

Abstract
During prion disease cellular prion protein (PrPC) is refolded into a pathogenic isoform (PrPSc) that accumulates in the central nervous system and causes neurodegeneration and death. We used immunofluorescence, quantitative cryo-immunogold EM and tomography to detect nascent, full-length PrPSc in the hippocampus of prion-infected mice from early pre-clinical disease stages onwards. Comparison of uninfected and infected brains showed that sites containing full-length PrPSc could be recognized in the neuropil by bright spots and streaks of immunofluorescence on semi-thin (200 nm) sections, and by clusters of cryo-immunogold EM labeling. PrPSc was found mainly on neuronal plasma membranes, most strikingly on membrane invaginations and sites of cell-to-cell contact, and was evident by 65 days postinoculation, or 54% of the incubation period to terminal disease. Both axons and dendrites in the neuropil were affected. We hypothesize that closely apposed plasma membranes provide a favourable environment for prion conversion and intercellular prion transfer. Only a small proportion of clustered PrP immunogold labeling was found at synapses, indicating that synapses are not targeted specifically in prion disease.
Keywords: prion, PrPSc neurodegeneration, cryo-immunogold EM, plasma membrane, synapse
Introduction
PrPC is a glycosylphosphatidylinositol (GPI)-anchored membrane glycoprotein that may undergo a template-assisted conformational change to form PrPSc in prion disease (Colby and Prusiner, 2011). Prion diseases are characterized by a long incubation period followed by a short clinical phase. Early pre-clinical signs in brain are increased PrPSc levels and astrocytosis, accompanied by subtle behavioural changes and loss of synapses and synaptic proteins (Masters et al., 1984; Jeffrey et al., 2000; Guenther et al., 2001; Cunningham et al., 2003; Siskova et al., 2009; Tamguney et al., 2009). However, the mechanisms responsible are unknown. In cultured cells, PrPC traffics to the plasma membrane before conversion to PrPSc takes place (Caughey and Raymond, 1991; Borchelt et al., 1992), but whether conversion occurs on the plasma membrane and/or in an endo-lysosomal compartment is unclear.
PrPSc tends to aggregate, and insoluble, protease-resistant forms can be isolated from prion-infected brains. However, protease-sensitive PrPSc oligomers are predominant in some types of prion disease (Safar et al., 1998; Safar et al., 2002; Pastrana et al., 2006) and are infectious (Sajnani et al., 2012). To investigate the subcellular location of PrPSc during the course of prion disease in mouse brain we used cryo-immunogold EM for optimal antigen and ultrastructural preservation. We also performed electron tomography on immunolabeled sections for a three-dimensional view of the labeled tissue. We previously reported cryo-immunogold EM localization of PrPC and PrPSc in hippocampus of late pre-clinical stage prion-infected mice. We employed antibodies recognizing PrPC specifically (F4-31) (Stanker et al., 2010) or a C-terminal epitope of PrP available in both PrPC and (undenatured) PrPSc (R2) (Peretz et al., 1997; Williamson et al., 1998). Quantification of the labeling indicated that F4-31-labeled PrPC levels were similar in uninfected and prion-infected mice, while R2 labeling increased after infection. R2 labeling density was 8-fold greater on plasma membrane and 24-fold greater on early/recycling endosome-like vesicles of small diameter neurites in infected mice compared to uninfected mice, indicating sites of PrPSc localization. Most of this labeling was trypsin sensitive (Godsave et al., 2008) indicative of oligomeric PrPSc rather than larger insoluble aggregates.
In neuroblastoma cells, PrPSc comprises predominantly full-length, mature GPI-anchored protein at conversion, with trimming of the N-terminus occurring within a few hours, probably in lysosomes (Caughey et al., 1991; Taraboulos et al., 1992). To investigate sites of PrPSc conversion, we therefore used an antibody (Saf32), which recognizes an epitope within the N-terminus of full length PrP (Safar et al., 2005a). Furthermore, Saf32 gave higher levels of labeling than R2 in prion-infected mouse brain and was more suitable for labeling at earlier stages of prion infection. At all stages examined, we observed a high proportion of Saf32-labeled PrPSc on extensive plasma membrane invaginations and at sites of cell-to-cell contact. Saf32-labeled PrPSc was only occasionally found close to synapses, indicating that synapto-toxicity is likely an indirect consequence of plasma membrane-associated pathology.
Methods
Animals and preparation of tissue
All mouse studies were carried out in accordance with the recommendations of the Public Health Services/National Institutes of Health Guide for the Care and Use of Laboratory animals. Using previously described procedures for prion infection (Carlson et al., 1989), wild-type FVB mice were injected intracerebrally with 30 μl of 1% brain homogenate from mice infected with Rocky Mountain Laboratory (RML) prions. Mice were routinely monitored for: 1) rigidity of the tail; 2) limb and truncal ataxia; 3) forelimb flexion instead of extension when suspended by the tail. Probable prion disease was diagnosed when at least two of these signs progressed over a period of a few days (Prusiner et al., 2004). Under the infection conditions described here, clinical prion disease is apparent at approximately 120 days. For immunolabeling quantification, two RML-injected wild-type mice were sacrificed at each of the following times after inoculation: 65 dpi, 85 dpi, 99 dpi. Two mice were also sacrificed at 104 dpi for R2 labeling. It is highly unlikely that any of the original inoculate remained in any of these brains. Efficient clearance of PrPSc from the brain has been shown to occur after i.c inoculation of hamsters (Safar et al., 2005a) and after suppression of PrP expression in prion-infected bigenic mice (Safar et al., 2005b). Prnp0/0 mice and FVB mice injected with normal brain homogenate were sacrificed at 116 dpi as controls. Saf32 labeling of Prnp0/0 mice inoculated with brain homogenate from uninfected or RML prion-infected mice and sacrificed at 116 dpi give similar low levels of background labeling (unpublished results). Mice were sacrificed and transcardially perfused with fixative containing 2% paraformaldehyde (PFA) and 0.2% glutaraldehyde, as described previously (Mironov et al., 2003). Brains were collected and postfixed in the same fixative for 2 to 24 hours, transferred to 1% PFA for 7 days and then stored in 0.5% PFA until use. Brain tissue blocks were embedded in gelatin, frozen in sucrose, and cryosectioned for immunolabeling as described previously (Godsave et al., 2008). Unless otherwise stated, sections were 200-nm–thick for immunofluorescence and 60–70-nm–thick for cryo-immunogold EM.
SDS-PAGE and Western blotting
SDS-PAGE and Western blotting were conducted essentially as described (Wille & Prusiner 1999). In brief, 10% (w/v) brain homogenates and PTA-precipitates (Safar et al., 1998) were mixed with equal volumes of SDS-sample buffer, boiled for 5 min and cooled on ice prior to loading of 50 μL samples onto 15% SDS-PAGE gels. The Western blots were incubated with anti-PrP recombinant Fab HuM-P (Safar et al., 2002) diluted 1:1,000. Following extensive washing, the blots were incubated with peroxidase-labeled anti-mouse or anti-rabbit Fc secondary antibody (Pierce Biotechnology, Rockford, IL) and developed by the enhanced chemiluminescence (ECL) system (Amersham Life Sciences, Arlington Heights, IL).
Immunolabeling
Brain tissue sections were immunolabeled for immunofluorescence, and for cryo-immunogold EM using Protein A gold to detect antibody binding, as described previously (Godsave et al., 2008). Primary antibodies used were as follows: Saf32, monoclonal anti-PrP N-terminal region (Spi-Bio, Montigny Le Bretonneux Cedex, France); R2, mouse Fab recognizing PrP C-terminal residues 225-231 (Peretz et al., 1997; Williamson et al., 1998); F4-31, monoclonal anti-PrPC-specific antibody (Godsave et al., 2008; Stanker et al., 2010); mouse anti-VAMP2, rabbit anti-VAMP2 and rabbit anti-vesicular glutamate transporter 1 (Synaptic Systems, Göttingen, Germany); rabbit anti-GABA (Immunostar, Hudson, WI, USA); rabbit anti-glutamine synthetase (Sigma-Aldrich). Rabbit anti-mouse IgG bridging antibody was from DAKOCytomation (Glostrup, Denmark). Protein A–gold conjugate was obtained from Utrecht Medical Center (The Netherlands). Fluorescent secondary antibodies, goat anti-mouse IgG coupled to Alexa Fluor 488 and goat anti-rabbit IgG coupled to texas red were from Molecular Probes (Eugene, OR, USA). Fluorescently labeled sections were examined using a Zeiss AxioObserver Z1 inverted microscope. Micrographs were taken using a Hamamatsu ORCA AG Black and White CCD-camera with Zeiss axiovision software, which adds false colours to the images. Sections for EM were generally collected on copper slot grids (EMS, Hatfield, PA, USA). Labeled grids were examined using a CM10 electron microscope (FEI, Eindhoven, Netherlands) at 80 kV.
Quantification
Strips of tissue spanning the stratum oriens, between the pyramidal layer and alveus, were scanned for the presence of clusters of at least four gold particles present in a cell process located within an area equivalent to 250 X 250 nm. Larger clusters extending over a wider area were counted once. Micrographs were made of all clusters present within a strip measuring 30-μm wide.
Electron tomography
Electron tomography was performed using a Tecnai 12 electron microscope (FEI, Eindhoven, Netherlands) at 120 kv on 200-nm Saf32-immunolabeled cryosections. Multiple tilt series were acquired in 2-degree increments, from −58° to +60°. Reconstructions were generated by weighted back projection using the IMOD software (Kremer et al., 1996).
Results
PrPSc isolated from prion-infected brain comprises both full length and N-terminally truncated forms
Western blotting of PrP in brain extracts from prion-infected mice typically gives several bands. These reflect both differences in glycosylation, and the presence of N-terminally truncated forms that are generated after conversion, e.g (Safar et al., 2005b). We performed Western blotting on brain samples from FVB mice infected with RML prions, using sodium phosphotungstate precipitation (Safar et al., 1998) and protease treatments to distinguish PrPC and PrPSc (D’Castro et al., 2010). The results indicated that the majority of PrPSc is N-terminally truncated, and that full length PrPSc is selectively enriched in newly formed PrPSc (Figure 1a–c).
Figure 1.

RML prions in FVB mice exist as a mixture of truncated and non-truncated molecules and nascent PrPSc can be labeled selectively by antibodies to the PrP N-terminus. For a–c, lane 1: unprocessed brain homogenate of RML-infected FVB brains (10%, w/v); lane 2: PTA-precipitate obtained from the brain homogenate in lane 1. (a) Preparation of RML prions without use of extraneous proteases. The brain homogenate and the subsequent PTA-precipitate contain full-length PrPSc and N-terminally truncated PrP 27-30. In addition to the N-terminal truncation, both truncated and non-truncated peptides present as di-, mono-, and un-glycosylated forms with overlapping band patterns. (b) Preparation of RML prions using pronase E. Again, the PTA-precipitate contains full-length PrPSc and N-terminally truncated PrP 27-30. Pronase E has been described to digest PrPC and other proteins, but to leave PrPSc and PrP 27-30 intact (D’Castro et al., 2010). (c) Preparation of RML prions using proteinase K. Here, the PTA-precipitate contains only the N-terminally truncated PrP 27-30, which presents as a combination of di-, mono-, and un-glycosylated peptides, resulting in a banding pattern of reduced complexity. Western blots were developed with the anti-PrP recombinant Fab HuM-P (Safar et al., 2002). Molecular mass markers indicating 15, 20, 30, and 40 kDa, respectively, are shown on the left. (d, e) Immunofluorescence labeling of serial cryo-sections (400 nm thick) from the 99 dpi prion-infected hippocampus stratum oriens with d) F4-31 PrPC-specific antibody and e) Saf32. Nuclei were stained with DAPI (blue). Note that F4-31 gives a characteristic ‘granular’ labeling pattern throughout the neuropil, while Saf32 shows more scattered spots and streaks of bright immunofluorescence. Bar in (e) represents 10 μm and also applies to (d).
PrPSc increases in stratum oriens during prion disease
For Saf32 localization of full length PrP in brain, we focused on the stratum oriens of the hippocampal CA1 region, where there is clear pathology in prion-infected mice (Godsave et al., 2008). The Saf32 antibody binds to both PrPC and PrPSc. However, comparison of Saf32 immunofluorescence with that of the F4-31 high affinity antibody specific for PrPC (Godsave et al., 2008; Stanker et al., 2010) indicated that bright spots and streaks of Saf32 labeling represented sites containing PrPSc (Figure 1d,e).
We then used Saf32 to localize PrPSc in several pre-clinical stages of prion disease. An early sign of prion-related pathology is the appearance of reactive astrocytes (Hwang et al., 2009; Tamguney et al., 2009), easily recognizable in the EM since they have many processes containing large numbers of intermediate filaments composed of GFAP. By EM we occasionally saw evidence for reactive astrocytes by 65 dpi. By 99 dpi, astrocytic gliosis was prominent, also by GFAP immunofluorescence (results not shown). To obtain an impression of the distribution of full-length PrP in this region as prion disease progressed, we performed immunofluorescence on cryo-sections that were 200-nm thick. This generated high-resolution light microscopy images with low autofluorescence. As with F4-31 labeling of prion infected hippocampus (Figure 1d), labeling of uninfected hippocampus with Saf32 antibody resulted in an evenly distributed “granular” pattern throughout the neuropil (Figure 2e). By contrast, additional bright spots and streaks of Saf32 immunofluorescence were found in prion-infected hippocampus at all stages examined (65, 85, 99 dpi; Figure 2). The thickness of the sections used (200 nm) is similar to the diameter of many small neurites in the neuropil, so streaks of immunofluorescence may indicate labeling of single cell processes. As expected, pyramidal cell bodies and radial dendrites in the stratum radiatum were unlabeled by all anti-PrP antibodies tested (Godsave et al., 2008) and Figure 2). At 65 dpi, when mice were still asymptomatic, increased Saf32 immunofluorescence was observed particularly in regions of the neuropil close to the alveus. There were also small patches of bright labeling in the alveus (Figure 2a). Foci of bright Saf32 immunofluorescence became increasingly widespread with advancing disease incubation time. In some cases, we saw broad bands or patches of fluorescent spots and streaks in the stratum oriens (Figure 2). We reasoned that this might reflect either the pattern of prion spread or a greater susceptibility of certain types of neurons to prion infection. However, we were unable to determine co-localization with several markers for different neuronal populations or cellular regions, including GABA (Figure 2a, inset i′ and data not shown); the synaptic vesicle marker, VAMP2 (Figure 3a–c); and a dendritic marker, MAP2 (not shown). Fig. 2b, inset ii′, shows an astroglial cell labeled with antibodies to glutamine synthetase in 85 dpi hippocampus. There is prominent Saf32 labeling surrounding this cell, but little co-labeling. Peri-astrocytic PrPSc has also been observed in prion-diseased human brain (Kovacs et al., 2005).
Figure 2. Saf32 immunofluorescence labeling of PrP in the hippocampal CA1 region.

Bottom left, a diagram of the hippocampus, with the area shown in (a–e) indicated by the box. (a–c) Composite images of prion-infected hippocampus at 65 dpi (a), 85 dpi (b) and 99 dpi (c). In (a) an area of higher labeling in the stratum oriens (boxed area i), is shown at higher magnification in i′, double-labeled with Saf32 (green) and anti-GABA (red). Co-labeling was not apparent. Patches of bright labeling are also visible in the alveus. In (b), area equivalent to box ii from a serial section, double-labeled with Saf32 (green) and anti-glutamine synthetase (red) is shown in ii′. Again, little co-labeling is evident. (d, e) As controls, hippocampal sections taken from a PrP-deficient mouse (KO; d) and an uninfected FVB mouse (Uninf; e). Saf32 gives an evenly distributed ‘granular’ pattern of labeling in the uninfected hippocampus (e). Nuclei were stained with DAPI (blue) in (a–e). Abbreviations: pyr, pyramidal layer; or, stratum oriens; alv, alveus; KO, Prnp0/0; CA1, CA2, CA3, regions of the hippocampus; DG, dentate gyrus. Bars represent 10 μm; bar in (e) also applies to (a–d).
Figure 3. Reduced immunofluorescence labeling of synaptic vesicle markers and lack of co-localization with Saf32 in prion disease.

(a–c) Sections of 99 dpi prion-infected hippocampal CA1 stratum oriens were labeled with rabbit anti-VAMP2 (red; a) and Saf32 (green; b), shown together in (c). Co-localization of VAMP2 and PrPSc was sparse. Immunofluorescence labeling of synaptic vesicle markers in uninfected mice (d–f) and from prion-infected mice at 99 dpi (g–i); sections were labeled with mouse anti-VAMP2 (green; d,g) and rabbit anti-vGlut1 (red; e,h), shown together in (f,i). In (d–i), one or two cells of the pyramidal layer are visible at the bottom of each image. Note the more disperse labeling with both antibodies in the prion-diseased hippocampus. In both uninfected and prion-infected hippocampus, there are co-labeled structures (yellow in f and i) and structures labeled only by VAMP2. Nuclei were counterstained with DAPI (blue). Bars, 10 μm and apply to panels in the same respective row.
Clusters of Saf32 cryo-immunogold EM labeling indicate sites containing PrPSc
To obtain information on the subcellular localization of full-length PrP in the stratum oriens, we compared Saf32 labeling of sections from prion-infected, and uninfected mice, using quantitative cryo-immunogold EM. As expected, Saf32 labeling of uninfected hippocampus showed disperse gold labeling mainly on plasma membranes (Figure 4a), consistent with previous results obtained with F4-31 and R2 (Godsave et al., 2008). In prion-infected hippocampus there were additionally many clusters of Saf32 immunogold labeling (Figures 4–7). For quantification purposes we defined a labeling cluster as at least four gold particles on a cell process within an area equivalent to 250 X 250 nm. We then compared the density of labeling clusters in defined areas of stratum oriens in uninfected and prion-deficient controls and at different stages of prion disease. Relatively few clusters were found in sections from one of the mice at 65 dpi. However, in all other prion-infected brains at least 90% of clusters, and all clusters of more than 5 gold particles, could be considered to be infection-specific (Table 1, Figures 4–7) and to indicate sites containing PrPSc. Since we cannot use proteinase K on cryo-sections for EM, we use the term PrPSc here to refer to both proteinase K-sensitive and -resistant forms.
Figure 4. Cryo-immunogold EM of cell surface and vesicular PrP in stratum oriens.

Saf32 cryo-immunogold EM labeling of PrP on the cell surface (a–e, h) and vesicles (f, g) in stratum oriens (a-g), and alveus (h). (a) Sparse labeling of PrPC (arrows) in neuropil of uninfected hippocampus. (b–h) Clusters of labeling indicate PrPSc at 65 dpi (b, c, h), 85 dpi (d), and 99 dpi (e, f, g). PrPSc can be seen on very small diameter processes in (b, e), and at cell-cell junctions (b and c arrowheads; d, e). (c) Cryo-section, 200 nm thick, grazing the surface of a process that runs horizontally across the image. Several other processes, which appear darker, are on top of this. The arrowhead indicates Saf32 labeling at a point of contact. In (d), the plasma membrane of one of the processes appears to be invaginating. Arrows in (h) indicate clusters of labeling. Abbreviations: Den, dendrite; Ax, axon; sv, synaptic vesicles; Li, lipid droplet. Bars, 200 nm.
Figure 7. Slice from a tomographic reconstruction of a Saf32-labeled invagination.
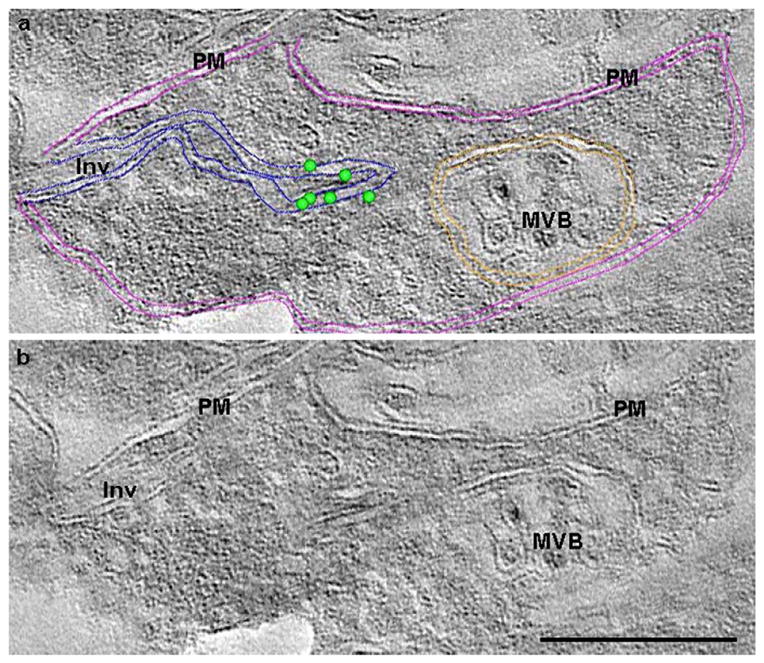
Slice from a tomographic reconstruction close to the surface of a 200-nm–thick tissue section showing Saf32 cryo-immunogold EM-labeled structures in hippocampal stratum oriens at 65 dpi. The reconstructed image is duplicated to show annotations (a) of features that are visible or can be inferred in (b). In (a) the locations of gold particles at the surface of the section are indicated by green dots. Coloured lines mark membranes: pink, cell surface; blue, invagination; orange, multivesicular body. The invaginating membranes join the plasma membrane at the cell surface on the left of the image. Immunogold labeling is present close to a point where the invaginating membranes converge. PM, plasma membrane; Inv, invaginating membranes; MVB, multivesicular body. Scale bar, 200 nm.
Table 1.
Clusters of Saf32 immunogold labeling are prion disease-related.
| Sample source | Gold particles in clusters (n) | Clusters (n) |
|---|---|---|
| 65 dpi | 122 | 22 |
| 85 dpi (20 μm) | 130 | 24 |
| BH | 8 | 2 |
| KO | 5 | 1 |
| 65 dpi | 40 | 8 |
| 85 dpi | 89 | 20 |
| 99 dpi | 141 | 27 |
| KO | 4 | 1 |
| 99 dpi | 103 | 21 |
| BH | 8 | 2 |
| KO | 0 | 0 |
Samples were obtained from FVB mice inoculated with RML prions at the indicated days postinoculation (dpi), and from PrP-knockout (KO) mice and FVB mice (BH) inoculated with normal brain homogenate at 116 dpi. Numbers of clusters of Saf32 labeling and the total number of gold particles in clusters were counted in a 30-μm–wide strip of stratum oriens. The results are grouped according to the batches of primary and secondary antibody used. Clusters of gold labeling were often found in infected brains with only small numbers of clusters observed in control hippocampal sections (BH and KO). Within experiments, the numbers of clusters per brain increased with disease incubation time. All labeling experiments involved different animals.
Saf32-labeled PrPSc is frequently present at sites of cell-to-cell contact
Using Saf32 for cryo-immunogold EM, the types of structure that showed clusters of labeling in stratum oriens were similar at all stages examined (Figure 6). We found most of the Saf32-labeled clusters on “small neurites”, defined as being less than 250 nm across (Fig. 4b, c, e), in agreement with our analysis of R2 labeled PrPSc at a late pre-clinical disease stage (Godsave et al., 2008). Labeling in the alveus was also often associated with small diameter cell processes (Fig. 4h). A much smaller proportion of clustered Saf32 labeling was found on larger axons and dendrites, and we failed to find Saf32-labeled PrPSc on processes characteristic for astrocytes, though we cannot exclude the possibility that some labeled processes were non-neuronal. Clustered Saf32 labeling was most frequently found at the cell surface. Interestingly, a very high proportion (80%) of this surface labeling was at sites of cell-to-cell contact (Figure 4b–e). Because we generally observed an increase in the space between cell processes during prion disease, the labeling between closely apposed membranes was noteworthy.
Figure 6. Quantification of clustered Saf32 labeling in infected hippocampus.
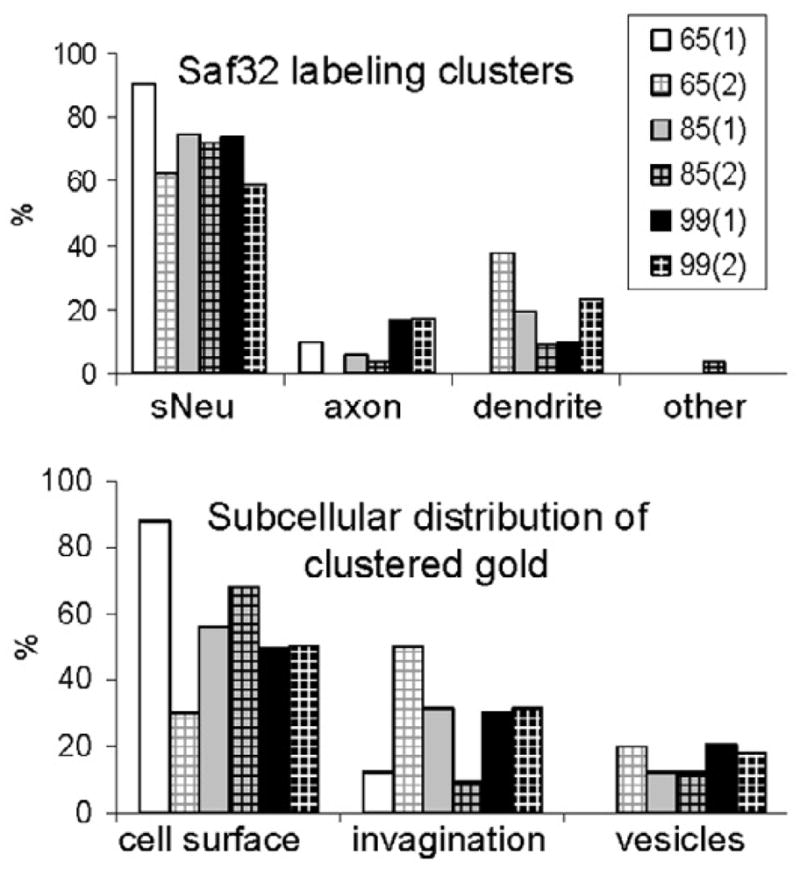
Quantification of Saf32 labeling, expressed as the proportional distribution of clustered gold particles in different types of neuronal structures (top) or subcellular compartments (bottom) at the timepoints indicated, in dpi. Numbers in parentheses indicate samples from different animals. “Other” indicates labeling in small vesicles of an astrocyte. Labeling on vesicles comprised 13% of the total clustered gold particles analyzed in infected hippocampus with 1.4% on synaptic vesicles. Abbreviations: sNeu, small neurite with diameter of <250 nm.
A much smaller proportion of clustered labeling was associated with small vesicles, most of which resembled early endocytic or recycling vesicles (Figure 4f–g and Figure 6). Generally, these clusters either comprised gold particles on several separate vesicles, and/or they included gold particles on nearby plasma membrane, although individual vesicles with several gold particles were also observed (Figure 4f). Occasionally, some of the gold particles in a cluster were found on synaptic vesicles (< 2% of clustered gold particles analysed). No clusters were observed in multivesicular bodies or lysosomes.
PrPSc is present on plasma membrane invaginations in axons and dendrites
Additionally, we found PrPSc clusters on several types of plasma membrane invagination. These included spiroplasma-like inclusions that were observed mainly in dendrites. Their membranes often had an electron-dense cytosolic coating (Figure 5b) and were sometimes branched, as described previously (Liberski et al., 2005; Ersdal et al., 2009; Jeffrey et al., 2011). We also observed PrP clusters on pairs of membranes running through the cytosol parallel to the long axis of (mainly) small neurites, both axonal and dendritic. We first observed these in stratum oriens close to the alveus at 65 dpi (Figure 5a), and they were subsequently found to be more widespread and present at all stages examined. The membranes had no visible coating and in some cases could be followed to the limiting membrane, indicating that they also represent plasma membrane invaginations (Figure 5a, c, d). Individual neuronal processes bearing clustered Saf32 labeling on membrane invaginations rarely contained labeled vesicles and were often surrounded by unlabeled processes, especially at early stages.
Figure 5. Cryo-immunogold EM labeling of PrPSc on membrane invaginations.

Hippocampal sections were taken from prion-infected mice at 65 dpi (a), 85 dpi (b), 99 dpi (c, d, e), 104 dpi (f). (a–d) Saf32 labeling of PrPSc on membrane invaginations in stratum oriens of prion-infected hippocampus. (a) Process with labeled paired membranes (black arrow) that continue to the surface membrane. The inset shows a higher magnification view of the process. The white arrow indicates another process with paired membranes labeled with two gold particles. (b) Dendrite, with labeled coated membranes (arrowheads) that extend to the plasma membrane. (c) Labeled paired membranes (arrowheads) in an axon terminal close to a synapse (arrow). (d) Saf32 labeling (15-nm gold) on a post-synaptic membrane invagination (arrowheads) and on paired membranes of an excitatory axon terminal (black arrows) co-labeled with anti-vGlut1 (10-nm gold). (e) Image showing degenerating axon terminal with no clustered Saf32 labeling on the axon or post-synaptic structures. Arrows indicate synapses. (f) Membrane invagination labeled with R2 antibody to the C-terminus of PrP. Abbreviations: Den, dendrite; Ax, axon; MVB, multivesicular body. Bars, 200 nm.
In a previous cryo-immunogold EM study using R2, labeled invaginations were not observed in prion-infected stratum oriens in the region close to the pyramidal layer that was investigated in detail (Godsave et al., 2008). However, after examining a wider area of stratum oriens, we also saw membrane invaginations labeled with R2 (Figure 5f). We would expect that R2 labels a larger population of PrPSc than Saf32, since Saf32 can only recognize N-terminally intact PrP. However, Saf32 gave higher levels of labeling than R2 (data not shown), raising the possibility that PrP antigens are differentially accessible in different forms of PrPSc.
We used electron tomography to obtain a more detailed image of Saf32-labeled invaginations. The gold labeling cannot penetrate into the tissue, and was therefore present on the surface of the section. Interestingly, clustered label was generally found either at a site of membrane convergence (Figure 7) or in a sharp curve of a membrane (not shown). Figure 7 shows a “slice” from a tomographic reconstruction from stratum oriens of a mouse at 65 dpi. The Saf32-labeled membranes could be traced out to the cell surface in the tomogram, showing that they represented invaginated plasma membrane.
PrPSc is found infrequently at synapses
Because synapto-pathology has been found to occur in prion disease, (reviewed in (Mallucci, 2009), we analyzed whether PrPSc was present near synapses in prion-infected mice, using Saf32 and evaluating 30 μm-wide strips of stratum oriens. Only five percent of labeling clusters were clearly associated with a synaptic bouton or a post-synaptic structure, generally in membrane invaginations (Figure 5c, d). Labeling could be found close to asymmetric glutamatergic synapses, and to GABAergic terminals (unlabeled by antibodies to vesicular glutamate transporter, Figure 5d). Degenerating dark axon terminals and synapses with high curvature were observed in prion-infected hippocampus by 99 dpi (Figure 5e), but they very rarely exhibited Saf32 labeling clusters.
As an indicator of changes in the numbers of functional synapses in prion disease, we performed immunofluorescence labeling with a general marker of synaptic vesicles (VAMP2) and of vesicles at glutamatergic synapses (vGlut1). As expected, vGlut1-positive processes were generally also labeled by antibodies to VAMP2. There were additional structures, presumably non-glutamatergic, that were only labeled by VAMP2 antibody. The results suggested that both GABAergic and glutamatergic synapses are affected by prion disease, as labeling with both markers became more disperse at 99 dpi compared to uninfected brains (Figure 3d–i). By contrast, in agreement with our cryo-immunogold EM observations, VAMP2 showed only minimal co-localization with Saf32 (Figure 3a–c).
Discussion
Clustered Saf32 immunogold labeling is a marker for PrPSc in hippocampus
Following PrPC to PrPSc conversion, N-terminal amino acids 23–89 of the mature protein are trimmed off in acidic late-endosomal compartments in cultured cells (Caughey et al., 1991; Taraboulos et al., 1992). Similarly, in FVB mice infected with RML prions (as used here), a majority of the PrPSc in brain extracts lacked the N-terminus, indicating rapid N-terminal trimming after conversion in vivo (Figure 1a–c). We therefore used Saf32, an antibody to the PrP N-terminus, to label nascent full-length PrPSc selectively. We used cryo-immunogold EM for subcellular localization of PrPSc in preference to other immuno-EM techniques, because of the superior membrane preservation, and the ability to localize PrPC as well as PrPSc at the subcellular level. This optimized our chances of detecting newly converted PrPSc. The Saf32 antibody labeled both PrPC and PrPSc, but PrPC labeling is disperse and its levels do not increase in prion-infected hippocampus, while those of total PrP (PrPC plus PrPSc) do increase (Godsave et al., 2008). Individual Saf32-labeled PrPC and PrPSc molecules were indistinguishable. However, at the three pre-clinical disease stages examined (65, 85, 99 dpi), clusters of cryo-immunogold EM labeling and bright foci of immunofluorescence were a good indicator of sites containing full-length PrPSc.
Nascent PrPSc is localized mainly on plasma membranes
Structures labeled preferentially by antibodies recognizing full-length, nascent PrPSc are candidate PrPC-to-PrPSc conversion sites. At all stages examined, most clustered Saf32 cryo-immunogold labeling indicating PrPSc was found on plasma membranes. This was either at the cell surface, often at sites of cell-to-cell contact, or on invaginations (Figure 6). Since the substrate for conversion, PrPC, is also found mostly on plasma membranes (Godsave et al., 2008), we suspect that this is the main site of PrPC-to-PrPSc conversion. Our data are based on one mouse model of prion disease. However, they are consistent with results from several previous immuno-EM studies of late pre-clinical and terminally diseased brain using other animals and prion strains (Jeffrey et al., 1992; Godsave et al., 2008; Ersdal et al., 2009; Jeffrey et al., 2011). Our data indicate further that closely apposed membranes may facilitate conversion or stabilize newly formed PrPSc. Our recent study of Peyer’s patches in mice orally inoculated with prions also strongly suggested that prion formation occurs on plasma membranes of follicular dendritic cells (Kujala et al., 2011). Furthermore, epitope-tagged PrPC was converted to PrPSc primarily on the plasma membrane of neuroblastoma cells exposed to exogenous PrPSc (Goold et al., 2011).
By contrast, studies using trafficking inhibitors in cultured cells provided evidence for PrPC-to-PrPSc conversion in recycling vesicles (Marijanovic et al., 2009). We have observed a proportion of Saf32 immunogold labeling in small vesicles resembling early endocytic or recycling vesicles. However, individual vesicles were generally not abundantly labeled, suggesting that they contained little nascent PrPSc and that endocytic vesicles are not the major site of conversion. Previously though, small vesicles showed the largest increases in R2 labeling density (gold per μm membrane) in hippocampus after prion infection (Godsave et al., 2008), possibly indicating trafficking of N-terminally truncated PrPSc.
PrPSc at intercellular junctions may indicate cell-to-cell spread
Several mechanisms for intercellular prion spread have been proposed, including transfer via membrane-bound subcellular particles such as exosomes, or membrane bridges (Fevrier et al., 2004; Gousset et al., 2009; Mattei et al., 2009). We saw evidence for exosomal release of PrP after transcytosis through gut epithelium in orally prion-inoculated mice (Kujala et al., 2011), but exocytosis of PrPSc was not apparent in hippocampus using either R2 (Godsave et al., 2008) or Saf32 antibodies. Other studies indicated that cells could become infected by contact with prion-infected cells, even if these were fixed (Kanu et al., 2002). We often observed Saf32 labeling on closely apposed plasma membranes and suspect that intercellular transfer of prions in the hippocampus may occur predominantly via cell-to-cell contact.
PrPSc does not target synapses specifically
Synapto-toxicity may be an important early event in several neurodegenerative diseases, including Alzheimer’s disease (Palop and Mucke, 2010; Schulz-Schaeffer, 2010). Synapse loss has also been observed long before the onset of overt clinical disease in stratum radiatum of prion-infected hippocampus. In mice intracerebrally infected with ME7 scrapie prions, decreases in synapse numbers were detected by 34% of the incubation period by quantitative electron microscopy, by which time abnormal PrP deposition was detectable in hippocampus by immunohistochemistry (Jeffrey et al., 2000; Siskova et al., 2009). Behavioural changes and a reduction in the level of a pre-synaptic marker were detected slightly later, but still clearly preceded the onset of neuronal cell loss (Cunningham et al., 2003). Electrophysiological changes also begin at an early pre-clinical stage in these mice (Chiti et al., 2006). The role that PrPSc plays in causing synapto-pathology is unclear, however. PrPSc has been described as co-localizing with synaptic markers using light microscopy (Siso et al., 2002; Kovacs et al., 2005) and immuno-EM (Fournier et al., 2000), and has been detected in synaptosomes (Bouzamondo-Bernstein et al., 2004). However, we and others did not find evidence for prions specifically targeting synapses in studies of hippocampus at late pre-clinical and terminal disease stages (Godsave et al., 2008; Ersdal et al., 2009). Here, we quantified immunogold EM labeling of nascent PrPSc in large areas spanning the stratum oriens, at several asymptomatic disease stages. Only 5% of clustered Saf32 labeling was clearly synapse-associated, with most of this in plasma membrane invaginations. Furthermore, many synapses with a degenerating axon terminal showed no Saf32 labeling clusters, either pre- or post-synaptically. A wider examination of R2 labeling at a late pre-clinical stage (104 dpi) was also consistent with this view (data not shown). It is possible that synapto-toxicity is mediated by a subpopulation of PrPSc not recognized by the antibodies used, or that prions cause a rapid removal of synapses. However, our results suggest that synapses are not the principal origin of prion pathology, but synapse integrity could be affected detrimentally by events at other locations.
PrPSc aggregation occurs on plasma membranes of both axons and dendrites
Electron microscopic evidence suggested that synapse loss or dysfunction is initiated axonally (Gray et al., 2009; Jeffrey et al., 2011), and axonal PrPSc has been described in brains of CJD cases using confocal laser scanning microscopy (Kovacs et al., 2005). However, several immuno-EM studies indicated that disease-associated PrP occurs preferentially on glial processes and dendrites (reviewed in (Jeffrey et al., 2011), where pathology including dendritic spine loss and development of varicosities also occurs before neuronal loss (Brown et al., 2001; Fuhrmann et al., 2007). Our results showed PrPSc on both axons and dendrites.
We also found examples of clustered Saf32 labeling close to both glutamatergic and non-glutamatergic terminals. Several studies reported that a subclass of GABAergic neurons is particularly vulnerable to neurodegeneration in prion disease (Ferrer et al., 1993; Guentchev et al., 1998; Guentchev et al., 1999). However, early loss of asymmetric glutamatergic synapses was found by quantitative EM analyses (Jeffrey et al., 2000; Siskova et al., 2009) and both glutamatergic and GABAergic terminals were affected in scrapie-infected hamster neocortex (Bouzamondo-Bernstein et al., 2004). Again, these results suggest that PrPSc does not target specific structures in the neuropil.
PrPSc-associated plasma membrane invaginations are an early feature of prion disease
Our results showed that plasma membrane invaginations with PrPSc aggregates in the fold are an early sign of prion pathology. They were not generally associated with other clear abnormalities. However, aggregation of a membrane-associated protein can have wide-ranging pathological effects, as illustrated by α-synuclein–associated toxicity in models of Parkinson’s disease (Auluck et al., 2010). Direct effects of PrPSc might include perturbed endocytosis, signaling, cell-to-cell contact, axonal transport, metal homeostasis, membrane trafficking, and membrane fluidity or potential (Crozet et al., 2008; Ermolayev et al., 2009; Ersdal et al., 2009; Dearmond and Bajsarowicz, 2010). Interestingly, prion infection of mice expressing PrP lacking a GPI anchor indicated that membrane association was necessary for maximal PrPSc toxicity. PrPSc amyloid built up in the brains of these mice, but they either showed no evidence of neurodegeneration or developed late-onset neurological dysfunction, depending on the PrP expression levels (Chesebro et al., 2005; Stöhr et al., 2011).
The cryo-immunogold EM results obtained in this study and previously (Godsave et al., 2008) show that subtle differences in the subcellular distributions of conformers of PrP can be revealed using antibodies to different PrP epitopes. Other neurodegenerative diseases involving protein aggregation are now thought to develop by a prion mechanism (Prusiner, 2012). Our approach may help to elucidate where misfolding occurs in these diseases.
In conclusion, our data indicate that prions form on plasma membranes in the neuropil, where they may elicit pathology with effects extending more distally. We suspect that intercellular prion spread occurs via cell-to-cell contact. Only a small proportion of PrPSc was found very close to synapses, indicating that synapses are not targeted directly by PrPSc in prion disease.
Acknowledgments
This work was supported by AntePrion EU FP6 LSHB-CT-2006-0190090 and Priority EU FP7 222887. We are grateful to Alex Rouvinski and Albert Taraboulos at the Hebrew University Hadassah Medical School in Jerusalem, and Pekka Kujala and other members of Peter Peters’ lab at NKI, for discussions and critical reading of the manuscript. We thank Nico Ong for help with EM photography, Diane Latawiec for assistance with tissue fixation and Hang Nguyen for editing the manuscript.
Footnotes
Disclosure statement: The authors declare no competing financial interests.
There are no actual or potential conflicts of interest for any of the authors in relation to this manuscript.
The data in this manuscript have not been previously published, have not been submitted elsewhere and will not be submitted elsewhere while under consideration at Neurobiology of Aging.
Appropriate approval and procedures were used concerning all animal experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Auluck PK, Caraveo G, Lindquist S. alpha-Synuclein: membrane interactions and toxicity in Parkinson’s disease. Annu Rev Cell Dev Biol. 2010;26:211–233. doi: 10.1146/annurev.cellbio.042308.113313. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Taraboulos A, Prusiner SB. Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem. 1992;267:16188–16199. [PubMed] [Google Scholar]
- Bouzamondo-Bernstein E, Hopkins SD, Spilman P, Uyehara-Lock J, Deering C, Safar J, Prusiner SB, Ralston HJ, 3rd, DeArmond SJ. The neurodegeneration sequence in prion diseases: evidence from functional, morphological and ultrastructural studies of the GABAergic system. J Neuropathol Exp Neurol. 2004;63:882–899. doi: 10.1093/jnen/63.8.882. [DOI] [PubMed] [Google Scholar]
- Brown D, Belichenko P, Sales J, Jeffrey M, Fraser JR. Early loss of dendritic spines in murine scrapie revealed by confocal analysis. Neuroreport. 2001;12:179–183. doi: 10.1097/00001756-200101220-00043. [DOI] [PubMed] [Google Scholar]
- Carlson GA, Westaway D, DeArmond SJ, Peterson-Torchia M, Prusiner SB. Primary structure of prion protein may modify scrapie isolate properties. Proc Natl Acad Sci U S A. 1989;86:7475–7479. doi: 10.1073/pnas.86.19.7475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, Raymond GJ. The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem. 1991;266:18217–18223. [PubMed] [Google Scholar]
- Caughey B, Raymond GJ, Ernst D, Race RE. N-terminal truncation of the scrapie-associated form of PrP by lysosomal protease(s): implications regarding the site of conversion of PrP to the protease-resistant state. J Virol. 1991;65:6597–6603. doi: 10.1128/jvi.65.12.6597-6603.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesebro B, Trifilo M, Race R, Meade-White K, Teng C, LaCasse R, Raymond L, Favara C, Baron G, Priola S, Caughey B, Masliah E, Oldstone M. Anchorless prion protein results in infectious amyloid disease without clinical scrapie. Science. 2005;308:1435–1439. doi: 10.1126/science.1110837. [DOI] [PubMed] [Google Scholar]
- Chiti Z, Knutsen OM, Betmouni S, Greene JR. An integrated, temporal study of the behavioural, electrophysiological and neuropathological consequences of murine prion disease. Neurobiol Dis. 2006;22:363–373. doi: 10.1016/j.nbd.2005.12.002. [DOI] [PubMed] [Google Scholar]
- Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3:a006833. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crozet C, Beranger F, Lehmann S. Cellular pathogenesis in prion diseases. Vet Res. 2008;39:44. doi: 10.1051/vetres:2008021. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Deacon R, Wells H, Boche D, Waters S, Diniz CP, Scott H, Rawlins JN, Perry VH. Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur J Neurosci. 2003;17:2147–2155. doi: 10.1046/j.1460-9568.2003.02662.x. [DOI] [PubMed] [Google Scholar]
- D’Castro L, Wenborn A, Gros N, Joiner S, Cronier S, Collinge J, Wadsworth JD. Isolation of proteinase K-sensitive prions using pronase E and phosphotungstic acid. PLoS One. 2010;5:e15679. doi: 10.1371/journal.pone.0015679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dearmond SJ, Bajsarowicz K. PrPSc accumulation in neuronal plasma membranes links Notch-1 activation to dendritic degeneration in prion diseases. Mol Neurodegener. 2010;5:6. doi: 10.1186/1750-1326-5-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermolayev V, Cathomen T, Merk J, Friedrich M, Hartig W, Harms GS, Klein MA, Flechsig E. Impaired axonal transport in motor neurons correlates with clinical prion disease. PLoS Pathog. 2009;5:e1000558. doi: 10.1371/journal.ppat.1000558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ersdal C, Goodsir CM, Simmons MM, McGovern G, Jeffrey M. Abnormal prion protein is associated with changes of plasma membranes and endocytosis in bovine spongiform encephalopathy (BSE)-affected cattle brains. Neuropathol Appl Neurobiol. 2009;35:259–271. doi: 10.1111/j.1365-2990.2008.00988.x. [DOI] [PubMed] [Google Scholar]
- Ferrer I, Casas R, Rivera R. Parvalbumin-immunoreactive cortical neurons in Creutzfeldt-Jakob disease. Ann Neurol. 1993;34:864–866. doi: 10.1002/ana.410340617. [DOI] [PubMed] [Google Scholar]
- Fevrier B, Vilette D, Archer F, Loew D, Faigle W, Vidal M, Laude H, Raposo G. Cells release prions in association with exosomes. Proc Natl Acad Sci U S A. 2004;101:9683–9688. doi: 10.1073/pnas.0308413101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fournier JG, Escaig-Haye F, Grigoriev V. Ultrastructural localization of prion proteins: physiological and pathological implications. Microsc Res Tech. 2000;50:76–88. doi: 10.1002/1097-0029(20000701)50:1<76::AID-JEMT11>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Mitteregger G, Kretzschmar H, Herms J. Dendritic pathology in prion disease starts at the synaptic spine. J Neurosci. 2007;27:6224–6233. doi: 10.1523/JNEUROSCI.5062-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godsave SF, Wille H, Kujala P, Latawiec D, DeArmond SJ, Serban A, Prusiner SB, Peters PJ. Cryo-immunogold electron microscopy for prions: toward identification of a conversion site. J Neurosci. 2008;28:12489–12499. doi: 10.1523/JNEUROSCI.4474-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goold R, Rabbanian S, Sutton L, Andre R, Arora P, Moonga J, Clarke AR, Schiavo G, Jat P, Collinge J, Tabrizi SJ. Rapid cell-surface prion protein conversion revealed using a novel cell system. Nat Commun. 2011;2:281. doi: 10.1038/ncomms1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J, Olivo-Marin JC, Mannel D, Zurzolo C. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol. 2009;11:328–336. doi: 10.1038/ncb1841. [DOI] [PubMed] [Google Scholar]
- Gray BC, Siskova Z, Perry VH, O’Connor V. Selective presynaptic degeneration in the synaptopathy associated with ME7-induced hippocampal pathology. Neurobiol Dis. 2009;35:63–74. doi: 10.1016/j.nbd.2009.04.001. [DOI] [PubMed] [Google Scholar]
- Guentchev M, Groschup MH, Kordek R, Liberski PP, Budka H. Severe, early and selective loss of a subpopulation of GABAergic inhibitory neurons in experimental transmissible spongiform encephalopathies. Brain Pathol. 1998;8:615–623. doi: 10.1111/j.1750-3639.1998.tb00188.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guentchev M, Wanschitz J, Voigtlander T, Flicker H, Budka H. Selective neuronal vulnerability in human prion diseases. Fatal familial insomnia differs from other types of prion diseases. Am J Pathol. 1999;155:1453–1457. doi: 10.1016/S0002-9440(10)65459-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenther K, Deacon RM, Perry VH, Rawlins JN. Early behavioural changes in scrapie-affected mice and the influence of dapsone. Eur J Neurosci. 2001;14:401–409. doi: 10.1046/j.0953-816x.2001.01645.x. [DOI] [PubMed] [Google Scholar]
- Hwang D, Lee IY, Yoo H, Gehlenborg N, Cho JH, Petritis B, Baxter D, Pitstick R, Young R, Spicer D, Price ND, Hohmann JG, Dearmond SJ, Carlson GA, Hood LE. A systems approach to prion disease. Mol Syst Biol. 2009;5:252. doi: 10.1038/msb.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey M, McGovern G, Siso S, Gonzalez L. Cellular and sub-cellular pathology of animal prion diseases: relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathol. 2011;121:113–134. doi: 10.1007/s00401-010-0700-3. [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Scott JR, Halliday WG. Infection specific prion protein (PrP) accumulates on neuronal plasmalemma in scrapie infected mice. Neurosci Lett. 1992;147:106–109. doi: 10.1016/0304-3940(92)90785-6. [DOI] [PubMed] [Google Scholar]
- Jeffrey M, Halliday WG, Bell J, Johnston AR, MacLeod NK, Ingham C, Sayers AR, Brown DA, Fraser JR. Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol Appl Neurobiol. 2000;26:41–54. doi: 10.1046/j.1365-2990.2000.00216.x. [DOI] [PubMed] [Google Scholar]
- Kanu N, Imokawa Y, Drechsel DN, Williamson RA, Birkett CR, Bostock CJ, Brockes JP. Transfer of scrapie prion infectivity by cell contact in culture. Curr Biol. 2002;12:523–530. doi: 10.1016/s0960-9822(02)00722-4. [DOI] [PubMed] [Google Scholar]
- Kovacs GG, Preusser M, Strohschneider M, Budka H. Subcellular localization of disease-associated prion protein in the human brain. Am J Pathol. 2005;166:287–294. doi: 10.1016/S0002-9440(10)62252-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kremer JR, Mastronarde DN, McIntosh JR. Computer visualization of three-dimensional image data using IMOD. J Struct Biol. 1996;116:71–76. doi: 10.1006/jsbi.1996.0013. [DOI] [PubMed] [Google Scholar]
- Kujala P, Raymond CR, Romeijn M, Godsave SF, van Kasteren SI, Wille H, Prusiner SB, Mabbott NA, Peters PJ. Prion uptake in the gut: identification of the first uptake and replication sites. PLoS Pathog. 2011;7:e1002449. doi: 10.1371/journal.ppat.1002449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberski PP, Streichenberger N, Giraud P, Soutrenon M, Meyronnet D, Sikorska B, Kopp N. Ultrastructural pathology of prion diseases revisited: brain biopsy studies. Neuropathol Appl Neurobiol. 2005;31:88–96. doi: 10.1111/j.1365-2990.2004.00595.x. [DOI] [PubMed] [Google Scholar]
- Mallucci GR. Prion neurodegeneration: starts and stops at the synapse. Prion. 2009;3:195–201. doi: 10.4161/pri.3.4.9981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marijanovic Z, Caputo A, Campana V, Zurzolo C. Identification of an intracellular site of prion conversion. PLoS Pathog. 2009;5:e1000426. doi: 10.1371/journal.ppat.1000426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters CL, Rohwer RG, Franko MC, Brown P, Gajdusek DC. The sequential development of spongiform change and gliosis of scrapie in the golden Syrian hamster. J Neuropathol Exp Neurol. 1984;43:242–252. doi: 10.1097/00005072-198405000-00003. [DOI] [PubMed] [Google Scholar]
- Mattei V, Barenco MG, Tasciotti V, Garofalo T, Longo A, Boller K, Lower J, Misasi R, Montrasio F, Sorice M. Paracrine diffusion of PrP(C) and propagation of prion infectivity by plasma membrane-derived microvesicles. PLoS One. 2009;4:e5057. doi: 10.1371/journal.pone.0005057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mironov A, Jr, Latawiec D, Wille H, Bouzamondo-Bernstein E, Legname G, Williamson RA, Burton D, DeArmond SJ, Prusiner SB, Peters PJ. Cytosolic prion protein in neurons. J Neurosci. 2003;23:7183–7193. doi: 10.1523/JNEUROSCI.23-18-07183.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010;13:812–818. doi: 10.1038/nn.2583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastrana MA, Sajnani G, Onisko B, Castilla J, Morales R, Soto C, Requena JR. Isolation and Characterization of a Proteinase K-Sensitive PrP(Sc) Fraction. Biochemistry. 2006;45:15710–15717. doi: 10.1021/bi0615442. [DOI] [PubMed] [Google Scholar]
- Peretz D, Williamson RA, Matsunaga Y, Serban H, Pinilla C, Bastidas RB, Rozenshteyn R, James TL, Houghten RA, Cohen FE, Prusiner SB, Burton DR. A conformational transition at the N terminus of the prion protein features in formation of the scrapie isoform. J Mol Biol. 1997;273:614–622. doi: 10.1006/jmbi.1997.1328. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Cell biology. A unifying role for prions in neurodegenerative diseases. Science. 2012;336:1511–1513. doi: 10.1126/science.1222951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB, Safar J, DeArmond SJ. Bioassays of prions. In: Prusiner SB, editor. Prion Biology and Diseases. 2. New York: Cold Spring Harbor Laboratory Press; 2004. pp. 143–186. [Google Scholar]
- Safar J, Wille H, Itri V, Groth D, Serban H, Torchia M, Cohen FE, Prusiner SB. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4:1157–1165. doi: 10.1038/2654. [DOI] [PubMed] [Google Scholar]
- Safar JG, Kellings K, Serban A, Groth D, Cleaver JE, Prusiner SB, Riesner D. Search for a prion-specific nucleic acid. J Virol. 2005a;79:10796–10806. doi: 10.1128/JVI.79.16.10796-10806.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar JG, DeArmond SJ, Kociuba K, Deering C, Didorenko S, Bouzamondo-Bernstein E, Prusiner SB, Tremblay P. Prion clearance in bigenic mice. J Gen Virol. 2005b;86:2913–2923. doi: 10.1099/vir.0.80947-0. [DOI] [PubMed] [Google Scholar]
- Safar JG, Scott M, Monaghan J, Deering C, Didorenko S, Vergara J, Ball H, Legname G, Leclerc E, Solforosi L, Serban H, Groth D, Burton DR, Prusiner SB, Williamson RA. Measuring prions causing bovine spongiform encephalopathy or chronic wasting disease by immunoassays and transgenic mice. Nat Biotechnol. 2002;20:1147–1150. doi: 10.1038/nbt748. [DOI] [PubMed] [Google Scholar]
- Sajnani G, Silva CJ, Ramos A, Pastrana MA, Onisko BC, Erickson ML, Antaki EM, Dynin I, Vazquez-Fernandez E, Sigurdson CJ, Carter JM, Requena JR. PK-sensitive PrP is infectious and shares basic structural features with PK-resistant PrP. PLoS Pathog. 2012;8:e1002547. doi: 10.1371/journal.ppat.1002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz-Schaeffer WJ. The synaptic pathology of alpha-synuclein aggregation in dementia with Lewy bodies, Parkinson’s disease and Parkinson’s disease dementia. Acta Neuropathol. 2010;120:131–143. doi: 10.1007/s00401-010-0711-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siskova Z, Page A, O’Connor V, Perry VH. Degenerating synaptic boutons in prion disease: microglia activation without synaptic stripping. Am J Pathol. 2009;175:1610–1621. doi: 10.2353/ajpath.2009.090372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siso S, Puig B, Varea R, Vidal E, Acin C, Prinz M, Montrasio F, Badiola J, Aguzzi A, Pumarola M, Ferrer I. Abnormal synaptic protein expression and cell death in murine scrapie. Acta Neuropathol. 2002;103:615–626. doi: 10.1007/s00401-001-0512-6. [DOI] [PubMed] [Google Scholar]
- Stanker LH, Serban AV, Cleveland E, Hnasko R, Lemus A, Safar J, DeArmond SJ, Prusiner SB. Conformation-dependent high-affinity monoclonal antibodies to prion proteins. J Immunol. 2010;185:729–737. doi: 10.4049/jimmunol.0902930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöhr J, Watts JC, Legname G, Oehler A, Lemus A, Nguyen HO, Sussman J, Wille H, DeArmond SJ, Prusiner SB, Giles K. Spontaneous generation of anchorless prions in transgenic mice. Proc Natl Acad Sci U S A. 2011;108:21223–21228. doi: 10.1073/pnas.1117827108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamguney G, Francis KP, Giles K, Lemus A, DeArmond SJ, Prusiner SB. Measuring prions by bioluminescence imaging. Proc Natl Acad Sci U S A. 2009;106:15002–15006. doi: 10.1073/pnas.0907339106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taraboulos A, Raeber AJ, Borchelt DR, Serban D, Prusiner SB. Synthesis and trafficking of prion proteins in cultured cells. Mol Biol Cell. 1992;3:851–863. doi: 10.1091/mbc.3.8.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wille H, Prusiner SB. Ultrastructural studies on scrapie prion protein crystals obtained from reverse micellar solutions. Biophys J. 1999;76:1048–1062. doi: 10.1016/S0006-3495(99)77270-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williamson RA, Peretz D, Pinilla C, Ball H, Bastidas RB, Rozenshteyn R, Houghten RA, Prusiner SB, Burton DR. Mapping the prion protein using recombinant antibodies. J Virol. 1998;72:9413–9418. doi: 10.1128/jvi.72.11.9413-9418.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]