H2O2 responses in the presence (1.5 Ca2+) or absence (0 Ca2+) of 1.5 mM extracellular Ca2+ (N = 4 for each). (G) Example of data showing effects of 1 mM hydrogen peroxide (H2O2) in the presence of 100 μM BD1063 compared with the vehicle control.
Figure S2 Lack of effect of Sig1R ligands on thapsigargin-evoked Ca2+ release in SVECs. Data were generated by measurement of intracellular Ca2+ in SVECs. (A–D) Example of data showing effects of 1 μM thapsigargin in the absence of extracellular Ca2 (0 Ca2+) but the presence of 100 μM BD1047 (A), 100 μM BD1063 (B), 100 μM 4-IBP (C) or 100 μM SKF10047 (d) compared with vehicle controls (N = 4 for each). The compounds were applied 30 min before testing of thapsigargin and were maintained throughout the experiments. (E) Mean data for the types of experiment illustrated in (A–D), showing analysis of the peak responses to thapsigargin (n/N = 3/14). 4-IBP data are shown separately because of the use of a different vehicle control in these experiments.
Figure S3 Effects of Sig1R ligands on TRPC5, TRPM2 and TRPM3 channels in HEK 293 cells. (A) Example of Ca2+ measurement data for TRPC5 cells, showing responses to 10 μM LPC in the presence of 100 μM BD1047 or the vehicle control (N = 6 for each). Also shown is the lack of effect of Gd3+ in cells lacking TRPC5 expression (Tet–). (B) Mean data of the type shown in Figure 2B but for maximum responses to 5 μM sphingosine-1-phosphate (S1P) (n/N = 3/18 for each condition). (C) Example of Ca2+ measurement data for TRPM2 cells, showing responses to 1 mM H2O2 in the presence of 100 μM BD1047 or the vehicle control (N = 8 for each). (D) Whole-cell voltage clamp data from TRPM3 cells showing current–voltage relationships (I-Vs) for the experiment shown in Figure 4D.
Figure S4 Absence of stimulatory effects of Sig1R ligands on TRPC5 or TRPM3. (A) Data were generated by Ca2+ measurement from HEK 293 cells induced to express TRPC5 (Tet+) or not induced to express TRPC5 (Tet–). Cells were exposed to 100 μM Gd3+ (n/N = 4/36) (a positive control), 100 μM SKF10047, 100 μM 4-IBP, 100 μM BD1063 or 100 μM BD1047 (n/N = 3/18). (B) Data were generated by Ca2+ measurement from HEK 293 cells induced to express TRPM3 (Tet+) or not induced to express TRPM3 (Tet–). Cells were exposed to 5 μM PregS (n/N = 4/32) (a positive control), 100 μM SKF10047, 100 μM 4-IBP, 100 μM BD1063 or 100 μM BD1047 (n/N = 3/18).
Figure S5 Validation of Sig1R depletion. (A) PCR products from SVECs transfected with scrambled siRNA (sc.si) or Sig1R siRNA (Sig1R.si). Expected product sizes were 196 (β-actin, i) and 222 (Sig1R, ii) base pairs (bp). The 100 bp DNA marker ladder is on the left (M) and an example of Sig1R PCR reaction without reverse transcriptase (–RT) is on the right. (B) As for (A) but mRNA was isolated from HEK 293 cells. (C) Western blot with anti-Sig1R antibody for HEK 293 cell proteins after transfection with scrambled (control) siRNA (sc.si) or Sig1R siRNA (Sig1R.si).
Figure S6 Supporting data suggesting lack of functional role of the Sig1R. (A) Mean Ca2+ measurement data for effects of sc.si (control) and Sig1R siRNA (Sig1R.si) on 10 μM histamine-evoked signals (n/N = 3/12). (B) Mean data for the effect of 50 μM BD1063 (30 min treatment) on 10 μM histamine-evoked Ca2+ signals in SVECs transfected with sc.si or Sig1R.si (n/N = 3/12 for each data set). Data are shown for the peak response to histamine and the response 5 min after the application of histamine. (C, D) Example of Ca2+ measurement data showing 10 μM LPC-evoked TRPC5 activity (C, N = 5) and 5 μM PregS-evoked TRPM3 activity (D, N = 4) in HEK 293 cells transfected with sc.si or Sig1R.si. (E) Mean data for the maximum responses to 5 μM PregS in TRPM3 cells treated with sc.si or Sig1R.si in the presence of 100 μM BD1047 or vehicle control (n/N = 3/16 for each condition).
Figure S7 Supporting data for Sig1R staining and localization of TRPC5-GFP and TRPM3-YFP channels. (A) Peptide specificity of endogenous Sig1R detection by anti-Sig1R antibody. Examples of images are shown for HEK 293 cells exposed to anti-Sig1R antibody (upper image) or anti-Sig1R antibody pre-adsorbed to its antigenic peptide (lower image). Positive labelling is shown by the red of the secondary antibody. The blue is from DAPI, the counter-nuclear stain. (B, C) Images show typical non-induced HEK 293 cells transiently expressing TRPC5-GFP (B) or TRPM3-YFP (C). Fluorescence from the GFP or YFP is shown in green. Cells were stained with anti-Sig1R antibody (red). Cells were pretreated with 100 μM SKF10047 (B) or BD1047 (C).
Figure S8 Comparison of the chemical structures of BD1063, 4-IBP, BD1047, resveratrol and diethylstilbestrol. Basic nitrogens are highlighted in red. (A) The lowest energy conformations of BD1063 (1) and 4-IBP (2) (chair, all equatorial) show a common structural motif, which is represented by preliminary pharmacophore model 3. (B) BD1047 (4) can be considered a ring-opened analogue of BD1063 (1) with significant conformational freedom, and one of its possible conformations is highly similar to that of BD1063. Being an analogue of the common metal chelator tetramethylethylenediamine (TMEDA, 6), BD1047 might also act as a ligand for metal ions (Mn+) such as Mg2+, Zn2+, Cu2+ and Ca2+ (5) in the same way as TMEDA (7). (C) In its lowest energy conformation, resveratrol (8) is flat because of conjugation of all π bonds. The ethyl groups of diethylstilbestrol (9) force rotation of its aromatic groups by approximately 90°, thereby preventing full conjugation. Thus, despite sharing a hydroxylated stilbene scaffold, resveratrol and diethylstilbestrol have markedly different 3D structures in their lowest energy conformations.
References
- Aird WC. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ Res. 2007;100:158–173. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- Bauer CC, Boyle JP, Porter KE, Peers C. Modulation of Ca2+ signalling in human vascular endothelial cells by hydrogen sulfide. Atherosclerosis. 2010;209:374–380. doi: 10.1016/j.atherosclerosis.2009.10.004. [DOI] [PubMed] [Google Scholar]
- Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer. 2008;8:592–603. doi: 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Cardiac calcium signalling. Biochem Soc Trans. 2003;31((Pt 5)):930–933. doi: 10.1042/bst0310930. [DOI] [PubMed] [Google Scholar]
- Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–529. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. Angiogenesis in life, disease and medicine. Nature. 2005;438:932–936. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- Chaudhuri P, Colles SM, Bhat M, Van Wagoner DR, Birnbaumer L, Graham LM. Elucidation of a TRPC6-TRPC5 channel cascade that restricts endothelial cell movement. Mol Biol Cell. 2008;19:3203–3211. doi: 10.1091/mbc.E07-08-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 2007;131:1047–1058. doi: 10.1016/j.cell.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Damann N, Voets T, Nilius B. TRPs in our senses. Curr Biol. 2008;18:R880–R889. doi: 10.1016/j.cub.2008.07.063. [DOI] [PubMed] [Google Scholar]
- Faehling M, Kroll J, Föhr KJ, Fellbrich G, Mayr U, Trischler G, et al. Essential role of calcium in vascular endothelial growth factor A-induced signaling: mechanism of the antiangiogenic effect of carboxyamidotriazole. FASEB J. 2002;16:1805–1807. doi: 10.1096/fj.01-0938fje. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- Ge R, Tai Y, Sun Y, Zhou K, Yang S, Cheng T, et al. Critical role of TRPC6 channels in VEGF-mediated angiogenesis. Cancer Lett. 2009;283:43–51. doi: 10.1016/j.canlet.2009.03.023. [DOI] [PubMed] [Google Scholar]
- Grimm C, Kraft R, Schultz G, Harteneck C. Activation of the melastatin-related cation channel TRPM3 by D-erythro-sphingosine. Mol Pharmacol. 2005;67:798–805. doi: 10.1124/mol.104.006734. [DOI] [PubMed] [Google Scholar]
- Hamdollah Zadeh MA, Glass CA, Magnussen A, Hancox JC, Bates DO. VEGF-mediated elevated intracellular calcium and angiogenesis in human microvascular endothelial cells in vitro are inhibited by dominant negative TRPC6. Microcirculation. 2008;15:605–614. doi: 10.1080/10739680802220323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecquet CM, Ahmmed GU, Vogel SM, Malik AB. Role of TRPM2 channel in mediating H2O2-induced Ca2+ entry and endothelial hyperpermeability. Circ Res. 2008;102:347–355. doi: 10.1161/CIRCRESAHA.107.160176. [DOI] [PubMed] [Google Scholar]
- Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–263. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- Jho D, Mehta D, Ahmmed G, Gao XP, Tiruppathi C, Broman M, et al. Angiopoietin-1 opposes VEGF-induced increase in endothelial permeability by inhibiting TRPC1-dependent Ca2+ influx. Circ Res. 2005;96:1282–1290. doi: 10.1161/01.RES.0000171894.03801.03. [DOI] [PubMed] [Google Scholar]
- Jiang LH, Gamper N, Beech DJ. Properties and therapeutic potential of transient receptor potential channels with putative roles in adversity: focus on TRPC5, TRPM2 and TRPA1. Curr Drug Targets. 2011;12:724–736. doi: 10.2174/138945011795378568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Cubbon RM, Wilson LA, Amer MS, McKeown L, Hou B, et al. Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ Res. 2011;108:1190–1198. doi: 10.1161/CIRCRESAHA.111.243352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Cao J, Luo J, Nilius B, Huang Y, Ambudkar IS, et al. Depletion of intracellular Ca2+ stores stimulates the translocation of vanilloid transient receptor potential 4-C1 heteromeric channels to the plasma membrane. Arterioscler Thromb Vasc Biol. 2010;30:2249–2255. doi: 10.1161/ATVBAHA.110.212084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Flemming R, Xu SZ, Perraud AL, Beech DJ. Critical intracellular Ca2+ dependence of transient receptor potential melastatin 2 (TRPM2) cation channel activation. J Biol Chem. 2003;278:11002–11006. doi: 10.1074/jbc.M210810200. [DOI] [PubMed] [Google Scholar]
- Maurice T, Su TP. The pharmacology of sigma-1 receptors. Pharmacol Ther. 2009;124:195–206. doi: 10.1016/j.pharmthera.2009.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnet FP. Sigma-1 receptor as regulator of neuronal intracellular Ca2+: clinical and therapeutic relevance. Biol Cell. 2005;97:873–883. doi: 10.1042/BC20040149. [DOI] [PubMed] [Google Scholar]
- Naylor J, Milligan CJ, Zeng F, Jones C, Beech DJ. Production of a specific extracellular inhibitor of TRPM3 channels. Br J Pharmacol. 2008;155:567–573. doi: 10.1038/bjp.2008.283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor J, Li J, Milligan CJ, Zeng F, Sukumar P, Hou B, et al. Pregnenolone sulphate- and cholesterol-regulated TRPM3 channels coupled to vascular smooth muscle secretion and contraction. Circ Res. 2010;106:1507–1515. doi: 10.1161/CIRCRESAHA.110.219329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naylor J, Al-Shawaf E, McKeown L, Manna PT, Porter KE, O'Regan D, et al. TRPC5 channel sensitivities to antioxidants and hydroxylated stilbenes. J Biol Chem. 2011;286:5078–5086. doi: 10.1074/jbc.M110.196956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Droogmans G. Ion channels and their functional role in vascular endothelium. Physiol Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- Olsson AK, Dimberg A, Kreuger J, Claesson-Welsh L. VEGF receptor signalling – in control of vascular function. Nat Rev Mol Cell Biol. 2006;7:359–371. doi: 10.1038/nrm1911. [DOI] [PubMed] [Google Scholar]
- Su TP, Hayashi T, Maurice T, Buch S, Ruoho AE. The sigma-1 receptor chaperone as an inter-organelle signaling modulator. Trends Pharmacol Sci. 2010;31:557–566. doi: 10.1016/j.tips.2010.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tagashira H, Bhuiyan S, Shioda N, Hasegawa H, Kanai H, Fukunaga K. Sigma1-receptor stimulation with fluvoxamine ameliorates transverse aortic constriction-induced myocardial hypertrophy and dysfunction in mice. Am J Physiol Heart Circ Physiol. 2010;299:H1535–H1545. doi: 10.1152/ajpheart.00198.2010. [DOI] [PubMed] [Google Scholar]
- Venkatachalam K, Montell C. TRP channels. Annu Rev Biochem. 2007;76:387–417. doi: 10.1146/annurev.biochem.75.103004.142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner TF, Loch S, Lambert S, Straub I, Mannebach S, Mathar I, et al. Transient receptor potential M3 channels are ionotropic steroid receptors in pancreatic beta cells. Nat Cell Biol. 2008;10:1421–1430. doi: 10.1038/ncb1801. [DOI] [PubMed] [Google Scholar]
- Wong CO, Sukumar P, Beech DJ, Yao X. Nitric oxide lacks direct effect on TRPC5 channels but suppresses endogenous TRPC5-containing channels in endothelial cells. Pflugers Arch. 2010;460:121–130. doi: 10.1007/s00424-010-0823-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S, Touyz RM. Reactive oxygen species and vascular remodelling in hypertension: still alive. Can J Cardiol. 2006;22:947–951. doi: 10.1016/s0828-282x(06)70314-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Inoue R, Morii T, Takahashi N, Yamamoto S, Hara Y, et al. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2:596–607. doi: 10.1038/nchembio821. [DOI] [PubMed] [Google Scholar]
- Zeng F, Xu SZ, Jackson PK, McHugh D, Kumar B, Fountain SJ, et al. Human TRPC5 channel activated by a multiplicity of signals in a single cell. J Physiol. 2004;559((Pt 3)):739–750. doi: 10.1113/jphysiol.2004.065391. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.