

Abstract
Background
The SH2-containing inositol-5′-phosphatase 1 (SHIP1) metabolizes PI(3,4,5)P3 to PI(3,4)P2. SHIP1-deficient mice exhibit progressive inflammation. Pharmacological activation of SHIP1 is emerging as a potential therapy for pulmonary inflammatory diseases. Here we characterize the efficacy of AQX-1125, a small-molecule SHIP1 activator currently in clinical development.
Experimental Approach
The effects of AQX-1125 were tested in several in vitro assays: on enzyme catalytic activity utilizing recombinant human SHIP1, on Akt phosphorylation in SHIP1-proficient and SHIP1-deficient cell lines, on cytokine release in murine splenocytes, on human leukocyte chemotaxis using modified Boyden chambers and on β-hexosaminidase release from murine mast cells. In addition, pharmacokinetic and drug distribution studies were performed in rats and dogs.
Results
AQX-1125 increased the catalytic activity of human recombinant SHIP1, an effect, which was absent after deletion of the C2 region. AQX-1125 inhibited Akt phosphorylation in SHIP1-proficient but not in SHIP1-deficient cells, reduced cytokine production in splenocytes, inhibited the activation of mast cells and inhibited human leukocyte chemotaxis. In vivo, AQX-1125 exhibited >80% oral bioavailability and >5 h terminal half-life.
Conclusions
Consistent with the role of SHIP1 in cell activation and chemotaxis, the SHIP1 activator AQX-1125 inhibits Akt phosphorylation, inflammatory mediator production and leukocyte chemotaxis in vitro. The in vitro effects and the pharmacokinetic properties of the compound make it a suitable candidate for in vivo testing in various models of inflammation.
Linked Article
This article is accompanied by Stenton et al., pp. 1519–1529 of this issue. To view this article visit http://dx.doi.org/10.1111/bph.12038
Keywords: SHIP1, inflammation, pulmonary, chemotaxis, PI3K, cell motility, phosphatidylinositol
Introduction
SH2-containing inositol-5′-phosphatase 1 (SHIP1) is an intracellular protein whose expression is primarily restricted to cells of the haematopoietic lineage. SHIP1 is a negative regulator of the PI3K pathway. SHIP1 suppresses intracellular signalling mediated by the product of PI3K, PI(3,4,5)P3 (PIP3), by catalysing the degradation of PIP3 to PI(3,4)P2 (PIP2), thus suppressing PIP3-mediated cellular effects. In addition to its catalytic function, SHIP1 can also act as a negative regulator of signalling through its interactions with other proteins, most notably Shc, LAT and Dok family members (Conde et al., 2011). Therefore, through its catalytic function and ability to act as an intracellular scaffolding protein, SHIP1 acts as a pluripotent regulator of haematopoietic cell function (Krystal, 2000; March and Ravichandran, 2002; Ooms et al., 2009; Parry et al., 2010).
The highly restricted expression pattern of SHIP1 in addition to its ability to act as a negative regulator of immune cell signalling makes it a highly attractive target for the development of anti-inflammatory therapeutics. In recent years, pharmacological activation of SHIP1 has emerged as a novel approach for the therapy of various inflammatory diseases. In 2005, Andersen and colleagues synthesized various pelorol analogues and demonstrated their effects as pharmacological activators of SHIP1 (Yang et al., 2005). Subsequent work identified the mode of action of these compounds, and demonstrated that they act as allosteric activators of SHIP1 (Ong et al., 2007). Furthermore, they provided the first experimental evidence for the anti-inflammatory activity of these compounds in vitro and in vivo (Ong et al., 2007). Subsequently, Kerr and colleagues (Brooks et al., 2010) highlighted the potential for SHIP1 modulation as a therapeutic tool in immunity and cancer.
Here we describe the in vitro pharmacological characterization of a novel, next-generation SHIP1 activating compound, AQX-1125 ((1S,3S,4R)-4-[(3aS,4R,5S,7aS)-4-(aminomethyl)-7a-methyl-1-methylidene-octahydro-1H-inden-5-yl]-3-(hydroxymethyl)-4-methylcyclohexan-1-ol; acetic acid salt), on enzyme activation, in cell-based models of inflammatory cell activation and in standard pharmacokinetic and distribution models in vivo, the basis for the in vivo efficacy and utility investigations subsequently (Stenton et al., in review).
Methods
SHIP1 enzyme activity assays
SHIP1 enzyme assays
6-His-tagged human SHIP1 (His-hSHIP1) and 6-His-tagged human SHIP1 with C2 domain deleted (His-hSHIP1ΔC2; cloned and expressed by SignalChem, Richmond, BC, Canada) enzymatic activity was assessed using a standard malachite green-based assay protocol using 50 μM 1,3,4,5-inositol–tetrakisphosphate (IP4) (Echelon Biosciences, Salt Lake City, UT, USA) as a substrate. The amount of inorganic phosphate released was assessed by adding BIOMOL GREEN™ reagent (Enzo Life Sciences, Farmingdale, NY, USA). To study enzyme kinetics using either IP4 or diC8-PI(3,4,5)P3 (diC8-PIP3) as substrates, enzyme activity over time was measured in the absence or presence of AQX-MN115 or AQX-1125 and varying concentrations of IP4 or diC8-PIP3. Phosphate released at each substrate concentration was plotted against time, and initial velocities were determined and plotted against IP4 or diC8-PIP3 concentration.
Scintillation proximity assay
Copper chelate (His-Tag) YSi SPA Scintillation Beads (GE Health Care, Piscataway, NJ, USA) were incubated with recombinant His-hSHIP1 and 10 μM of [14C]-AQX-1125 (American Radiolabeled Chemicals Inc., St Louis, MO, USA). The amount of bead-associated radioactivity was quantified in a Wallac TriLux plate scintillation counter (PerkinElmer, Waltham, MA, USA).
Akt activation assays
Cell culture and primary cell isolation
MOLT-4 and Jurkat T-ALL cells (ATCC, Manassas, VA, USA) were cultured in RPMI 1640 containing 10% FBS and 1% penicillin/streptomycin. Murine splenocytes were obtained from adult, female C57BL/6 mice. Splenic B cells were obtained by CD19 positive selection with CD19 microbeads using an AutoMACS system (Miltenyi Biotec, Bergisch Gladbach, Germany). Cells from the column flow through were the CD19-negative, T-cell-enriched fractions. Splenic B (85% CD19 positive) and T (75% CD3 positive) cell purities were assessed by flow cytometry with CD19 and CD3 cell surface markers.
Akt phosphorylation assays in MOLT-4 and Jurkat T-ALL cells or splenic cells
MOLT-4 and Jurkat T-ALL cells were cultured in serum-free RPMI overnight and were then treated with AQX-1125 for 30 min followed by insulin-like growth factor-1 (IGF-1) stimulation for 1 h (Peprotech, Rocky Hill, NJ, USA). Freshly isolated splenic T-cells were equilibrated in serum-free RPMI for 30 min before AQX-1125 treatment for 30 min followed by anti-CD3/CD28 stimulation for 30 min. Cells were lysed and supernatants were collected. Akt phosphorylation at S473 in each sample was determined by Western blotting or using the Akt[pS473] Singleplex Luminex (Life Technologies Inc., Carlsbad, CA, USA) assay based on manufacturer's instructions.
Western blotting
Approximately 15–20 μg of total protein from each sample was separated on a 4–12% Tris-Glycine gel (Life Technologies Inc.). Proteins were transferred to a nitrocellulose membrane (Life Technologies Inc.) and the membrane was blocked before probing with primary antibodies overnight at 4°C. The following antibodies were used: mouse anti-SHIP1 (P1C1; Santa Cruz, Santa Cruz, CA), rabbit anti-pAkt (S473; 193H12), rabbit anti-Akt and rabbit anti-β-actin (Cell Signalling, Boston, MA, USA). The membrane was then incubated with appropriate HRP-linked secondary antibodies. Target proteins on the membrane were detected with ECL solution (GE Healthcare, Waukesha, WI, USA) and exposed to film.
Splenic cell cytokine release, BrdU incorporation and LDH release
Isolated splenocytes were pretreated with vehicle or compound at various concentrations for 30 min and stimulated for 72 h with anti-CD3/CD28. Supernatant was collected and assayed using a kit to detect LDH (Roche, Basel, Switzerland) and for cytokine release using the Cytokine Mouse 10-plex Luminex Panel kit (Life Technologies). The proliferation of anti-CD3/CD28 stimulated splenocytes was assessed by measuring BrdU incorporation using a colorimetric ELISA (Roche) and 2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt (XTT) metabolism of anti-CD3/CD28 stimulated isolated splenocytes was measured as described (Roehm et al., 1991).
β-hexosaminidase release from mouse bone marrow-derived mast cells (BMMCs)
Bone marrow cells were obtained from 4- to 8-week-old SHIP1+/+ and SHIP1−/− mice (obtained from Dr. Gerry Krystal, University of British Columbia, Vancouver, Canada) and BMMCs were prepared as described (Ong et al., 2007). Mast cells were sensitized overnight with anti-DNP IgE (SPE-7). Cells were then washed and incubated in the presence of vehicle control (media only) or AQX-1125 (60 μM) prior to stimulation with the indicated concentration of DNP-human serum albumin (DNP-HSA), followed by the measurement of β-hexosaminidase from the culture supernatant (Naal et al., 2004).
Chemotaxis assays in human blood leukocytes
Leukocyte isolation
Peripheral blood mononuclear cells (PBMCs) were isolated from human buffy coats (San Diego Blood Bank) using Ficoll-Paque (GE Healthcare, Piscataway, NJ) density centrifugation. Monocytes were obtained from PBMC by plastic adherence and the nonadherent fraction was used for T-cell enrichment using a nylon wool column. B cells were obtained by negative selection (StemCell Technologies, Vancouver, BC, Canada). Neutrophils were obtained from the buffy coats by dextran sedimentation.
Chemotaxis assay
Chemotaxis was assessed using a modified Boyden chamber. The final concentrations of chemokines were 100 ng mL−1 monocyte chemotactic protein-1 (MCP-1) for monocytes, 10 nM IL-8 and 5 nM growth-regulated alpha protein (GRO-α) for neutrophils, 1 μg mL−1 BCA-1 for B cells, 50 nM MIP-1α for non-activated T-cells and 50 nM I-TAC or 20 nM IP-10 for IL-2 activated T-cells (all chemokines from Peprotech). Chemokine receptor specific inhibitors (at a final concentration): 10 μM C-C chemokine receptor type 2 (CCR2) (MK-0812), 10 μM C-X-C chemokine receptor (CXCR)1/2 (SCH527123), 10 μg mL−1 anti-hCXCR5 (MAB190, R&D Systems, Minneapolis, MN, USA), 10 μg mL−1 anti-hCCR1 (eBioScience, San Diego, CA, USA) and 10 μg mL−1 anti-hCXCR3 (MAB160, R&D Systems) were used as positive controls for monocytes, neutrophils, B cells, non-activated T-cells and IL-2 activated T-cells, respectively. Monocyte and neutrophil migration was assessed by counting the number of adherent cells present on the basal surface of the membranes. The migration of B cells and T-cells were determined by counting the cells in the bottom chambers.
Selectivity/specificity screening of AQX-1125
The selectivity and specificity of AQX-1125 was tested in a number of commercially available screens. AQX-1125 (0.01–30 μM) was evaluated for potential off-target effects through the CEREP (Celle l'Evescault, Le bois l'Evêque, France) Diversity Profile (competitive GPCR binding assays, enzyme assays and phosphatase enzyme assays). Additional selectivity testing was conducted whereby the in vitro binding affinity of AQX-1125 (10 μM) to 439 human kinases and three pathogen kinases was evaluated in the Ambit scanMAX™ competitive binding assay (Ambit Biosciences, San Diego, CA, USA). AQX-1125 (0.001–30 μM) was tested in six agonist (androgen receptor, oestrogen receptor α, oestrogen receptor β, glucocorticoid receptor, mineralocorticoid receptor and progesterone receptor) and seven antagonist (androgen receptor, oestrogen receptor α, oestrogen receptor β, oestrogen-related receptor α, glucocorticoid receptor, mineralocorticoid receptor and progesterone receptor) GeneBLAzer® assays for potential agonist or antagonist activity using the Invitrogen Cell-Based Nuclear Receptor SelectScreen and fluorescence resonance energy transfer (Lifetechnologies Inc., Grand Island, NY, USA).
In vivo animal studies
All in vivo animal study protocols were approved by local ethics committees. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al., 2010).
Pharmacokinetic analysis of AQX-1125 in rats and dogs
Female Sprague-Dawley rats and beagle dogs (male and female) were treated with AQX-1125 in saline by bolus i.v. injection (at 1 mL kg−1) or by oral gavage (at 5 mL kg−1). Blood was sampled 0 to 48 h post-dose and plasma isolated. Plasma concentrations of AQX-1125 were quantified using a validated HPLC-MS/MS method. Pharmacokinetic parameters were determined by non-compartmental analysis using Model 200 of WinNonlin Version 1.4 (Pharsight, Mountain View, CA, USA). The bioavailability (F) was estimated using (oral AUC0-∞/i.v. AUC0-∞) × (i.v. dose/oral dose).
Measurement of tissue distribution of AQX-1125 in rats
Male Sprague-Dawley rats were treated with a single dose of 10 mg kg−1 [14C]-AQX-1125 (20 μCi mg−1) by bolus i.v. injection. At the indicated time points, the animals were killed and frozen by immersion in a hexane/dry-ice bath, embedded in an aqueous suspension of 2% (w/v) carboxymethylcellulose, frozen and sectioned using a cryomicrotome. Whole-body sections (40 μm thick) were taken in the sagittal plane at various levels of interest. Sections were exposed to a 14C-sensitive phospho-imaging plate (Fuji Biomedical, Stamford, CT, USA) for 4 days. The concentrations of radioactivity were expressed as the μmol equivalents of [14C]-AQX-1125 per gram sample.
In a separate set of studies, the lung tissue concentration of AQX-1125 was compared to the plasma concentration 4 or 24 h after a single oral administration of AQX-1125 (30 mg kg−1, at 5 mL kg−1) to male Sprague-Dawley rats. AQX-1125 content of the blood and the lungs was analysed by HPLC-MS/MS.
Compound synthesis and lipophilicity
AQX-1125 was prepared from commercially available 5-dehydroepiandrosterone as described (Raymond et al., 2011). AQX-1125 was radiolabelled with carbon-14 to a specific activity of 55 mCi mmol−1 by the American Radiolabeled Chemicals Inc. (St Louis, MO, USA). AQX-MN115 was used as a control (Yang et al., 2005; Andersen et al., 2007). Lipophilicity measurements of the compounds were conducted as described (Liu et al., 2008).
Statistical analysis
Statistical analyses were performed by anova followed by Dunnett's post hoc test. Probability values of P < 0.05 were considered statistically significant.
Results
Characterization of the small-molecule SHIP1 activator, AQX-1125
Screening of a focused library of small molecules identified several novel activators of SHIP1, two of which are shown in Table 1. AQX-1125 and AQX-MN115 activated the enzyme by 20% and 77% respectively. AQX-MN115 and AQX-1125 underwent additional detailed kinetic enzymatic analysis. The results of these studies showed that the compounds induce a concentration-dependent decrease in the KM of the enzyme, without affecting the turnover constant (kcat) and the maximum velocity (Vmax) values, resulting in an increase in the kcat/KM ratio. These effects were more pronounced for AQX-MN115 than for AQX-1125 (Figure 1, Table 2); however, based on its drug-like properties, AQX-1125 was selected for additional investigation. These observations are consistent with the hypothesis that allosteric SHIP1 activators enhance enzyme efficiency, possibly by enhancing substrate binding to the enzyme.
Table 1.
Structures and activities of a selected group of SHIP1 activators
| Compound structure | logPoct | Mean ± SEM % SHIP1 activation at 300 μM | |
|---|---|---|---|
| AQX-MN115 | 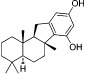 |
6.8 | 77 ± 17% |
| AQX-1125 | 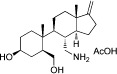 |
0.6a | 20 ± 5% |
Lipophilicity measurements (experimental logP or log D, pH 7.4) and SHIP1 activation (% activation at 300 μM) are shown.
logD at pH 7.4.
Figure 1.
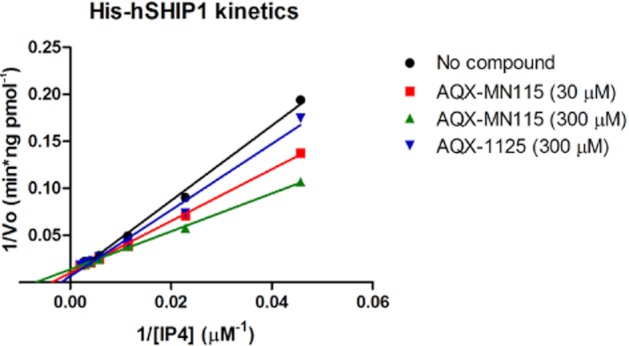
His-hSHIP1 enzyme kinetics in the presence of AQX-MN115 or AQX-1125. The reciprocal of his-hSHIP1 initial velocities were plotted against the reciprocal of the indicated concentrations of IP4 in the presence of various concentrations of AQX-1125 or AQX-MN115. Data show representative results of three independent experiments.
Table 2.
Effect of AQX-MN115 or AQX-1125 on SHIP1 kinetic enzymatic parameters
| Control | AQX-MN115 (30 μM) | AQX-MN115 (300 μM) | AQX-1125 (300 μM) | |
|---|---|---|---|---|
| kcat | 3.5 ± 0.4 | 3.4 ± 0.4 | 3.1 ± 0.2 | 3.4 ± 0.3 |
| KM | 214 ± 21 | 159 ± 2 | 129 ± 15 | 180 ± 9 |
| Vmax | 18 ± 2 | 17 ± 2 | 15 ± 1 | 17 ± 2 |
| kcat/KM | 0.017 ± 0.004 | 0.021 ± 0.003 | 0.024 ± 0.005 | 0.019 ± 0.003 |
SHIP1 activation by AQX-1125 involves a direct interaction and requires an intact C2 domain
To investigate regions of the SHIP1 enzyme required for AQX-1125 activity, we compared the abilities of AQX-1125 and AQX-MN115 to activate wild-type versus C2-deficient SHIP1 (SHIP1ΔC2) enzyme. The activating effect of AQX-1125 was 28% at 100 μM in the native enzyme but no effect of AQX-1125 was observed when the SHIP1 ΔC2 enzyme was used (Figure 2A). Similar findings were seen with AQX-MN115 (Figure 2B), which induced 60% activation in wild-type enzyme at 100 μM, but only 18% activation in SHIP1ΔC2. AQX-1125 was determined to bind directly to SHIP1 using a scintillation proximity assay (SPA). SPA beads were coated with either the full-length SHIP1 enzyme or control protein (BSA) prior to incubation with [14C]-AQX-1125. In the presence of SHIP1 and 10 μM [14C]-AQX-1125, there was an increase of 258 ± 29 cpm (n = 3), over background, indicative of a direct binding interaction.
Figure 2.
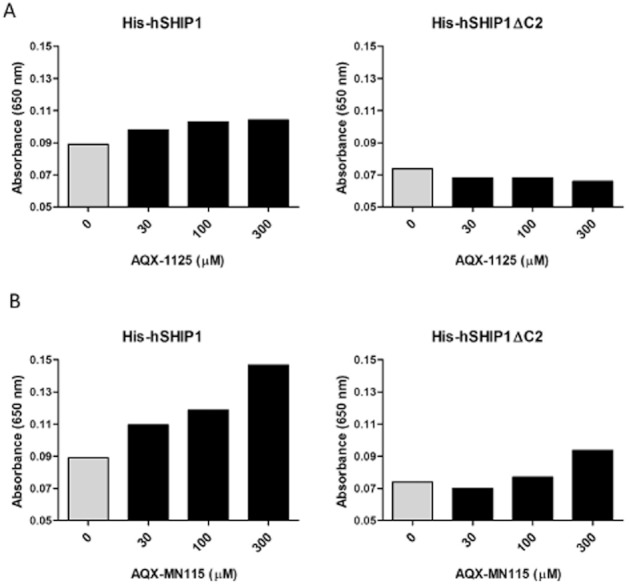
SHIP1 activators require the C2 domain of SHIP1. The effects of (A) AQX-1125 and (B) AQX-MN115 on SHIP1 activity were compared in wild-type human His-SHIP1 (His-hSHIP1) and human His-SHIP1 with a deletion of the C2 domain (His-hSHIP1 ΔC2) using the BIOMOL GREEN enzyme assay with 50 μM of IP4 as the substrate. Data show representative results (mean) of two independent experiments.
AQX-1125 inhibits Akt activation in MOLT-4, but not in Jurkat cells
It was hypothesized that pharmacological SHIP1 activation would result in an inhibition of Akt phosphorylation. In order to test this hypothesis, the effect of AQX-1125 on IGF-1-mediated phospho-Akt (S473) was compared in the SHIP1-proficient MOLT-4 cell line and the SHIP1-deficient Jurkat cell line. AQX-1125 induced a concentration-dependent decrease in Akt phosphorylation in MOLT-4 cells, while it failed to affect Akt phosphorylation in Jurkat cells (Figure 3). The degree of the inhibition of Akt activation by AQX-1125 in MOLT-4 cells was determined and quantified by densitometry. At 0.1 μM AQX-1125 the inhibition amounted to an average of 34%, while at 10 μM the inhibition amounted to an average of 82% in two independent experiments. Similarly, AQX-1125 exerted a dose-dependent inhibition of Akt phosphorylation in SHIP+/+ mouse splenic lymphocytes, with a maximal inhibitory effect at 30 μM amounting to 73 ± 4% and 82 ± 58% in T-cells and B cells, respectively (n = 3) as measured by Luminex analysis.
Figure 3.
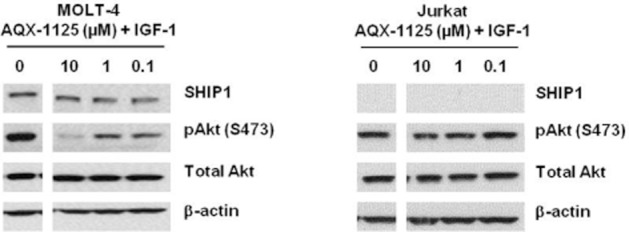
AQX-1125 inhibits the phosphorylation of Akt (S473) in MOLT-4 but not Jurkat cells. The effect of AQX-1125 on the level of pAkt (S473) in serum starved and IGF-1 stimulated MOLT-4 and Jurkat cells were examined. Data show representative results of three independent experiments.
AQX-1125 inhibits cytokine release in mixed splenocytes
The effect of AQX-1125 on cytokine release from splenocytes stimulated with anti-CD3/CD28 was investigated. AQX-1125 induced a concentration-dependent decrease in the production of multiple pro-inflammatory mediators in this system (Figure 4A, B), without affecting cell viability (LDH release; Figure 4C). Interestingly, the reduction in cytokine production was not uniform across all cytokines studied, with the greatest inhibitory effect on GM-CSF and the production of IL-4 being inhibited the least. Furthermore, no significant inhibition was observed for the anti-inflammatory cytokine IL-10 (Figure 4A).
Figure 4.
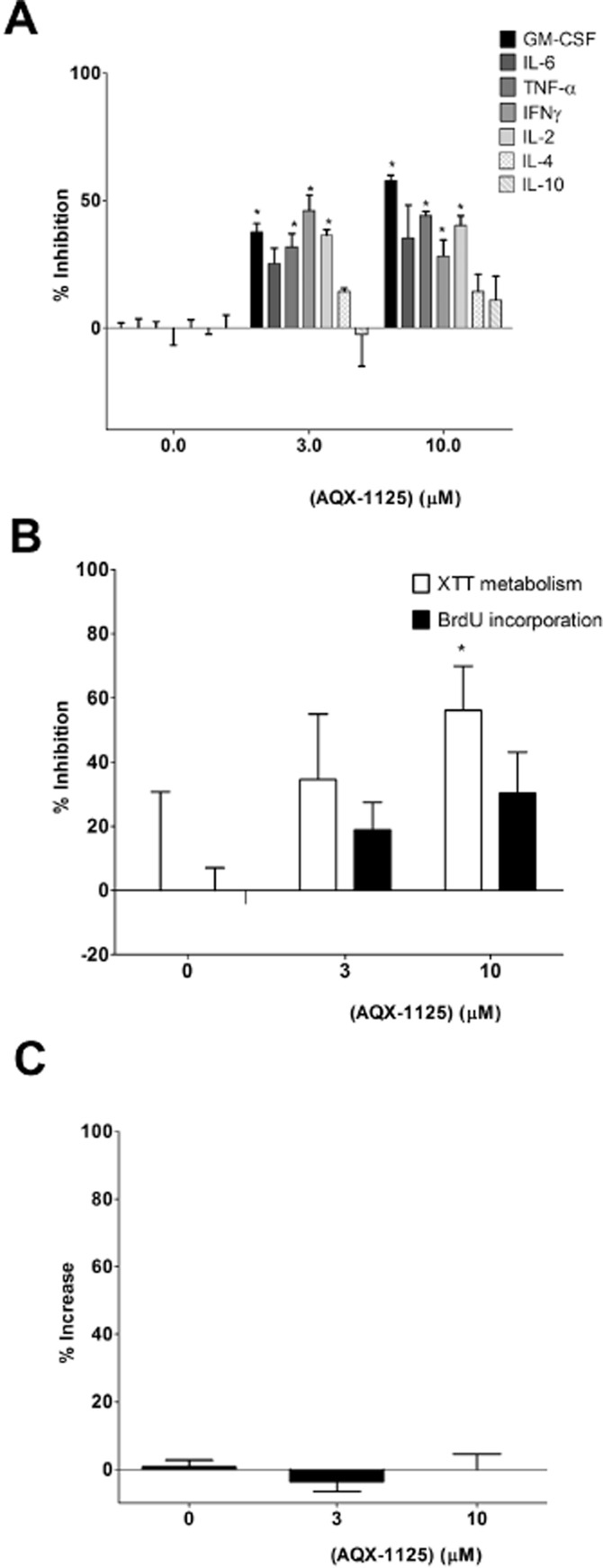
Anti-inflammatory effect of AQX-1125 in murine splenocytes. Efficacy of AQX-1125 on (A) anti-CD3/CD28 stimulated splenocyte cytokine release, (B) BrdU incorporation and XTT metabolism, indices of cell proliferation, and (C) LDH release, an index of cell death. Data (mean ± SEM) are presented as % inhibition of stimulation (A and B) or as % increase (C). *P < 0.05 denotes significant effect of AQX-1125, compared to vehicle control. Data are shown as mean ± SEM of n = 3 independent experiments.
AQX-1125 inhibits β-hexosaminidase release in mouse BMMCs
The effect of AQX-1125 was next tested in BMMCs from SHIP1+/+ and SHIP1−/− mice. As shown in Figure 5, and consistent with prior data (Ong et al., 2007; Haddon et al., 2009), BMMCs from SHIP1−/− mice had markedly higher IgE-mediated β-hexosaminidase release than SHIP+/+ BMMCs (60% maximal release, as opposed to 15% maximal release). AQX-1125 significantly decreased β-hexosaminidase release from SHIP+/+ but not from the SHIP1-deficient BMMCs (Figure 5).
Figure 5.
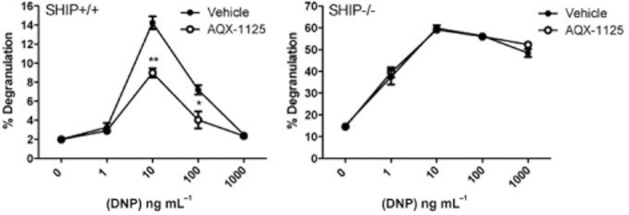
AQX-1125 inhibits degranulation of SHIP1+/+ but not SHIP1−/− mast cells. Sensitized SHIP1+/+ and SHIP1−/− BMMCs were pre-incubated with 60 μM of AQX-1125 for 30 min prior to degranulation with a range of DNP-HSA concentrations. Degranulation was assessed by measuring β-hexosaminidase release in the culture supernatant and the percent degranulation was calculated as the amount of β-hexosaminidase present with DNP-HSA divided by the total β-hexosaminidase present in the cells upon lysis. Data (mean ± SEM, n = 3) show results of three independent experiments. ** denotes P < 0.01, and * denotes P < 0.05 compared to vehicle controls.
AQX-1125 inhibits chemotaxis assays in human mononuclear cells
As shown in Figure 6 using data from four independent donors, AQX-1125 concentration dependently and significantly (P < 0.05) inhibited monocyte chemotaxis to MCP-1. Moreover, AQX-1125 dose dependently inhibited chemotaxis of most cell types at low micromolar concentrations independent of the chemotactic stimulus (Table 3).
Figure 6.
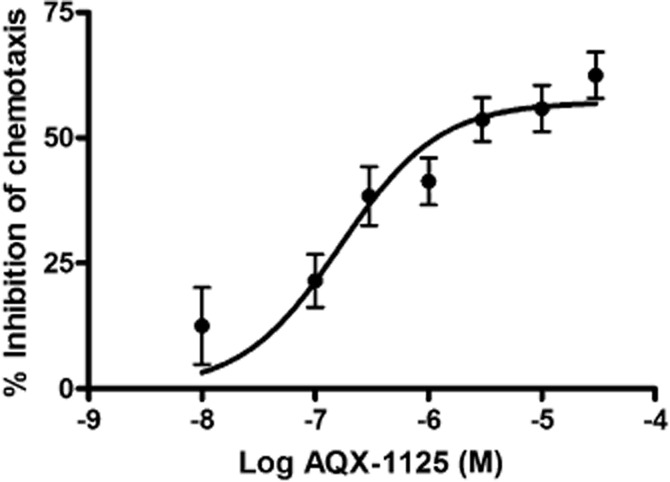
AQX-1125 inhibits monocyte chemotaxis to MCP-1. Treatment of enriched human monocytes with AQX-1125 resulted in a dose-dependent inhibition of chemotaxis to MCP-1. Data represent mean ± SEM (n = 4).
Table 3.
Effect of AQX-1125 on leukocyte chemotaxis
| Inhibition of chemotaxis by AQX-1125 in human leukocyte subsets | |||||
|---|---|---|---|---|---|
| Cell type | Chemokine | Chemokine receptor | Average EC50 of AQX-1125 (μM)a | Average maximal inhibition (%) | Number of donors |
| Monocytes | MCP-1 | CCR2 | 0.260 ± 0.14 | 67 ± 12 | 4 |
| B cells | BCA-1 | CXCR5 | 0.002 ± 0.001 | 48 ± 18 | 4 |
| Activated T-cells | IP-10 | CXCR3 | 0.01 ± 0.01 | 57 ± 3.8 | 3 |
| Activated T-cells | I-TAC | CXCR3 | 0.240 ± 0.17 | 57 ± 5.0 | 2 |
| Resting T-cells | MIP-1α | CCR1 | 0.760 ± 1.16 | 66 ± 40 | 3 |
| Neutrophils | GRO-α | CXCR2 | 0.050 ± 0.00 | 64 | 1 |
| Neutrophils | IL-8 | CXCR1/2 | 0.120 ± 0.01 | 52 ± 27 | 2 |
Data are expressed as percent inhibition of chemotaxis in the presence of AQX-1125 compared to the vehicle control.
Selectivity/specificity studies with AQX-1125
Prior to progressing into extensive in vivo studies with AQX-1125, a range of selectivity and specificity investigations were conducted to probe AQX-1125 for other potential molecular targets. In an in vitro pharmacology receptor binding and enzyme activity screen, AQX-1125 (3 μM) showed no significant effect (84 targets in the Cerep Diversity Profile). AQX-1125 (0.01–30 μM) was further evaluated and shown to have no effect on functional androgen, oestrogen, oestrogen-related receptor α, glucocorticoid, mineralocorticoid and progesterone receptor assays. Moreover, the in vitro binding affinity of AQX-1125 (10 μM) to 439 human kinases and three pathogen kinases was also evaluated in the Ambit scanMAX™ competitive binding assay. AQX-1125 showed no significant effect on any of these kinase targets.
Pharmacokinetic analysis of AQX-1125 in rats and dogs
The pharmacokinetic profile of AQX-1125 has been evaluated in the rat and dog. The calculated non-compartmental pharmacokinetic parameters following a single-dose administration are summarized in Table 4. In female Sprague-Dawley rats, the single-dose pharmacokinetics of AQX-1125 showed that the increases in maximal plasma concentration (Cmax) and AUC0–∞ were dose-proportional at the lower end of the dosing regimen and greater than dose proportional at the higher doses. The oral bioavailability of AQX-1125 in rats was 66 and 85% at 10 and 30 mg kg−1 respectively. The single-dose pharmacokinetics of AQX-1125 were also evaluated at 10 or 30 mg kg−1, administered by oral gavage to male beagle dogs. The increase in exposure was approximately dose proportional for a threefold increase in oral dose from 10 to 30 mg kg−1, a 2.8-fold increase in mean Cmax and a 3.6-fold increase in mean AUC0–∞. Oral bioavailability of AQX-1125 in dogs was 88 and 104% at 10 and 30 mg kg−1 respectively.
Table 4.
Pharmacokinetics of AQX-1125 in rats and dogs
| Species | n | Route | Dose (mg kg−1) | Tmax (h) | Cmax (μmol L−1) | t1/2 (h) | AUC0-∞ (h × μmol L−1) | AUC0-∞/dose (h × μmol L−1) mg−1 kg−1 | F |
|---|---|---|---|---|---|---|---|---|---|
| Rat | 4 | IV | 1 | 0.083 | 0.448 | 5.6 | 0.833 | 0.833 | 1.00 |
| 4 | PO | 10 | 1.000 | 0.830 | 5.2 | 5.473 | 0.547 | 0.66 | |
| 4 | PO | 30 | 1.167 | 3.825 | 4.5 | 21.179 | 0.706 | 0.85 | |
| Dog | 3 | IV | 1 | 0.083 | 1.341 | 11.9 | 4.871 | 4.871 | 1.00 |
| 3 | PO | 10 | 0.833 | 4.940 | 9.4 | 42.835 | 4.284 | 0.88 | |
| 3 | PO | 30 | 2.000 | 13.679 | 8.4 | 152.861 | 5.095 | 1.05 |
Measurement of tissue distribution of AQX-1125 in rats
The tissue distribution of AQX-1125 was studied in male Sprague-Dawley rats (n = 7) given a single i.v. administration of 10 mg kg−1 [14C]-AQX-1125 free base (10.0 μCi mg−1 in 0.9% saline). At 1, 4, 12, 24, 48, 72 and 120 h post-dose, tissue concentrations of [14C]-AQX-1125 were determined by quantitative whole-body autoradiography. The [14C]-AQX-1125-derived radioactivity was widely distributed after a single i.v. dose and most tissues had concentrations that were higher than blood at all time points. The highest levels of radioactivity in tissues excluding the excretory organs were found in the choroid plexus, seminal vesicles, pancreas, stomach gastric mucosa and pituitary gland. The Cmax of [14C]-AQX-1125-derived radioactivity in most tissues was found at 1 and 4 h post-dose (21 and 19 of 41 tissues respectively). Analysis of cardiac blood showed a Cmax at 1 h (0.62 μg equiv g−1) and concentrations in blood became below the limit of quantification by 72 h. All tissues exhibited decreased concentrations starting at 12 h post-dose. At 120 h post-dose, 24 of the 41 tissues had concentrations above the lower limit of quantification, ranging from 1.57 μg equiv g−1 (pituitary) to 0.04 μg equiv g−1 (thymus), with seven tissues having values sixfold greater than the lower limit of quantification (seminal vesicle, periosteum, spleen, liver, testes, epididymis and pituitary gland). Representative autography pictures are shown in Figure 7, while Table 5 presents the tissue concentrations of AQX 1125-derived radioactivity at Cmax observed following a single 10 mg kg−1 i.v. administration.
Figure 7.
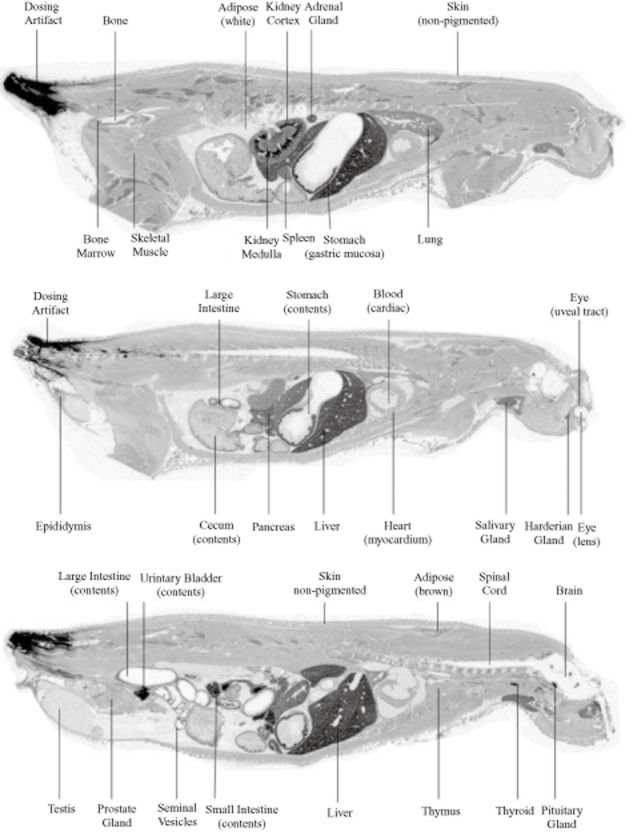
Distribution of AQX-1125 in rats. Representative whole-body autoradiogram of a 40 μm thick sagittal section of a rat treated with 10 mg kg−1 [14C]-AQX-1125 administered by an i.v. bolus injection and processed at 4 h.
Table 5.
Tissue distribution of AQX-1125 in the rat
| Tissue concentration (μmol equiv kg−1 tissue) | ||||
|---|---|---|---|---|
| 1 h | 4 h | 12 h | 24 h | |
| Blood | 1.9 | 0.6 | 0.2 | 0.2 |
| Large intestinal contents | 0.1 | 28.0 | 443.5 | 98.9 |
| Urinary bladder contents | 236.3 | 139.2 | 17.6 | 10.3 |
| Small intestinal contents | 183.5 | 175.1 | 39.2 | 4.1 |
| Cecum contents | ND | 63.2 | 125.8 | 19.8 |
| Kidney medulla | 60.4 | 22.0 | 6.7 | 3.4 |
| Choroid plexus | 54.2 | 58.4 | 18.6 | 6.8 |
| Seminal vesicle | 22.0 | 56.9 | 24.6 | 11.6 |
| Pancreas | 48.4 | 56.3 | 17.0 | 2.9 |
| Stomach (gastric mucosa) | 47.3 | 51.6 | 9.3 | 4.1 |
| Pituitary gland | 50.1 | 40.5 | 31.7 | 29.8 |
| Liver | 48.2 | 34.8 | 26.0 | 13.5 |
| Large intestine | 43.8 | 40.0 | 33.4 | 6.7 |
| Thyroid | 43.1 | 35.4 | 9.0 | 3.0 |
| Harderian gland | 33.5 | 40.4 | 21.4 | 18.8 |
| Spleen (high) | ND | 40.2 | 18.5 | 14.1 |
| Small intestine | 29.0 | 38.8 | 22.7 | 0.7 |
| Lung | 36.7 | 24.5 | 4.8 | 1.7 |
| Adrenal gland | 36.3 | 22.6 | 6.3 | 2.4 |
| Kidney cortex | 32.1 | 12.0 | 4.4 | 2.9 |
| Salivary gland | 29.7 | 22.0 | 5.6 | 1.4 |
| Periosteum | 28.5 | 28.4 | 10.7 | 3.9 |
| Spleen | 27.3 | 14.8 | 6.6 | 2.4 |
| Adipose (brown) | 21.8 | 18.1 | 4.6 | 1.4 |
| Bone marrow | 21.0 | 10.8 | 4.0 | 0.7 |
| Uveal tract | 16.8 | 18.2 | 8.4 | 4.8 |
| Lymph node | 15.3 | 14.3 | 3.6 | 2.8 |
| Spleen (low) | ND | 14.6 | 4.4 | 1.3 |
| Prostate gland | 12.6 | 8.4 | 3.6 | 2.4 |
| Thymus | 8.2 | 11.6 | 3.5 | 1.7 |
| Cecum | ND | 10.8 | 3.0 | 1.4 |
| Heart | 10.6 | 4.5 | 1.3 | 0.4 |
| Skin | 7.7 | 8.5 | 1.6 | 0.6 |
| Skeletal muscle | 8.4 | 4.2 | 1.0 | 0.3 |
| Epididymis | 7.1 | 6.4 | 6.7 | 6.4 |
| Urinary bladder | 5.5 | 5.7 | 0.7 | 0.6 |
| Testis | 1.8 | 3.1 | 2.8 | 3.2 |
| Seminal fluid | 0.4 | 2.7 | 0.8 | 0.7 |
| Adipose (white) | 1.4 | 1.2 | 0.1 | BLQ |
| Bone | 1.3 | 1.2 | 0.3 | BLQ |
| Stomach (contents) | 0.6 | 0.4 | BLQ | BLQ |
| Spinal cord | 0.1 | 0.2 | BLQ | BLQ |
| Eye (lens) | 0.1 | 0.2 | BLQ | BLQ |
| Brain (cerebrum) | 0.1 | 0.1 | BLQ | BLQ |
| Brain (cerebellum) | BLQ | 0.1 | BLQ | BLQ |
| Brain (medulla) | BLQ | BLQ | BLQ | BLQ |
In addition, the concentration of AQX 1125 in the plasma and lungs was evaluated following oral AQX-1125 administration (30 mg kg−1) in 0.9% saline to male Sprague-Dawley rats. Blood and lung samples were collected at 4 and 24 h post-dose and plasma and tissue concentrations were determined by LC-MS/MS. High tissue concentrations of AQX-1125 were detected, as compared to plasma concentrations, at each time point studied (Figure 8). The lung/plasma concentration ratio of the compound was approximately 30-fold at 4 h post-dose.
Figure 8.
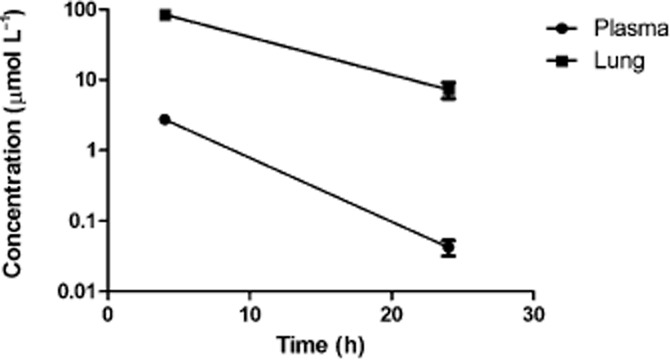
AQX-1125 has a long terminal half-life and yields high concentrations in the lung after oral dosing. Comparison of the concentration of AQX 1125 in plasma and lungs made, following repeat oral AQX-1125 administration (30 mg kg−1) in male Sprague-Dawley rats. Data represent mean ± SEM (n = 4).
Discussion
Herein we describe the identification and characterization of AQX-1125, a next-generation small-molecule activator of SHIP1 currently in clinical development. SHIP1 represents an attractive alternative approach to modulation of PI3K pathway signalling due to its highly restricted expression pattern. Conceptually, SHIP1 modulators may be viewed as re-directors of phosphatidylinositol phosphate (PIP) signalling by driving PIP3 to PI(3,4)P2, a mediator, which has its own signalling abilities and biological roles (Ma et al., 2008; Sasaki et al., 2010), whereas PI3K inhibitors terminate PIP signalling by preventing the formation of PIP3. Thus, with the pharmacological inhibition of PI3K having been the focus of intense efforts in the development of anti-inflammatory treatments, and the subsequent limited success, the activation of SHIP1 represents a novel therapeutic approach to the development of anti-inflammatory compounds.
AQX-1125 was found to directly bind to SHIP1, but failed to activate the SHIP1 enzyme when the C2 domain was deleted, consistent with the first-generation SHIP1 activator AQX-MN100 (Ong et al., 2007). In kinetic assays, the presence of AQX-1125 decreased the KM resulting in an increase in the ratio of kcat/KM. Taken together, these data support the hypothesis that AQX-1125 induces a conformational change of the enzyme, resulting in better substrate binding and enzyme efficiency. The target specificity of AQX-1125 was further validated in both the 89 target CEREP Diversity panels and the 442 kinase Ambit scanMAX competitive binding assay where it did not show any significant effects on any of the targets tested.
SHIP1 is a negative regulator of many inflammatory signalling processes of the immune system. It has documented roles in controlling vital cell functions such as development, proliferation, activation, cytokine secretion and migration (Kerr, 2011). Downstream from SHIP1 is Akt. In the absence of SHIP1, Akt signalling is enhanced in myeloid and NK cells (Liu et al., 1999) and when SHIP1 is transfected into cell lines that do not normally express this enzyme, Akt phosphorylation is markedly decreased (Freeburn et al., 2002; Baran et al., 2003; Horn et al., 2004). AQX-1125 was able to decrease the phosphorylation of Akt in multiple cell types, consistent with its function as a SHIP1 activator. This effect was absent in the SHIP1-deficient Jurkat cell line. Since Akt acts as a ‘master switch’ of various cellular responses (inflammatory responses, proliferation, motility), it is conceivable that the functional responses to AQX-1125 discussed in the subsequent sections result, at least in part, from the inhibition of cellular Akt activity.
AQX-1125 at 3 μM exerted a concentration-dependent reduction in the production of GM-CSF, TNF-α, IFNγ and IL-2 secretion in in vitro activated murine splenocytes without a corresponding reduction in IL-10 and IL-4. This is consistent with earlier findings using the first-generation SHIP1activator, AQX-MN100, which inhibited LPS-mediated TNFα release from macrophages (Ong et al., 2007). It is also consistent with data from the literature in which SHIP1−/− macrophages secrete enhanced level of proinflammatory mediatiors (TNF-α, IL-6, MCP-1, IFN-β) as compared to SHIP1+/+ cells (Gabhann et al., 2010; Keck et al., 2010). Furthermore, SHIP1−/− NK cells have been shown to secrete higher levels of IFNγ, which is reduced by overexpression of SHIP1. Taken together, these findings provide further evidence of the anti-inflammatory effects of SHIP1 activation.
Several lines of evidence support an evolutionarily conserved role for the localized production of PIPs in cell motility and directional migration towards chemoattractants (Logan and Mandato, 2006). Accordingly, SHIP1 has been shown to regulate chemotaxis and introduction of constitutively active SHIP1 into SHIP1-deficient cells abrogates the chemotactic response (Kim et al., 1999; Sattler et al., 2001; Mancini et al., 2002; Wain et al., 2005; Nishio et al., 2007). The current findings confirm that pharmacological SHIP1 activation is a potent inhibitor of leukocyte chemotaxis. Consistent with the role of PIPs and SHIP1 in cellular signalling, the inhibitory effect of AQX-1125 on chemotaxis was independent of the stimulus used to induce chemotaxis, and was largely independent of the cell type studied. Based on this data and several lines of studies showing that PIPs regulate actin reorganization at the leading edge in migrating cells, and that SHIP1 modulates cytoskeletal actin reorganization (Lesourne et al., 2005; Harris et al., 2011; Liu et al., 2011; Rahman et al., 2011), we hypothesize that the inhibition of chemotaxis by the SHIP1 activator, AQX-1125, is one of its modes of anti-inflammatory action.
In summary, we describe herein the identification and characterization of a novel class of pharmacological SHIP1 activators. AQX-1125 has emerged as a compound with good oral bioavailability, minimal metabolism, long terminal half-life and an ability to yield high concentrations in the lung. Therefore, this compound was subjected to in-depth in vitro and in vivo investigations. These in vivo efficacy studies are outlined in part 2 of the current series of articles (Stenton et al., 2012).
Acknowledgments
The help of Dr. Marta Guarna, Mr. Shannon Lentz and Ms. Haley Roberts with the analysis of data subsets is appreciated. In addition, the authors wish to acknowledge Bio-Quant Inc. (a subsidiary of Apricus Biosciences, San Diego, CA) for their expertise in the work outlined herein. We are also grateful for Dr. Gerald Krystal (University of British Columbia) for providing wild-type and SHIP1−/− mice for these studies.
Glossary
Abbreviations
- BMMC
bone marrow-derived mast cell
- CCR2
C-C chemokine receptor type 2
- Cmax
maximal plasma concentration
- CXCR
C-X-C chemokine receptor
- GRO
growth-regulated alpha protein
- His-hSHIP1ΔC2
6-His-tagged human SHIP1 with C2 domain deleted
- His-hSHIP1
6-His-tagged human SHIP1
- IGF-1
insulin-like growth factor-1
- IP4
1,3,4,5-inositol tetrakisphosphate
- kcat
turnover constant
- MCP-1
monocyte chemotactic protein-1
- PBMC
peripheral blood mononuclear cells
- PIP
phosphatidylinositol phosphate
- PIP3
phosphatidylinositol −3,4,5-trisphosphate
- SHIP1
SH2-containing inositol-5′-phosphatase 1
- Vmax
maximum velocity
- XTT
2,3-bis-(2-methoxy-4-nitro-5-sulfophenyl)-2H-tetrazolium-5-carboxanilide inner salt
Conflict of interest
All authors of this article are employees, former employees, consultants or stockholders of Aquinox Pharmaceuticals, a for-profit organization involved in the development of small-molecule SHIP1 activators as potential human therapeutic agents.
References
- Andersen R, Nodwell M, Mui A. SHIP1 modulator compounds. 2007. WO 2007/147251 A1.
- Baran CP, Tridandapani S, Helgason CD, Humphries RK, Krystal G, Marsh CB. The inositol 5′-phosphatase SHIP-1 and the Src kinase Lyn negatively regulate macrophage colony-stimulating factor-induced Akt activity. J Biol Chem. 2003;278:38628–38636. doi: 10.1074/jbc.M305021200. [DOI] [PubMed] [Google Scholar]
- Brooks R, Fuhler GM, Iyer S, Smith MJ, Park MY, Paraiso KHT, et al. SHIP1 inhibition increases immunoregulatory capacity and triggers apoptosis of hematopoietic cancer cells. J Immunol. 2010;184:3582–3589. doi: 10.4049/jimmunol.0902844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde C, Gloire G, Piette J. Enzymatic and non-enzymatic activities of SHIP-1 in signal transduction. Biochem Pharmacol. 2011;10:1320–1334. doi: 10.1016/j.bcp.2011.05.031. [DOI] [PubMed] [Google Scholar]
- Freeburn RW, Wright KL, Burgess SJ, Astoul E, Cantrell DA, Ward SG. Evidence that SHIP-1 contributes to phosphatidylinositol 3,4,5-trisphosphate metabolism in T lymphocytes and can regulate novel phosphoinositide 3-kinase effectors. J Immunol. 2002;169:5441–5450. doi: 10.4049/jimmunol.169.10.5441. [DOI] [PubMed] [Google Scholar]
- Gabhann JN, Higgs R, Brennan K, Thomas W, Damen JE, Ben Larbi N, et al. Absence of SHIP-1 results in constitutive phosphorylation of tank-binding kinase 1 and enhanced TLR3-dependent IFN-beta production. J Immunol. 2010;184:2314–2320. doi: 10.4049/jimmunol.0902589. [DOI] [PubMed] [Google Scholar]
- Haddon DJ, Antignano F, Hughes MR, Blanchet MR, Zbytnuik L, Krystal G, et al. SHIP1 is a repressor of mast cell hyperplasia, cytokine production, and allergic inflammation in vivo. J Immunol. 2009;183:228–236. doi: 10.4049/jimmunol.0900427. [DOI] [PubMed] [Google Scholar]
- Harris SJ, Parry RV, Foster JG, Blunt MD, Wang A, Marelli-Berg F, et al. Evidence that the lipid phosphatase SHIP-1 regulates T lymphocyte morphology and motility. J Immunol. 2011;186:4936–4945. doi: 10.4049/jimmunol.1002350. [DOI] [PubMed] [Google Scholar]
- Horn S, Endl E, Fehse B, Weck MM, Mayr GW, Jücker M. Restoration of SHIP activity in a human leukemia cell line downregulates constitutively activated phosphatidylinositol 3-kinase/Akt/GSK-3beta signaling and leads to an increased transit time through the G1 phase of the cell cycle. Leukemia. 2004;18:1839–1849. doi: 10.1038/sj.leu.2403529. [DOI] [PubMed] [Google Scholar]
- Keck S, Freudenberg M, Huber M. Activation of murine macrophages via TLR2 and TLR4 is negatively regulated by a Lyn/P13K module and promotedby SHIP1. J Immunol. 2010;184:5809–5818. doi: 10.4049/jimmunol.0901423. [DOI] [PubMed] [Google Scholar]
- Kerr WG. Inhibitor and activator: dual functions for SHIP in immunity and cancer. Ann N Y Acad Sci. 2011;1217:1–17. doi: 10.1111/j.1749-6632.2010.05869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CH, Hangoc G, Cooper S, Helgason CD, Yew S, Humphries RK, et al. Altered responsiveness to chemokines due to targeted disruption of SHIP. J Clin Invest. 1999;104:1751–1759. doi: 10.1172/JCI7310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal G. Lipid phosphatases in the immune system. Semin Immunol. 2000;12:397–403. doi: 10.1006/smim.2000.0222. [DOI] [PubMed] [Google Scholar]
- Lesourne R, Fridman WH, Daëron M. Dynamic interactions of Fc gamma receptor IIB with filamin-bound SHIP1 amplify filamentous actin-dependent negative regulation of Fc epsilon receptor I signaling. J Immunol. 2005;174:1365–1373. doi: 10.4049/jimmunol.174.3.1365. [DOI] [PubMed] [Google Scholar]
- Liu C, Miller H, Hui KL, Grooman B, Bolland S, Upadhyaya A, et al. A balance of Bruton's tyrosine kinase and SHIP activation regulates B cell receptor cluster formation by controlling actin remodeling. J Immunol. 2011;187:230–239. doi: 10.4049/jimmunol.1100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Sasaki T, Kozieradzki I, Wakeham A, Itie A, Dumont DJ, et al. SHIP1 is a negative regulator of growth factor receptor mediated PKB/Akt activation and myeloid cell survival. Genes Dev. 1999;13:786–791. doi: 10.1101/gad.13.7.786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Hefesha H, Tanaka H, Scriba G, Fahr A. Lipophilicity measurement of drugs by reversed phase HPLC over wide pH range using an alkaline-resistant silica-based stationary phase, XBridgeTM Shield RP18. Chem Pharm Bull. 2008;56:1417–1422. doi: 10.1248/cpb.56.1417. [DOI] [PubMed] [Google Scholar]
- Logan MR, Mandato CA. Regulation of the actin cytoskeleton by PIP2 in cytokinesis. Biol Cell. 2006;98:377–388. doi: 10.1042/BC20050081. [DOI] [PubMed] [Google Scholar]
- Ma K, Cheung SM, Marshall AJ, Duronio V. PI(3,4,5)P3 and PI(3,4)P2 levels correlate with PKB/akt phosphorylation at Thr308 and Ser473, respectively; PI(3,4)P2 levels determine PKB activity. Cell Signal. 2008;20:684–694. doi: 10.1016/j.cellsig.2007.12.004. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancini A, Koch A, Wilms R, Tamura T. The SH2-containing inositol 5-phosphatase (SHIP)-1 is implicated in the control of cell-cell junction and induces dissociation and dispersion of MDCK cells. Oncogene. 2002;21:1477–1484. doi: 10.1038/sj.onc.1205224. [DOI] [PubMed] [Google Scholar]
- March ME, Ravichandran K. Regulation of the immune response by SHIP. Semin Immunol. 2002;14:37–47. doi: 10.1006/smim.2001.0340. [DOI] [PubMed] [Google Scholar]
- Naal RMZG, Tabb J, Holowka D, Baird B. In situ measurement of degranulation as a biosensor based on RBL-2H3 mast cells. Biosens Bioelectron. 2004;20:791–796. doi: 10.1016/j.bios.2004.03.017. [DOI] [PubMed] [Google Scholar]
- Nishio M, Watanabe K, Sasaki J, Taya C, Takasuga S, Iizuka R, et al. Control of cell polarity and motility by the PtdIns(3,4,5)P3 phosphatase SHIP1. Nat Cell Biol. 2007;9:36–44. doi: 10.1038/ncb1515. [DOI] [PubMed] [Google Scholar]
- Ong CJ, Ming-Lum A, Nodwell M, Ghanipour A, Yang L, Williams DE, et al. Small-molecule agonists of SHIP1 inhibit the phosphoinositide 3-kinase pathway in hematopoietic cells. Blood. 2007;110:1942–1949. doi: 10.1182/blood-2007-03-079699. [DOI] [PubMed] [Google Scholar]
- Ooms LM, Horan KA, Rahman P, Seaton G, Gurung R, Kethesparan DS, et al. The role of the inositol polyphosphate 5-phosphatases in cellular function and human disease. Biochem J. 2009;419:29–49. doi: 10.1042/BJ20081673. [DOI] [PubMed] [Google Scholar]
- Parry RV, Harris SJ, Ward SG. Fine tuning T lymphocytes: a role for the lipid phosphatase SHIP-1. Biochim Biophys Acta. 2010;1804:592–597. doi: 10.1016/j.bbapap.2009.09.019. [DOI] [PubMed] [Google Scholar]
- Rahman P, Huysmans RD, Wiradjaja F, Gurung R, Ooms LM, Sheffield DA, et al. Silencer of Death Domains (SODD) inhibits skeletal muscle and Kidney Enriched Inositol 5-Phosphatase (SKIP) and regulates phosphoinositide 3-kinase (PI3K)/Akt signaling to the actin cytoskeleton. J Biol Chem. 2011;286:29758–29770. doi: 10.1074/jbc.M111.263103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond JR, Han K, Zhou Y, He Y, Noren B, Yee JGK. Indene derivatives as pharmaceutical agents. 2011. EP 2 277 848 A1.
- Roehm NW, Rodgers GH, Hatfield SM, Glasebrook AL. An improved colorimetric assay for cell proliferation and viability utilizing the tetrazolium salt XTT. J Immunol Methods. 1991;142:257–265. doi: 10.1016/0022-1759(91)90114-u. [DOI] [PubMed] [Google Scholar]
- Sasaki J, Kofuji S, Itoh R, Momiyama T, Takayama K, Murakami H, et al. The PtdIns(3,4)P2 phosphatase INPP4A is a suppressor of excitotoxic neuronal death. Nature. 2010;465:497–501. doi: 10.1038/nature09023. [DOI] [PubMed] [Google Scholar]
- Sattler M, Verma S, Pride YB, Salgia R, Rohrschneider LR, Griffin JD. SHIP1, an SH2 domain containing polyinositol-5-phosphatase, regulates migration through two critical tyrosine residues and forms a novel signaling complex with DOK1 and CRKL. J Biol Chem. 2001;276:2451–2458. doi: 10.1074/jbc.M006250200. [DOI] [PubMed] [Google Scholar]
- Stenton GR, Mackenzie LF, Tam P, Cross JL, Harwig C, Raymond J, et al. Characterization of AQX-1125, a small molecule SHIP1 activator. Part 2. Efficacy studies in pulmonary inflammation models in vivo. Br J Pharmacol. 2012;168:1519–1529. doi: 10.1111/bph.12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wain CM, Westwick J, Ward SG. Heterologous regulation of chemokine receptor signaling by the lipid phosphatase SHIP in lymphocytes. Cell Signal. 2005;17:1194–1202. doi: 10.1016/j.cellsig.2004.12.009. [DOI] [PubMed] [Google Scholar]
- Yang L, Williams DE, Mui A, Ong C, Krystal G, van Soest R, et al. Synthesis of pelorol and analogues: activators of the inositol 5-phosphatase SHIP. Org Lett. 2005;7:1073–1076. doi: 10.1021/ol047316m. [DOI] [PubMed] [Google Scholar]