

Abstract
Background and Purpose
To evaluate the ability of cannabidiolic acid (CBDA) to reduce nausea and vomiting and enhance 5-HT1A receptor activation in animal models.
Experimental Approach
We investigated the effect of CBDA on (i) lithium chloride (LiCl)-induced conditioned gaping to a flavour (nausea-induced behaviour) or a context (model of anticipatory nausea) in rats; (ii) saccharin palatability in rats; (iii) motion-, LiCl- or cisplatin-induced vomiting in house musk shrews (Suncus murinus); and (iv) rat brainstem 5-HT1A receptor activation by 8-hydroxy-2-(di-n-propylamino)tetralin (8-OH-DPAT) and mouse whole brain CB1 receptor activation by CP55940, using [35S]GTPγS-binding assays.
Key Results
In shrews, CBDA (0.1 and/or 0.5 mg·kg−1 i.p.) reduced toxin- and motion-induced vomiting, and increased the onset latency of the first motion-induced emetic episode. In rats, CBDA (0.01 and 0.1 mg·kg−1 i.p.) suppressed LiCl- and context-induced conditioned gaping, effects that were blocked by the 5-HT1A receptor antagonist, WAY100635 (0.1 mg·kg−1 i.p.), and, at 0.01 mg·kg−1 i.p., enhanced saccharin palatability. CBDA-induced suppression of LiCl-induced conditioned gaping was unaffected by the CB1 receptor antagonist, SR141716A (1 mg·kg−1 i.p.). In vitro, CBDA (0.1–100 nM) increased the Emax of 8-OH-DPAT.
Conclusions and Implications
Compared with cannabidiol, CBDA displays significantly greater potency at inhibiting vomiting in shrews and nausea in rats, and at enhancing 5-HT1A receptor activation, an action that accounts for its ability to attenuate conditioned gaping in rats. Consequently, CBDA shows promise as a treatment for nausea and vomiting, including anticipatory nausea for which no specific therapy is currently available.
Keywords: cannabidiolic acid, 5-HT1A receptor, rat, shrew, conditioned gaping, anticipatory nausea, taste reactivity, toxin-induced vomiting, motion-induced vomiting, emesis
Introduction
The cannabis plant is a natural source of at least 70 compounds known collectively as phytocannabinoids, and there is convincing evidence that one of these, cannabidiol (CBD; Figure 1), can suppress nausea and vomiting. CBD can reduce vomiting in Suncus murinus (house musk shrew) produced by nicotine, cisplatin or lithium chloride (LiCl, Kwiatkowska et al., 2004; Parker et al., 2004; Rock et al., 2011; 2012), although not by motion (Cluny et al., 2008). It can also reduce the establishment of conditioned gaping reactions (Grill and Norgren, 1978a,b) elicited by a LiCl-paired flavour, a model of nausea-induced behaviour in rats (see Parker and Limebeer, 2008 for review). Unlike conditioned taste avoidance, which can be produced by rewarding drugs as well as by emetic drugs, conditioned gaping reactions are only produced in rats by drugs that induce vomiting in emetic species, such as shrews (Parker, 2003; Parker et al., 2008). In addition, in a rodent model of anticipatory nausea evident in chemotherapy patients returning to the treatment-paired context, CBD (unlike traditional anti-emetics) effectively suppresses the expression of conditioned gaping elicited by LiCl-paired contextual cues (Rock et al., 2008).
Figure 1.

Structures of CBD and CBDA.
It has also been found in a phase II clinical trial that Sativex, a medicine that contains the phytocannabinoids, Δ9-tetrahydrocannabinol (Δ9-THC) and CBD, was both effective in reducing the incidence of chemotherapy-induced nausea and vomiting, and well tolerated by patients (Duran et al., 2010). It should be noted, however, that the log dose–response curves for the anti-emetic effects produced by CBD in house musk shrews are biphasic. Thus, CBD suppresses acute cisplatin-induced vomiting at 5 mg·kg−1, but potentiates it at 40 mg·kg−1 (Kwiatkowska et al., 2004). Similarly, acute vomiting elicited by LiCl is suppressed by low doses of CBD (5–10 mg·kg−1), whereas higher doses (20–40 mg·kg−1) of this phytocannabinoid act to facilitate LiCl-induced vomiting, rather than to reduce its expression (Parker et al., 2004). This narrow range of CBD efficacy may limit its clinical use as an anti-emetic.
Several findings that we recently made (Rock et al., 2012) support the hypothesis that CBD attenuates toxin-induced vomiting in shrews and signs of nausea in rats through indirect agonism of 5-HT1A receptors located in the brainstem. First, each of these effects of CBD can be prevented by the administration of a selective 5-HT1A receptor antagonist, either WAY100135 or WAY100635. Second, CBD displays significant potency at enhancing the ability of the selective 5-HT1A receptor agonist, 8-OH-DPAT, to stimulate [35S]GTPγS binding to rat brainstem membranes. Third, when co-administered with 8-OH-DPAT, CBD suppresses LiCl-induced signs of nausea in rats in an apparently synergistic manner. It is also noteworthy that a number of other in vivo effects of CBD seem to be 5-HT1A receptor mediated and that the log dose–response curve of CBD for the production of these effects is bell shaped (Mishima et al., 2005; Campos and Guimarães, 2008; Zanelati et al., 2010; Gomes et al., 2011; Maione et al., 2011). As to the ability of CBD to reduce signs of toxin-induced nausea in rats, we have postulated that this results from CBD-induced enhancement of the activation of somatodendritic 5-HT1A receptors in the dorsal raphe nucleus, as activation of these receptors is expected to reduce the release of nausea-inducing 5-HT in forebrain regions (Rock et al., 2012).
We have shown previously that CBD can also target CB1 receptors with significant potency. For example, in experiments performed with mouse whole brain membranes, we found that CBD (1 μM) can antagonize the cannabinoid receptor agonist, CP55940, in the [35S]GTPγS-binding assay with an apparent KB of 79 nM. This value is close to the concentration (100 nM) at which CBD increases the maximal effect (Emax) of 8-OH-DPAT in rat brainstem membranes (Rock et al., 2012), but significantly below the Ki value of CBD (4.9 μM) for its displacement of CP55940 from specific binding sites in mouse whole brain membranes (Thomas et al., 2007). In view of the ability of CBD to interact with CB1 receptors, it is also noteworthy that its ability to suppress vomiting in house musk shrews is not blocked by the selective cannabinoid CB1 receptor antagonist, SR141716A (Parker et al., 2004).
CBD is formed in cannabis from an acidic precursor, cannabidiolic acid (CBDA; Potter et al., 2008), the isolation from cannabis and structure of which was reported in 1965 (Mechoulam and Gaoni, 2010). In harvested cannabis, CBDA (Figure 1) gradually loses its carboxyl group to form CBD, a process that can be greatly accelerated by heating or burning cannabis, as happens when it is smoked. Currently, much more is known about the pharmacological actions of CBD, which are numerous (reviewed in Pertwee, 2008; Fernández-Ruiz et al., 2012), than about those of CBDA. There is already evidence, however, that CBDA shares the ability of CBD to activate the transient receptor potential (TRP) cation channels, TRPV1 and TRPA1, and to antagonize TRPM8 (De Petrocellis et al., 2008; 2011). Importantly, however, CBDA produces these effects with significantly less potency than CBD. Indeed, the concentration ranges at which CBD targets all of these cation channels, except TRPV1, overlap with the concentration (100 nM) at which it increases the Emax of 8-OH-DPAT in rat brainstem membranes (Rock et al., 2012). At 1 μM, both CBD and CBDA have also been shown to produce a significant downward shift in log concentration–response curve of the GPR55 agonist, L-α-lysophosphatidylinositol, for its stimulation of ERK1/2 phosphorylation in human GPR55-transfected HEK293 cells (Anavi-Goffer et al., 2012). Furthermore, CBDA has been found to affect the contractility of gastrointestinal tissue of house musk shrews in vitro, as indicated by its ability, at 10 μM, to reduce both the magnitude of contractions induced by carbachol or by electrical field stimulation and the tension of intestinal segments that had been pre-contracted with potassium chloride (Cluny et al., 2011). In addition, Takeda et al. (2008) have reported that CBDA is a selective inhibitor of COX-2, an enzyme expressed by cells undergoing inflammation; more recently, however, Ruhaak et al. (2011) found that CBDA did not inhibit this enzyme, prompting a need for further research.
The present investigation sought to determine whether CBDA can (i) inhibit LiCl-induced conditioned gaping to a flavour and to a context in rats in a 5-HT1A receptor-dependent manner; (ii) prevent toxin-induced vomiting in house musk shrews; and (iii) enhance activation of 5-HT1A receptors in rat brainstem membranes. We also investigated whether CBDA can suppress motion-induced vomiting in shrews. Our overall objective was to explore the possibility that CBDA might inhibit vomiting and conditioned gaping and enhance 5-HT1A receptor activation with greater potency or selectivity, and/or over a wider dose range, than CBD. Some of the results described in this paper have been presented to the International Cannabinoid Research Society (Bolognini et al., 2012).
Methods
Animals for in vivo experiments
Animal procedures complied with the Canadian Council on Animal Care and the National Institutes of Health guidelines or the UK Animals Act (Scientific Procedures) 1986. The protocols were approved by the Institutional Animal Care Committee, which is accredited by the Canadian Council on Animal Care or by the University of Bradford animal ethics committee. Toxin-induced emesis experiments were performed with male (36.5–45.8 g) and female (18.9–29.3 g) house musk shrews (S. murinus), aged 87–815 days at the time of testing, that had been bred and raised at the University of Guelph. They were single housed in cages in a colony room at an ambient temperature of 22°C on a 10/14 h light-dark schedule (lights off at 19:00 h). Shrews were tested during their light cycle. Motion-induced emesis experiments were performed with male (60–80 g) house musk shrews (S. murinus) that had been bred and raised at the University of Bradford colony. They were housed up to three to a cage at 22°C on a 10.5/13.5 h light-dark schedule with lights off at 19:00 h (on at 08:30 h). All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Naïve male Sprague–Dawley rats, obtained from Charles River Laboratories (St Constant, Quebec), were used for assessment of anti-nausea-like behaviour. They were single housed in shoebox cages, subjected to an ambient temperature of 21°C and a 12/12 h light-dark schedule (lights off at 08:00 h), and maintained on food and water ad libitum. Their body weights ranged from 264 to 430 g on the day of conditioning.
Drugs and materials for in vivo experiments
Samples of CBDA extracted from cannabis were provided by GW Pharmaceuticals (Porton Down, Wiltshire, UK), dimethyl sulfoxide (DMSO) by Sigma-Aldrich (Poole, Dorset, UK), and ethanol, Cremophor, LiCl, cisplatin and WAY100635 by Sigma (St Louis, MO, USA). In both the LiCl- and cisplatin-induced emesis experiments, performed in Guelph with shrews, and the LiCl-induced gaping experiments, performed in Guelph with rats, CBDA was prepared in a vehicle (VEH) consisting of a 1:1:18 mixture of ethanol, Cremophor and saline (SAL) and was administered i.p. in a volume of 2 mL·kg−1. This VEH and injection volume were also used for SR141716A, which was administered to rats i.p. at a dose (1 mg·kg−1) expected to reverse the effects of CB1 agonists (e.g. Vlachou et al., 2003), without potentiating the aversive effects of LiCl (Parker et al., 2003). In the motion-induced emesis experiments, performed in Bradford with shrews, CBDA was prepared in a VEH of 2% DMSO in distilled water and administered i.p. in a volume of 10 mL·kg−1. LiCl was prepared as a 0.15 M solution with sterile water and administered i.p. in a volume of 60 mL·kg−1 (390 mg·kg−1) to shrews (see Parker et al., 2004) and in a volume of 20 mL·kg−1 (127.2 mg·kg−1) to rats. Cisplatin was prepared as a 1 mg·mL−1 solution in SAL and was administered to shrews i.p., in a volume of 20 mL·kg−1 (20 mg·kg−1). WAY100635 (0.1 mg·mL−1) was prepared in SAL and administered to rats i.p in a volume of 1 mL·kg−1 (0.1 mg·kg−1).
In vivo procedures
Effect of CBDA on LiCl- or cisplatin-induced vomiting in house musk shrews
Shrews were transferred from the colony room to an empty cage in the experimental room that contained four meal worms. After 15 min they were injected with CBDA or VEH. In experiment A, they received an injection of either CBDA, at a dose of 0.05 mg·kg−1 (n = 8), 0.1 mg·kg−1 (n = 8), 0.5 mg·kg−1 (n = 6), or 5 mg·kg−1 (n = 8), or VEH (n = 6), followed 45 min later by an injection of LiCl (390 mg·kg−1). In experiment B, shrews were injected with 0.5 mg·kg−1 CBDA (n = 8), a dose found to be effective in experiment A, or with VEH (n = 8), 45 min before receiving an injection of cisplatin (20 mg·kg−1). Shrews were then immediately put individually into an observation chamber for 45 min in the LiCl experiment or for 70 min in the cisplatin experiment. The Plexiglas observation chambers (22.5 × 26 × 20 cm) were placed on a table with a clear glass top. A mirror beneath the chamber facilitated viewing of the ventral surface of each shrew and hence of all vomiting episodes. The frequency of these episodes was counted by an observer blind to the experimental conditions.
Effect of CBDA on LiCl-induced conditioned gaping to a flavour in rats
All rats were surgically implanted with an intraoral cannula under isoflurane anaesthesia as described by Limebeer et al. (2010). Following recovery from surgery (at least 3 days), each rat was subjected to an adaptation trial for which it was placed in the taste reactivity chamber with its cannula attached to an infusion pump (Model KDS100; KD Scientific, Holliston, MA, USA) for fluid delivery. The taste reactivity chambers were made of clear Plexiglas (22.5 × 26 × 20 cm) and placed on a table with a clear glass top. A mirror beneath each chamber facilitated viewing of the ventral surface of the rat and hence of any orofacial responses. Water was infused into the intraoral cannula of each rat for 2 min at a rate of 1 mL·min−1. On the day following this adaptation trial, the rats were subjected to a conditioning trial in which they received i.p. pretreatment injections of VEH (n = 12) or of one of four doses of CBDA (0.01 mg·kg−1, n = 8; 0.1 mg·kg−1, n = 12; 0.5 mg·kg−1, n = 7; 5.0 mg·kg−1, n = 8). Forty-five min after the pretreatment injection, each rat was individually placed in the taste reactivity chamber and intraorally infused with 0.1% saccharin solution for 2 min at a rate of 1 mL·min−1 while any orofacial responses were observed using a mirror located beneath the chambers. These responses were monitored using a video camera (Sony DCR-HC48; Henry's Cameras, Waterloo, ON, Canada) fire wired into a computer. Immediately after the saccharin infusion, all rats were injected with 20 mL·kg−1 of 0.15 M LiCl and returned to their home cage.
Seventy two hr later, all the rats were tested drug-free. They were again intraorally infused with 0.1% saccharin solution at a rate of 1 mL·min−1 over a 2 min period, during which their orofacial responses were video recorded. Rats were then returned to their home cages. The videotapes were later scored (at ½ speed) by an observer blind to the experimental conditions using ‘The Observer’ (Noldus Information Technology, Inc., Leesburg, VA, USA) for the behaviours of gaping (large openings of the mouth and jaw, with lower incisors exposed) and of tongue protrusions.
Conditioned taste avoidance was assessed in a single bottle test. Rats were water restricted at 16:00 h. The following morning, a single bottle containing 0.1% saccharin was placed in each cage at 09:00 h. Saccharin consumption was measured 30, 120 and 360 min later.
Effect of WAY100635 on CBDA-induced suppression of LiCl-induced conditioned gaping to a flavour in rats
The rats were treated exactly as in the LiCl-induced conditioned gaping experiments described in the previous section, except that they received an injection of either WAY100635 or SAL 15 min prior to the pretreatment injection of CBDA (0.1 mg·kg−1) or VEH. Rats were subjected to one or other of the following treatments (n = 12): SAL-VEH, SAL-CBDA, WAY100635-VEH and WAY100635-CBDA.
Effect of SR141716A on CBDA-induced suppression of LiCl-induced conditioned gaping to a flavour in rats
The rats were treated exactly as in the experiment with WAY100635 described in the previous section, except that they received an injection of either SR141716A (1 mg·kg−1) or VEH 15 min prior to the pretreatment injection of CBDA (0.1 mg·kg−1) or VEH. Rats were subjected to one or other of the following treatments: SAL-VEH (n = 12), SAL-CBDA (n = 12), SR141716A-VEH (n = 6) or SR141716A-CBDA (n = 6).
Effect of WAY100635 on the expression of CBDA-induced suppression of LiCl-induced conditioned gaping to a context in rats
A distinctive context was created by exposing rats to location, visual and tactile cues different from those to which they were being subjected in their home cage environment. This was achieved by placing a ‘contextual’ conditioning chamber in a dark room next to a 25 W light source. This chamber was made of black opaque Plexiglas, but was in all other respects identical to the one used in the LiCl-induced conditioned gaping experiments. The rats underwent four conditioning trials, during which the contextual chamber was paired with 127 mg·kg−1 LiCl. In every conditioning trial, each rat was injected with LiCl and immediately placed in the distinctive context for a 30 min period. This procedure was repeated four times, with a 72 h interval between each conditioning trial. For the test trial, rats were randomly assigned to one of four treatment groups (n = 10): SAL-CBDA, WAY100635-CBDA, SAL-VEH, WAY100635-VEH. CBDA or VEH was administered 15 min after SAL or WAY100635. Forty-five min later, rats were individually placed in the contextual chamber for a period of 5 min during which their orofacial responses were video recorded using a mirror located beneath the chamber.
Effect of CBDA on motion-induced emesis in shrews
The induction and quantification of the emetic response to motion have been described previously (Cluny et al., 2008). Briefly, six linked transparent compartments [100 mm (width) × 150 mm (length) × 150 mm (height)] are placed on a track that moves horizontally at a frequency of 1 Hz and amplitude of 40 mm. This motion induces emetic episodes, each of which consists of a bout of retching that involves strong repeated abdominal contractions accompanied by wide opening of the mouth and, initially, also by the passage of matter from the upper gastrointestinal tract. In this investigation, shrews (n = 6–18) were administered CBDA (0.02, 0.1 or 0.5 mg·kg−1) or VEH i.p. They were then immediately placed individually in a compartment of the shaker and monitored over a 45 min period for any signs of emesis and for any other overt behavioural changes. Motion was then applied to the compartments for 10 min during which the number of emetic episodes and the latency of onset of the first of these episodes were noted. If no emetic episodes were observed, the latency of onset was recorded as 600 s. Animals were observed for a further 2 min before being returned to their housing cage.
Analysis of in vivo data
Values have been expressed as means and variability as SEM. In the house musk shrew studies, the effect of CBDA on emesis was analysed using an independent t-test or one-way anova followed by a Bonferroni or Dunnett's multiple comparison post hoc test. In the rat studies, the number of gapes or hedonic reactions was analysed using a one-way anova and planned comparison tests. Statistical comparisons of the amounts of saccharin consumed during conditioned taste avoidance tests by different groups of rats were made by using either a 5 × 3 mixed factors anova, or in the experiments with WAY100635, a 4 × 3 mixed factors anova. For all analyses, P-values <0.05 were considered significant.
Animals for in vitro experiments
Brainstem tissue was obtained from adult male Lister hooded rats, ranging from 7 to 9 weeks of age, and purchased from Harlan UK, Ltd. (Blackthorn, UK). Mouse whole brain membranes were obtained from adult male MF1 mice purchased from Harlan UK, Ltd. All animal care and experimental procedures complied with the UK Animals (Scientific Procedures) Act, 1986, and associated guidelines for the use of experimental animals.
Drugs and materials for in vitro experiments
CBDA, extracted from Cannabis sativa, was provided by GW Pharmaceuticals. 8-OH-DPAT HBr and CP55940 were supplied by Tocris (Bristol, UK). [35S]GTPγS (1250 Ci·mmol−1), [3H]8-OH-DPAT (135.2 Ci·mmol−1) and [3H]CP555940 (160 Ci·mmol−1) were obtained from PerkinElmer Life Sciences, Inc. (Boston, MA, USA), GTPγS and adenosine deaminase from Roche Diagnostic (Indianapolis, IN, USA), and GDP and DMSO from Sigma-Aldrich.
In vitro procedures
Preparation of cell membranes from rat brainstem and mouse whole brain
Rat brainstem tissue was homogenized in ice-cold Choi lysis buffer (Tris–HCl 20 mM, sucrose 0.32 M, EDTA 0.2 mM, EGTA 0.5 mM, pH 7.5) containing Roche© protease inhibitor cocktail (1:40 v/v; Roche Diagnostics, Mannheim, Germany) and PMSF (1 mM). The homogenate was centrifuged at 13 500× g for 15 min and the resulting pellet was kept at −80°C for at least 2 h. The pellet was then resuspended in a buffer (Tris–HCl 50 mM; EDTA 1.0 mM; MgCl2 3.0 mM; pH 7.4), homogenized and stored at −80°C (Rock et al., 2012). Mouse whole brain membranes were prepared as described by Thomas et al. (2004).
[35S]GTPγS-binding assays
These assays were carried out with rat brainstem or mouse whole brain membranes (100 and 5 μg protein per well respectively), GTPγS-binding buffer (50 mM Tris–HCl; 50 mM Tris-Base; 5 mM MgCl2; 1 mM EDTA; 100 mM NaCl; 1 mM dithiothreitol; 0.1% BSA), 0.1 nM [35S]GTPγS and 30 μM GDP, in a final volume of 500 μL (Thomas et al., 2005). Membranes were pre-incubated for 30 min at 30°C with 0.5 U·mL−1 adenosine deaminase (200 U·mL−1) to remove any endogenous adenosine. Binding was initiated by the addition of [35S]GTPγS. Non-specific binding was measured in the presence of 30 μM GTPγS. Assays were performed at 30°C for 60 min. The reaction was terminated by the addition of ice-cold Tris-binding buffer and vacuum filtration using a 24-well sampling manifold (Brandel Cell Harvester; Brandel, Inc., Gaithersburg, MD, USA) and Whatman GF/B glass fibre filters that had been soaked in wash buffer at 4°C for 24 h. Each reaction tube was washed three times with a 4 mL aliquot of buffer. The filters were oven dried for 60 min and then placed in 5 mL of scintillation fluid (Ultima Gold XR; PerkinElmer, Buckinghamshire, UK). Radioactivity was quantified by liquid scintillation spectrometry.
Radioligand displacement assays
Assays were carried out with [3H]8-OH-DPAT or [3H]CP55940 and Tris-binding buffer (50 mM Tris–HCl, 50 mM Tris-Base, 0.1% BSA; pH 7.4), total assay volume of 500 μL, using the filtration procedure described previously by Ross et al. (1999). Binding was initiated by the addition of rat brainstem membranes (500 μg protein per well), for experiments with [3H]8-OH-DPAT, or of mouse whole brain membranes (33 μg protein per well), for experiments with [3H]CP55940. All assays were performed at 37°C for 60 min before termination by addition of ice-cold Tris-binding buffer and vacuum filtration using a 24-well sampling manifold (Brandel Cell Harvester) and Brandel GF/B filters that had been soaked in wash buffer at 4°C for at least 24 h. Each reaction well was washed six times with a 1.2 mL aliquot of Tris-binding buffer. The filters were oven dried for 60 min and then placed in 5 mL of scintillation fluid (Ultima Gold XR). Radioactivity was quantified by liquid scintillation spectrometry. Specific binding was defined as the difference between the binding that occurred in the presence and absence of 1 μM unlabelled 8-OH-DPAT or CP55940. The concentration of [3H]8-OH-DPAT and [3H]CP55940 used in these displacement assays was 0.7 nM. Compounds under investigation were stored at −20°C as stock solutions of 10 mM in DMSO, the VEH concentration in all assay wells being 0.1% DMSO. The binding parameters for [3H]CP55940, determined by fitting data from saturation-binding experiments to a one-site saturation plot using GraphPad Prism (GraphPad Software, Inc., San Diego, CA, USA), were 2336 fmol·mg−1 protein (Bmax) and 2.31 nM (Kd) (Thomas et al., 2004).
Analysis of in vitro data
Values have been expressed as means and variability as SEM or as 95% confidence limits. The concentration of 8-OH-DPAT or CBDA that produced a 50% displacement of [3H]8-OH-DPAT or [3H]CP55940 from specific binding sites (IC50 values) was calculated using GraphPad Prism and the corresponding Ki value of CP55940 was calculated using the equation of Cheng and Prusoff (1973). Values for EC50, Emax and SEM or 95% confidence limits of these values have been calculated by non-linear regression analysis using the equation for a sigmoidal dose–response curve (GraphPad Prism). The apparent dissociation constant (KB) value of CBDA for its antagonism of CP55940 in the [35S]GTPγS-binding assay has been calculated by Schild analysis (Graph Pad Prism). Mean values were compared with zero by column statistics analysis using the Wilcoxon signed rank test (GraphPad Prism 5.0). P-values <0.05 were considered significant.
Results
CBDA reduces LiCl- and cisplatin-induced emesis in shrews
CBDA reduced LiCl-induced emesis at doses of 0.1 and 0.5 mg·kg−1, and cisplatin-induced emesis at a dose of 0.5 mg·kg−1. A one-way anova of the number of vomiting episodes elicited by LiCl among the pretreatment groups revealed a significant effect of group, F(4, 31) = 7.4; P < 0.001 (Figure 2A). Subsequent Bonferroni post hoc comparison tests revealed that shrews pretreated with 0.5 mg·kg−1 (P < 0.01) or 0.1 mg·kg−1 (P < 0.05) CBDA vomited less frequently than those pretreated with VEH. Similarly, as shown in Figure 2B, an independent t-test indicated that shrews given cisplatin and pretreated with CBDA vomited significantly less than VEH-treated controls, F(1,13) = 9.2, P < 0.01. When administered by themselves, neither CBDA nor VEH produced emesis during the 45 min pretreatment period before toxin administration (data not shown).
Figure 2.

Effect of CBDA (0.05, 0.1, 0.5 or 5.0 mg·kg−1) or VEH administered i.p. 45 min prior to toxin administration. The number of emetic episodes in shrews treated with LiCl (A) or cisplatin (B) was measured. Each bar represents the mean ± SEM (n = 6–8). The asterisks indicate a significant difference from the VEH-treated control animals (*P < 0.05; **P < 0.01, one-way anova).
CBDA reduces LiCl-induced conditioned gaping to a flavour in rats
Planned comparison tests showed that CBDA significantly reduced LiCl-induced gaping relative to VEH-pretreated controls at doses of 0.01 and 0.1 mg·kg−1 (P < 0.02), although not at 0.5 or 5 mg·kg−1 (Figure 3A), a one-way anova indicating a significant effect of dose, F(4, 42) = 4.8; P = 0.003. The mean number of tongue protrusions elicited during the conditioning trial by 0.1% saccharin in groups of rats pretreated with various doses of CBDA is presented in Figure 3B. A one-way anova of the tongue protrusion data revealed a significant effect of dose, F(4, 46) = 3.8; P = 0.01. Planned comparisons indicated that at a dose of 0.01 mg·kg−1, CBDA significantly enhanced saccharin hedonics (tongue protrusions) relative to VEH controls as well as to groups pretreated with 0.5 or 5.0 mg·kg−1 CBD (P < 0.02).
Figure 3.
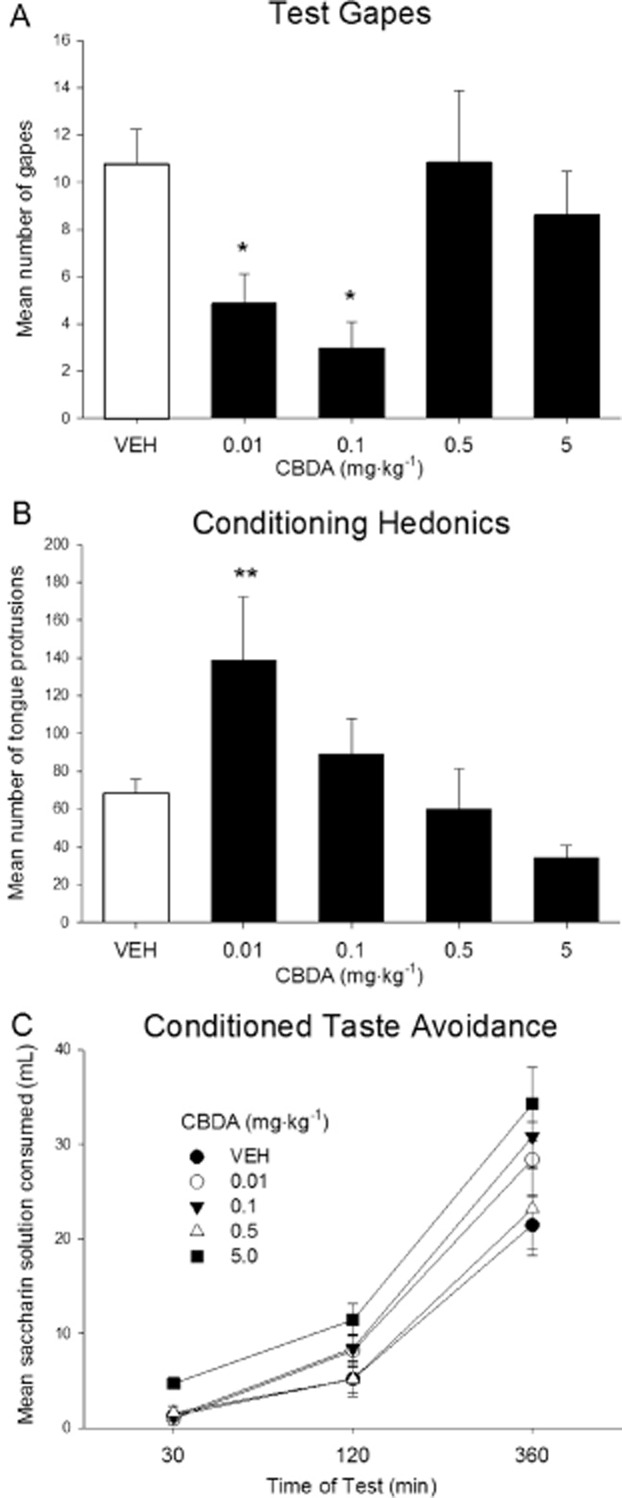
Effect of CBDA (0.01, 0.1, 0.5, 5.0 mg·kg−1) or VEH administered i.p. to rats 45 min prior to LiCl. The number of conditioned gaping responses was measured during the test trial (A).The number of tongue protrusions was measured during the conditioning trial (B). Each bar represents the mean ± SEM (n = 7–12). The cumulative amount of saccharin solution consumed (mL ± SEM) during a one-bottle consumption test was measured at 30, 120 and 360 min after introduction of the bottle to fluid-restricted rats (C). The asterisks indicate a significant difference from the VEH-treated control animals (*P < 0.02; **P < 0.01; one-way anova).
It is unlikely that CBDA attenuated conditioned gaping in rats through a direct effect on learning, because neither of the gape-attenuating doses of CBDA (0.01 and 0.1 mg·kg−1) interfered with LiCl-induced conditioned taste avoidance, a behavioural effect that is not dependent on a nauseating treatment (e.g. Parker et al., 2008). The mean amounts of saccharin consumed during the conditioned taste avoidance test at 30, 120 and 360 min by groups of rats treated with VEH or with 0.01, 0.1, 0.5 or 5.0 mg·kg−1 CBDA are presented in Figure 3C. A 5 × 3 mixed factors anova revealed a significant effect of both time of test, F(2,84) = 237.5; P < 0.01, and group, F(4, 42) = 2.9; P = 0.03. Planned comparison tests revealed that the group of rats receiving 5.0 mg·kg−1 CBDA drank significantly more saccharin than the VEH-treated group (P < 0.02). No other significant differences between treatments were detected and there was no group by time interaction (P > 0.05).
Effect of CBDA on LiCl-induced conditioned gaping to a flavour in rats pretreated with WAY100635 or SR141716A
The ability of CBDA (0.1 mg·kg−1) to suppress LiCl-induced conditioned gaping in rats was abolished by pretreatment with the 5-HT1A antagonist, WAY100635, at a dose of 0.1 mg·kg−1 (Figure 4A). A one-way anova revealed a significant effect of pretreatment, F(3, 44) = 8.9; P < 0.001. Planned comparison tests revealed that the SAL-CBDA group displayed fewer gapes than all other groups. Groups pretreated with WAY100635 prior to CBDA did not display suppressed conditioned gaping reactions relative to SAL-VEH. No differences between any of the groups of rats used for this set of experiments were detected during the taste reactivity conditioning trial.
Figure 4.

Effect of WAY100635 (0.1 mg·kg−1) or SAL administered i.p. 15 min prior to CBDA (0.1 mg·kg−1 i.p.) or VEH in LiCl-treated rats. The number of conditioned gaping responses was measured during the test trial (A). Each bar represents the mean number of conditioned gaping responses ± SEM (n = 12). The cumulative amount of saccharin solution consumed (mL ± SEM) during a one-bottle consumption test was measured at 30, 120 and 360 min after introduction of the bottle to fluid-restricted rats (B). Individual one-way anovas for each time point indicate that there is no significant difference between any of the four pretreatment groups (P > 0.05). The asterisks indicate a significant difference from the SAL-VEH-treated control animals (***P < 0.001; one-way anova).
The mean amount of saccharin consumed during the conditioned taste avoidance test at 30, 120 and 360 min is presented in Figure 4B. A 4 × 3 mixed factors anova revealed a significant effect of time of test, F(2,88) = 298.2; P < 0.01, and a group by time interaction, F(6,88) = 2.5; P = 0.03, but no main effect of group. Individual one-way anovas for each time point indicate that only at the 360 min time point did the VEH-CBDA group drink marginally more saccharin than all the other groups. However, individual one-way anovas for each time point indicate that this apparent difference is not statistically significant (P = 0.055) and, indeed, that at none of the time points are there statistically significant differences between any of the groups.
In contrast to WAY100635, SR141716A (1 mg·kg−1) did not attenuate the suppression of LiCl-induced conditioned gaping produced by CBDA (Figure 5A). A one-way anova revealed a significant effect of pretreatment group, F(3,32) = 9.4; P < 0.001. Planned comparisons indicated that both the VEH-CBDA and the SR141716A-CBDA groups gaped significantly less than the VEH-VEH-treated controls (P < 0.002). No differences between any of the groups of rats used in these experiments were detected during the taste reactivity conditioning trial. The mean amounts of saccharin consumed during the conditioned taste avoidance test at 30, 120 and 360 min are shown in Figure 5B. A 4 × 3 mixed factors anova revealed a significant effect of time of test, F(2,64) = 225.6; P < 0.001, but no significant interaction or pretreatment effect (P > 0.05).
Figure 5.

Effect of SR141716A (SR; 1 mg·kg−1) or VEH administered i.p. 15 min prior to CBDA (0.1 mg·kg−1 i.p.) or VEH in LiCl-treated rats. The number of conditioned gaping responses was measured during the test trial (A). Each bar represents the mean number of these responses ± SEM (n = 6–12). The cumulative amount of saccharin solution consumed (mL ± SEM) during a one-bottle consumption test was measured at 30, 120 and 360 min after introduction of the bottle to fluid-restricted rats (B). The asterisks indicate a significant difference from the VEH-treated control animals (**P < 0.002; one-way anova).
WAY100635 prevents the expression of CBDA-induced suppression of context-induced conditioned gaping (anticipatory nausea) in rats
CBDA (0.1 mg·kg−1) interfered with the nausea-inducing effects of the context previously paired with LiCl, and this effect was blocked by pretreatment with the 5-HT1A antagonist WAY100635. The mean number of gapes during the test trial is presented in Figure 6. A one-way anova revealed a significant effect of pretreatment, F(3, 36) = 3.7; P = 0.02. Planned comparison tests revealed that the SAL-CBDA group displayed fewer gapes than all other groups. Groups pretreated with WAY100635 prior to CBDA did not display suppressed conditioned gaping reactions relative to SAL-VEH.
Figure 6.
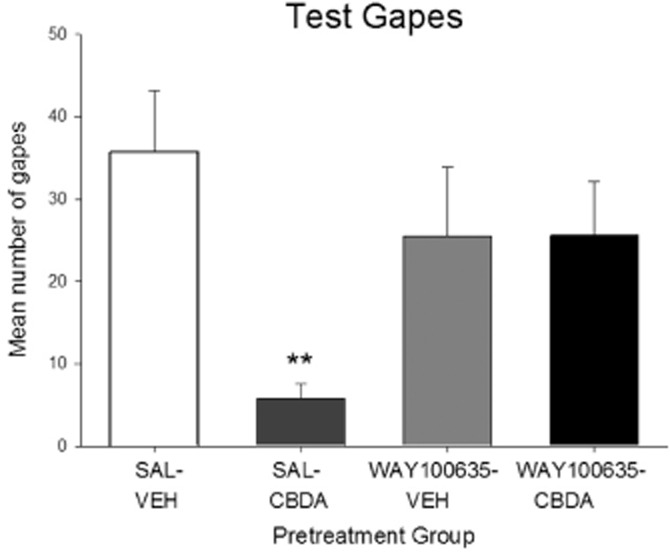
Effect of WAY100635 (0.1 mg·kg−1) or SAL administered i.p. on the ability of CBDA (0.1 mg·kg−1 i.p.) to suppress the expression of gaping in rats to a context previously paired with LiCl (a model of anticipatory nausea). Each bar represents the mean number of conditioned gaping responses ± SEM (n = 10). The asterisks indicate a significant difference from the SAL-VEH-treated control animals (**P < 0.01; one-way anova).
CBDA reduces emesis in shrews induced by motion
CBDA (0.1 and 0.5 mg·kg−1) can both reduce the number of emetic episodes and increase the latency of onset of emesis in shrews in response to motion (Figure 7A,B). This it does in a dose-dependent manner. Thus, there is a significant difference between treatment groups in the number of motion-induced emetic episodes, F(3,32) = 8.677; P = 0.0002. Dunnett's post hoc analysis revealed that CBDA (0.1 mg·kg−1, P < 0.01; 0.5 mg·kg−1, P < 0.001) reduced the number of motion-induced emetic episodes compared with VEH-treated controls (Figure 7A). anova also revealed a significant difference in the latency of onset of emesis between treatment groups, F(3,32) = 8.530; P = 0.0003. Post hoc analysis showed that CBDA (0.1 mg·kg−1, P < 0.05; 0.5 mg·kg−1, P < 0.001) increased the latency to the onset of the first motion-induced emetic episode compared with VEH-treated controls (Figure 7B). No emetic episodes were observed in the 45 min observation period between the administration of CBDA or VEH and the initiation of the motion stimulus.
Figure 7.

Effect of CBDA (0.02, 0.1, 0.5 mg·kg−1) or VEH administered i.p. to shrews 45 min prior to the application of motion. The number of emetic episodes (A) and latency of onset to the first emetic episode (B) induced by a 10 min horizontal motion stimulus (frequency: 1 Hz; amplitude: 40 mm) was measured in shrews. Each bar represents the mean ± SEM, n = 5–15. The asterisks indicate a significant difference from the VEH-treated control animals analysed using one-way anova followed by Dunnett's post hoc test (*P < 0.05; **P < 0.01; ***P < 0.001).
CBDA enhances the ability of a 5-HT1A receptor agonist to stimulate [35S]GTPγS binding to rat brainstem membranes
As shown in Figure 8 and Table 1, CBDA produced significant increases in the Emax of 8-OH-DPAT at concentrations of 0.1, 1.0, 10 and 100 nM, although not at 0.01 nM or 1 μM. These increases in Emax were accompanied by increases in the mean EC50 values of CBDA. However, none of these increases were statistically significant (P > 0.05; Table 1). By itself, CBDA did not produce any significant stimulation of [35S]GTPγS binding to rat brainstem membranes at concentrations ranging from 0.01 nM to 1 μM (P > 0.05; n = 6). Its mean Emax in this assay, with 95% confidence limits shown in parentheses, was found to be 5.7% (–0.9 and 12.3%). CBDA did not share the ability of 8-OH-DPAT to displace [3H]8-OH-DPAT from rat brainstem membranes at concentrations in the nanomolar range. Thus, CBDA concentrations of 0.1, 1, 10, 100 and 1000 nM did not cause mean values for % displacement of [3H]8-OH-DPAT to rise significantly above zero (column statistics analysis; n = 8). The IC50 and Emax values of 8-OH-DPAT for its displacement of [3H]8-OH-DPAT with 95% confidence limits shown in parentheses were 4.8 nM (3.2 and 7.5 nM; n = 8) and 102.1% (95.9 and 108.3%) respectively.
Figure 8.

Effect of CBDA (0.01, 0.1, 1.0, 10, 100, 1000 nM) on 8-OH-DPAT-induced stimulation of [35S]GTPγS binding to rat brainstem membranes. Symbols represent mean values ± SEM (n = 6 or 7). Mean Emax and EC50 values for 8-OH-DPAT in panels (A)–(F) that were determined in the presence of VEH (DMSO) or CBDA, together with the 95% confidence limits of these values, are listed in Table 1.
Table 1.
Effects of various concentrations of CBDA on mean EC50 and Emax values of 8-OH-DPAT for its stimulation of [35S]GTPγS binding to rat brainstem membranes
| Pretreatment | Mean EC50 (nM) | 95% confidence limits (nM) | Mean Emax (%) | 95% confidence limits (%) | n |
|---|---|---|---|---|---|
| Vehicle | 5.0 | 1.6 and 15.0 | 29.1 | 24.6 and 33.7 | 7 |
| 0.01 nM CBDA | 10.4 | 3.6 and 30.1 | 31.3 | 26.7 and 35.9 | 7 |
| Vehicle | 11.4 | 5.0 and 25.9 | 29.2 | 25.1 and 33.3 | 7 |
| 0.1 nM CBDA | 43.9 | 21.9 and 88.1 | 45.1* | 39.8 and 50.3 | 7 |
| Vehicle | 6.0 | 1.8 and 20.3 | 26.8 | 22.2 and 31.4 | 6 |
| 1.0 nM CBDA | 15.5 | 4.0 and 59.7 | 45.1* | 36.7 and 53.4 | 6 |
| Vehicle | 12.6 | 5.3 and 30.0 | 29.1 | 25.2 and 33.0 | 7 |
| 10 nM CBDA | 43.7 | 21.1 and 90.2 | 46.8* | 40.9 and 52.8 | 7 |
| Vehicle | 9.5 | 4.1 and 21.9 | 22.9 | 19.9 and 25.8 | 7 |
| 100 nM CBDA | 26.3 | 13.4 and 51.6 | 37.1* | 33.0 and 41.2 | 7 |
| Vehicle | 7.8 | 3.1 and 19.8 | 31.7 | 26.7 and 36.7 | 6 |
| 1000 nM CBDA | 18.3 | 5.0 and 67.4 | 28.8 | 22.6 and 35.0 | 6 |
The 95% confidence limits of this mean value do not overlap with those of the mean value in the previous row, indicating it to be significantly greater than the mean value obtained from experiments with vehicle-treated membranes (P < 0.05). See also Figure 8.
CBDA can block cannabinoid CB1 receptors
CBDA displaced [3H]CP55940 from specific binding sites on mouse whole brain membranes and antagonized stimulation of [35S]GTPγS binding to these membranes induced by the selective cannabinoid receptor agonist, CP55940 (Figure 9A,B). These mean Ki and KB values, with their 95% confidence limits shown in parentheses, are 0.9 μM (0.7 and 1.3 μM) and 1.8 μM (0.6 and 5.5 μM) respectively. CBDA neither stimulated nor inhibited [35S]GTPγS binding to mouse whole brain membranes when administered by itself at concentrations ranging from 1 nM to 10 μM (n = 10). CBDA concentrations of 1, 10, 100, 1000 and 10 000 nM did not alter [35S]GTPγS binding to a value that was significantly different from zero (column statistics analysis; P > 0.05), and the mean Emax of CBDA in this assay was not significantly greater than zero (Figure 9C; P > 0.05).
Figure 9.

Panel (A) Effect of CBDA on specific binding of [3H]CP55940 to mouse whole brain membranes (n = 6). Its mean Ki value with the 95% confidence limits of this value shown in parentheses is 0.9 μM (0.7 and 1.3 μM). Panel (B) Effect of 10 μM CBDA on CP55940-induced stimulation of [35S]GTPγS binding to mouse whole brain membranes. The mean EC50 and Emax values of CP55940 with their 95% confidence limits shown in parentheses are 12.4 nM (4.3 and 35.8 nM; n = 8) and 64.5% (52.5 and 76.5%), respectively, in the presence of VEH (DMSO), and 66.6 nM (27.0 and 164 nM; n = 8) and 63.3% (53.7 and 72.9%), respectively, in the presence of 10 μM CBDA. The mean apparent KB value of CBDA for this antagonism with its 95% confidence limits shown in parentheses is 1.8 μM (0.6 and 5.5 μM). Panel (C) Effect of CBDA on [35S]GTPγS binding to mouse whole brain membranes (n = 16). None of the five mean values shown are significantly different from zero (column statistics analysis; P > 0.05).
Discussion
The results we obtained in this investigation revealed a number of pharmacological similarities between CBDA and CBD. Thus, as we found previously for CBD (Rock et al., 2008; 2012), CBDA appears (i) to suppress LiCl- and cisplatin-induced vomiting in shrews; (ii) to reduce the establishment of LiCl-induced conditioned gaping elicited by a flavour and the expression of LiCl-induced conditioned gaping elicited by a context (anticipatory nausea) in rats; (iii) to enhance the ability of 8-OH-DPAT to stimulate [35S]GTPγS binding to rat brainstem membranes without displaying any detectable activity in this assay in the absence of 8-OH-DPAT; and (iv) to display a bell-shaped log dose–response curve for the production of the second and third of these effects. We also found that like the inhibitory effect of CBD on LiCl-induced conditioned gaping in rats (Rock et al., 2012), the production of such inhibition by CBDA could be abolished by the 5-HT1A receptor-selective antagonist, WAY100635. In contrast, however, CBDA-induced suppression of LiCl-induced conditioned gaping in rats was not attenuated by the CB1 receptor inverse agonist/antagonist SR141716A, suggesting that this effect is not CB1 mediated. Finally, again as found previously for CBD (Rock et al., 2012), concentrations of CBDA that enhanced 8-OH-DPAT-induced stimulation of [35S]GTPγS binding to rat brainstem membranes failed to displace [3H]8-OH-DPAT from such membranes. Taken together, these findings support the hypothesis that, as we have suggested previously for CBD (Rock et al., 2012), CBDA inhibits nausea-induced behaviour in rats by somehow enhancing the activation of 5-HT1A receptors. It would be interesting to investigate the possibility that these are somatodendritic 5-HT1A autoreceptors that are known to be located within the dorsal raphe nucleus and to reduce the firing of 5-HT afferents when activated, for example, by establishing whether CBDA shares the ability of CBD to inhibit nausea-induced behaviour in rats when injected directly into that region of the brain (Rock et al., 2012).
We also detected some pharmacological differences between CBDA and CBD. First, we found that at doses of 0.1 and 0.5 mg·kg−1 i.p., CBDA reduced vomiting in shrews when this was induced by motion. In contrast, we have found previously that CBD does not produce such an effect, at least when it is administered at doses ranging from 0.5 to 40 mg·kg−1 i.p. (Cluny et al., 2008). Second, we found that CBDA displays markedly greater potency than CBD (Rock et al., 2012) at enhancing 8-OH-DPAT-induced stimulation of [35S]GTPγS binding to rat brainstem membranes, and that this enhancement was induced over a much wider range of concentrations by CBDA than by CBD. More specifically, whereas this effect was produced by CBD at 100 nM but not at 10 nM or 1 μM (Rock et al., 2012), it was produced by CBDA at concentrations ranging from 0.1 to 100 nM. Third, we found that these CBDA-induced increases in the Emax of 8-OH-DPAT were not accompanied by an upward shift in the log concentration–response curve of this 5-HT1A receptor agonist of the sort we found previously to be produced by 100 nM CBD (Rock et al., 2012). Whether there is a concentration of CBDA, for example, somewhere between 0.1 and 100 nM, that does produce an upward shift of this kind remains to be established.
In addition, CBDA differs from CBD in the manner in which it appears to target the cannabinoid CB1 receptor. Thus, in contrast to CBD (Thomas et al., 2007), CBDA seemed to produce a dextral but not a downward shift in the log concentration–response curve of CP55940 in mouse whole brain membranes, and its mean apparent KB value for this antagonism does not differ significantly from its mean Ki value for the displacement of [3H]CP55940 (Results). CBDA also differs from CBD (Thomas et al., 2007), first, by failing to alter [35S]GTPγS binding to mouse whole brain membranes, at concentrations ranging from 1 nM to 10 μM (Results), and second (Figure 8), by increasing the Emax of 8-OH-DPAT in rat brainstem membranes at a concentration (0.1–100 nM) well below those at which it antagonizes CP55940 in mouse whole brain membranes, as indicated by the apparent KB value of CBDA for this antagonism (1.8 μM). These data suggest that CBDA does not possess significant activity as either an agonist or an inverse agonist at the CB1 receptor even at a concentration that is 10 000-fold higher than a concentration (0.1 nM) at which it can enhance the activation of 5-HT1A receptors by 8-OH-DPAT.
CBDA undergoes slow non-enzymatic decarboxylation to CBD, a process that can be accelerated by heat (Potter et al., 2008). This prompts a need for further research directed at investigating the extent to which any of the effects we observed following CBDA administration in vivo or in vitro were due partly or wholly to CBD or any other compounds that may have been formed from CBDA by enzymatic or non-enzymatic processes. In the meantime, however, it is noteworthy that because of the differences we detected between these two phytocannabinoids (see previous two paragraphs), it is unlikely that CBD played a major role in the production of the effects we observed after CBDA administration, particularly in those experiments in which CBDA displayed higher potency than CBD.
It will also be important to seek out the mechanism(s) by which CBDA induces its apparent enhancement of 5-HT1A receptor activation. Such research should be directed initially at establishing whether, at concentrations in the submicromolar range, CBDA can interact directly with 5-HT1A receptors to enhance activation of this kind. This could be investigated by carrying out in vitro experiments with 5-HT1A-transfected cells that, in contrast to brain tissue, do not express other types of receptor. These experiments could be performed using not only the [35S]GTPγS-binding assay, but also a second in vitro assay in which, for example, the measured response is inhibition of forskolin-stimulated cyclic AMP production. It would also be of interest to explore the possibility that CBDA affects 5-HT1A receptor activation in brain tissue indirectly, by interacting with one or more other pharmacological targets that are functionally linked to this receptor.
It is important to note that CBDA selectively interfered with the production of LiCl-induced conditioned gaping reactions, but spared LiCl-induced conditioned taste avoidance; therefore, it did not interfere with learning per se (see Parker et al., 2008 for review). Furthermore, a low dose of CBDA (0.01 mg·kg−1 i.p.) also enhanced saccharin palatability unconditionally, as indicated by our finding that rats injected with this dose displayed more saccharin-elicited tongue protrusions (hedonic reactions) than controls during conditioning (Figure 3B). This suggests that this dose of CBDA may have a hedonic or an appetite-enhancing effect, as has been found for Δ9-THC (Jarrett et al., 2005). It would be interesting, therefore, to establish whether low doses of CBDA enhance feeding or modify depression-like behaviours in other paradigms, such as the forced swim test.
The ability of CBDA to reduce both motion- and toxin-induced vomiting is noteworthy, as compounds such as CBD and 5-HT3 receptor antagonists that are effective in reducing toxin-induced vomiting often fail to reduce signs of motion sickness (Stott et al., 1989; Levine et al., 2000; Cluny et al., 2008). This is not the case with CBDA, as it suppressed both toxin- and motion-induced vomiting in shrews. Toxin-induced vomiting involves the area postrema (Horn et al., 2007; De Jonghe and Horn, 2009), a structure that is not essential for motion-induced vomiting (Wilpizeski et al., 1986; Lang et al., 1999), whereas motion-induced vomiting requires a functional vestibular system (Johnson et al., 1962; 1999; Money and Friedberg, 1964; Kennedy et al., 1965; Reason, 1978; Wilpizeski et al., 1986; Cheung et al., 1991) and is thought to result from a sensory mismatch between vestibular, visual and non-vestibular information from sensory inputs. Furthermore, when saccharin is paired with vestibular stimulation, rats display a conditioned gaping reaction (like that induced by LiCl) when re-exposed to the saccharin flavour (Cordick et al., 1999), and vestibular lesions selectively abolish motion-, but not LiCl-induced conditioned gaping reactions (Ossenkopp et al., 2003). These published findings indicate that the neuronal pathways that mediate the inhibitory effect of CBDA on motion-induced vomiting are most probably not the same as those that mediate its inhibitory effect on toxin-induced vomiting. It is also noteworthy that the suppressive effect of 8-OH-DPAT on motion-induced emesis has been found not to be reversed by pretreatment with WAY100635, even at the rather high dose of 1 mg·kg−1 i.p. (Javid and Naylor, 2006), suggesting that this suppressive effect is not 5-HT1A receptor mediated. It is possible, therefore, that the suppressive effect of CBDA on motion-induced vomiting is also induced through one or more 5-HT1A receptor-independent mechanisms.
The current first-line therapy for treatment of acute and delayed chemotherapy-induced nausea and vomiting is the combination of 5-HT3 receptor antagonists (e.g. ondansetron), NK1 receptor antagonists (e.g. aprepitant) and dexamethasone (e.g. Poli-Bigelli et al., 2003). Although this therapy is effective in controlling acute and delayed vomiting, it is somewhat less effective in controlling acute and delayed nausea (e.g. de Boer-Dennert et al., 1997; Poli-Bigelli et al., 2003; Meiri et al., 2007). However, in patients who experience anticipatory nausea, there is no specific therapy available (Nesse et al., 1980; Morrow et al., 1998; Ballatori and Roila, 2003; Hickok et al., 2003; Foubert and Vaessen, 2005); indeed, the most commonly prescribed treatment for this side effect is a non-specific anxiolytic (Malik et al., 1995). Anticipatory nausea remains an area of unmet clinical need in cancer therapy. As current anti-emetics, such as the 5-HT3 antagonist ondansetron, are not effective in reducing anticipatory nausea once it develops in rats (Limebeer et al., 2008) and humans (Morrow et al., 1998), CBDA's ability to reduce the expression of anticipatory nausea is important. Other cannabinoids such as CBD (Rock et al., 2008), Δ9-THC, (Limebeer et al., 2006), and the fatty acid amide hydrolase inhibitor, URB597 (Rock et al., 2008), have also been shown to be effective in reducing anticipatory nausea in rats, highlighting a unique therapeutic potential for cannabinoids.
In conclusion, this investigation has shown for the first time that CBDA can potently suppress signs of nausea in rats in a 5-HT1A receptor-dependent manner, and that it can increase the ability of the 5-HT1A receptor agonist, 8-OH-DPAT, to stimulate [35S]GTPγS binding to rat brainstem membrane, again with high potency. We also found that CBDA potently inhibits toxin- and motion-induced vomiting in shrews, although any involvement of 5-HT1A receptors in this inhibition has yet to be investigated. The cannabinoid receptor agonist, Marinol (synthetic Δ9-THC), is currently indicated to treat nausea and vomiting in patients who have failed to respond adequately to conventional anti-emetics (e.g. Meiri et al., 2007). Δ9-THC (3–10 mg·kg−1) has been shown to reduce vomiting in house musk shrews (Kwiatkowska et al., 2004), whereas doses of CBDA as low as 0.5 mg·kg−1 were effective in this study. We found that, in contrast to Δ9-THC (Pertwee, 2008), CBDA does not seem to activate CB1 receptors in vitro even at the rather high concentration of 10 μM. When taken together, our findings support the hypothesis that CBDA can suppress signs of nausea and vomiting in a CB1 receptor-independent manner, raising the possibility that if used to ameliorate nausea or vomiting in the clinic, it would not trigger any CB1 receptor-mediated tolerability or abuse liability problems. Importantly, these findings suggest that CBDA could be developed as a potent and selective treatment for nausea and vomiting, and in particular for the treatment of anticipatory nausea, a symptom with no specific therapy currently available. Our findings also raise the possibility that compared with CBD (Mishima et al., 2005; Campos and Guimarães, 2008; Zanelati et al., 2010; Gomes et al., 2011; Maione et al., 2011), CBDA may display greater potency, efficacy or selectivity at ameliorating signs of cerebral infarction, pain, anxiety and depression in an apparent 5-HT1A receptor-dependent manner in animal models, and so possibly also in the clinic.
Acknowledgments
This research was supported by grants from GW Pharmaceuticals to R. G. P., L. A. P. and F. A. J., from the National Institute on Drug Abuse to R. G. P. (DA-03672), and from Natural Sciences and Engineering Council of Canada (NSERC-92057) to L. A. P. The authors would like to thank Linda Groocock for management of the shrew colony at the University of Guelph, and the University of Bradford for holding and maintaining its shrew colony.
Glossary
Abbreviations
- 5-HT1A
5-hydroxytryptamine1A
- 8-OH-DPAT
8-hydroxy-2-(di-n-propylamino)tetralin
- CBD
cannabidiol
- CBDA
cannabidiolic acid
- DMSO
dimethyl sulfoxide
- LiCl
lithium chloride
- SAL
saline
- SR141716A
N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride
- VEH
vehicle
- Δ9-THC
Δ9-tetrahydrocannabinol
Conflict of interest
The research performed in Guelph and Aberdeen was funded by GW Research Ltd, UK. CS and MD are employees of GW Research, Ltd., UK.
References
- Anavi-Goffer S, Baillie G, Irving AJ, Gertsch J, Greig IR, Pertwee RG, et al. Modulation of L-α-lysophosphatidylinositol/GPR55 mitogen-activated protein kinase (MAPK) signaling by cannabinoids. J Biol Chem. 2012;287:91–104. doi: 10.1074/jbc.M111.296020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballatori E, Roila F. Impact of nausea and vomiting on quality of life in cancer patients during chemotherapy. Health Qual Life Outcomes. 2003;1:46. doi: 10.1186/1477-7525-1-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer-Dennert M, de Wit R, Schmitz PIM, Djontono J, v Beurden V, Stoter G, et al. Patient perceptions of the side-effects of chemotherapy: the influence of 5HT3 antagonists. Br J Cancer. 1997;76:1055–1061. doi: 10.1038/bjc.1997.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolognini D, Rock EM, Cascio MG, Parker L, Pertwee RG. Cannabidiolic acid behaves as a highly potent enhancer of 5-HT1A receptor activation both in vitro and in vivo. 2012. p. 43. 22nd Annual Symposium of the Cannabinoids. International Cannabinoid Research Society.
- Campos AC, Guimarães FS. Involvement of 5HT1A receptors in the anxiolytic-like effects of cannabidiol injected into the dorsolateral periaqueductal gray of rats. Psychopharmacology. 2008;199:223–230. doi: 10.1007/s00213-008-1168-x. [DOI] [PubMed] [Google Scholar]
- Cheng Y-C, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- Cheung BS, Howard IP, Money KE. Visually-induced sickness in normal and bilaterally labyrinthine-defective subjects. Aviat Space Environ Med. 1991;62:527–531. [PubMed] [Google Scholar]
- Cluny NL, Naylor RJ, Whittle BA, Javid FA. The effects of cannabidiol and tetrahydrocannabinol on motion-induced emesis in Suncus murinus. Basic Clin Pharmacol Toxicol. 2008;103:150–156. doi: 10.1111/j.1742-7843.2008.00253.x. [DOI] [PubMed] [Google Scholar]
- Cluny NL, Naylor RJ, Whittle BA, Javid FA. The effects of cannabidiolic acid and cannabidiol on contractility of the gastrointestinal tract of Suncus murinus. Arch Pharm Res. 2011;34:1509–1517. doi: 10.1007/s12272-011-0913-6. [DOI] [PubMed] [Google Scholar]
- Cordick N, Parker LA, Ossenkopp KP. Rotation-induced conditioned rejection in the taste reactivity test. Neuroreport. 1999;10:1557–1559. doi: 10.1097/00001756-199905140-00030. [DOI] [PubMed] [Google Scholar]
- De Jonghe BC, Horn CC. Chemotherapy agent cisplatin induces 48-h Fos expression in the brain of a vomiting species, the house musk shrew (Suncus murinus. Am J Physiol Regul Integr Comp Physiol. 2009;296:R902–R911. doi: 10.1152/ajpregu.90952.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Petrocellis L, Vellani V, Schiano-Moriello A, Marini P, Magherini PC, Orlando P, et al. Plant-derived cannabinoids modulate the activity of transient receptor potential channels of ankyrin type-1 and melastatin type-8. J Pharmacol Exp Ther. 2008;325:1007–1015. doi: 10.1124/jpet.107.134809. [DOI] [PubMed] [Google Scholar]
- De Petrocellis L, Ligresti A, Moriello AS, Allarà M, Bisogno T, Petrosino S, et al. Effects of cannabinoids and cannabinoid-enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol. 2011;163:1479–1494. doi: 10.1111/j.1476-5381.2010.01166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran M, Pérez E, Abanades S, Vidal X, Saura C, Majem M, et al. Preliminary efficacy and safety of an oromucosal standardized cannabis extract in chemotherapy-induced nausea and vomiting. Br J Clin Pharmacol. 2010;70:656–663. doi: 10.1111/j.1365-2125.2010.03743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Ruiz J, Sagredo O, Pazos MR, García C, Pertwee R, Mechoulam R, et al. Cannabidiol for neurodegenerative disorders: important new clinical applications for this phytocannabinoid? Br J Clin Pharmacol. 2012 doi: 10.1111/j.1365-2125.2012.04341.x. doi: 10.1111/j.1365-2125.2012.04341.x (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foubert J, Vaessen G. Nausea: the neglected symptom? Eur J Oncol Nurs. 2005;9:21–32. doi: 10.1016/j.ejon.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Gomes FV, Resstel LBM, Guimarães FS. The anxiolytic-like effects of cannabidiol injected into the bed nucleus of the stria terminalis are mediated by 5-HT1A receptors. Psychopharmacology. 2011;213:465–473. doi: 10.1007/s00213-010-2036-z. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Norgren R. Chronically decerebrate rats demonstrate satiation but not bait shyness. Science. 1978a;201:267–269. doi: 10.1126/science.663655. [DOI] [PubMed] [Google Scholar]
- Grill HJ, Norgren R. The taste reactivity test. I. Mimetic responses to gustatory stimuli in neurologically normal rats. Brain Res. 1978b;143:263–279. doi: 10.1016/0006-8993(78)90568-1. [DOI] [PubMed] [Google Scholar]
- Hickok JT, Roscoe JA, Morrow GR, King DK, Atkins JN, Fitch TR. Nausea and emesis remain significant problems of chemotherapy despite prophylaxis with 5-hydroxytryptamine-3 antiemetics. Cancer. 2003;97:2880–2886. doi: 10.1002/cncr.11408. [DOI] [PubMed] [Google Scholar]
- Horn CC, Ciucci M, Chaudhury A. Brain Fos expression during 48 h after cisplatin treatment: neural pathways for acute and delayed visceral sickness. Auton Neurosci. 2007;132:44–51. doi: 10.1016/j.autneu.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarrett MM, Limebeer CL, Parker LA. Effect of Δ9-tetrahydrocannabinol on sucrose palatability as measured by the taste reactivity test. Physiol Behav. 2005;86:475–479. doi: 10.1016/j.physbeh.2005.08.033. [DOI] [PubMed] [Google Scholar]
- Javid FA, Naylor RJ. The effect of the 5-HT1A receptor agonist, 8-OH-DPAT, on motion-induced emesis in Suncus murinus. Pharmacol Biochem Behav. 2006;85:820–826. doi: 10.1016/j.pbb.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Johnson WH, Meek J, Graybiel A. Effects of labyrinthectomy on canal sickness in squirrel monkey. Ann Otol Rhinol Laryngol. 1962;71:289–298. doi: 10.1177/000348946207100201. [DOI] [PubMed] [Google Scholar]
- Johnson WH, Sunahara FA, Landolt JP. Importance of the vestibular system in visually induced nausea and self-vection. J Vestib Res. 1999;9:83–87. [PubMed] [Google Scholar]
- Kennedy RS, Graybiel A, McDonough RC, Beckwith FD. Symptomatology under storm conditions in the North Atlantic in control subjects and in persons with bilateral labyrinthine defects. 1965. pp. 1–10. NSAM-928. Res Rep US Nav Sch Aviat Med. [PubMed]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwiatkowska M, Parker LA, Burton P, Mechoulam R. A comparative analysis of the potential of cannabinoids and ondansetron to suppress cisplatin-induced emesis in the Suncus murinus (house musk shrew) Psychopharmacology. 2004;174:254–259. doi: 10.1007/s00213-003-1739-9. [DOI] [PubMed] [Google Scholar]
- Lang IM, Sarna SK, Shaker R. Gastrointestinal motor and myoelectric correlates of motion sickness. Am J Physiol. 1999;277:G642–G652. doi: 10.1152/ajpgi.1999.277.3.G642. [DOI] [PubMed] [Google Scholar]
- Levine ME, Chillas JC, Stern RM, Knox GW. The effects of serotonin (5-HT3) receptor antagonists on gastric tachyarrhythmia and the symptoms of motion sickness. Aviat Space Environ Med. 2000;71:1111–1114. [PubMed] [Google Scholar]
- Limebeer CL, Hall G, Parker LA. Exposure to a lithium-paired context elicits gaping in rats: a model of anticipatory nausea. Physiol Behav. 2006;88:398–403. doi: 10.1016/j.physbeh.2006.04.014. [DOI] [PubMed] [Google Scholar]
- Limebeer CL, Krohn JP, Cross-Mellor S, Litt DE, Ossenkopp KP, Parker LA. Exposure to a context previously associated with nausea elicits conditioned gaping in rats: a model of anticipatory nausea. Behav Brain Res. 2008;187:33–40. doi: 10.1016/j.bbr.2007.08.024. [DOI] [PubMed] [Google Scholar]
- Limebeer CL, Vemuri VK, Bedard H, Lang ST, Ossenkopp KP, Makriyannis A, et al. Inverse agonism of cannabinoid CB1 receptors potentiates LiCl-induced nausea in the conditioned gaping model in rats. Br J Pharmacol. 2010;161:336–349. doi: 10.1111/j.1476-5381.2010.00885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maione S, Piscitelli F, Gatta L, Vita D, De Petrocellis L, Palazzo E, et al. Non-psychoactive cannabinoids modulate the descending pathway of antinociception in anaesthetized rats through several mechanisms of action. Br J Pharmacol. 2011;162:584–596. doi: 10.1111/j.1476-5381.2010.01063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik IA, Khan WA, Qazilbash M, Ata E, Butt A, Khan MA. Clinical efficacy of lorazepam in prophylaxis of anticipatory, acute and delayed nausea and vomiting induced by high-doses of cisplatin – a prospective randomized trial. Am J Clin Oncol. 1995;18:170–175. doi: 10.1097/00000421-199504000-00017. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mechoulam R, Gaoni Y. The isolation and structure of cannabinolic, cannabidiolic and cannabigerolic acids. Tetrahedron. 1965;21:1223–1229. doi: 10.1016/0040-4020(65)80064-3. [DOI] [PubMed] [Google Scholar]
- Meiri E, Jhangiani H, Vredenburgh JJ, Barbato LM, Carter FJ, Yang HM, et al. Efficacy of dronabinol alone and in combination with ondansetron versus ondansetron alone for delayed chemotherapy-induced nausea and vomiting. Curr Med Res Opin. 2007;23:533–543. doi: 10.1185/030079907x167525. [DOI] [PubMed] [Google Scholar]
- Mishima K, Hayakawa K, Abe K, Ikeda T, Egashira N, Iwasaki K, et al. Cannabidiol prevents cerebral infarction via a serotonergic 5-hydroxytryptamine1A receptor-dependent mechanism. Stroke. 2005;36:1071–1076. doi: 10.1161/01.STR.0000163083.59201.34. [DOI] [PubMed] [Google Scholar]
- Money KE, Friedberg J. The role of the semicircular canals in causation of motion sickness and nystagmus in the dog. Can J Physiol Pharmacol. 1964;42:793–801. doi: 10.1139/y64-089. [DOI] [PubMed] [Google Scholar]
- Morrow GR, Roscoe JA, Kirshner JJ, Hynes HE, Rosenbluth RJ. Anticipatory nausea and vomiting in the era of 5-HT3 antiemetics. Support Care Cancer. 1998;6:244–247. doi: 10.1007/s005200050161. [DOI] [PubMed] [Google Scholar]
- Nesse RM, Carli T, Curtis GC, Kleinman PD. Pretreatment nausea in cancer chemotherapy: a conditioned response? Psychosom Med. 1980;42:33–36. doi: 10.1097/00006842-198001000-00004. [DOI] [PubMed] [Google Scholar]
- Ossenkopp KP, Parker LA, Limebeer CL, Burton P, Fudge MA, Cross-Mellor SK. Vestibular lesions selectively abolish body rotation-induced, but not lithium-induced, conditioned taste aversions (oral rejection responses) in rats. Behav Neurosci. 2003;117:105–112. [PubMed] [Google Scholar]
- Parker LA. Taste avoidance and taste aversion: evidence for two different processes. Learn Behav. 2003;31:165–172. doi: 10.3758/bf03195979. [DOI] [PubMed] [Google Scholar]
- Parker LA, Limebeer CL. Cannabinoids in the management of nausea and vomiting. In: Köfalvi A, editor. Cannabinoids and the Brain. New York: Springer; 2008. pp. 259–273. [Google Scholar]
- Parker LA, Mechoulam R, Schlievert C, Abbott L, Fudge ML, Burton P. Effects of cannabinoids on lithium-induced conditioned rejection reactions in a rat model of nausea. Psychopharmacology. 2003;166:156–162. doi: 10.1007/s00213-002-1329-2. [DOI] [PubMed] [Google Scholar]
- Parker LA, Kwiatkowska M, Burton P, Mechoulam R. Effect of cannabinoids on lithium-induced vomiting in the Suncus murinus (house musk shrew) Psychopharmacology. 2004;171:156–161. doi: 10.1007/s00213-003-1571-2. [DOI] [PubMed] [Google Scholar]
- Parker LA, Rana SA, Limebeer CL. Conditioned nausea in rats: assessment by conditioned disgust reactions, rather than conditioned taste avoidance. Can J Exp Psychol. 2008;62:198–209. doi: 10.1037/a0012531. [DOI] [PubMed] [Google Scholar]
- Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: Δ9-tetrahydrocannabinol, cannabidiol and Δ9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153:199–215. doi: 10.1038/sj.bjp.0707442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli-Bigelli S, Rodrigues-Pereira J, Carides AD, Ma GJ, Eldridge K, Hipple A, et al. Addition of the neurokinin 1 receptor antagonist aprepitant to standard antiemetic therapy improves control of chemotherapy-induced nausea and vomiting. Results from a randomized, double-blind, placebo-controlled trial in Latin America. Cancer. 2003;97:3090–3098. doi: 10.1002/cncr.11433. [DOI] [PubMed] [Google Scholar]
- Potter DJ, Clark P, Brown MB. Potency of Δ9-THC and other cannabinoids in cannabis in England in 2005: implications for psychoactivity and pharmacology. J Forensic Sci. 2008;53:90–94. doi: 10.1111/j.1556-4029.2007.00603.x. [DOI] [PubMed] [Google Scholar]
- Reason JT. Motion sickness adaptation: a neural mismatch model. J R Soc Med. 1978;71:819–829. doi: 10.1177/014107687807101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rock EM, Limebeer CL, Mechoulam R, Piomelli D, Parker LA. The effect of cannabidiol and URB597 on conditioned gaping (a model of nausea) elicited by a lithium-paired context in the rat. Psychopharmacology. 2008;196:389–395. doi: 10.1007/s00213-007-0970-1. [DOI] [PubMed] [Google Scholar]
- Rock EM, Goodwin JM, Limebeer CL, Breuer A, Pertwee RG, Mechoulam R, et al. Interaction between non-psychotropic cannabinoids in marihuana: effect of cannabigerol (CBG) on the anti-nausea or anti-emetic effects of cannabidiol (CBD) in rats and shrews. Psychopharmacology. 2011;215:505–512. doi: 10.1007/s00213-010-2157-4. [DOI] [PubMed] [Google Scholar]
- Rock EM, Bolognini D, Limebeer CL, Cascio MG, Anavi-Goffer S, Fletcher PJ, et al. Cannabidiol, a non-psychotropic component of cannabis, attenuates vomiting and nausea-like behaviour via indirect agonism of 5-HT1A somatodendritic autoreceptors in the dorsal raphe nucleus. Br J Pharmacol. 2012;165:2620–2634. doi: 10.1111/j.1476-5381.2011.01621.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross RA, Gibson TM, Stevenson LA, Saha B, Crocker P, Razdan RK, et al. Structural determinants of the partial agonist-inverse agonist properties of 6′-azidohex-2′-yne-Δ8-tetrahydrocannabinol at cannabinoid receptors. Br J Pharmacol. 1999;128:735–743. doi: 10.1038/sj.bjp.0702836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruhaak LR, Felth J, Karlsson PC, Rafter JJ, Verpoorte R, Bohlin L. Evaluation of the cyclooxygenase inhibiting effects of six major cannabinoids isolated from Cannabis sativa. Biol Pharm Bull. 2011;34:774–778. doi: 10.1248/bpb.34.774. [DOI] [PubMed] [Google Scholar]
- Stott JR, Barnes GR, Wright RJ, Ruddock CJ. The effect on motion sickness and oculomotor function of GR 38032F, a 5-HT3-receptor antagonist with anti-emetic properties. Br J Clin Pharmacol. 1989;27:147–157. doi: 10.1111/j.1365-2125.1989.tb05345.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda S, Misawa K, Yamamoto I, Watanabe K. Cannabidiolic acid as a selective cyclooxygenase-2 inhibitory component in cannabis. Drug Metab Dispos. 2008;36:1917–1921. doi: 10.1124/dmd.108.020909. [DOI] [PubMed] [Google Scholar]
- Thomas A, Ross RA, Saha B, Mahadevan A, Razdan RK, Pertwee RG. 6″-Azidohex-2″-yne-cannabidiol: a potential neutral, competitive cannabinoid CB1 receptor antagonist. Eur J Pharmacol. 2004;487:213–221. doi: 10.1016/j.ejphar.2004.01.023. [DOI] [PubMed] [Google Scholar]
- Thomas A, Stevenson LA, Wease KN, Price MR, Baillie G, Ross RA, et al. Evidence that the plant cannabinoid Δ9-tetrahydrocannabivarin is a cannabinoid CB1 and CB2 receptor antagonist. Br J Pharmacol. 2005;146:917–926. doi: 10.1038/sj.bjp.0706414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A, Baillie GL, Phillips AM, Razdan RK, Ross RA, Pertwee RG. Cannabidiol displays unexpectedly high potency as an antagonist of CB1 and CB2 receptor agonists in vitro. Br J Pharmacol. 2007;150:613–623. doi: 10.1038/sj.bjp.0707133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachou S, Nomikos GG, Panagis G. WIN 55,212-2 decreases the reinforcing actions of cocaine through CB1 cannabinoid receptor stimulation. Behav Brain Res. 2003;141:215–222. doi: 10.1016/s0166-4328(02)00370-4. [DOI] [PubMed] [Google Scholar]
- Wilpizeski CR, Lowry LD, Goldman WS. Motion-induced sickness following bilateral ablation of area postrema in squirrel monkeys. Laryngoscope. 1986;96:1221–1225. doi: 10.1002/lary.1986.96.11.1221. [DOI] [PubMed] [Google Scholar]
- Zanelati TV, Biojone C, Moreira FA, Guimarães FS, Joca SRL. Antidepressant-like effects of cannabidiol in mice: possible involvement of 5-HT1A receptors. Br J Pharmacol. 2010;159:122–128. doi: 10.1111/j.1476-5381.2009.00521.x. [DOI] [PMC free article] [PubMed] [Google Scholar]