

Abstract
We evaluated the hypothesis that activation of endogenous angiotensin-converting enzyme (ACE) 2 would improve cardiac dysfunction induced by diabetes. Ten days after diabetes induction (streptozotocin, 50mg/kg, i.v.), male Wistar rats were treated with the ACE2 activator 1-[[2-(dimethylamino)ethyl]amino]-4-(hydroxymethyl)-7-[[(4-methylphenyl)sulfonyl]oxy]-9H-xanthen-9-one (XNT, 1mg/kg/day, gavage) or saline (control) for 30 days. Echocardiography was performed to analyze the cardiac function and kinetic fluorogenic assays were used to determine cardiac ACE and ACE2 activities. Cardiac ACE2, ACE, Mas receptor, AT1 receptor, AT2 receptor and collagen type I and III mRNA and ACE2, ACE, Mas, AT1 receptor, AT2 receptor, ERK1/2, Akt, AMPK-α and AMPK-β1 protein were measured by qRT-PCR and western blotting techniques, respectively. Histological sections of hearts were analyzed to evaluate the presence of hypertrophy and fibrosis. Diabetic animals presented hyperglycemia and diastolic dysfunction along with cardiac hypertrophy and fibrosis. XNT treatment prevented further increase in glycemia and improved the cardiac function, as well as the hypertrophy and fibrosis. These effects were associated with increases in cardiac ACE2/ACE ratios (activity: ~26%; mRNA: ~113%; and protein: ~188%) and with a decrease in AT1 receptor expression. Additionally, XNT inhibited ERK1/2 phosphorylation and prevented changes in AMPK-α and AMPK-β1 expression. XNT treatment did not induce any significant change in AT2 receptor and Akt expression. These results indicate that activation of intrinsic cardiac ACE2 by oral XNT treatment protects the heart against diabetes-induced dysfunction through mechanisms involving ACE, ACE2, ERK1/2, AMPK-α and AMPK-β1 modulation.
Keywords: Cardiomyopathy, ACE, Angiotensin-(1-7), Angiotensin II, AMP-activated protein kinases, MAP kinases
1. Introduction
Diabetic patients might present a peculiar cardiovascular entity, the diabetic cardiomyopathy, characterized by diastolic and systolic dysfunction without coronariopathy and hypertension [1, 2]. Cardiovascular diseases (CVD) are the most important cause of death in the diabetic population and diabetes increases two- to fourfold the risk of CVD [3]. Indeed, hyperglycemia is one of the metabolic and homeostatic abnormalities that increase the cardiovascular mortality in patients with diabetes [4, 5].
It has been well-known that hyperactivity of the Angiotensin-converting enzyme (ACE)/Angiotensin (Ang) II/AT1 receptor axis of the Renin-angiotensin System (RAS) is associated with the establishment and progression of CVD and diabetes [6, 7]. On the other hand, recent studies have reported that ACE2 holds beneficial cardiovascular actions, such as anti-hypertensive [8], anti-fibrotic [9, 10], anti-oxidant [11], anti-inflammatory and anti-atherosclerotic [12, 13] effects. This enzyme is an important member of the RAS since it catalyzes the hydrolysis of the C-terminal residue of Ang II to produce the cardioprotective peptide Ang-(1-7) [14–16]. Thus, the axis composed by ACE2, Ang-(1-7) and Mas, the receptor for this peptide, plays a critical protective role in balancing the deleterious effects of the ACE/Ang II/AT1 receptor axis.
It is consensus that blockage of the ACE/Ang II/AT1 receptor axis improves morbidity, mortality and cardiovascular events in patients with CVD or high-risk diabetes [17–21]. However, the need for new therapies emerges due to ACE inhibitor intolerance and no consistent proof of specific cardiovascular protection by blockage of AT1 receptor that exceeds or efficiently synergizes the effect of ACE inhibition [22, 23]. Recently, activation of the ACE2/Ang-(1-7)/Mas axis has been considered a suitable approach for modulating the pathological effects of ACE/Ang II/AT1 receptor axis hyperactivity in CVD. In fact, ACE2 is a key regulator of the heart function [24] and its deficiency mimics or enhances the cardiac dysfunction induced by ACE/Ang II/AT1 receptor axis hyperactivity [9, 13]. Genetic and pharmacological manipulation of ACE2 has been demonstrated to be an important strategy to treat diabetes. However, these studies have focused on organs other than the heart such as kidney and pancreas [25–27]. For instance, Bindom et al. [25] reported an improvement in fasting glycemia, glucose tolerance and islet insulin content along with an enhancement of beta cells viability in type 2 diabetic mice overexpressing ACE2 [25].
To explore the therapeutic potential of ACE2, we have identified small-molecule ACE2 activators based on the virtual screening of its structure [28]. One of these compounds is the 1-[(2-dimethylamino)ethylamino]-4-(hydroxymethyl)-7-[(4-methylphenyl)sulfonyl oxy]-9Hxanthene-9-one (XNT). Acute administration of XNT induced a dose-dependent hypotensive response in spontaneously hypertensive rats (SHR) while chronic treatment with XNT improved the cardiac function and reversed the cardiac and renal fibrosis in SHR [28]. Furthermore, XNT prevented the increase in right ventricular systolic pressure and hypertrophy in a monocrotaline-induced pulmonary hypertension model [29] and attenuated the thrombus formation in SHR [12]. Thus, since ACE2 causes beneficial end-organ outcomes in diabetes and reverses hypertension-induced cardiac fibrosis, we hypothesized that activation of endogenous ACE2 using XNT would produce therapeutic outcomes in diabetes-induced cardiac dysfunction. To test this hypothesis, physiological, histological and molecular analyses were conducted to evaluate the effects of chronic oral XNT treatment in diabetic cardiomyopathy.
2. Material and methods
2.1. Materials
The ACE2 activator, XNT, was synthesized by Alchem Laboratories Corp. (Alachua, FL, USA). Fluorogenic substrates for ACE2 (catalog ID: ES007) and ACE (catalog ID: ES005) were obtained from R&D Systems (Minneapolis, MN, USA). The following antibodies were used in the western blotting assays: ACE2 (Santa Cruz sc20998, Santa Cruz, CA, USA), ACE (Millipore CD143, Billerica, MA, USA), Mas (Alomone AAR-013, Jerusalem, Israel), AT1 receptor (Santa Cruz sc1173, Santa Cruz, CA, USA), AT2 receptor (Alomone AAR-012, Jerusalem, Israel), ERK1/2 (phosphorylated and total: Cell Signaling #9106, Danvers, MA, USA and Santa Cruz sc93/sc154, Santa Cruz, CA, USA, respectively), Akt (phosphorylated and total: Cell Signaling #9271 and #9272, Danvers, MA, USA, respectively), AMPK-α (phosphorylated and total: Cell Signaling #2535 and #2603, Danvers, MA, USA, respectively), AMPK-β1 (phosphorylated and total: Cell Signaling #4181 and #4150, Danvers, MA, USA, respectively) and GAPDH (Sigma G8795, St. Louis, MO, USA). The following TaqMan® probes (Roche Indianapolis, IN, USA) were utilized in the RT-PCR assays: ACE2, Rn01416923-m1; ACE, Rn00561094-m1; Mas, Rn00562673-s1; AT1 receptor, Rn02132799-s1; AT2 receptor, Rn00560677-s1; collagen type I, Rn00801649-g1; and collagen type III, Rn01437683-m1.
2.2. Methods
2.2.1. Animals, diabetes induction and XNT treatment
All experimental protocols were performed in accordance with the Federal University of Minas Gerais (Brazil) and the University of Florida Institutional Animal Care and Use Committees, which are in compliance with the NIH guidelines. The experiments were performed in male Wistar rats (180–200g) obtained from CEBIO-UFMG (Belo Horizonte, MG, Brazil) and from Charles River Laboratories (Wilmington, MA, USA). They were housed in a light/dark cycle (12h/12h) room with standard rat chow and water ad libitum. Briefly, the rats were fasted (~16h), anesthetized (ketamine:xylazine, 60:6mg/kg, i.p.) and injected with streptozotocin (STZ; 50mg/kg i.v., Sigma; St. Louis, MO, USA) to induce diabetes. Control non-diabetic (CTL) rats were injected with ~0.2mL of sodium citrate buffer (10mmol/L, pH 4.5). Ten days after diabetes induction, the rats were assessed for blood glucose levels (Accu-Chek®; Roche, IN, USA). The animals with fasting blood glucose concentration over 126mmol/L were considered diabetic. The treatment with XNT (1mg/kg/day, gavage) or vehicle (saline pH 2–2.5; equivalent volume, gavage) was initiated ten days after diabetes induction and conducted for 30 days. The dose of 1mg/kg/day of XNT for oral administration was based on pilot experiments performed to determine the lowest amount of XNT able to induce cardiovascular effects. After testing the doses of 0.6mg/kg/day and 1mg/kg/day, we chose the dose of 1mg/kg/day based on the effects observed [30]. We have demonstrated in previous studies that XNT is able to activate ACE2 both in vitro and in vivo [10, 28].
2.2.2. Echocardiographic analysis
Transthoracic echocardiographic examination was performed using an Acuson Cypress™ machine equipped with an 8-MHz linear-array transducer (Siemens; Munich, Germany). The rats were anesthetized with a ketamine/xylazine mixture (60:6mg/kg, i.p.) (CTL: n=8; STZ: n=7; STZ+XNT: n=6). Left ventricular systolic and diastolic functions were assessed by ejection fraction (EF) and mitral inflow pulsed-wave Doppler, respectively. Three measurements were performed: 1) initial - at diabetes induction (day 0 - D0); 2) intermediate - ~28 days after diabetes induction (day 28 - D28); and 3) final - at the end of the experiments (day 40 - D40). Two-dimensional guided M-mode imaging at the papillary muscle level was used to measure the left ventricular end-systolic (LVESD) and end-diastolic (LVEDD) diameters and posterior wall thickness (LVPWT) during diastole. The EF was calculated from the M-mode echocardiogram using the equation: EF(%)=[(LVEDD3−LVESD3)/LVEDD3]×100. Mitral inflow pulsed-wave Doppler velocity was recorded from the apical four-chamber view. All analyses were performed in a blinded way by the same echocardiographist and included morphological and functional parameters. Furthermore, to evaluate if the anesthesia used in our protocol could interfere in the parameters, we adjusted the data to heart rate and no significant differences were observed (data not shown).
2.2.3. Histological analysis
Heart beat was stopped in diastole using 10% KCl (i.v.). The hearts were fixed in 4% Bouin and stained with Hematoxylin & Eosin for cell morphometry (CTL: n=4; STZ: n=5; STZ+XNT: n=4) or with Picrosirius Red for fibrosis (CTL: n=3; STZ: n=3; STZ+XNT: n=4). Three sections (5μm) from each animal were visualized in a light microscope (BX41®; Olympus, Center Valley, PA, USA), photographed (Q-Color3™; Olympus, Center Valley, PA, USA) under x400 magnification and analyzed using the ImageJ software (http://rsbweb.nih.gov/ij/). Cardiomyocyte diameter of the left ventricular wall (~100 cardiomyocytes for each animal) was measured across the region corresponding to the nucleus. Only cardiomyocytes cut longitudinally with nuclei and cellular limits visible were considered for analysis. Cardiac interstitial fibrosis of the left ventricle was measured by area percentage analysis. All analyses were performed in a blinded way by the same researcher.
2.2.4. Insulin sensitivity test
Insulin sensitivity test was performed in overnight fed rats two days before the end of the treatment (day 38 - D38). After intraperitoneal injection of insulin (0.75 U/kg body weight; Sigma, MO, USA), tail-blood samples were taken at 0, 15, 30, 60 and 90 minutes for measurement of blood glucose levels (Accu-Chek®; Roche, IN, USA).
2.2.5. ACE and ACE2 activities
Enzymatic activity was measured in a microplate reader (BioTekSynergy™ 2; BioTek, Winooski, VT, USA), as previously described [28]. Briefly, left ventricle samples (ACE: 30μg and ACE2: 60μg; n=4) were homogenized in a buffer composed by 75mmol/L Tris-HCl pH 7.5, 1mol/L NaCl and 0.5mmol/L ZnCl2. All assays were performed in duplicate at pH 7.4 in a reaction mixture containing: 50μmol/L substrate, 5mol/L NaCl, 75mmol/L Tris-HCl and 0.5μmol/L ZnCl2. Samples were read every minute for 4h immediately after the addition of the fluorogenic peptide substrates at 37°C. Background fluorescence readings were obtained from reactions without tissue samples and the final enzymatic activity of the samples was corrected by the obtained background value.
2.2.6. Real-time RT-PCR
Quantitative real-time RT-PCR (qPCR) was used to evaluate ACE2 (n=4), ACE (n=4), Mas (n=4), AT1 receptor (n=4), AT2 receptor (n=3), collagen type I (n=3) and collagen type III (n=4) mRNA levels in left ventricles. Total RNA was obtained using RNAqueous-4-PCR kit (Applied Biosystems/Ambion; Austin, TX, USA) and 150ng (ACE, ACE2, Mas, AT1 receptor and collagens) or 600ng (AT2 receptor) of total RNA were reverse transcribed using the iScript™cDNA synthesis kit (Bio-Rad; Hercules, CA, USA) in 20μL of reaction mixture. The cDNA samples were amplified by qPCR using an ABI Prism® 7000 Detection system (Applied Biosystems, Foster City, CS, USA). Gene expression was quantified using the comparative Ct (threshold cycle) method [31] with GAPDH as an endogenous control.
2.2.7. Western blotting analysis
After the end of the XNT treatment, left ventricles were harvested and Western blotting technique was performed to quantify the protein density of ACE2, ACE, Mas, AT1 receptor, AT2 receptor, ERK1/2, Akt, AMPK-α and AMPK-β1. Briefly, proteins of the left ventricle were isolated, separated by electrophoresis, transferred to a nitrocellulose membrane and probed with one of the following primary antibodies: ACE2 (n=3; 100μg; 1:1000), ACE (n=3; 100μg; 1:1000), Mas (CTL: n=3; STZ: n=3; STZ+XNT: n=4; 100μg; 1:1000), AT1 receptor (n=3; 15μg; 1:400), AT2 receptor (n=3; 50μg; 1:400), ERK1/2 (n=3; 100μg; phosphorylated 1:1000 and total 1:5000), Akt (n=3; 50μg; phosphorylated 1:1000 and total 1:1000), AMPK-α (n=3; 100μg; phosphorylated 1:250 and total 1:500) and AMPK-β1 (CTL: n=3; STZ: n=3; STZ+XNT: n=4; 100μg; phosphorylated 1:1000 and total 1:2000). To obtain an endogenous control for protein quantification, the membranes were stripped and re-probed with GAPDH antibody (1:20000 or 1:30000) for ACE2, ACE, Mas, AT1 receptor and AT2 receptor or with the total ERK1/2, Akt, AMPK-α and AMPK-β1 antibodies. Protein bands were detected with Amershan™ ECL Plus Western Blotting Detection Reagents (GE Healthcare, Waukesha, WI, USA), their densities were acquired using the Molecular Imager® GS-800™ calibrated densitometer and quantified using the Quantity One 1-D analysis software (Bio-Rad, Hercules, CA, USA).
2.2.8. Statistical analysis
The data are expressed as mean ± S.E.M. and P<0.05 was considered statistically significant. Statistical analyses were performed using Student’s t-test, ANOVA or Kruskal-Wallis analysis followed by the Newman-Keuls, Bonferroni or Dunn’s post tests, as indicated in the Figure legends.
3. Results
3.1. Efficacy of the oral XNT treatment to increase left ventricular ACE2 activity
Firstly, we evaluated the effect of the oral XNT treatment during 30 days on cardiac ACE2 activity since in previous studies we have shown that XNT causes an increase in ACE2 activity in vitro [28]. XNT administration increased the activity of ACE2 in hearts by ~65% in normal rats (CTL: 0.43±0.08 vs. XNT: 0.71±0.03 ΔRFU/Δmin/μg; P<0.05).
3.2. Effects of XNT on cardiac dysfunction induced by diabetes
Given the pronounced cardiac ACE2 activation reached by the oral XNT treatment, we subsequently tested the effects of XNT on cardiac dysfunction observed in diabetic animals. The echocardiographic analysis revealed that diabetic rats presented diastolic dysfunction evidenced by decreases in the peak early (E) to atrial/late peak (A) velocity of the transmitral inflow ratio (E/A ratio) (Figs. 1A, 1B and 1C). The XNT treatment reduced these changes in the pattern of the E/A ratio (Fig. 1C). Indeed, we observed mitral regurgitation only in control diabetic animals (Fig. 1B). Along with the diastolic dysfunction, diabetic animals also showed a slight, but significant, decrease in the EF. The XNT treatment efficiently prevented the alteration in the EF (Fig. 1D). However, no significant differences in LVEDD and LVESD were observed between the diabetic and diabetic+XNT groups (Figs. 1E and 1F). Importantly, these findings were not accompanied by changes in systemic arterial pressure (data not shown).
FIGURE 1.
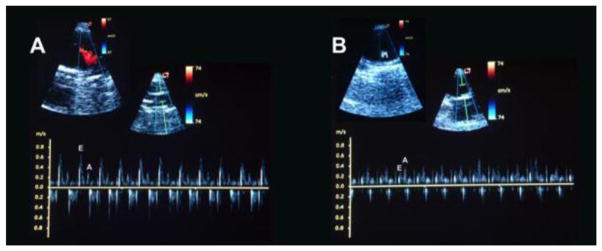
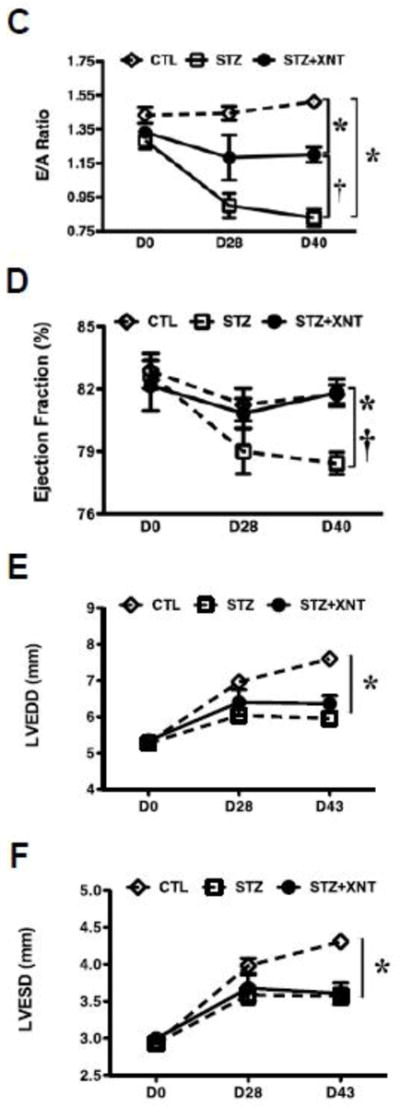
Effects of ACE2 activation on cardiac function of diabetic animals. Representative transmitral pulsed-wave Doppler of a (A) control animal, displaying normal early (E) and late (A) mitral valve inflow waves and of a (B) diabetic animal treated with saline, showing inversion of the E and A waves. At the left upper side of the panel B it is possible to observe mitral regurgitation. (C) Peak velocity of E and A waves (E/A) ratio of the mitral valve inflow; (D) ejection fraction (%); (E) left ventricular end diastolic diameter (LVEDD, mm); and (F) left ventricular end systolic diameter (LVESD, mm) of control (CTL, n=8) and diabetic animals treated with saline (STZ, n=7) or XNT (STZ+XNT, n=6). The data are shown as mean ± S.E.M. *P<0.05 vs. CTL and †P<0.05 vs. STZ. Two-way ANOVA followed by the Bonferroni post test.
3.3. Effects of XNT on cardiac remodeling induced by diabetes
The histological analysis showed that the treatment with XNT prevented the cardiac hypertrophy observed in diabetic rats (Figs. 2A and 2B). A similar effect was viewed when the cardiac fibrosis was analyzed, i.e. the XNT administration decreased the fibrosis induced by diabetes (Figs. 2C and 2D). Also, diabetes caused a significant increase in the expression of collagen type III in the heart and this effect was reduced by the XNT treatment (Fig. 2E). On the other hand, no significant changes were seen in the collagen type I expression among any of the groups (Fig. 2F).
FIGURE 2.
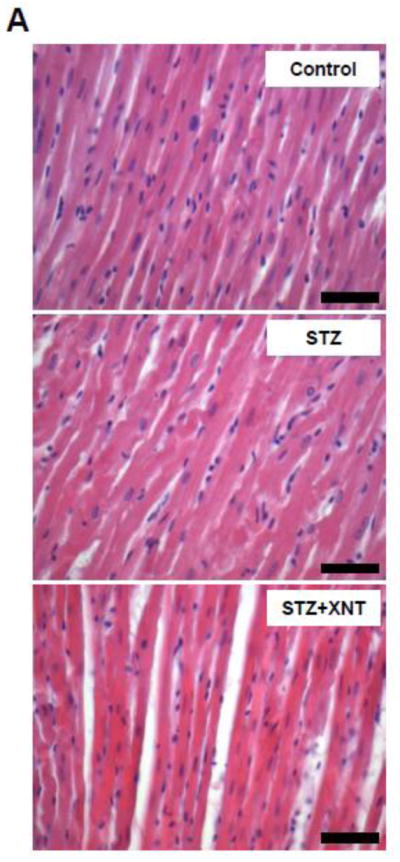

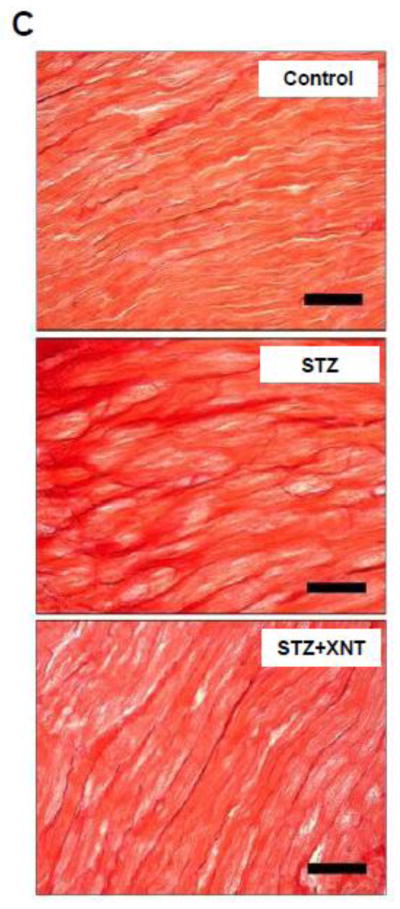
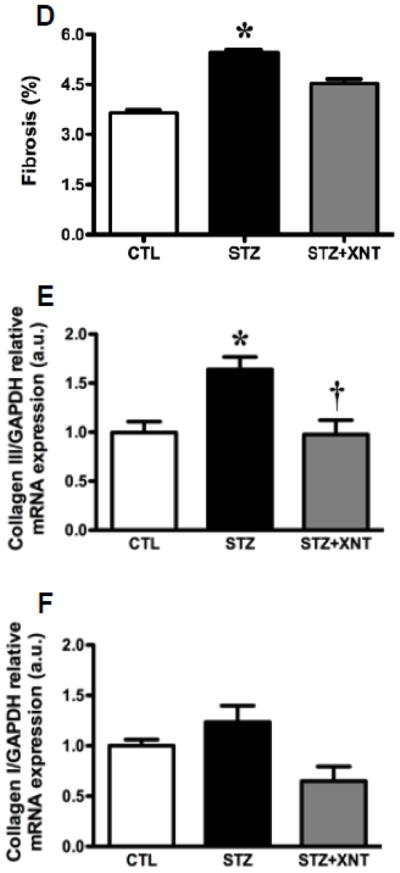
Effects of XNT on the cardiac remodeling induced by diabetes. (A) Representative photomicrographs of cardiomyocyte diameters of control animals, diabetic rats treated with saline (STZ) and diabetic animals treated with XNT (STZ+XNT). H&E (scale bar = 40 μm). (B) Cardiomyocyte diameter quantification (μm) (CTL: n=4; STZ: n=5; STZ+XNT: n=4). (C) Representative photomicrographs of left ventricular interstitial fibrosis of control animals, diabetic rats treated with saline (STZ) and diabetic animals treated with XNT (STZ+XNT). Intense red color indicates fibrosis. Picrosirius-Red staining (scale bar = 40 μm). (D) Quantification of the interstitial fibrosis (%) (CTL: n=3; STZ: n=3; STZ+XNT: n=4). mRNA expression of collagen type (E) III and (F) I in control animals, diabetic rats treated with saline (STZ) and diabetic animals treated with XNT (STZ+XNT). Collagen type III (n=4) and collagen type I (n=3). The data are shown as mean ± S.E.M. *P<0.05 vs. CTL and †P<0.05 vs. STZ. One-way ANOVA followed by the Newman-Keuls post test or Kruskal-Wallis analysis followed by the Dunn’s post test.
3.4. Modulation of the cardiac RAS induced by XNT in diabetic animals
To investigate the mechanisms underlying the effects of XNT on diabetic cardiomyopathy, we analyzed the balance between ACE and ACE2. Oral treatment with XNT increased the ACE2 activity, as well as the mRNA expression in hearts of diabetic animals. No significant changes were observed in the ACE2 protein expression among any of the groups (Table 1). On the other hand, ACE2 activation decreased the ACE mRNA and protein expression in diabetic rats. No significant changes were observed in the ACE activity among any of the groups (Table 1). More important, we evaluated the ratios between the two ACEs. As expected, diabetic animals presented a decreased ACE2/ACE protein ratio (~76%) and XNT efficiently increased this ratio by ~188%. The ACE2/ACE activity ratio was reduced by ~14% in diabetic rats and XNT administration increased it by ~26% when compared with diabetic animals. In terms of mRNA expression, the ratio between ACE2 and ACE was increased in diabetic rats (~34%) and a further increase of ~113% was induced by XNT administration (Table 1).
TABLE 1.
Effects of XNT on the cardiac activity and expression of ACE2 and ACE.
| Activity (ΔRFU/min/μg) | mRNA (a.u.) | Protein (a.u.) | |||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| CTL | STZ | STZ+XNT | CTL | STZ | STZ+XNT | CTL | STZ | STZ+XNT | |
|
|
|||||||||
| ACE2 | 0.49±0.07 | 0.54±0.06 | 0.76±0.06*† | 1.00±0.06 | 1.42±0.18 | 2.34±0.41* | 0.34±0.05 | 0.24±0.03 | 0.41±0.11 |
| ACE | 1.11±0.18 | 1.42±0.26 | 1.58±0.21 | 1.00±0.05 | 1.05±0.03 | 0.82±0.04*† | 0.087±0.002 | 0.26±0.03* | 0.150±0.003† |
| ACE2/ACE | 0.44±0.10 | 0.38±0.08 | 0.48±0.07 | 1.00±0.07 | 1.34±0.18 | 2.85±0.52*† | 3.93±0.55 | 0.95±0.17* | 2.73±0.71 |
The data are shown as mean ± S.E.M. Activity: ACE2 (n=4, 60μg), ACE (n=4, 30μg). mRNA: ACE2 and ACE (n=4). Protein: ACE2 and ACE (n=3, 1:1000), GAPDH (1:30000). Please see the representative blots in the Supplementary data.
P<0.05 vs. Control (CTL) group and
P<0.05 vs. Diabetes (STZ) group. Kruskal-Wallis analysis followed by the Dunn’s post test or One-way ANOVA followed by the Newman-Keuls post test (activity).
Additionally, the expression of AT1 receptor, AT2 receptor and Mas was examined. We observed that diabetes caused a trend toward enhancing the AT1 receptor mRNA expression along with a significant increase in AT1 receptor protein expression. These effects were absent in diabetic animals treated with XNT (Table 2). A robust increase without any significant change in protein expression was observed in Mas mRNA expression in diabetic rats. This effect was reverted by XNT administration (Table 2). Furthermore, diabetic animals presented an augmentation in the AT2 receptor protein expression when compared with control rats and XNT did not interfere in this effect (Table 2). Regarding the AT2 receptor mRNA expression, we observed RT-PCR amplification only in diabetic animals treated or not with XNT and any significant difference was viewed between these two groups (data not shown). In keeping with the beneficial effects of XNT on the ACE2/ACE ratios, diabetic animals treated with saline presented a reduction of ~44% in the Mas/AT1 receptor protein expression ratio while an increase of ~78% in the Mas/AT1 receptor protein expression ratio was observed in diabetic animals treated with XNT. Regarding the mRNA expression, the ratio between Mas and AT1 receptor increased in diabetic rats treated or not with XNT (~125%) (Table 2).
TABLE 2.
Effects of XNT on the cardiac expression of Ang II and Ang-(1-7) receptors.
| mRNA (a.u.) | Protein (a.u.) | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| CTL | STZ | STZ+XNT | CTL | STZ | STZ+XNT | |
|
|
||||||
| Mas | 1.00±0.20 | 3.11±0.12* | 1.84±0.19 | 0.12±0.01 | 0.12±0.01 | 0.11±0.01 |
| AT1R | 1.00±0.17 | 1.38±0.20 | 0.82±0.04† | 0.17±0.02 | 0.30±0.02* | 0.16±0.02† |
| Mas/AT1R | 1.00±0.26 | 2.25±0.34* | 2.24±0.33* | 0.72±0.11 | 0.40±0.04 | 0.71±0.11 |
| AT2R | - | - | - | 0.04±0.01 | 0.15±0.01* | 0.15±0.03* |
The data are shown as mean ± S.E.M. mRNA: Mas and AT1 receptor (n=4), AT2 receptor (n=3). Protein: Mas [1:1000, Control (CTL): n=3; Diabetes (STZ): n=3; Diabetes+XNT (STZ+XNT): n=4]; AT1 receptor (1:400; n=4), AT2 receptor (1:400; n=3). Please see the representative blots in the Supplementary data.
P<0.05 vs. Control (CTL) group and
P<0.05 vs. STZ group. One-way ANOVA followed by the Newman-Keuls post test.
3.5. Effects of XNT on hyperglycemia, AMPK and MAPK expression in diabetic rats
Since AMPK play a role in glucose homeostasis and in cardiac performance, we explored the putative involvement of these kinases in the cardiac protection triggered by XNT in diabetes. Diabetic animals presented hyperglycemia and XNT significantly prevented a further increase in blood glucose levels in diabetic animals after 30 days of treatment (Fig. 3A). Interestingly, diabetic rats treated with XNT presented an increased sensitivity to insulin. This effect was evident 90 minutes after the administration of insulin (Fig. 3B). Also, phosphorylation of the catalytic AMPK isoform (AMPK-α) was reduced while the regulatory AMPK isoform (AMPK-β1) was augmented in hearts of diabetic rats. The XNT treatment prevented these effects on both AMPK isoforms (Figs. 4A and 4B).
FIGURE 3.

Effects of diabetes and XNT treatment on glucose regulation. (A) Fasting blood glucose levels (mg/dL) after 10 and 40 days (D10 and D40, respectively) of diabetes induction. CLT (n=15, white diamond), STZ (n=11, white rectangle) and STZ+XNT (n=16, black circle). (B) Insulin sensitivity in control and diabetic rats treated or not with XNT (STZ: n=11; STZ+XNT: n=8). The data are shown as mean ± S.E.M. *P<0.05 vs. CLT and †P<0.05 vs. STZ. Two-way ANOVA followed by the Bonferroni post test.
FIGURE 4.

Effects of XNT on cardiac AMPK expression in diabetic rats. (A) AMPK-α (total: 1:250, phosphorylated: 1:500; n=3) and (B) AMPK-β1 (total: 1:1000, phosphorylated: 1:2000; CTL: n=3; STZ: n=3; STZ+XNT: n=4). The data are shown as mean ± S.E.M. *P<0.05 vs. CLT and †P<0.05 vs. STZ. One-way ANOVA followed by the Newman-Keuls post test.
Finally, we investigated the left ventricular expression of mitogen activated protein kinases (ERK1/2 and Akt) in diabetic rats treated with XNT. ACE2 activation reduced the increased phosphorylation of ERK1/2 observed in diabetic animals (Fig. 5A). On the other hand, no significant changes were viewed in Akt expression among any of the groups (Fig. 5B).
FIGURE 5.
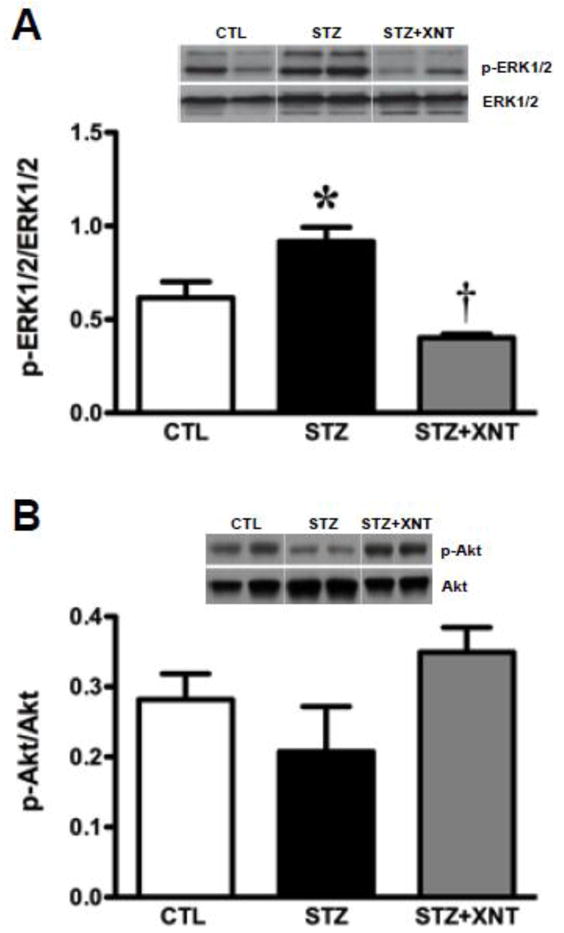
Effects of XNT on cardiac protein kinases expression in diabetic rats. (A) ERK1/2 (total: 1:1000, phosphorylated: 1:5000; n=3) and (B) Akt (total and phosphorylated 1:1000; n=3). The data are shown as mean ± S.E.M. *P<0.05 vs. CTL and †P<0.05 vs. STZ. One-way ANOVA followed by the Newman-Keuls post test.
4. Discussion
The ACE2 is an important regulator of the cardiac function [24] and several studies have shown that this enzyme induces beneficial outcomes in CVD and diabetic nephropathy [9–13, 26, 27]. Despite the relevance of the diabetic cardiomyopathy, the role of ACE2 in cardiac function and structure in diabetic animals has not been fully investigated. Thus, in the current study we addressed the effects of an ACE2 activator on the cardiac functional and structural changes induced by diabetes.
The most significant findings of this study are that oral administration of an ACE2 activator produced beneficial outcomes in diabetes-induced cardiomyopathy and these effects were independent of decreases in plasma glucose levels. The use of a chronic treatment with the ACE2 activator, XNT, prevented the diastolic dysfunction induced by diabetes. An important cause of diastolic dysfunction is the structural remodeling of the heart due to increased myocardial stiffness [32]. On this way, along with the improvement in the cardiac function, the XNT inhibited hypertrophy, fibrosis and the increase in collagen type III mRNA expression induced by diabetes. Also, the XNT treatment modulated the ACE2 and ACE expression and the phosphorylation of ERK1/2. Indeed, additionally to the protective role in the cardiac function, several studies have shown that ACE2 plays a role in the maintenance of the heart structure. ACE2 activation prevented cardiac fibrosis and its replacement in ACE2 knockout mice attenuated the Ang II-induced adverse myocardial remodeling through inhibition of ERK1/2 [9, 10, 28]. In hearts, ERK1/2 is increased after four weeks of diabetes induction and it induces ventricular hypertrophy along with pathological remodeling, chamber dilation and diastolic dysfunction [33–35]. In fact, we have demonstrated that the anti-fibrotic effect of chronic administration of XNT in spontaneous hypertensive rats is associated with a decrease in ERK1/2 phosphorylation [10]. Therefore, ACE2 activation may contribute to the maintenance of the heart function and its structural integrity by balancing the activity of the ACE/Ang II/AT1 receptor and ACE2/Ang-(1-7)/Mas axes of the RAS, as well as by inhibiting the ERK1/2 activity. One limitation of our study is that we did not measure the plasma and cardiac Ang II and Ang-(1-7) levels. However, it is important to note that qualitative immunohistochemical analysis showed that Ang-(1-7) immunostaining is increased in hearts of rats treated with XTN [10].
It has been suggested that ACE2 holds a role in glucose homeostasis [25]. Hyperglycemia induces myocardium damage through RAS activation, oxidative stress, calcium homeostasis alteration and structural changes due to glycation end product formation, hypertrophy and fibrosis [36–38]. We found that XNT prevented further increase in glycemia in diabetic animals. This effect might be involved in the cardioprotection observed in XNT-treated rats. Thus, activation of pancreatic ACE2 could favorably influence the heart, probably by regulating the pancreatic beta cells function and improving the metabolism of the heart. This premise is supported by the recent data of Bindom et al. [25], who showed that overexpression of ACE2 in the pancreas of db/db mice increases islet insulin content and beta cell proliferation, reduces beta cell apoptosis and improves fasting glycemia. It is important to note that because XNT prevented further increase in blood glucose levels and improved the insulin sensitivity in diabetic animals, this certainly contributed to the limitation of the development of cardiovascular damages. However, this was not the main mechanism underlying the beneficial effects of XNT since the blood glucose levels were still very high as 300 mg/dL in XNT-treated diabetic rats. Unfortunately, we did not investigate the pancreatic mechanisms involved in the XNT effects. Thus, future studies are needed to clarify the effects of XNT on glucose metabolism and insulin synthesis and degradation.
As an important cellular energy sensor and regulator of the metabolic homeostasis, activation of AMP-activated protein kinases (AMPK) leads to shifting towards an energy-producing state from an energy-consuming state [39, 40]. AMPK is a heterotrimeric protein consisting of α (catalytic), β and γ (regulatory) subunits [41–43]. It has been shown that AMPK can mediate GLUT4-dependent and insulin-independent muscular glucose uptake and increase fatty acid oxidation. In addition, its activity is inhibited in presence of high glucose concentration [44–48]. In hearts, inhibition of AMPK is involved in diabetic cardiomyopathy development and its activation can prevent post-ischemic cardiac dysfunction [49–51]. We demonstrated that diabetic animals treated with saline showed a decrease in the AMPK-α phosphorylation along with an increased AMPK-β1 phosphorylation. Administration of XNT prevented these changes. Thus, it is tempting to suggest that ACE2 modulates AMPK activity improving cardiac metabolic imbalance and resulting in beneficial outcomes in diabetes-induced cardiac dysfunction.
Nowadays, there are few studies evaluating the biological effects of small molecule ACE2 activators. We have consistently demonstrated that these molecules are potent activators of ACE2 both in vitro, as well as in vivo [10, 12, 28, 29]. However, it is pertinent to note that off-target effects of XNT on these beneficial outcomes cannot be ruled out at the present time. In fact, XNT treatment induced an increase in ACE2 activity and mRNA expression, but no changes in protein expression, indicating that changes in the post-transcriptional control of the ACE2 gene expression may be induced by XNT.
5. Conclusion
In this study, we demonstrated a functional and structural protection in hearts of diabetic animals promoted by XNT, an ACE2 activator. This cardioprotection involved the enhancement of the ACE2/ACE and Mas/AT1 receptor ratios and it was associated with reduction of the ERK1/2 phosphorylation, prevention of changes in AMPK-α and AMPK-β1 expression and prevention of cardiac hypertrophy and fibrosis. Thus, ACE2 activators could be supplemented with antihyperglycemic drugs as a combined therapy for the control of end-organ damages caused by diabetes. Our findings provide a promising therapeutic strategy to treat diabetic cardiomyopathy by oral XNT.
Supplementary Material
Highlights.
We tested if XNT, an ACE2 activator, would improve diabetes-induced heart dysfunction
Diabetic rats showed heart dysfunction/hypertrophy/fibrosis which were blunted by XNT
ACE2 activation also increased cardiac ACE2/ACE ratio and decreased AT1 expression
Inhibition of ERK and modulation of AMPK-α and β1 are also involved in these effects
Activation of intrinsic ACE2 protects the heart against diabetes-induced dysfunction
Acknowledgments
This work was supported in part by grants from Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG-Brazil), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq-Brazil) and Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES-Brazil). This work was also supported by NIH grants HL56921 and HL33610.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. 1972;30:595–602. doi: 10.1016/0002-9149(72)90595-4. [DOI] [PubMed] [Google Scholar]
- 2.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007;115:3213–3223. doi: 10.1161/CIRCULATIONAHA.106.679597. [DOI] [PubMed] [Google Scholar]
- 3.Skyler JS, Bergenstal R, Bonow RO, Buse J, Deedwania P, Gale EA, Howard BV, Kirkman MS, Kosiborod M, Reaven P, Sherwin RS. Intensive glycemic control and the prevention of cardiovascular events: implications of the ACCORD, ADVANCE, and VA Diabetes Trials: a position statement of the American Diabetes Association and a Scientific Statement of the American College of Cardiology Foundation and the American Heart Association. J Am Coll Cardiol. 2009;53:298–304. doi: 10.1016/j.jacc.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 4.Mazzone T, Chait A, Plutzky J. Cardiovascular disease risk in type 2 diabetes mellitus: insights from mechanistic studies. Lancet. 2008;371:1800–1809. doi: 10.1016/S0140-6736(08)60768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Selvin E, Marinopoulos S, Berkenblit G, Rami T, Brancati FL, Powe NR, Golden SH. Meta-analysis: glycosylated hemoglobin and cardiovascular disease in diabetes mellitus. Ann Intern Med. 2004;141:421–431. doi: 10.7326/0003-4819-141-6-200409210-00007. [DOI] [PubMed] [Google Scholar]
- 6.Probstfield JL, O’Brien KD. Progression of cardiovascular damage: the role of renin-angiotensin system blockade. Am J Cardiol. 2010;105:10A–20A. doi: 10.1016/j.amjcard.2009.10.006. [DOI] [PubMed] [Google Scholar]
- 7.Ruggenenti P, Cravedi P, Remuzzi G. The RAAS in the pathogenesis and treatment of diabetic nephropathy. Nat Rev Nephrol. 2010;6:319–330. doi: 10.1038/nrneph.2010.58. [DOI] [PubMed] [Google Scholar]
- 8.Wysocki J, Ye M, Rodriguez E, Gonzalez-Pacheco FR, Barrios C, Evora K, Schuster M, Loibner H, Brosnihan KB, Ferrario CM, Penninger JM, Batlle D. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: prevention of angiotensin II-dependent hypertension. Hypertension. 2010;55:90–98. doi: 10.1161/HYPERTENSIONAHA.109.138420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhong J, Basu R, Guo D, Chow FL, Byrns S, Schuster M, Loibner H, Wang XH, Penninger JM, Kassiri Z, Oudit GY. Angiotensin-converting enzyme 2 suppresses pathological hypertrophy, myocardial fibrosis, and cardiac dysfunction. Circulation. 2010;122:717–728. doi: 10.1161/CIRCULATIONAHA.110.955369. [DOI] [PubMed] [Google Scholar]
- 10.Ferreira AJ, Shenoy V, Qi Y, Fraga-Silva RA, Santos RAS, Katovich MJ, Raizada MK. Angiotensin-converting enzyme 2 activation protects against hypertension-induced cardiac fibrosis involving extracellular signal-regulated kinases. Exp Physiol. 2011;96:287–294. doi: 10.1113/expphysiol.2010.055277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gwathmey TM, Pendergrass KD, Reid SD, Rose JC, Diz DI, Chappell MC. Angiotensin-(1-7)-angiotensin-converting enzyme 2 attenuates reactive oxygen species formation to angiotensin II within the cell nucleus. Hypertension. 2010;55:166–171. doi: 10.1161/HYPERTENSIONAHA.109.141622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fraga-Silva RA, Sorg BS, Wankhede M, Dedeugd C, Jun JY, Baker MB, Li Y, Castellano RK, Katovich MJ, Raizada MK, Ferreira AJ. ACE2 activation promotes antithrombotic activity. Mol Med. 2010;16:210–215. doi: 10.2119/molmed.2009.00160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thomas MC, Pickering RJ, Tsorotes D, Koitka A, Sheehy K, Bernardi S, Toffoli B, Nguyen-Huu TP, Head GA, Fu Y, Chin-Dusting J, Cooper ME, Tikellis C. Genetic Ace2 deficiency accentuates vascular inflammation and atherosclerosis in the ApoE knockout mouse. Circ Res. 2010;107:888–897. doi: 10.1161/CIRCRESAHA.110.219279. [DOI] [PubMed] [Google Scholar]
- 14.Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res. 2000;87:E1–E9. doi: 10.1161/01.res.87.5.e1. [DOI] [PubMed] [Google Scholar]
- 15.Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem. 2000;275:33238–33243. doi: 10.1074/jbc.M002615200. [DOI] [PubMed] [Google Scholar]
- 16.Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–14843. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 17.SOLVD. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. The SOLVD Investigators. N Engl J Med. 1991;325:293–302. doi: 10.1056/NEJM199108013250501. [DOI] [PubMed] [Google Scholar]
- 18.Yusuf S, Sleight P, Pogue J, Bosch J, Davies R, Dagenais G. Effects of an angiotensin-converting-enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–153. doi: 10.1056/NEJM200001203420301. [DOI] [PubMed] [Google Scholar]
- 19.Cohn JN, Tognoni G. A randomized trial of the angiotensin-receptor blocker valsartan in chronic heart failure. N Engl J Med. 2001;345:1667–1675. doi: 10.1056/NEJMoa010713. [DOI] [PubMed] [Google Scholar]
- 20.Pfeffer MA, Swedberg K, Granger CB, Held P, McMurray JJ, Michelson EL, Olofsson B, Ostergren J, Yusuf S, Pocock S. Effects of candesartan on mortality and morbidity in patients with chronic heart failure: the CHARM-Overall programme. Lancet. 2003;362:759–766. doi: 10.1016/s0140-6736(03)14282-1. [DOI] [PubMed] [Google Scholar]
- 21.Cowan BR, Young AA, Anderson C, Doughty RN, Krittayaphong R, Lonn E, Marwick TH, Reid CM, Sanderson JE, Schmieder RE, Teo K, Wadham AK, Worthley SG, Yu CM, Yusuf S, Jennings GL. Left ventricular mass and volume with telmisartan, ramipril, or combination in patients with previous atherosclerotic events or with diabetes mellitus (from the ONgoing Telmisartan Alone and in Combination With Ramipril Global Endpoint Trial [ONTARGET]) Am J Cardiol. 2009;104:1484–1489. doi: 10.1016/j.amjcard.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 22.Schindler C. ACE-inhibitor, AT1-receptor-antagonist, or both? A clinical pharmacologist’s perspective after publication of the results of ONTARGET. Ther Adv Cardiovasc Dis. 2008;2:233–248. doi: 10.1177/1753944708094309. [DOI] [PubMed] [Google Scholar]
- 23.Chrysant SG. Current status of dual Renin Angiotensin aldosterone system blockade for the treatment of cardiovascular diseases. Am J Cardiol. 2010;105:849–852. doi: 10.1016/j.amjcard.2009.11.044. [DOI] [PubMed] [Google Scholar]
- 24.Crackower MA, Sarao R, Oudit GY, Yagil C, Kozieradzki I, Scanga SE, Oliveira-dos-Santos AJ, da Costa J, Zhang L, Pei Y, Scholey J, Ferrario CM, Manoukian AS, Chappell MC, Backx PH, Yagil Y, Penninger JM. Angiotensin-converting enzyme 2 is an essential regulator of heart function. Nature. 2002;417:822–828. doi: 10.1038/nature00786. [DOI] [PubMed] [Google Scholar]
- 25.Bindom SM, Hans CP, Xia H, Boulares AH, Lazartigues E. Angiotensin I-converting enzyme type 2 (ACE2) gene therapy improves glycemic control in diabetic mice. Diabetes. 2010;59:2540–2548. doi: 10.2337/db09-0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oudit GY, Liu GC, Zhong J, Basu R, Chow FL, Zhou J, Loibner H, Janzek E, Schuster M, Penninger JM, Herzenberg AM, Kassiri Z, Scholey JW. Human recombinant ACE2 reduces the progression of diabetic nephropathy. Diabetes. 2010;59:529–538. doi: 10.2337/db09-1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu CX, Hu Q, Wang Y, Zhang W, Ma ZY, Feng JB, Wang R, Wang XP, Dong B, Gao F, Zhang MX, Zhang Y. Angiotensin-converting enzyme (ACE) 2 overexpression ameliorates glomerular injury in a rat model of diabetic nephropathy: a comparison with ACE inhibition. Mol Med. 2011;17:59–69. doi: 10.2119/molmed.2010.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hernandez Prada JA, Ferreira AJ, Katovich MJ, Shenoy V, Qi Y, Santos RAS, Castellano RK, Lampkins AJ, Gubala V, Ostrov DA, Raizada MK. Structure-based identification of small-molecule angiotensin-converting enzyme 2 activators as novel antihypertensive agents. Hypertension. 2008;51:1312–1317. doi: 10.1161/HYPERTENSIONAHA.107.108944. [DOI] [PubMed] [Google Scholar]
- 29.Ferreira AJ, Shenoy V, Yamazato Y, Sriramula S, Francis J, Yuan L, Castellano RK, Ostrov DA, Oh SP, Katovich MJ, Raizada MK. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med. 2009;179:1048–1054. doi: 10.1164/rccm.200811-1678OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Murça TM, Almeida TC, Raizada MK, Ferreira AJ. Chronic Activation of Endogenous Angiotensin-Converting Enzyme 2 Protects Diabetic Rats from Cardiovascular Autonomic Dysfunction. Exp Physiol. 2012 doi: 10.1113/expphysiol.2011.063461. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 32.Zile MR, Brutsaert DL. New concepts in diastolic dysfunction and diastolic heart failure: Part II: causal mechanisms and treatment. Circulation. 2002;105:1503–1508. doi: 10.1161/hc1202.105290. [DOI] [PubMed] [Google Scholar]
- 33.Naito Z, Takashi E, Xu G, Ishiwata T, Teduka K, Yokoyama M, Yamada N, Sugisaki Y, Asano G. Different influences of hyperglycemic duration on phosphorylated extracellular signal-regulated kinase 1/2 in rat heart. Exp Mol Pathol. 2003;74:23–32. doi: 10.1016/s0014-4800(03)80005-9. [DOI] [PubMed] [Google Scholar]
- 34.de Boer RA, Pokharel S, Flesch M, van Kampen DA, Suurmeijer AJ, Boomsma F, van Gilst WH, van Veldhuisen DJ, Pinto YM. Extracellular signal regulated kinase and SMAD signaling both mediate the angiotensin II driven progression towards overt heart failure in homozygous TGR(mRen2)27. J Mol Med (Berl) 2004;82:678–687. doi: 10.1007/s00109-004-0579-3. [DOI] [PubMed] [Google Scholar]
- 35.Zheng M, Dilly K, Dos Santos Cruz J, Li M, Gu Y, Ursitti JA, Chen J, Ross J, Jr, Chien KR, Lederer JW, Wang Y. Sarcoplasmic reticulum calcium defect in Ras-induced hypertrophic cardiomyopathy heart. Am J Physiol Heart Circ Physiol. 2004;286:H424–H433. doi: 10.1152/ajpheart.00110.2003. [DOI] [PubMed] [Google Scholar]
- 36.Singh VP, Le B, Khode R, Baker KM, Kumar R. Intracellular angiotensin II production in diabetic rats is correlated with cardiomyocyte apoptosis, oxidative stress, and cardiac fibrosis. Diabetes. 2008;57:3297–3306. doi: 10.2337/db08-0805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pekiner B, Ulusu NN, Das-Evcimen N, Sahilli M, Aktan F, Stefek M, Stolc S, Karasu Ç. In vivo treatment with stobadine prevents lipid peroxidation, protein glycation and calcium overload but does not ameliorate Ca2+-ATPase activity in heart and liver of streptozotocin-diabetic rats: comparison with vitamin E. Biochim Biophys Acta. 2002;1588:71–78. doi: 10.1016/s0925-4439(02)00141-2. [DOI] [PubMed] [Google Scholar]
- 38.van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, Ijsselmuiden AJ, Schalkwijk CG, Bronzwaer JG, Diamant M, Borbely A, van der Velden J, Stienen GJ, Laarman GJ, Niessen HW, Paulus WJ. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. 2008;117:43–51. doi: 10.1161/CIRCULATIONAHA.107.728550. [DOI] [PubMed] [Google Scholar]
- 39.Carling D. The AMP-activated protein kinase cascade-a unifying system for energy control. Trends Biochem Sci. 2004;29:18–24. doi: 10.1016/j.tibs.2003.11.005. [DOI] [PubMed] [Google Scholar]
- 40.Zhang BB, Zhou G, Li C. AMPK: an emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009;9:407–416. doi: 10.1016/j.cmet.2009.03.012. [DOI] [PubMed] [Google Scholar]
- 41.Stapleton D, Mitchelhill KI, Gao G, Widmer J, Michell BJ, Teh T, House CM, Fernandez CS, Cox T, Witters LA, Kemp BE. Mammalian AMP-activated protein kinase subfamily. J Biol Chem. 1996;271:611–614. doi: 10.1074/jbc.271.2.611. [DOI] [PubMed] [Google Scholar]
- 42.Thornton C, Snowden MA, Carling D. Identification of a novel AMP-activated protein kinase beta subunit isoform that is highly expressed in skeletal muscle. J Biol Chem. 1998;273:12443–12450. doi: 10.1074/jbc.273.20.12443. [DOI] [PubMed] [Google Scholar]
- 43.Hoehn KL, Turner N, Swarbrick MM, Wilks D, Preston E, Phua Y, Joshi H, Furler SM, Larance M, Hegarty BD, Leslie SJ, Pickford R, Hoy AJ, Kraegen EW, James DE, Cooney GJ. Acute or chronic upregulation of mitochondrial fatty acid oxidation has no net effect on whole-body energy expenditure or adiposity. Cell Metab. 2010;11:70–76. doi: 10.1016/j.cmet.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Long YC, Barnes BR, Mahlapuu M, Steiler TL, Martinsson S, Leng Y, Wallberg-Henriksson H, Andersson L, Zierath JR. Role of AMP-activated protein kinase in the coordinated expression of genes controlling glucose and lipid metabolism in mouse white skeletal muscle. Diabetologia. 2005;48:2354–2364. doi: 10.1007/s00125-005-1962-5. [DOI] [PubMed] [Google Scholar]
- 45.Ruderman NB, Saha AK, Kraegen EW. Minireview: malonyl CoA, AMP-activated protein kinase, and adiposity. Endocrinology. 2003;144:5166–5171. doi: 10.1210/en.2003-0849. [DOI] [PubMed] [Google Scholar]
- 46.Itani SI, Saha AK, Kurowski TG, Coffin HR, Tornheim K, Ruderman NB. Glucose autoregulates its uptake in skeletal muscle: involvement of AMP-activated protein kinase. Diabetes. 2003;52:1635–1640. doi: 10.2337/diabetes.52.7.1635. [DOI] [PubMed] [Google Scholar]
- 47.da Silva Xavier G, Leclerc I, Varadi A, Tsuboi T, Moule SK, Rutter GA. Role for AMP-activated protein kinase in glucose-stimulated insulin secretion and preproinsulin gene expression. Biochem J. 2003;371:761–774. doi: 10.1042/BJ20021812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Witczak CA, Sharoff CG, Goodyear LJ. AMP-activated protein kinase in skeletal muscle: from structure and localization to its role as a master regulator of cellular metabolism. Cell Mol Life Sci. 2008;65:3737–3755. doi: 10.1007/s00018-008-8244-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Daniels A, van Bilsen M, Janssen BJ, Brouns AE, Cleutjens JP, Roemen TH, Schaart G, van der Velden J, van der Vusse GJ, van Nieuwenhoven FA. Impaired cardiac functional reserve in type 2 diabetic db/db mice is associated with metabolic, but not structural, remodelling. Acta Physiol (Oxf) 2010;200:11–22. doi: 10.1111/j.1748-1716.2010.02102.x. [DOI] [PubMed] [Google Scholar]
- 50.Li YJ, Wang PH, Chen C, Zou MH, Wang DW. Improvement of mechanical heart function by trimetazidine in db/db mice. Acta Pharmacol Sin. 2010;31:560–569. doi: 10.1038/aps.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Paiva MA, Rutter-Locher Z, Goncalves LM, Providencia LA, Davidson SM, Yellon DM, Mocanu MM. Enhancing AMPK activation during ischemia protects the diabetic heart against reperfusion injury. Am J Physiol Heart Circ Physiol. 2011;300:H2123–H2134. doi: 10.1152/ajpheart.00707.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.