

Abstract
Background and Purpose
Given the importance of VEGF and haem oxygenase (HO)-1 in wound healing, the present study tested the hypothesis that CKD712, a synthetic tetrahydroisoquinoline alkaloid, activated VEGF production through the induction of HO-1 in human dermal fibroblasts (HDFs) and in mouse skin to stimulate wound healing.
Experimental Approach
Using HDFs, the effects of CKD712 on the production of VEGF and migration were evaluated. The mechanisms responsible were investigated using various signal inhibitors and small interfering RNA techniques. The ability of CKD712 to promote wound healing was also investigated in full-thickness skin-wounded mice.
Key Results
CKD712 treatment of HDFs increased VEGF production and accelerated migration, which was antagonized by anti-VEGF antibodies. Both an AMPK inhibitor (compound C) anda HO-1 activity inhibitor (SnPPIX) but not inhibitors of MAPKs, PI3K and PKC reduced the production of VEGF by CKD712. Interestingly, SnPPIX inhibited HO-1 expression but not p-AMPK, whereas compound C inhibited both p-AMPK and HO-1 induction by CKD712. Moreover, CKD712 decreased HO-1 expression without affecting the expression of p-AMPK by siHO-1 transfection, but it failed to induce HO-1 in siAMPKα1-transfected cells, suggesting that AMPK is involved in HO-1 induction by CKD712 in HDFs. Also, CKD712 shortened the time of wound closure in an SnPPIX-sensitive manner in a full-thickness skin-wounded mouse model.
Conclusion and Implications
CKD712 accelerated cutaneous wound healing, at least in part, by the production of VEGF through HO-1 induction in HDFs and mouse skin.
Keywords: wound closure, haem oxygenase-1, AMPK, human dermal fibroblast, mice, cutaneous wound
Introduction
The repair of wounds is a complex process that involves the tight coordination of various types of cells and specific cytokines. The molecular and cellular mechanisms of wound healing are notably similar to those involved in general tissue repair processes, such as those occurring after myocardial infarction, spinal cord injury and visceral injuries (Gurtner et al., 2008). The indispensible component of healing is the formation of new blood vessels by the process known as angiogenesis. VEGF plays an important role in angiogenesis during the proliferative phase of wound repair (Singer and Clark, 1999; Grazul-Bilska et al., 2003), and it appears that macrophages reinforce inflammatory responses and also secrete VEGF, ultimately promoting angiogenesis. In addition, neovascularization is essential for the synthesis, deposition and organization of the molecules that are essential for wound healing processes. These molecules include components of the extracellular matrix (ECM), TGF-β, EGF and PDGF, and those derived from macrophages to cause fibroblast infiltration. Under the direction of TGF-β and PDGF, fibroblasts are phenotypically converted into myofibroblasts possessing α-smooth muscle actin. Thus, myofibroblasts align themselves along the borders of the ECM to generate a constrictive force that facilitates wound closure.
It has been suggested that haem oxygenase (HO)-1 has two different roles during inflammation: (i) an anti-inflammatory action to inhibit leukocyte infiltration and (ii) the promotion of a VEGF-driven, non-inflammatory, angiogenesis that facilitates tissue repair (Bussolati and Mason, 2006). Recent investigation suggests that the induction of HO-1 is necessary for efficient wound closure and neovascularization in cutaneously wounded mice (Grochot-Przeczek et al., 2010). This enzyme, originally described as a stress-responsive protein that is induced by stimulants, such as inflammatory cytokines, heat shock, heavy metals and oxidants; it also degrades haem to iron, biliverdin and carbon monoxide (CO). Biliverdin is subsequently converted to bilirubin by biliverdin reductase, and the increased level of iron stimulates the synthesis of ferritin, a cytoprotective and antioxidant protein. Thus, the HO-1 system has antiapoptotic, antioxidant and immunomodulatory functions under various conditions, and these functions have effects in the wounded tissues (Platt and Nath, 1998; Hanselmann et al., 2001; Kampfer et al., 2001; Grochot-Przeczek et al., 2009).
(S)-1-α-naphthylmethyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (CDK712) (Figure 1) is a synthetic alkaloid that exhibits a variety of pharmacological actions, including the induction of HO-1 in various cells. For example, CKD712 reduced rat myocardial ischaemia-reperfusion injury (Jin et al., 2009) and improved the survival rate of septic mice due to HO-1 induction (Tsoyi et al., 2008). Furthermore, HO-1 has been reported to promote angiogenesis and mediate the angiogenic response induced by VEGF (Angermayr et al., 2006; Abraham and Kappas, 2008).
Figure 1.
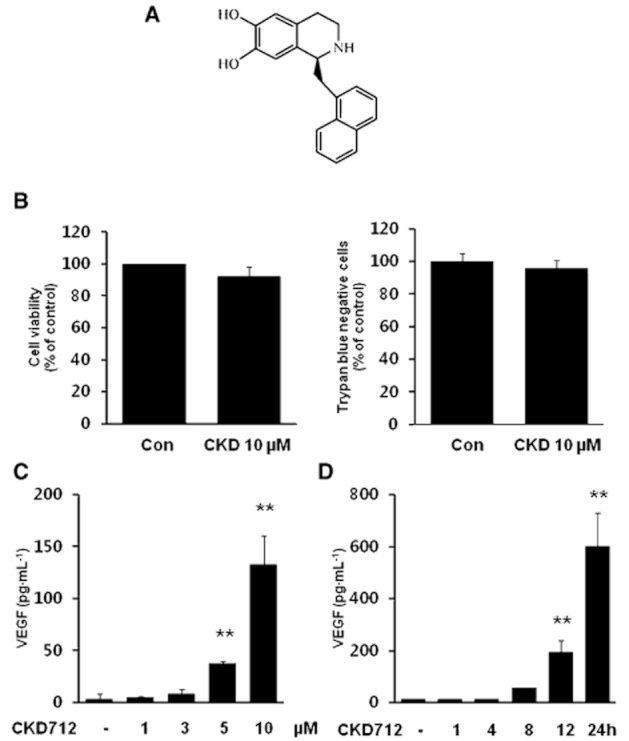
Chemical structure of CKD712 and its effect on the induction of VEGF in HDFs. (S)-1-α-naphthylmethyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline, CKD712 (A). The cytotoxicity of CKD712 was determined using the MTT and Trypan blue assays (B). The cells were treated with CKD712 (1, 3, 5 and 10 μM) for 12 h (C) or incubated with CKD712 (10 μM) for 1, 4, 8, 12 and 24 h (D). After the incubation, samples of the culture media were collected and subjected to ELISA for VEGF. The data are presented as the mean ± SD of three independent experiments. **P < 0.01, significantly different from control.
Given the importance of VEGF and HO-1 in wound healing, it is of great interest to investigate whether CKD712 produces VEGF in human dermal fibroblasts (HDFs) through HO-1 to facilitate the wound healing processes. Therefore, the hypothesis that CKD712 promotes wound closure by producing VEGF in HDFs through the induction of HO-1 was tested. In addition, an in vivo experiment was performed to ascertain whether the administration of CKD712 accelerates wound closure in a HO-1 sensitive manner in punch-inflicted full-thickness skin wounds in mice.
Methods
Cell culture
HDF cells were obtained from Dr Ham SA, Gyeongsang National University, Korea and were cultured in DMEM supplemented with 25 mM N-(2-hydroxyethyl)piperazine-N-2-ethanesulphonic acid, 100 U·mL−1 penicillin, 100 μg·mL−1 streptomycin and 10% heat-inactivated FCS and were grown in plastic tissue culture dishes in a humidified 5% CO2 atmosphere at 37°C.
Cell viability
Cell viability was determined colorimetrically using the MTT and Trypan blue assays. Cells at the exponential phase were seeded at 1 × 104 cells per well in 24-well plates. After different treatments, 20 μL of a 5 mg·mL−1 MTT solution was added to each well (0.1 mg per well), and the wells were incubated for 4 h. The supernatants were aspirated, and the formazan crystals formed in each well were dissolved in 200 μL of dimethyl sulphoxide for 30 min at 37°C; the optical density at 570 nm was measured using a Microplate Reader (Bio-Rad, Hercules, CA). All of the chemical exposures were performed in 96-well tissue culture plates for the purpose of chemical dilutions. The cells were placed in a humidified 5% CO2 incubator for 24 h at 37°C. After incubation, cell viability was assessed with the Trypan blue exclusion test (Life Technologies, Carlsbad, CA, USA) by manually counting the cells using a haemocytometer. Briefly, 10 μL of a 0.5% solution of the dye was added to 100 μL of treated cells (1.0 × 105 cells·mL−1). The suspension was later applied to a haemocytometer, and both viable (transparent) and nonviable (blue) cells were counted. A minimum of 200 cells were counted for each data point in a total of eight microscopic fields.
ELISA assays
The ELISA assay for VEGF was performed according to the manufacturer's protocol. The VEGF concentration was determined in conditioned media.
Western blot analysis
A whole-cell lysate was treated with a buffer containing 0.5% SDS, 1% Nonidet P-40, 1% sodium deoxycholate, 150 mM NaCl, 50 mM Tris–Cl (pH 7.5) and protease inhibitors. The protein concentration of each sample was then determined using a BCA protein assay kit (Pierce, Rockford, IL). To detect p-AMPK, AMPK, p-ACC, ACC, β-actin and HO-1, the whole-cell lysates were analysed by electrophoresis in polyacrylamide gels of different percentages, depending on the size of the protein of interest. The gels were then transferred to PVDF membranes by semi-dry electrophoretic transfer at 15 V for 60 to 75 min. The PVDF membranes were then blocked overnight at 4°C in 5% BSA. The cells were incubated with primary antibodies diluted 1:500 in Tris/buffered saline/Tween 20 containing 5% BSA overnight at 4°C and were incubated with the secondary antibodies at room temperature for 1 h. The signals were detected using ECL.
Cell migration assay
Cells were grown to full confluence and treated with 8 μg·mL−1 mitomycin C in culture media for 2 h to prevent proliferation. The cells were washed with PBS, scratched with a pipette tip, and incubated in fresh media with CKD712 and other chemicals. After incubation for the periods indicated, the cells were fixed and stained with Trypan blue. The fixed cells were observed using a microscope, and the number of cells that migrated across the regions of the wound's edge was counted.
Transfection of the cells with small interfering RNA (siRNA)
siRNAs against human HO-1, AMPK-α1 and scramble siRNA were purchased from Santa Cruz Biotechnology, and transient transfections were performed using the Superfect® transfection reagent from QIAGEN (Hilden, Germany). The cells were incubated with the siRNAs at a concentration of 100 nM for 16 h in serum-free media. After incubation, the transfected cells were treated with CKD712.
Wound healing study
All animal care and experimental procedures were approved by the Institutional Animal Care Committee of Gyeongsang National University. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (McGrath et al., 2010). A total of 24 animals were used in the work described here. Male mice (Balb/c 8 weeks, 30–35 g) were randomly divided into four groups. Two full-thickness 5 mm punch wounds were inflicted on the dorsal surface of each mouse, with the area being shaved prior to the procedure. The mice were injected i.p. for 12 days with saline (n = 6) or CKD712 (5 mg·kg−1, n = 6; 10 mg·kg−1, n = 6) or CKD712 (10 mg·mL−1) plus SnPPIX (10 mg·kg−1, n = 6) immediately after wounding (day 0). Photographs were taken from day 0 to day 12 to measure the wound areas.
Data analysis
The data are expressed as the mean ± SD of the results obtained from n number of replicate treatments. The differences between the data sets were assessed using a one-way anova followed by Newman–Keuls tests. The Kaplan–Meier method was used to compare the differences in the mortality rates between the groups. P < 0.05 was accepted as statistically significant.
Materials
CKD712 was obtained from the Chong Kun Dang Pharmaceutical Co. (Chun-An, Korea). Signal inhibitors SB203580 (p38 MAPK inhibitor), SP600125 (JNK inhibitor), PD98059 (ERK inhibitor), LY294001 (PI3K inhibitor), wortmannin (PI3K inhibitor), Gö6976 (PKC inhibitor), AG 490 (PKC inhibitor) and compound C (AMPK inhibitor) were obtained from Calbiochem (San Diego, CA). The haem oxygenase inhibitor, SnPPIX (tin protoporphyrin), anti-β-actin and anti-HO-1 antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Anti-phospho-AMPKα (Thr172), anti-AMPKα, anti-phospho-acetyl-CoA carboxylase (ACC) and anti-ACC antibodies were purchased from Cell Signaling Technology (Beverly, MA). ELISA kits – antibody and recombinant – for human VEGF were procured from R&D Systems (Abingdon, UK). The enhanced chemiluminescence (ECL) Western blotting detection reagent was obtained from Amersham (Buckinghamshire, UK). All of the other chemicals, including haemin, were supplied by Sigma-Aldrich (St. Louis, MO).
Results
CKD712 induces VEGF in a time- and concentration-dependent manner
As shown in Figure 1B, 10 μM CKD712 did not induce cytotoxicity as measured in either the MTT or Trypan blue assay. To determine the optimal concentration and time of CKD712 required to activate VEGF production, the cells were incubated with different concentrations of CKD712 (1, 3, 5 and 10 μM) for 12 h (Figure 1C) or with 10 μM CKD712 for 0, 1, 4, 8, 12 and 24 h (Figure 1D). CKD712 significantly increased VEGF production in a dose- and time-dependent fashion in HDFs.
CKD712 enhances migration through VEGF
The migration of fibroblasts from the area surrounding tissues into wound sites is an important process during wound healing. To elucidate the role of CKD712 on the cellular migration of HDFs, a scratch assay model was performed in the presence of mitomycin C, which blocks proliferation. CKD712 increased HDF cell migration in a time- and concentration-dependent manner (Figure 2A and B). In addition, the rate of migration was also proportional to the incubation time. Next, we assessed whether VEGF plays a role in the CKD712-mediated increase in cell migration. As shown in Figure 2C and D, treatment with anti-VEGF antibodies greatly inhibited the cell migration induced by CKD712, whereas recombinant VEGF remarkably increased the cell migration compared with the control, indicating that CKD712 enhances the cell migration of HDFs, at least in part, by the production of VEGF.
Figure 2.
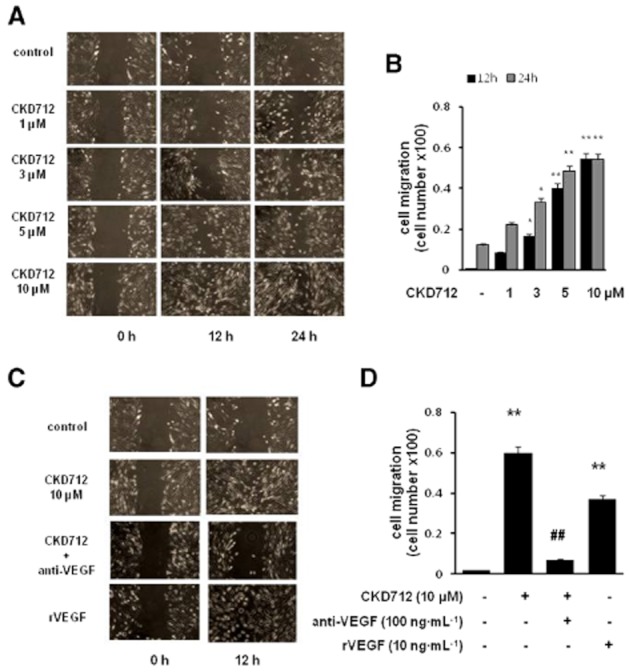
Involvement of VEGF in CKD712-mediated cell migration and its effect on the migration of HDFs. Confluent cells were treated with mitomycin C (8 μg·mL−1) for 2 h. After washing with PBS, the cells were wounded by scraping, and fresh medium was added. (A,B) Cells were treated with CKD712 at the indicated concentrations for 12 or 24 h and then cell migration was assessed. (C, D) Cell migration was also assessed in HDF cells stimulated with recombinant VEGF (10 ng·mL−1) or with CKD712 (10 μM) in presence or absence of anti-VEGF antibody (100 ng·mL−1). Representative photographs from three independent experiments are shown with similar results. *P < 0.05, **P < 0.01, significantly different from control; ##P < 0.01, significantly different from CKD712.
Inhibition of AMPK or HO-1 attenuates the production of VEGF and cell migration induced by CKD712
To investigate the mechanisms underlying the CKD712-activated production of VEGF, the cells were pretreated with various signal inhibitors, and the induction of VEGF by CKD712 was investigated. Figure 3A shows that both tin protoporphyrin (SnPPIX, an inhibitor of haem oxygenases) and compound C (an inhibitor of AMPK) significantly reduced the VEGF induction by CKD712. As shown in Figure 3B and C, the increases in VEGF induced by CKD712 were significantly reduced by the presence of SnPPIX (Figure 3B) and compound C (Figure 3C); however, SnPPIX or compound C alone did not affect VEGF production. To confirm that these pharmacological inhibitors have inhibitory effects on the cell migration induced by CKD712, a cell migration assay was performed, as described in the Methods. As shown in Figure 3D and E, the increase in the migration rate due to CKD712 was significantly diminished by both SnPPIX and compound C, whereas SnPPIX or compound C alone did not affect migration. Collectively, these data suggest that the signals for HO-1 and/or AMPK may regulate induction of VEGF by CKD712, thus influencing the migration rate.
Figure 3.
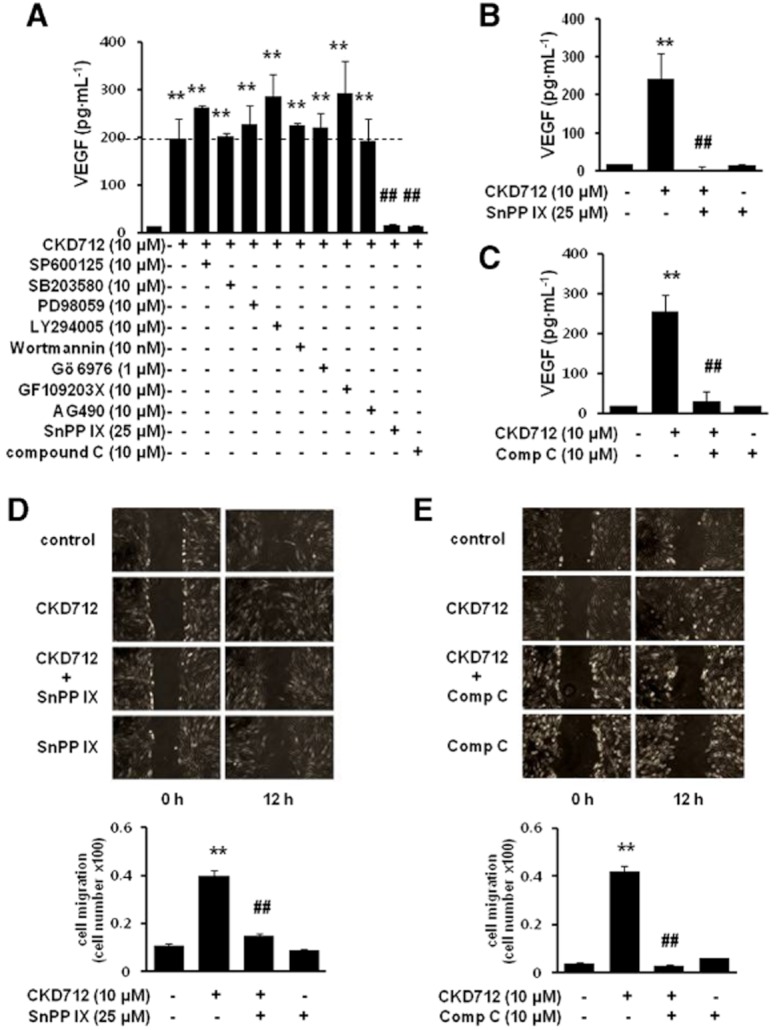
Dependence on HO-1/AMPK signalling of the production of VEGF by CKD712 and the effect of SnPPIX and compound C on the cell migration induced by CKD712. The cells were pretreated with various inhibitors and then treated with CKD712. After incubation, samples of the culture media were collected and subjected to ELISA for VEGF (A–C). Confluent cells were treated with mitomycin C (8 μg·mL−1) for 2 h. After washing with PBS, the cells were wounded by scraping, and fresh medium was added. The cells were incubated with CKD712 (10 μM) in the presence or absence of SnPPIX (25 μM) (D) or compound C (10 μM) (E) for 12 h. The data are presented as the mean ± SD of three independent experiments. **P < 0.01, significantly different from control; ##P < 0.01, significantly different from CKD712.
CKD712 activates AMPK, which induces HO-1 expression
Because the production of VEGF and cell migration induced by CKD712 was dependent on HO-1 and AMPK signals, we investigated whether CKD712 induced HO-1 or AMPK. As shown in Figure 4A and B, CKD712 induced HO-1 expression, the phosphorylation of AMPK and its downstream target, ACC, in a time- and concentration-dependent manner. When we found that CKD712 activated AMPK to induce HO-1 expression, we used an inhibitor of HO-1, SnPPIX. This inhibitor decreased HO-1 expression, but not p-AMPK, following CKD712. In contrast, compound C significantly inhibited both p-AMPK and HO-1 expression after CKD712, indicating that the AMPK signal induced by CKD712 governs HO-1 expression (Figure 3C and D).
Figure 4.

Effect of CKD712 on the HO-1/AMPK protein levels in HDFs. The cells were treated with CKD712 (1, 3, 5 and 10 μM) for 12 h (A) or incubated with CKD712 (10 μM) for different time periods (1, 4, 8, 12 and 24 h) (B). After incubation, the cells were harvested for Western blotting to detect HO-1, p-AMPK and p-ACC. The cells were treated with CKD712 (10 μM), with or without SnPPIX (25 μM) or compound C (10 μM) and incubated for 12 h (C) or 24 h (D). After incubation, the cells were lysed and analysed by Western blotting. The bands of the blots are representative of three independent experiments, with similar results. The data are presented as the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01, significantly different from control; ##P < 0.01, significantly different from CKD712 alone.
Effect of siRNA transfection on HO-1, AMPK and VEGF production induced by CKD712
siRNA approaches were used to confirm the role of AMPK and HO-1 in CKD712-mediated VEGF release. As shown in Figure 5A and D, the expression of the corresponding protein (HO-1 in A and AMPK in D) was significantly diminished by the transfection with the corresponding siRNA. Although the phosphorylation level of AMPK was not affected by the transfection with siHO-1RNA (Figure 5B), the expression of HO-1 was significantly reduced in the siAMPK-α1-transfected cells (Figure 5E). Interestingly, the increase in VEGF due to CKD712 was significantly reduced in the cells transfected with siHO-1 or siAMPK-α1 (Figure 5C and F). The latter findings suggest that CKD712 induced VEGF release through activation of a AMPK/HO-1 pathway.
Figure 5.
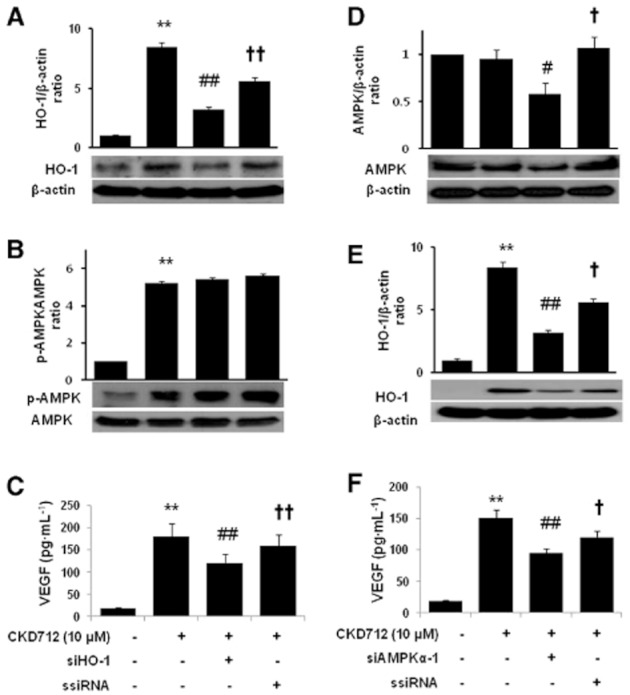
Effect of siRNA transfection on the CKD712-mediated expression of HO-1 and AMPK and VEGF production in HDFs. After transfection with HO-1- or AMPK-siRNA, the cells were treated with CKD712 (10 μM) for 24 h (A) or 12 h (B). Then cells were harvested and analysed by Western blotting for HO-1 (A, E) or AMPK (B, D). The cells were treated with CKD712 (10 μM) for 12 h, and culture medium samples were collected and subjected to ELISA for VEGF (C, F). The data are presented as the mean ± SD of three independent experiments. Significance compared with the control, **P < 0.01, *P < 0.05, significantly different from control; ##P < 0.01, significantly different from CKD712 #P < 0.05; ††P < 0.01, †P < 0.05, significantly different from CKD712 + siRNA.
Effect of siRNA transfection on HO-1, AMPK and VEGF production by haemin
We used haemin, a well-known HO-1 inducer, to determine whether the AMPK-dependent HO-1 induction was a specific action of CKD712 or a common characteristic of HO-1 inducers. As shown in the left panel of Figure 6, haemin also increased the expression of HO-1 (a) and p-AMPK (b) and increased VEGF production (c). However, when transfected with siHO-1 RNA, the expression of HO-1 and production of VEGF were significantly reduced, but the expression of p-AMPK was not reduced. As shown in the right panel of Figure 6, AMPK down-regulation through siRNA attenuated the effects of haemin on AMPK (d) and HO-1 (e) expression and VEGF release (f). These findings also suggested that HO-1 induction by haemin depends on the activity of AMPK.
Figure 6.
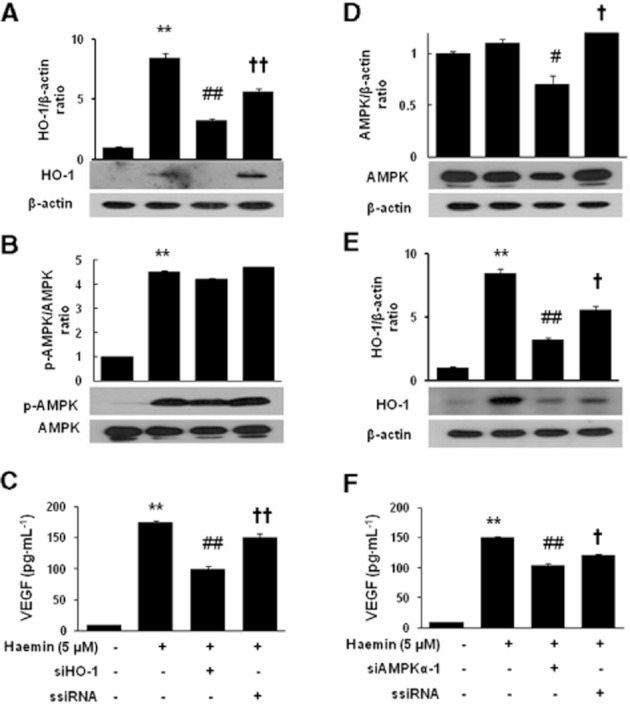
Effect of siRNA transfection on the expression of HO-1, AMPK and VEGF production induced by haemin in HDFs. After transfection with the corresponding siRNA, the cells were treated with haemin (15 μM) for 24 h (A) or 12 h (B). The cells were harvested and analysed by Western blotting for HO-1 (A, E) or for AMPK (B, D). The cells were treated with haemin (15 μM) for 12 h, and culture medium samples were collected and subjected to ELISA for VEGF (C, F). The data are presented as the mean ± SD of three independent experiments. **P < 0.01, *P < 0.05, significantly different from control; ##P < 0.01, #P < 0.05, significantly different from haemin; ††P < 0.01, †P < 0.05, significantly different from haemin + corresponding siRNA.
CKD712 promotes cutaneous wound healing process
To confirm that the in vitro effect of CKD712 in the promotion of wound healing could be applied in vivo, an animal wound model was used, as described in the Methods. Figure 7 shows that the CKD712-injected groups demonstrated significantly accelerated wound closure after injury compared with those of the saline group. Furthermore, the accelerated wound closure by CKD712 was significantly delayed by SnPPIX, indicating increased HO-1 activity plays a major role.
Figure 7.
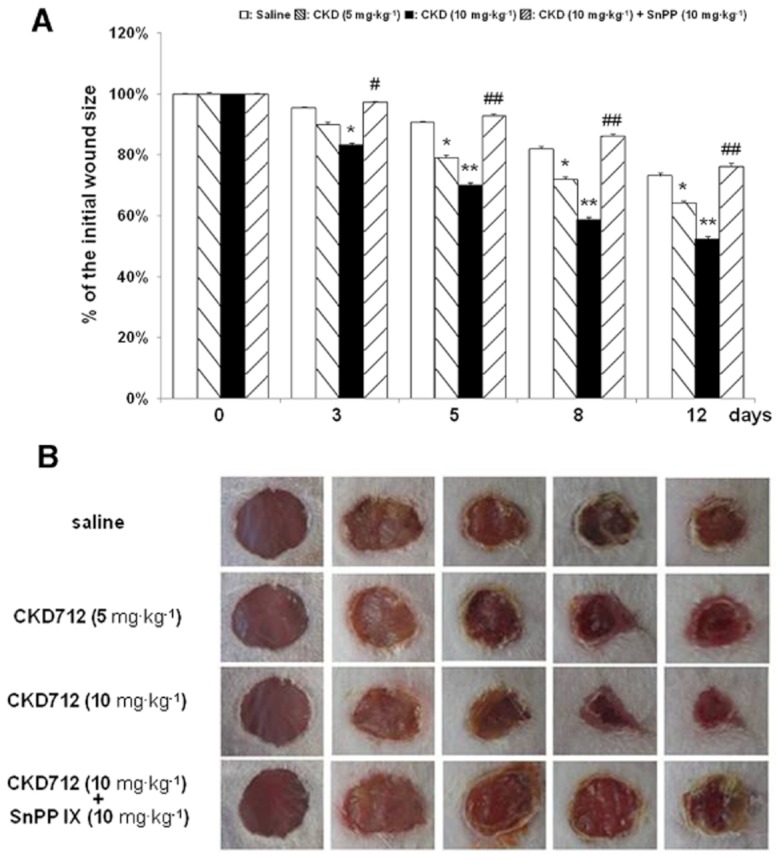
The wound closure induced by CKD712 is antagonized by SnPPIX in an animal wound model. Two full-thickness 5-mm punch wounds were inflicted on the dorsal surface of each mouse, and the mice were injected (i.p) with saline, CKD712 (5 mg·kg−1), CKD712 (10 mg·kg−1) or CKD712 (10 mg·kg−1 plus SnPPIX (10 mg·kg−1). The time course of the changes in the wound size with different treatments in mice (A). Images of a representative mouse from each group taken on post-injury days 0, 3, 5, 8 and 12 are shown (B). The wound sizes at the indicated time points in topically treated mice. (n = 6 mice per group) **P < 0.01, *P < 0.05 significantly different from saline; ##P < 0.01, #P < 0.05, significantly different from CKD712.
Discussion
The present study shows that CKD712 induced HO-1, which in turn stimulated VEGF release and accelerated HDF cell migration. In addition, the administration of CKD712 significantly shortened the time of wound closure in a full-thickness skin-wounded mouse model in an SnPPIX-sensitive manner. Previous studies have demonstrated the importance of HO-1 in regulating the synthesis and activity of VEGF (Dulak et al., 2002; Jozkowicz et al., 2002; Cisowski et al., 2005). Furthermore, HO-1 is known to have pro-angiogenic activity in human pancreatic cancer and a rat hind-limb ischaemia model (Sunamura et al., 2003; Suzuki et al., 2003). Anti-inflammatory, cytoprotective, cell proliferative and pro- migration properties of HO-1 have been reported (Platt and Nath, 1998). Accordingly, we hypothesized that the expression of HO-1 may play a role in the process of wound healing, so we tested whether CKD712 might promote wound closure by stimulating the production of VEGF in HDFs. We demonstrated that CKD712 increased VEGF release in HDFs in a time- and concentration-dependent manner and accelerated HDF cell migration as assessed by a wound scratch assay. VEGF stimulates pro-angiogenic properties of endothelial cells and is also established as a key regulator of angiogenesis (Herzog et al., 2011; Wang et al., 2011b). Here we showed that recombinant VEGF stimulated HDF cell migration, thus confirming previous reports of a role of VEGF in the process of HDF cell recruitment during wound healing (Froget et al., 2003). Incubation of HDF cells with an anti-VEGF antibody reduced the effect of CKD712 in HDF cell migration, indicating that VEGF mediated, at least in part, the effect of CKD712 in accelerating wound closure.
In terms of the underlying mechanisms, activation of MAPKs, PKC and PI3K are involved in VEGF production in human cells (Herrmann et al., 2011; Xue et al., 2011; Zhang et al., 2011; Choe et al., 2012) and these systems might be involved in CKD712-induced VEGF production in HDFs. Therefore, HDFs were pretreated with several signalling inhibitors to assess their effects on CKD712-induced production of VEGF. Although the inhibitors of MAPKs, PI3K and PKC did not affect the production of VEGF in response to CKD712, both the AMPK inhibitor (compound C) and the HO-1 inhibitor (SnPPIX) did reduce this production. This finding was compatible with earlier reports of the activation of HO-1 or AMPK stimulating VEGF output in many different types of cells (Morita et al., 2009; Kato et al., 2010; Lin et al., 2011; Miyake et al., 2011). Mediation of the production of VEGF by CKD712 either through an AMPK or HO-1 signal was confirmed by the scratch assay, which showed that both SnPPIX and compound C decreased the migration rate induced by CKD712. However, it remained unclear whether the activation of AMPK and HO-1 were independent or inter-dependent events. In order to address this question, we investigated the relationship between the two signalling pathways. CKD712 induced both HO-1 expression and AMPK phosphorylation in a concentration- and time-dependent manner but the activation of AMPK preceded the expression of HO-1. Thus, activation of AMPK peaked at 12 h, a result that correlated with the activation of p-ACC, a downstream effector of AMPK. In contrast, HO-1 expression increased at 12 h, reaching a peak at 24 h, suggesting that AMPK may lact upstream of HO-1. In support of this sequence, AICAR, an activator of AMPK, is known to induce HO-1 in endothelial cells (Liu et al., 2011b). In addition, the present results showed that SnPPIX inhibited HO-1 expression, but not p-AMPK levels, after the addition of CKD712. In contrast, compound C inhibited both p-AMPK and HO-1 after CKD712 treatment, indicating that the AMPK signal might control HO-1 induction. Although compound C is widely used as an AMPK inhibitor and is a useful tool for the investigation of AMPK-related signals, it also exhibits AMPK-independent effects (Liu et al., 2011a). We also recently reported that compound C, independently of AMPK, inhibited the NF-κB activity stimulated by TNF-α and decreased adhesion molecules in endothelial cells (Kim et al., 2011). Because compound C does not appear to possess sufficient selectivity to serve as a reliable investigational tool for an AMPK-dependent event, we used an siRNA transfection technique to confirm the involvement of AMPK. Transfection with siHO-1 decreased HO-1 induction without affecting AMPK phosphorylation, whereas CKD712 failed to induce HO-1 induction in the siAMPKα1-transfected cells, supporting the notion that AMPK modulates CKD712-mediated induction of HO-1. The use of haemin, a known HO-1 inducer, further confirmed that AMPK modulates HO-1 expression in HDFs a result consistent with earlier findings that haemin (15 mg·kg−1) induced AMPK expression in the soleus muscle of rats (Ndisang et al., 2009). In addition, up-regulation of HO-1 by CoPP (0.05 mg·kg−1), another HO-1 inducer, increased the levels of both the p-Akt and p-AMPK protein in streptozotocin-diabetic rat aorta (Sambuceti et al., 2009), suggesting that HO-1 inducers (haemin, CoPP) also increased AMPK expression in vivo. The present study did not elucidate the mechanisms of how AMPK activation causes HO-1 expression in HDFs. However, it is possible that a Nrf2/antioxidant-responsive element signalling pathway, as reported in endothelial cells (Liu et al., 2011b), or an Akt or p38 MAPK signalling system, as suggested in vascular smooth muscle cells (Wang et al., 2011a), is involved. This sequence of events may not operate in all cells as AMPK did not act upstream of HO-1 expression in endothelial cells (Wu et al. 2012) and HO-1 overexpression in animal models of diabetes was essential for the resultant increase in p-AMPK, p-Akt, peNOS, and NO bioavailability (Abraham and Kappas, 2008). Lastly, we attempted to test whether CKD712 shortens the time of wound closure in vivo. Full-thickness skin wounding is a useful and widely used animal model to investigate wound repair (Grochot-Przeczek et al., 2009). I.p. administration of CKD712 significantly accelerated wound healing in mice compared with the control (saline), effects reduced by SnPPIX, confirming in vitro data suggesting that CKD712 activity depends on HO-1 activation.
In summary, we have investigated whether CKD712 promotes wound closure through VEGF via the induction of HO-1 in HDF cells and in full-thickness wounded mouse skin. We clearly showed that CKD712 promoted cell migration by the production of VEGF due to the induction of HO-1 in HDFs, and the signalling mechanism for the induction of HO-1 by CKD712 involves AMPK activation. We propose that the facilitated wound closure may involve other beneficial effects of CKD712 in addition to VEGF production. Thus, we conclude that CKD712 accelerates cutaneous wound healing by the production of VEGF through induction of HO-1.
Acknowledgments
This work was supported by grant from NRF (03-2010-0298).
Glossary
Abbreviations
- ACC
acetyl-CoA carboxylase
- CKD712
(S)-1-α-naphthylmethyl-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline
- ECL
enhanced chemiluminescence
- ECM
extracellular matrix
- HDF
human dermal fibroblasts
- HO-1
haem oxygenase-1
- siRNA
small interfering RNA
- SnPP IX
tin protoporphyrin
- THI
tetrahydroisoquinoline
Conflict of interest
The authors state no conflicts of interest.
References
- Abraham NG, Kappas A. Pharmacological and clinical aspects of heme oxygenase. Pharmacol Rev. 2008;60:79–127. doi: 10.1124/pr.107.07104. [DOI] [PubMed] [Google Scholar]
- Angermayr B, Mejias M, Gracia-Sancho J, Garcia-Pagan JC, Bosch J, Fernandez M. Heme oxygenase attenuates oxidative stress and inflammation, and increases VEGF expression in portal hypertensive rats. J Hepatol. 2006;44:1033–1039. doi: 10.1016/j.jhep.2005.09.021. [DOI] [PubMed] [Google Scholar]
- Bussolati B, Mason JC. Dual role of VEGF-induced heme-oxygenase-1 in angiogenesis. Antioxid Redox Signal. 2006;8:1153–1163. doi: 10.1089/ars.2006.8.1153. [DOI] [PubMed] [Google Scholar]
- Choe JY, Lee SJ, Park SH, Kim SK. Tacrolimus (FK506) inhibits interleukin-1beta-induced angiopoietin-1, Tie-2 receptor, and vascular endothelial growth factor through down-regulation of JNK and p38 pathway in human rheumatoid fibroblast-like synoviocytes. Joint Bone Spine. 2012;79:137–143. doi: 10.1016/j.jbspin.2011.03.018. [DOI] [PubMed] [Google Scholar]
- Cisowski J, Loboda A, Jozkowicz A, Chen S, Agarwal A, Dulak J. Role of heme oxygenase-1 in hydrogen peroxide-induced VEGF synthesis: effect of HO-1 knockout. Biochem Biophys Res Commun. 2005;326:670–676. doi: 10.1016/j.bbrc.2004.11.083. [DOI] [PubMed] [Google Scholar]
- Dulak J, Jozkowicz A, Foresti R, Kasza A, Frick M, Huk I, et al. Heme oxygenase activity modulates vascular endothelial growth factor synthesis in vascular smooth muscle cells. Antioxid Redox Signal. 2002;4:229–240. doi: 10.1089/152308602753666280. [DOI] [PubMed] [Google Scholar]
- Froget S, Barthelemy E, Guillot F, Soler C, Coudert MC, Benbunan M, et al. Wound healing mediator production by human dermal fibroblasts grown within a collagen-GAG matrix for skin repair in humans. Eur Cytokine Netw. 2003;14:60–64. [PubMed] [Google Scholar]
- Grazul-Bilska AT, Johnson ML, Bilski JJ, Redmer DA, Reynolds LP, Abdullah A, et al. Wound healing: the role of growth factors. Drugs Today. 2003;39:787–800. doi: 10.1358/dot.2003.39.10.799472. [DOI] [PubMed] [Google Scholar]
- Grochot-Przeczek A, Lach R, Mis J, Skrzypek K, Gozdecka M, Sroczynska P, et al. Heme oxygenase-1 accelerates cutaneous wound healing in mice. Plos ONE. 2009;4:e5803. doi: 10.1371/journal.pone.0005803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grochot-Przeczek A, Dulak J, Jozkowicz A. Heme oxygenase-1 in neovascularisation: a diabetic perspective. Thromb Haemost. 2010;104:424–431. doi: 10.1160/TH09-12-0825. [DOI] [PubMed] [Google Scholar]
- Gurtner GC, Werner S, Barrandon Y, Longaker MT. Wound repair and regeneration. Nature. 2008;453:314–321. doi: 10.1038/nature07039. [DOI] [PubMed] [Google Scholar]
- Hanselmann C, Mauch C, Werner S. Heme oxygenase-1: a novel player in cutaneous wound repair and psoriasis? Biochem J. 2001;353:459–466. doi: 10.1042/0264-6021:3530459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann JL, Weil BR, Abarbanell AM, Wang Y, Poynter JA, Manukyan MC, et al. IL-6 and TGF-alpha co-stimulate mesenchymal stem cell vascular endothelial growth factor production by ERK-, JNK-, and PI3K-mediated mechanisms. Shock. 2011;35:512–516. doi: 10.1097/SHK.0b013e31820b2fb9. [DOI] [PubMed] [Google Scholar]
- Herzog B, Pellet-Many C, Britton G, Hartzoulakis B, Zachary IC. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signalling via FAK Tyr407 phosphorylation. Mol Biol Cell. 2011;22:2766–2776. doi: 10.1091/mbc.E09-12-1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin YC, Lee YS, Kim YM, Seo HG, Lee JH, Kim HJ, et al. (S)-1-(alpha-naphthylmethyl)-6,7-dihydroxy-1,2,3,4-tetrahydroisoquinoline (CKD712) reduces rat myocardial apoptosis against ischemia and reperfusion injury by activation of phosphatidylinositol 3-kinase/Akt signaling and anti-inflammatory action in vivo. J Pharmacol Exp Ther. 2009;330:440–448. doi: 10.1124/jpet.108.150342. [DOI] [PubMed] [Google Scholar]
- Jozkowicz A, Huk I, Nigisch A, Weigel G, Weidinger F, Dulak J. Effect of prostaglandin-J(2) on VEGF synthesis depends on the induction of heme oxygenase-1. Antioxid Redox Signal. 2002;4:577–585. doi: 10.1089/15230860260220076. [DOI] [PubMed] [Google Scholar]
- Kampfer H, Kolb N, Manderscheid M, Wetzler C, Pfeilschifter J, Frank S. Macrophage-derived heme-oxygenase-1: expression, regulation, and possible functions in skin repair. Mol Med. 2001;7:488–498. [PMC free article] [PubMed] [Google Scholar]
- Kato K, Tokuda H, Adachi S, Matsushima-Nishiwaki R, Natsume H, Yamakawa K, et al. AMP-activated protein kinase positively regulates FGF-2-stimulated VEGF synthesis in osteoblasts. Biochem Biophys Res Commun. 2010;400:123–127. doi: 10.1016/j.bbrc.2010.08.024. [DOI] [PubMed] [Google Scholar]
- Kim YM, Kim MY, Kim HJ, Roh GS, Ko GH, Seo HG, et al. Compound C independent of AMPK inhibits ICAM-1 and VCAM-1 expression in inflammatory stimulants-activated endothelial cells in vitro and in vivo. Atherosclerosis. 2011;219:57–64. doi: 10.1016/j.atherosclerosis.2011.06.043. [DOI] [PubMed] [Google Scholar]
- Lin HH, Lai SC, Chau LY. Heme oxygenase-1/carbon monoxide induces vascular endothelial growth factor expression via p38 kinase-dependent activation of Sp1. J Biol Chem. 2011;286:3829–3838. doi: 10.1074/jbc.M110.168831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XM, Peyton KJ, Shebib AR, Wang H, Durante W. Compound C stimulates heme oxygenase-1 gene expression via the Nrf2-ARE pathway to preserve human endothelial cell survival. Biochem Pharmacol. 2011a;82:371–379. doi: 10.1016/j.bcp.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XM, Peyton KJ, Shebib AR, Wang H, Korthuis RJ, Durante W. Activation of AMPK stimulates heme oxygenase-1 gene expression and human endothelial cell survival. Am J Physiol Heart Circ Physiol. 2011b;300:H84–H93. doi: 10.1152/ajpheart.00749.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyake M, Fujimoto K, Anai S, Ohnishi S, Kuwada M, Nakai Y, et al. Heme oxygenase-1 promotes angiogenesis in urothelial carcinoma of the urinary bladder. Oncol Rep. 2011;25:653–660. doi: 10.3892/or.2010.1125. [DOI] [PubMed] [Google Scholar]
- Morita K, LAee MS, Her S. Possible relation of hemin-induced HO-1 expression to the upregulation of VEGF and BDNF mRNA levels in rat C6 glioma cells. J Mol Neurosci. 2009;38:31–40. doi: 10.1007/s12031-008-9156-5. [DOI] [PubMed] [Google Scholar]
- Ndisang JF, Lane N, Jadhav A. The heme oxygenase system abates hyperglycemia in Zucker diabetic fatty rats by potentiating insulin-sensitizing pathways. Endocrinology. 2009;150:2098–2108. doi: 10.1210/en.2008-0239. [DOI] [PubMed] [Google Scholar]
- Platt JL, Nath KA. Heme oxygenase: protective gene or Trojan horse. Nat Med. 1998;4:1364–1365. doi: 10.1038/3947. [DOI] [PubMed] [Google Scholar]
- Sambuceti G, Morbelli S, Vanella L, Kusmic C, Marini C, Massollo M, et al. Diabetes impairs the vascular recruitment of normal stem cells by oxidant damage, reversed by increases in pAMPK, heme oxygenase-1, and adiponectin. Stem Cells. 2009;27:399–407. doi: 10.1634/stemcells.2008-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341:738–746. doi: 10.1056/NEJM199909023411006. [DOI] [PubMed] [Google Scholar]
- Sunamura M, Duda DG, Ghattas MH, Lozonschi L, Motoi F, Yamauchi J, et al. Heme oxygenase-1 accelerates tumor angiogenesis of human pancreatic cancer. Angiogenesis. 2003;6:15–24. doi: 10.1023/a:1025803600840. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Iso-o N, Takeshita S, Tsukamoto K, Mori I, Sato T, et al. Facilitated angiogenesis induced by heme oxygenase-1 gene transfer in a rat model of hind limb ischemia. Biochem Biophys Res Commun. 2003;302:138–143. doi: 10.1016/s0006-291x(03)00114-1. [DOI] [PubMed] [Google Scholar]
- Tsoyi K, Kim HJ, Shin JS, Kim DH, Cho HJ, Lee SS, et al. HO-1 and JAK-2/STAT-1 signals are involved in preferential inhibition of iNOS over COX-2 gene expression by newly synthesized tetrahydroisoquinoline alkaloid, CKD712, in cells activated with lipopolysacchride. Cell Signal. 2008;20:1839–1847. doi: 10.1016/j.cellsig.2008.06.012. [DOI] [PubMed] [Google Scholar]
- Wang JS, Ho FM, Kang HC, Lin WW, Huang KC. Celecoxib induces heme oxygenase-1 expression in macrophages and vascular smooth muscle cells via ROS-dependent signalling pathway. Naunyn Schmiedebergs Arch Pharmacol. 2011a;383:159–168. doi: 10.1007/s00210-010-0586-6. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zang QS, Liu Z, Wu Q, Maass D, Dulan G, et al. Regulation of VEGF induced endothelial cell migration by mitochondrial reactive oxygen species. Am J Physiol Cell Physiol. 2011b;301:C695–C704. doi: 10.1152/ajpcell.00322.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dong Y, Song P, Zou MH. Activation of the AMP-activated protein kinase (AMPK) by nitrated lipids in endothelial cells. Plos One. 2012;7:e31056. doi: 10.1371/journal.pone.0031056. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Xue Y, Li NL, Yang JY, Chen Y, Yang LL, Liu WC. Phosphatidylinositol 3′-kinase signalling pathway is essential for Rac1-induced hypoxia-inducible factor-1(alpha) and vascular endothelial growth factor expression. Am J Physiol Heart Circ Physiol. 2011;300:H2169–H2176. doi: 10.1152/ajpheart.00970.2010. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Yu D, Yin D, Wang Z. Activation of PI3K/mTOR signalling pathway contributes to induction of vascular endothelial growth factor by hCG in bovine developing luteal cells. Anim Reprod Sci. 2011;125:42–48. doi: 10.1016/j.anireprosci.2011.03.002. [DOI] [PubMed] [Google Scholar]