

Abstract
Hereditary motor and sensory disorders of the peripheral nerve form one of the most common groups of human genetic diseases collectively called Charcot–Marie–Tooth (CMT) neuropathy. Using linkage analysis in a three generation kindred, we have mapped a new locus for X-linked dominant CMT to chromosome Xp22.11. A microsatellite scan of the X chromosome established significant linkage to several markers including DXS993 (Zmax = 3.16; θ = 0.05). Extended haplotype analysis refined the linkage region to a 1.43-Mb interval flanked by markers DXS7110 and DXS8027. Whole exome sequencing identified a missense mutation c.G473A (p.R158H) in the pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) gene. The change localized within the 1.43-Mb linkage interval, segregated with the affected phenotype and was excluded in ethnically matched control chromosomes. PDK3 is one of the four isoenzymes regulating the pyruvate dehydrogenase complex (PDC), by reversible phosphorylation, and is a nuclear-coded protein located in the mitochondrial matrix. PDC catalyzes the oxidative decarboxylation of pyruvate to acetyl CoA and is a key enzyme linking glycolysis to the energy-producing Krebs cycle and lipogenic pathways. We found that the R158H mutation confers enzyme hyperactivity and binds with stronger affinity than the wild-type to the inner-lipoyl (L2) domain of the E2p chain of PDC. Our findings suggest a reduced pyruvate flux due to R158H mutant PDK3-mediated hyper-phosphorylation of the PDC as the underlying pathogenic cause of peripheral neuropathy. The results highlight an important causative link between peripheral nerve degeneration and an essential bioenergetic or biosynthetic pathway required for the maintenance of peripheral nerves.
INTRODUCTION
Charcot–Marie–Tooth (CMT) neuropathy is a group of degenerative disorders of human peripheral nerve affecting both motor and sensory neurons. This genetically and clinically heterogeneous syndrome is the most commonly inherited neuromuscular disorder affecting 1 in 2500 people (1). The CMT phenotype is characterized by progressive weakness and atrophy of distal muscles, high arched feet (pes cavus) and loss of deep tendon reflexes. X-linked CMT (CMTX) accounts for up to 15% of CMT. Currently, there are five known loci reported: CMTX1 (OMIM 304040), CMTX2 (OMIM 302801), CMTX3 (OMIM 302802), CMTX4 (Cowchock syndrome; OMIM 310490) and CMTX5 (OMIM 311070). CMTX1 is X-linked dominant and the most common form of CMTX. It is caused by mutations in gap junction beta 1/connexin32 (GJB1/Cx32) located on chromosome Xq13.1 (2). The loci for recessive CMTX (CMTX2 and CMTX3) were mapped to chromosome Xp22.2 and Xq26-28, respectively. Mental retardation was present in the family mapping to the CMTX2 locus and spasticity with pyramidal signs was observed in families mapping the CMTX3 locus (3). Cowchock syndrome (CMTX4) was mapped to chromosome Xq24-26 and is characterized by early onset polyneuropathy sensorineural hearing loss and mental retardation (4,5). The CMTX5 locus mapping to Xq21.32-q24 is associated with deafness and optic neuropathy and is caused by mutations in the phosphoribosyl pyrophosphate synthetase 1 gene (PRPS1) (6).
A proven approach for identifying genes is the use of whole exome sequencing (WES). The seminal papers describing the strategy of targeting the protein coding regions for exome sequencing is a testament to the power of this approach for identifying mutations in Mendelian disorders (7,8). By using a combination linkage analysis and exome sequencing, this approach is expediting the discovery of genes for CMT. Recent success for using exome analysis as a strategy for CMT gene discovery has provided a proof of principle for CMT (9) and the report of autosomal-dominant mutations in the ubiquitin ligase gene LRSAM1 (10) and the cytoplasmic dynein heavy chain 1, DYNC1H1 (11). We previously reported a family with X-linked dominant CMT that was negative for mutations in the coding region of the GJB1/Cx32 gene (12). In this study, we have used a combination of linkage analysis and WES to map a new locus for CMTX in this family and identify the causative gene.
RESULTS
Clinical history and neurologic examination
Nineteen members of the kindred underwent a clinical history and neurologic examination and 16 had neurophysiologic studies (Fig. 1A) as described previously (13). Two subjects were not available for clinical evaluation (II:3 and III:11). Five affected males and eight carrier females from two generations were identified (males mean age 33.0 ± 17.3 years; females 43.9 ± 16.7 years at assessment). Affected males were more severely affected than females (mean Charcot-Marie-Tooth Neuropathy Score, CMTNS, 11.5 ± 3.3 in males, 2.0 ± 2.2 in females) (14). Males had symptom onset in the first 13 years of life (mean 6.7 ± 4.5 years) with foot deformity and/or gait abnormalities. One male had mild delay in motor development, walking at 19 months of age. Three had foot/ankle surgery in their teens. Disease progression was gradual. Males in the older generation (aged 49 and 54 years) had greater disability than those in the younger generation. All remained ambulant and engaged in employment or study. Carrier females were asymptomatic or very mildly affected. Two developed mild foot deformity in their mid-teens, two noted hand tremor in their early forties and four were asymptomatic (although two of these had hand tremor on neurologic examination). Clinical features are summarized in Table 1 and Supplementary Material, Table S1.
Figure 1.

(A) Pedigree structure of X-linked (CMTX6) kindred. Squares indicate males, circles indicate females. Solid symbols denote affected individuals and open symbols denote unaffected family members. Internal dots indicate obligate gene carriers. Diagonal lines through symbols denote deceased individuals. The letters below individual identifiers denote the following: ‘c’ clinical examination; ‘g’ genotype examination; ‘c/e’ clinical and electrophysiological examination; ‘c/e/g’ clinical, electrophysiological and genotype examination. (B) Clinical findings in X-linked CMT (CMTX6). Affected male IV:2 aged 15 years, with mild lower limb muscle wasting (top left panel), and mild wasting of the hand intrinsic muscles (bottom panel). Affected male III:15 aged 49 years, with severe muscle wasting below the knees, and pes cavus (top right panel).
Table 1.
Clinical features of pedigree
| Subject | Age at onset | Age at visit | Foot deformity | Age at foot/ankle surgery | LL wasting level, severity | Abnormal gait | Difficulty heel walking | Ankle reflexes | LL pinprick sensation | LL vibration sensation | Ankle DFa | Ankle PFa | Ankle eversiona | Hand intrinsic wasting | Hand tremor | CMTNS |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Affected males | ||||||||||||||||
| IV:5 | 9 | 21.2 | Cavus | 15 years | Mid calf, moderate | Yes | Some | Absent | Reduced | Reduced | 3 | 4 | 3 | None | yes | 12 |
| IV:6 | 1.5 | 20.0 | Cavus | 14 years | Mid calf, moderate | Yes | Yes | Absent | Normal | Reduced | 4.5 | 5 | 4.5 | None | yes | 7 |
| III:15 | 6 | 49.2 | Cavus | None | Below knee, severe | Yes | Yes | Absent | Reduced | Reduced | 0 | 3.5 | 1 | Mild | no | np |
| IV:2 | 13 | 19.9 | Cavus | 17 years | Mid calf, moderate | Yes | Yes | Absent | Reduced | Reduced | 3 | 4.5 | 3.5 | Mild | yes | 12 |
| III:12 | 4 | 54.4 | Cavus | None | Mid calf, severe | Yes | Yes | Absent | Reduced | Reduced | 0 | 0 | 0 | None | yes | 15 |
| Carrier females | ||||||||||||||||
| III:10 | 13 | 56.8 | Cavus | 13 years | Foot, moderate | Yes | No | Normal | Reduced | Reduced | 5 | 5 | 4 | Mild | yes | 6 |
| IV:7 | ASx | 16.8 | None | None | None | No | No | Normal | Normal | Normal | 5 | 5 | 5 | None | yes | np |
| III:8 | 40 | 49.5 | None | None | Foot, mild | No | Yes | Absent | Normal | Reduced | 5 | 5 | 4.5 | Mild | yes | 3 |
| III:3 | 15 | 50.1 | Cavus | None | Foot, mild | No | No | Hyporeflexic | Normal | Normal | 5 | 5 | 5 | Mild | yes | 1 |
| III:5 | ASx | 59.5 | None | None | None | No | No | Absent | Normal | Normal | 5 | 5 | 5 | Mild | no | 0 |
| IV:8 | ASx | 18.1 | None | None | None | No | No | Normal | Normal | Normal | 5 | 5 | 5 | None | yes | np |
| III:4 | 41 | 51.6 | None | None | None | No | No | Normal | Normal | Normal | 5 | 5 | 5 | Mild | yes | 1 |
| III:14 | ASx | 48.7 | None | None | Foot, mild | No | No | Absent | Normal | Normal | 5 | 5 | 4.5 | Mild | no | 1 |
All males had progressive moderate-to-severe wasting below the knees (Fig. 1B), a steppage gait, absent ankle reflexes and distal lower limb weakness and sensory abnormalities. Although routine manual muscle testing revealed minimal weakness of the hand intrinsic muscles, hand dynamometry detected reduced grip strength in all affected males (mean z-score −5.0). Hand dexterity was also reduced as measured by the 9-hole peg test (Supplementary Material, Table S1). Cranial nerve abnormalities and proximal weakness were not present in any family members. All carrier females in the older generation had subtle abnormalities on neurologic examination, including mild wasting of hand intrinsic muscles, a mild postural hand tremor and absent or reduced ankle reflexes. Two teenage obligate carrier females (IV:7 and IV:8) had a normal neurologic examination apart from a very mild postural hand tremor. Hand-held dynamometry of distal muscle strength was more sensitive than manual muscle testing, indicating reduced grip strength and ankle dorsiflexion and plantar flexion strength in 67, 67 and 100%, respectively, of the six carrier females tested. Hand dexterity was also reduced in half of the females tested (Supplementary Material, Table S1).
Electromyography findings are presented (Table 2). The neurophysiologic phenotype is a predominantly axonal motor and sensory polyneuropathy with variable mild conduction slowing. Males had moderate-to-severe abnormalities, with loss of lower extremity responses after the age of 14–15 years. Three carrier females had a mild sensorimotor axonal neuropathy. Three other carrier females (III:3, III:4, III:5) had borderline nerve conduction studies with mild neurogenic changes on electromyography, while two obligate carrier females with minimal findings on neurologic examination declined neurophysiologic testing.
Table 2.
Neurophysiologic findings
| |
Motor |
Sensory |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Median |
Ulnar |
Tibial |
Common peroneal |
Median |
Ulnar |
Sural |
Superfical peroneal |
||||||||||
| Subject | Age | CMAP | MNCV | CMAP | MNCV | CMAP | MNCV | CMAP | MNCV | SNAP | SNCV | SNAP | SNCV | SNAP | SNCV | SNAP | SNCV |
| IV:5 | 21.2 | 4.1 | 61 | 8.1 | 47.7 | 0 | 0 | 0 | 0 | 0 | 0 | ||||||
| IV:5 | 14.1 | 6.5 | 47 | 8.9 | 51.9 | 0 | 0 | 0 | 0 | ND | ND | ||||||
| IV:6 | 20.0 | 6.9 | 59.8 | 6.3 | 53.3 | 0 | 0 | 0 | 0 | 0 | ND | ||||||
| IV:6 | 11.6 | 9.4 | 57.6 | 9.2 | 63.3 | 2.8 | 43.8 | 2.2 | 43.5 | 19.5 | 58 | 8.2 | 60.2 | 0 | ND | ||
| IV:2 | 19.9 | 7.1 | 49 | 6.7 | 52 | 0 | 0 | 0 | 0 | 0 | 0 | ||||||
| IV:2a | 15.8 | 7 | 54 | 5.8 | 47.7 | 0 | 0 | 0 | NDb | 0 | 0 | ||||||
| III:12a | 54.4 | 3.5 | 38.6 | 4.5 | 39 | 0 | 0 | 0 | 0 | 0 | 0 | ||||||
| III:10 | 56.8 | 13.1 | 54.8 | 8.7 | 58.1 | 3.6 | NR | 3.3 | 43.8 | 3.4 | 44.3 | 11 | 59.5 | 0 | 0 | ||
| III:10 | 49.8 | 12.1 | 54.8 | 11.9 | 62.8 | 0.6 | 44.9 | ND | ND | ND | ND | ND | |||||
| III:8 | 49.5 | 8.4 | 43 | 7.5 | 51.2 | 7.9 | 44.6 | 3.3 | 50.8 | 0 | 11 | 47.7 | 0 | 0 | |||
| III:3a | 50.1 | 6.9 | 63.1 | 11 | 55.2 | 16 | 59.1 | 5.7 | 58 | 9.2 | 76.2 | 15 | 68.6 | 14 | 52.1 | 11 | 63.4 |
| III:4a | 51.6 | 6.4 | 59.1 | 12.5 | 63.6 | 14.1 | 55.2 | 4.5 | 58 | 9.7 | 55.8 | 14 | 54.3 | 8.3 | 48 | 5.9 | 47.8 |
| III:5a | 59.5 | 5 | 47.2 | 10.7 | 63.1 | 14.9 | NR | 5.9 | 48.3 | 9.7 | 74 | 23 | 62.9 | 12 | 50 | 1.9 | 40.7 |
| III:14 | 48.7 | 5 | 61.8 | 12 | 58.6 | 14.4 | NR | 2.3 | 73.2 | 0 | 14 | 65.9 | 0 | 0 | |||
Abnormal values are in bold type.
Age, age at nerve conduction studies in years; CMAP, compound muscle action potential, amplitude in mV; MNCV = motor nerve conduction velocity in m/sec; ND, not done; NR, not recorded; Pt =patient; SNAP = sensory nerve action potential, amplitude in μV; SNCV = sensory nerve conduction velocity in m/sec. Sensory responses are antidromic.
aElectromyography findings.
IV:2 aged 15.8 years—Tibialis anterior–fibrillation potentials, moderate neurogenic activation pattern with polyphasic high amplitude motor unit action potentials.
III:12—Tibialis anterior–no spontaneous activity. No units under voluntary control recorded.
III:3, III:4, III:5—Tibialis anterior—Mildly neurogenic activation pattern, with high amplitude motor unit action potentials.
bRadial sensory response present: SNAP 4.1 μV (normal > 10).
Sound detection thresholds were within normal limits for all female participants and three of the four males. One male had moderate sensorineural hearing loss. On auditory brainstem response (ABR) assessment all female carriers had normal conduction velocities (ABR wave I–V interpeak latency) bilaterally (mean latency: 4.2 ± 0.2 ms). In contrast, affected males showed significantly impaired neural conduction (mean: 4.8 ± 0.5 ms). Interpeak latencies for three of four males were prolonged, with most of the delay between waves I–III, indicative of demyelinating changes in the auditory brainstem peripheral to the cochlear nucleus (15). There was a trend towards lower ABR wave V amplitudes in the male subjects (V/I ratio: M: 1.4 ± 0.3; F: 2.0 ± 0.7, P = 0.08), consistent with an axonopathy (16).
Linkage analysis
A total of 21 individuals were available for genotyping including 7 affected males and 7 obligate female carriers (Fig. 1A). The proband (IV:5) had previously been tested and found negative for mutations in the coding region of the GJB1/Cx32 gene (12). In this study, we sequenced the 5′ UTR and promoter region of the GJB1/Cx32 gene using previously described methods (17) and no pathogenic variants were identified. We then performed an in house X chromosome scan with 31 microsatellite markers using established protocols (18). Significant two-point LOD scores (Fig. 2A and Supplementary Material, Table S2) were obtained for the markers DXS1214 (Z = 2.3; θ = 0.0), DXS1068 (Z = 2.13; θ = 0.1) and DXS993 (Z = 3.16; θ = 0.05). The markers showing significant LOD scores did not correspond to chromosomal regions of the known CMTX loci (Fig. 2A). We then tested additional markers (DXS7110, DXS8027 and DXS1003) to further refine the CMTX6 locus. Significant LOD scores for DXS7110 and DXS8027 confirmed the linkage region to Xp22.11 (Supplementary Material, Table S2). A haplotype segregating with the disease in affected males and carrier females was observed between the markers DXS1226 and DXS1003. Two affected males and three obligate carrier females showed a recombination. The affected males (IV:5 and III:15) and obligate female carriers (III:10 and IV:7) showed a recombination between the markers DXS7110 and DXS8027 (Fig. 2B). The obligate female carriers (III:3 and III:4) carried the haplotype spanning the markers DXS7110 and DXS8027. The combined information from all recombinant individuals refined the CMTX6 locus to a 1.43-Mb interval flanked by the markers DXS7110 and DXS8027.
Figure 2.

(A) Maximum LOD score obtained for each marker tested in X chromosome scan plotted against the location (Mb) of each marker. Markers showing significant LOD scores (DXS1214, DXS1068 and DXS993) are shown. The location axis is orientated from Xpter (left) to Xqter (right). The known CMTX loci are shown in relation to their location along the X chromosome. (B) Extended haplotype analysis of markers from chromosome Xp22.11. The haplotype segregating with the disease is boxed. The markers are ordered from the telomere (top) to the centromere (bottom) of chromosome Xp. The flanking markers defining the CMTX6 1.43-Mb interval are DXS7110 and DXS8027.
Exome sequencing identifies a PDK3 missense mutation
WES was performed on four individuals (two affected males: IV:5 and III:11; one normal male IV:4 and a married in male III:9) from the family. Our strategy for WES variant filtering (Fig. 3A) identified a single non-synonymous base change at position chrX:24521596 (hg19). The variant mapped within the 1.43-Mb CMTX6 interval and occurred in the pyruvate dehydrogenase kinase isoenzyme 3 gene (PDK3 [MIM602526; Genbank accession number NM_001142386]). A transition mutation c.G473A in exon 4 predicted a p.R158H amino acid substitution. The haplotype data showed the proximal flanking marker (DXS8027) is intragenic to the PDK3 gene (located in the intron between exons 4 and 5) and is 15.15 kb downstream of the exonic PDK3 variant. A total of 16 genes, including the PDK3 gene, map within the interval and were captured with the Illumina TrueSeq Capture Kit. We assessed the coverage of all targeted coding bases for genes in the CMTX6 interval (including PDK3) and determined an average coverage of 99% at 1X reads to 85.8% at 10X reads for the individuals sequenced (Supplementary Material, Table S3). To confirm the PDK3 missense mutation, Sanger sequencing was performed to validate the variant in an affected male, a female obligate carrier and an unaffected individual (Fig. 3B). The DNA melt profiles of these individuals were then used as positive controls for phenotype grouping and segregation analysis with established protocols for a high resolution melt (HRM) genotyping assay (18). Full segregation of the variant with the affected phenotype was demonstrated and the variant was excluded in 1200 ethnically matched control chromosomes (Supplementary Material, Fig. S1A). The variant occurred at a highly conserved residue in the PDK3 protein (Supplementary Material, Fig. S1B) and the missense mutation is predicted to be damaging by both SIFT (19) and PolyPhen2 (20). We also performed two-point linkage analysis of the c.G473A mutation with an ‘A’ allele frequency of 1/1200 (based on the exclusion in control chromosomes). A maximum LOD score of 5.41 at zero recombination (Supplementary Material, Table S2) demonstrated highly significant linkage to the CMTX6 locus.
Figure 3.

(A) Filtering strategy applied to variants and indels detected for individuals undergoing WES. Solid squares denote affected males; open squares denote unaffected males. (B) Sequence traces for an affected male, carrier female and normal male for p.R158H. An asterisk denotes the base position resulting in the missense mutation that segregates with CMT in the family. The GenBank sequences NM_001142386 and NP_001135858.1 were used as the reference sequences for PDK3 cDNA and PDK3 protein respectively. The mutation was designated on the basis of numbering the A in the ATG translation initiation site as +1.
Screening for PDK3 mutations
To investigate the frequency of mutations in the PDK3 gene, 62 families with probable CMTX that had been excluded for the CMT1A duplication and GJB1 mutations underwent PDK3 gene screening by HRM analysis. Variants c.341A>C (p.K114T) and c.1001A>C (p.Y334S) in two unrelated families were identified. Both variants are likely to be rare polymorphisms as there was no segregation with the affected phenotype in the respective families (data not shown). Our findings suggest that PDK3 gene mutations may account for 1–2% of cases (1 in 63 families) in our cohort. Screening more CMTX families to identify additional mutations would further validate PDK3 as a causative gene for CMTX.
Pyruvate dehydrogenase kinase 3 (PDK3) isoenzyme
Pyruvate dehydrogenase kinase isoenzyme 3 (PDK3) is one of the four PDK isoenzymes, which negatively regulates the activity of pyruvate dehydrogenase complex (PDC) by reversible phosphorylation (21). In mitochondria, PDC catalyzes the oxidative decarboxylation of pyruvate to give rise to acetyl-CoA, linking glycolysis to the energy-producing Krebs cycle and lipogenic pathways. The human PDC is a 9.5 × 106-Da macromolecular machine organized around a structural core formed by 60 combined transacetylase (E2p) and E3-binding protein (E3BP) subunits, referred to as the E2p/E3BP core. To the 60-meric E2p/E3BP core, multiple copies of pyruvate dehydrogenase (E1p), dihydrolipoamide dehydrogenase (E3), pyruvate dehydrogenase kinase (PDK) and pyruvate dehydrogenase phosphatase (PDP) are attached (22). The phosphorylation of specific serine residues in E1p by PDK results in the inactivation of PDC, whereas dephosphorylation by PDP restores PDC activity (21). PDK isoenzymes exhibit tissue-specific expression; PDK1 is detected in heart, pancreatic islets and skeletal muscles; PDK2 is expressed in all tissues; PDK3 is present in testes, kidney and brain; and PDK4 is abundant in heart, skeletal muscle, kidney and pancreatic islets (23). Using commercially available cDNA libraries and published primers (24) we have also shown expression of PDK3 in human spinal cord, skeletal muscle, foetal brain and confirmed expression in adult whole brain (Supplementary Material, Fig. S2).
The dimeric forms of PDKs are the biologically functional entities (23,25,26) PDK dimers are recruited to the PDC by preferentially binding to the inner-lipoyl (L2) domain of the E2p chain (27). Binding of L2 to PDKs requires the covalently attached lipoyl group at the lysine 173 residue of L2 (28). The activity of PDKs is stimulated upon binding to E2p, but the response to the binding of different isoforms to isolated L2 differs. Both PDK2 and PDK3 are robustly activated by the E2p/E3BP core; and the bulk of this activation in PDK3 (but not PDK2) can be achieved using isolated L2 (25,29).
The gain-of-function R158H mutation confers hyperactivity in PDK3
To determine effects of the R158H missense mutation on PDK3 activity, both wild-type and the mutant PDK3 were expressed as maltose-binding protein (MBP) fusions. The presence of the N-terminal MBP moiety in the MBP-PDK3 protein is essential to maintain the insoluble PDK3 in solution. Kinase activity was assayed without (basal activity) and with (+E2p/E3BP) the 60-meric core of PDC (30). When E1p was used as substrate, R158H PDK3 is ∼5-fold more active than the wild-type kinase in either basal activity or E2p/E3BP core-enhanced activity (Fig. 4). Both wild-type and R158H PDK3 kinase activity assayed in the presence of E2p/E3BP showed similar 9- to 10-fold stimulation over basal activity. The results indicate the R158H mutation identified is a gain-of-function mutation that results in hyperactivity in PDK3.
Figure 4.

Enzyme activity of wild-type and mutant R158H PDK3 assayed with E1p as substrate. Recombinant human PDK3 with its N-terminus fused to maltose-binding protein (MBP-PDK3) were expressed and purified. Both basal activity without the E2p/E3BP core of PDC (in open bars) and E2p/E3BP core-enhanced PDK3 activity (+E2pE3BP, in shaded bar) were measured. Enzyme activity for the N120H/D121N PDK3 double mutant was assayed with E1p and measured under basal (open bars) and core enhanced (shaded bars) conditions.
PDK activity is inhibited by pyruvate, the α-keto acid substrate of PDC, and provides a feed-forward mechanism for pyruvate oxidation through an increased PDC flux (31). Table 3 shows that wild-type PDK2 and PDK4 are sensitive to pyruvate inhibition, with half maximal inhibition concentrations (IC50) of 1.0 and 0.5 mm, respectively. In contrast, both wild-type and R158H PDK3 exhibit similar IC50 values of 131 and 141 mm, respectively, which are two orders-of-magnitude higher than those for PDK2 and PDK4. The results confirm that PDK3 is least sensitive to inhibition by pyruvate or its analog dichloroacetate among the four PDK isoforms (23).
Table 3.
Half maximal concentrations for inhibition of wild-type and mutant PDK isoforms by pyruvate
| PDK isoforms | PDK3 |
PDK2 | PDK4 | |
|---|---|---|---|---|
| WT | R158H | WT | WT | |
| IC50, mm | 131 | 141 | 1.0 | 0.5 |
Wild-type and mutant PDK isoforms were assayed for E2p/E3BP-enhanced kinase activity (in triplicate) with E1p as substrate in the presence of increasing pyruvate concentrations. Half-maximal concentrations (IC50) for inhibition of PDK by pyruvate were obtained by plotting % PDK activity against increasing log concentrations of pyruvate.
Altered binding affinities of R158H PDK3 for nucleotides and L2
We also assessed the binding affinities of PDK3 R158H to nucleotides and the inner lipoyl domain (L2) of the E2 subunit. The L2 domain is the anchoring site for PDK3 on the PDC scaffold. The binding of ATP and the L2 domain to wild-type and mutant PDK3 was measured by isothermal titration calorimetry (ITC) as described previously (30). The R158H PDK3 showed a 10-fold higher dissociation constant (Kd) for ATP binding (27.9 µm) than the wild-type (2.9 µm) (Table 4). The reduced affinity for nucleotides of R158H PDK3 facilitates the removal of product inhibition by ADP, resulting in increased kinase activity (30). In addition, the R158H mutation causes a significant increase in binding affinity for L2, compared with wild-type, with Kd values of 0.24 and 1.6 µm for the mutant and wild-type PDK3, respectively. The result suggests a tighter association of the mutant PDK3 and the E2p/E3BP core of PDC than wild-type.
Table 4.
Dissociation constants for L2 and ATP binding to wild-type and R158H PDK3
| Ligand |
WT | R158H |
|---|---|---|
| Dissociation constant (Kd), µm | ||
| ATP | 2.9 | 27.9 |
| L2 | 1.6 | 0.24 |
DISCUSSION
In the present study, using a combination of linkage analysis and WES, we have mapped a new locus for an X-linked dominant form of CMT (CMTX6) and identified a mutation in the PDK3 gene which lies within the defined 1.43-Mb linkage interval. Molecular validation has shown the R158H PDK3 mutation fully segregates in the family and is absent in ethnically matched control chromosomes as well as published databases.
Structurally, like all other PDK isoenzymes, each PDK3 monomer (Fig. 5A) consists of two distinct domains: the N-terminal and the C-terminal domains. In the N-terminal domain, also known as the regulatory domain, an allosteric inhibitor site for pyruvate or dichloroacetate is located in the center of a four-helix bundle made up of helices α3, α5, α6 and α8. Also positioned in the N-terminal domain is the lipoyl-binding site for L2 binding formed by residues from helices α2, α3 and α8 (30). The ATP/ADP-binding site is located in the C-terminal domain, also known as the catalytic domain. The putative substrate binding cleft for the phosphorylation loop of E1p is situated between helices α7/α8 and helices α10/α12 (32). Two different conformations were observed in the available PDK3 crystal structures, namely the open (active) conformation and the closed (inactive) conformations, which are suggested to exist in equilibrium (Fig. 5B) (30,32). A key difference between the two conformations is the position of D121 in the middle of the continuous α6/α7 helix. In the closed conformation, D121 swings ∼180° toward the α8 helix, which unwinds one complete turn of the helix α7, resulting in a narrower active-site cleft that restricts the access of the E1b substrate (Fig. 5B, right panel). The apparently unstable unwound helix α7 is maintained by ionic interactions between negatively charged D121 and two positively charged residues R158 and R162. In contrast, the open conformation shows a short kink segment consisting of residues N120 and D121 between helices α6 and α7 (Fig. 5B, left panel). The open confirmation, which lacks the unwinding of α7, provides a wider active-site cleft that facilitates the E1b access and removes product inhibition by ADP, resulting in higher kinase activity than the closed conformation (30). A dynamic equilibrium between the open and the closed conformations is thought to be functionally important in the PDK3 catalytic cycle and its migration over the PDC scaffold (32).
Figure 5.
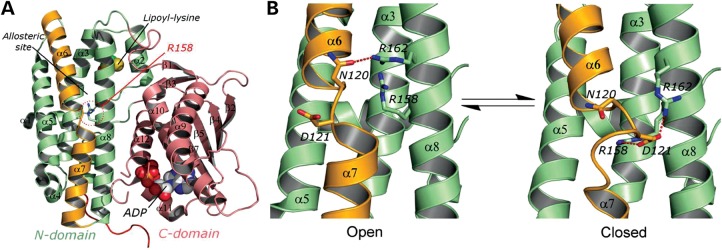
(A) Monomeric structure of PDK3 (PDB code: 1Y8O) is shown as ribbon models (the N-terminal domain in green, the C-terminal domain in pink, and the α6/7 helix in orange); the lipoyl-lysine residue of L2 bound to the N-terminal domain and ADP bound to the C-terminal domain are shown as space-filling models. R158 residue is shown in the stick model. (B) The close-up view of the N-terminal domain of PDK3 in equilibrium between the open conformation (PDB code: 1Y8O) and the closed conformation (molecule B of PBD code: 2PNR). The α6/7 helix (in orange) is subjected to significant iterative unwinding/winding in the equilibrium. The red dash line indicates hydrogen bonding or ionic interactions.
For the R158H PDK3 mutant, the change from an arginine to a histidine in position 158 likely weakens ionic interactions with D121 and reduces stability of the closed conformation, shifting the equilibrium toward the open conformation in the mutant R158H PDK3 (Fig. 5B, left panel). The resulting lower energy barrier to be overcome for alternating between the two conformations during a catalytic cycle renders the mutant PDK3 a significantly faster turnover enzyme than its wild-type counterpart. To support the pivotal role of R158 in mediating the catalytic cycle, N120 that coordinates to R162 in the open conformation and D121 that interacts with R158 and R162 in the close conformation (Fig. 5B) were changed to a histidine and an asparagine, respectively. The N120H/D121N PDK3 double mutant shows 2.1-fold higher basal and 1.34 higher E2p/E3BP-dependent activities than those of wild-type PDK3 (Fig. 4). These results support the notion that ionic interactions between R158, D121 and R162 are essential for maintaining the closed conformation, and that a less stable closed conformation by disrupting these salt bridges results in higher PDK3 activity.
The pathology of the R158H mutation and the subsequent biochemical consequences of overactivity of this enzyme are not clear at present. PDK3 was shown previously to be only expressed in the testes, kidney and brain (23). In this study, we confirmed expression in the brain and also showed expression in spinal cord and in skeletal muscle. It is therefore conceivable that the gain-of-function mutant PDK3 is expressed in both the motor and sensory nerves. The mutant PDK3 binds more tightly to the L2 domain of the PDC core than wild-type, which suggests that mutant PDK3 could pre-empt other wild-type PDK isoforms on the E2/E3BP core of PDC. In addition, the mutant PDK3 is insensitive to the inhibition by pyruvate, similar to wild-type PDK3. We postulate these combined effects of R158H PDK3 are likely to lock PDC in a predominantly phosphorylated inactive state leading to impaired ATP production and or lactate accumulation.
Peripheral neuropathy is common in mitochondrial disorders, occurring either as the primary clinical manifestation, e.g. CMT2A and CMT4A/2K due to mutations in MFN2 and GDAP1, respectively, or as part of a more complex phenotype, e.g. POLG1-related ataxia neuropathy spectrum disorders (33). Cellular energy depletion is likely the underlying cause of the peripheral neuropathy, although lactate accumulation may also contribute. PDC deficiency, which usually manifests in infancy with developmental delay, hypotonia, seizures and abnormal cerebral neuroimaging, is associated with peripheral neuropathy in 7% of cases (34). A relapsing chronic axonal neuropathy phenotype has also been seen in some patients (35).
Reduced PDC activity has previously been detected in fibroblast lines from several unrelated and genetically undefined CMT patients (36). However, the issues of variable phosphorylation of PDC due to cell culture and assay conditions was not addressed (37). Due to the tissue specific expression of the PDK3 protein, the usefulness of patient fibroblasts is limited as other isoforms of PDK (PDK1 and PDK2) also negatively regulate PDC activity in this tissue. In patients with the R158H mutation, the clinical phenotype is much milder than that seen in PDC deficiency, and is isolated to the peripheral nerves. Further studies on the expression, pathology and downstream biochemical consequences of the R158H mutant PDK3 in neuronal cell models are needed to shed further light on the etiology of this form of X-linked CMT.
MATERIALS AND METHODS
Patient samples
Patient ascertainment and collection of blood samples for analysis were collected with informed consent according to protocols approved by the Sydney Local Health District, Concord Hospital and Royal Children's Hospital Melbourne Human Research Ethics Committees. Genomic DNA was extracted from whole blood using the PureGene kit (Qiagen) according to manufacturer's recommendations. Members of the kindred underwent a clinical history, neurologic examination and neurophysiologic studies previously described (13). When necessary, additional information was obtained from medical records. Affected individuals underwent additional assessments including: ankle flexibility (38), hand dexterity [9-hole peg test (39) and functional dexterity test (40)] and testing of grip strength and ankle movements with hand-held dynamometry (41,42). The Charcot-Marie-Tooth Neuropathy Score (CMTNS) was calculated for affected subjects (14). Four affected males and three carrier females also underwent audiometric assessment (16).
Linkage and haplotype analysis
Two point linkage analysis was performed using the MLINK program from the Linkage package (version 5.1) (43) in the Fastlink implementation (version 4.1p) (44). Fully penetrant X-linked inheritance was assumed with a disease allele frequency of 0.0001. Marker allele frequencies were set at 1/n, where n is the number of observed alleles. Extended haplotypes of family members were constructed according to the order of the Rutgers combined linkage-physical map of the human genome (45) and based on the minimal number of intermarker recombinations.
Whole exome sequencing
WES was outsourced to Axeq Technologies (South Korea). DNA (2.5 µg) was used to prepare sequencing libraries using the Illumina TrueSeq Kit. The captured DNA was sequenced as paired end reads (100 bp) on an HiSeq 2000 sequencer (Illumina, San Diego, CA, USA). Sequence reads were mapped to the Human Feb. 2009 (GRCh37/hg19) assembly using BWA software (46). Sequence variants including single-nucleotide variations (SNVs) and indels were called using SAMtools (47). SNVs were annotated using dbSNP 132, and 1000 genomes SNP call release (20101109, 628 individuals). Sanger sequencing of PCR amplicons was outsourced for sequence validation using BigDye Terminator Cycle Sequencing protocols at the ACRF Facility, Garvan Institute of Medical Research (Australia).
Mutation screening and genotyping
Mutation analysis was performed using HRM protocols established in our laboratory (17). PCR amplicons for mutation scanning were designed to cover the coding exons and flanking intronic sequences. Primers for PDK3 screening were designed using the Light Scanner Primer Design Software (version 1.0.R.84 Idaho Technology) and are available on request. Melt Acquisition was performed on a 96-well Light Scanner (Idaho Technology) and the data analyzed with Light Scanner Call-IT 2.0 (Version 2.0.0.1331). Amplicons of differential melt curves were sequenced using BigDye Terminator Cycle Sequencing protocols at the ACRF Facility, Garvan Institute of Medical Research. HRM genotyping assays for segregation analysis and genotyping normal controls were performed as previously described with melt amplicons being designed to amplify 50–100 bp either side of the PDK3 mutation (48).
Expression analysis
Commercially available cDNA libraries (Clontech) for spinal cord, fetal brain, adult whole brain and skeletal muscle were used for expression analysis of PDK3. An aliquot of each library (1 µl) was amplified using Immomix Master Mix (Bioline) and size fractionated on a 1% w/v agarose gel. Hyperladder IV (Bioline) was used as the size marker standard. The previously published primers designed amplified a 100 bp PCR product specific for the PDK3 isoenzyme cDNA (24).
Vector construction and protein expression
Recombinant human PDK3 with its N-terminus fused to maltose-binding protein (MBP-PDK3), and the inner-lipoyl domain (L2) of human PDC were expressed and purified as described previously (30). Mutations in MBP-PDK3 were introduced using the QuickChange site-directed mutagenesis system from Stratagene (La Jolla, CA, USA) according to manufacturer's instructions.
Assay for kinase activity
Kinase activity was assayed by monitoring the incorporation of 32P form [γ-32P]ATP into the E1p component of PDC as previously reported (49). Briefly, the reaction mixture (25 μl in total volume) contained the following: 20 mm Tris–HCl (pH 7.5), 60 mm KCl, 2 mm dithiothreitol, 5 mm MgCl2 and 25 μg of bovine serum albumin, 0.05–0.2 μm PDK3, 4 μm the E1p heterotetramer, with or without 100 nm E2pE3BP (based on the 60-meric core). The phosphorylation reaction was initiated by adding [γ-32P]ATP (specific activity 200–300 cpm/pmol) to the reaction mixture to a final concentration of 0.4 mm. At 1 or 2 min, the reaction was terminated by the addition of 20% trichloroacetic acid (150 µl) and 50 mm sodium pyrophosphate. Upon centrifugation, the pellet was washed three times with 10% trichloroacetic acid. Microcentrifuge tubes without the caps were transferred to 5-ml scintillation vials containing 3 ml of scintillation mixture, and radioactivity incorporated into substrate was counted. The specific activities of PDK3 were calculated as moles of 32P incorporated/moles of kinase/min or min−1.
Determination of dissociation constants by ITC
The wild-type and mutant MBP-PDK3 proteins were exhaustively dialyzed against 2 l of dialysis buffer containing 50 mm Tris–HCl, pH 7.5, and 50 mm KCl, 5% glycerol. L2 or ATP solutions (300–400 µm) were placed in the titration syringe and injected in 10-ml increments into the reaction cell containing 1.4 ml of 30–40 µm MBP-PDK3 at 15°C in a VP-ITC microcalorimeter (MicroCal, Northampton, MA, USA). Dissociation constants (Kd) were deduced from the binding isotherms using the Origin 7 program for ITC. The concentrations of MBP-PDK3 and L2 were determined by measuring A280 and using calculated molar extinction coefficients (M−1 cm−1) of 111 230 and 10 810, respectively.
WEB RESOURCES
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org/
dbSNP, http://www.ncbi.nlm.nih.gov/snp/
Exome Variant Server, NHLBI Exome Sequencing Project, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UCSC Genome Browser, http://genome.ucsc.edu/
PolyPhen 2, http://genetics.bwh.harvard.edu/pph2/
SIFT, http://sift.jcvi.org/
SAMtools, http://samtools.sourceforge.net/
SUPPLEMENTARY MATERIAL
NOTE ADDED IN PROOF
The authors wish to acknowledge the identification of the CMTX4 Cowchock gene which was published during the editorial process of this manuscript. The AIFM1 gene was pubished by Rinaldi et al. (2012) Am. J. Hum. Genet., 91, 1095–1102.
FUNDING
This work is supported by the National Health and Medical Research Council (NHMRC) Project Grant APP1007705 awarded to G.A.N. and M.L.K. E.M.Y. is supported by a NHMRC Biomedical Postgraduate Research Scholarship. M.M.R. receives grant support from NHMRC under The Centres of Research Excellence scheme (APP1031893). The Murdoch Childrens Research Institute is supported by the Victorian Government's Operational Infrastructure Support Program. D.T.C. is supported by the National Institutes of Health Grants DK62306, DK26758 and DK92921, and the Welch Foundation Grant I-1286.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to family members participating in this study.
Conflict of Interest statement. None declared.
REFERENCES
- 1.Fowler W.M., Jr., Abresch R.T., Koch T.R., Brewer M.L., Bowden R.K., Wanlass R.L. Employment profiles in neuromuscular diseases. Am. J. Phys. Med. Rehabil. 1997;76:26–37. doi: 10.1097/00002060-199701000-00006. [DOI] [PubMed] [Google Scholar]
- 2.Bergoffen J., Scherer S.S., Wang S., Scott M.O., Bone L.J., Paul D.L., Chen K., Lensch M.W., Chance P.F., Fischbeck K.H. Connexin mutations in X-linked Charcot-Marie-Tooth disease. Science. 1993;262:2039–2042. doi: 10.1126/science.8266101. [DOI] [PubMed] [Google Scholar]
- 3.Ionasescu V.V., Trofatter J., Haines J.L., Summers A.M., Ionasescu R., Searby C. Heterogeneity in X-linked recessive Charcot-Marie-Tooth neuropathy. Am. J. Hum. Genet. 1991;48:1075–1083. [PMC free article] [PubMed] [Google Scholar]
- 4.Cowchock F.S., Duckett S.W., Streletz L.J., Graziani L.J., Jackson L.G. X-linked motor-sensory neuropathy type-II with deafness and mental retardation: a new disorder. Am. J. Med. Genet. 1985;20:307–315. doi: 10.1002/ajmg.1320200214. [DOI] [PubMed] [Google Scholar]
- 5.Priest J.M., Fischbeck K.H., Nouri N., Keats B.J. A locus for axonal motor-sensory neuropathy with deafness and mental retardation maps to Xq24-q26. Genomics. 1995;29:409–412. doi: 10.1006/geno.1995.9987. [DOI] [PubMed] [Google Scholar]
- 6.Kim H.J., Sohn K.M., Shy M.E., Krajewski K.M., Hwang M., Park J.H., Jang S.Y., Won H.H., Choi B.O., Hong S.H., et al. Mutations in PRPS1, which encodes the phosphoribosyl pyrophosphate synthetase enzyme critical for nucleotide biosynthesis, cause hereditary peripheral neuropathy with hearing loss and optic neuropathy (CMTX5) Am. J. Hum. Genet. 2007;81:552–558. doi: 10.1086/519529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E., et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ng S.B., Buckingham K.J., Lee C., Bigham A.W., Tabor H.K., Dent K.M., Huff C.D., Shannon P.T., Jabs E.W., Nickerson D.A., et al. Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 2010;42:30–35. doi: 10.1038/ng.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Montenegro G., Powell E., Huang J., Speziani F., Edwards Y.J., Beecham G., Hulme W., Siskind C., Vance J., Shy M., Zuchner S. Exome sequencing allows for rapid gene identification in a Charcot-Marie-Tooth family. Ann. Neurol. 2011;69:464–470. doi: 10.1002/ana.22235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weterman M.A., Sorrentino V., Kasher P.R., Jakobs M.E., van Engelen B.G., Fluiter K., de Wissel M.B., Sizarov A., Nurnberg G., Nurnberg P., et al. A frameshift mutation in LRSAM1 is responsible for a dominant hereditary polyneuropathy. Hum. Mol. Genet. 2012;21:358–370. doi: 10.1093/hmg/ddr471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weedon M.N., Hastings R., Caswell R., Xie W., Paszkiewicz K., Antoniadi T., Williams M., King C., Greenhalgh L., Newbury-Ecob R., Ellard S. Exome sequencing identifies a DYNC1H1 mutation in a large pedigree with dominant axonal Charcot-Marie-Tooth disease. Am. J. Hum. Genet. 2011;89:308–312. doi: 10.1016/j.ajhg.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yiu E.M., Geevasinga N., Nicholson G.A., Fagan E.R., Ryan M.M., Ouvrier R.A. A retrospective review of X-linked Charcot-Marie-Tooth disease in childhood. Neurology. 2011;76:461–466. doi: 10.1212/WNL.0b013e31820a0ceb. [DOI] [PubMed] [Google Scholar]
- 13.Yiu E.M., Burns J., Ryan M.M., Ouvrier R.A. Neurophysiologic abnormalities in children with Charcot-Marie-Tooth disease type 1A. J. Peripher. Nerv. Syst. 2008;17:236–241. doi: 10.1111/j.1529-8027.2008.00182.x. [DOI] [PubMed] [Google Scholar]
- 14.Shy M.E., Blake J., Krajewski K., Fuerst D.R., Laura M., Hahn A.F., Li J., Lewis R.A., Reilly M. Reliability and validity of the CMT neuropathy score as a measure of disability. Neurology. 2005;64:1209–1214. doi: 10.1212/01.WNL.0000156517.00615.A3. [DOI] [PubMed] [Google Scholar]
- 15.Rance G., Kearns L.S., Tan J., Gravina A., Rosenfeld L., Henley L., Carew P., Graydon K., O'Hare F., Mackey D.A. Auditory function in individuals within Leber's hereditary optic neuropathy pedigrees. J. Neurol. 2012;259:542–550. doi: 10.1007/s00415-011-6230-7. [DOI] [PubMed] [Google Scholar]
- 16.Rance G., Ryan M.M., Bayliss K., Gill K., O'Sullivan C., Whitechurch M. Auditory function in children with Charcot-Marie-Tooth disease. Brain. 2012;135:1412–1422. doi: 10.1093/brain/aws085. [DOI] [PubMed] [Google Scholar]
- 17.Kennerson M., Nicholson G., Kowalski B., Krajewski K., El-Khechen D., Feely S., Chu S., Shy M., Garbern J. X-linked distal hereditary motor neuropathy maps to the DSMAX locus on chromosome Xq13.1-q21. Neurology. 2009;72:246–252. doi: 10.1212/01.wnl.0000339483.86094.a5. [DOI] [PubMed] [Google Scholar]
- 18.Kennerson M.L., Nicholson G.A., Kaler S.G., Kowalski B., Mercer J.F., Tang J., Llanos R.M., Chu S., Takata R.I., Speck-Martins C.E., et al. Missense mutations in the copper transporter gene ATP7A cause X-linked distal hereditary motor neuropathy. Am. J. Hum. Genet. 2010;86:343–352. doi: 10.1016/j.ajhg.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ng P.C., Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–3814. doi: 10.1093/nar/gkg509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adzhubei I.A., Schmidt S., Peshkin L., Ramensky V.E., Gerasimova A., Bork P., Kondrashov A.S., Sunyaev S.R. A method and server for predicting damaging missense mutations. Nat. Methods. 2010;7:248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Reed L.J., Damuni Z., Merryfield M.L. Regulation of mammalian pyruvate and branched-chain alpha-keto acid dehydrogenase complexes by phosphorylation-dephosphorylation. Curr. Top. Cell Regul. 1985;27:41–49. doi: 10.1016/b978-0-12-152827-0.50011-6. [DOI] [PubMed] [Google Scholar]
- 22.Reed L.J. A trail of research from lipoic acid to alpha-keto acid dehydrogenase complexes. J. Biol. Chem. 2001;276:38329–38336. doi: 10.1074/jbc.R100026200. [DOI] [PubMed] [Google Scholar]
- 23.Bowker-Kinley M.M., Davis W.I., Wu P., Harris R.A., Popov K.M. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem. J. 1998;329(Pt 1):191–196. doi: 10.1042/bj3290191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Degenhardt T., Saramaki A., Malinen M., Rieck M., Vaisanen S., Huotari A., Herzig K.H., Muller R., Carlberg C. Three members of the human pyruvate dehydrogenase kinase gene family are direct targets of the peroxisome proliferator-activated receptor beta/delta. J. Mol. Biol. 2007;372:341–355. doi: 10.1016/j.jmb.2007.06.091. [DOI] [PubMed] [Google Scholar]
- 25.Baker J.C., Yan X., Peng T., Kasten S., Roche T.E. Marked differences between two isoforms of human pyruvate dehydrogenase kinase. J. Biol. Chem. 2000;275:15773–15781. doi: 10.1074/jbc.M909488199. [DOI] [PubMed] [Google Scholar]
- 26.Korotchkina L.G., Patel M.S. Site specificity of four pyruvate dehydrogenase kinase isoenzymes toward the three phosphorylation sites of human pyruvate dehydrogenase. J. Biol. Chem. 2001;276:37223–37229. doi: 10.1074/jbc.M103069200. [DOI] [PubMed] [Google Scholar]
- 27.Liu S., Baker J.C., Roche T.E. Binding of the pyruvate dehydrogenase kinase to recombinant constructs containing the inner lipoyl domain of the dihydrolipoyl acetyltransferase component. J. Biol. Chem. 1995;316:793–800. doi: 10.1074/jbc.270.2.793. [DOI] [PubMed] [Google Scholar]
- 28.Radke G.A., Ono K., Ravindran S., Roche T.E. Critical role of a lipoyl cofactor of the dihydrolipoyl acetyltransferase in the binding and enhanced function of the pyruvate dehydrogenase kinase. Biochem. Biophys. Res. Commun. 1993;190:982–991. doi: 10.1006/bbrc.1993.1146. [DOI] [PubMed] [Google Scholar]
- 29.Roche T.E., Hiromasa Y., Turkan A., Gong X., Peng T., Yan X., Kasten S.A., Bao H., Dong J. Essential roles of lipoyl domains in the activated function and control of pyruvate dehydrogenase kinases and phosphatase isoform 1. Eur. J. Biochem. 2003;270:1050–1056. doi: 10.1046/j.1432-1033.2003.03468.x. [DOI] [PubMed] [Google Scholar]
- 30.Kato M., Chuang J.L., Tso S.C., Wynn R.M., Chuang D.T. Crystal structure of pyruvate dehydrogenase kinase 3 bound to lipoyl domain 2 of human pyruvate dehydrogenase complex. EMBO J. 2005;24:1763–1774. doi: 10.1038/sj.emboj.7600663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roche T.E., Hiromasa Y. Pyruvate dehydrogenase kinase regulatory mechanisms and inhibition in treating diabetes, heart ischemia, and cancer. Cell Mol. Life Sci. 2007;64:830–849. doi: 10.1007/s00018-007-6380-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Devedjiev Y., Steussy C.N., Vassylyev D.G. Crystal structure of an asymmetric complex of pyruvate dehydrogenase kinase 3 with lipoyl domain 2 and its biological implications. J. Mol. Biol. 2007;370:407–416. doi: 10.1016/j.jmb.2007.04.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Menezes M.P., Ouvrier R.A. Peripheral neuropathy associated with mitochondrial disease in children. Dev. Med. Child Neurol. 2012;54:407–414. doi: 10.1111/j.1469-8749.2012.04271.x. [DOI] [PubMed] [Google Scholar]
- 34.Patel K.P., O'Brien T.W., Subramony S.H., Shuster J., Stacpoole P.W. The spectrum of pyruvate dehydrogenase complex deficiency: clinical, biochemical and genetic features in 371 patients. Mol. Genet Metab. 2012;106:385–394. doi: 10.1016/j.ymgme.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barnerias C., Saudubray J.M., Touati G., de L.P., Dulac O., Ponsot G., Marsac C., Brivet M., Desguerre I. Pyruvate dehydrogenase complex deficiency: four neurological phenotypes with differing pathogenesis. Dev. Med. Child Neurol. 2010;52:e1–e9. doi: 10.1111/j.1469-8749.2009.03541.x. [DOI] [PubMed] [Google Scholar]
- 36.Williams L.L. Pyruvate oxidation in Charcot-Marie-Tooth disease. Neurology. 1979;29:1492–1498. doi: 10.1212/wnl.29.11.1492. [DOI] [PubMed] [Google Scholar]
- 37.Sheu K.F., Hu C.W., Utter M.F. Pyruvate dehydrogenase complex activity in normal and deficient fibroblasts. J. Clin. Invest. 1981;67:1463–1471. doi: 10.1172/JCI110176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Khan K., Roberts P., Nattrass C., Bennell K., Mayes S., Way S., Brown J., McMeeken J., Wark J. Hip and ankle range of motion in elite classical ballet dancers and controls. Clin. J. Sport Med. 1997;7:174–179. doi: 10.1097/00042752-199707000-00004. [DOI] [PubMed] [Google Scholar]
- 39.Oxford G.K., Vogel K.A., Le V., Mitchell A., Muniz S., Vollmer M.A. Adult norms for a commercially available Nine Hole Peg Test for finger dexterity. Am. J. Occup. Ther. 2003;57:570–573. doi: 10.5014/ajot.57.5.570. [DOI] [PubMed] [Google Scholar]
- 40.Aaron D.H., Jansen C.W. Development of the Functional Dexterity Test (FDT): construction, validity, reliability, and normative data. J. Hand Ther. 2003;16:12–21. doi: 10.1016/s0894-1130(03)80019-4. [DOI] [PubMed] [Google Scholar]
- 41.Burns J., Redmond A., Ouvrier R., Crosbie J. Quantification of muscle strength and imbalance in neurogenic pes cavus, compared to health controls, using hand-held dynamometry. Foot Ankle Int. 2005;26:540–544. doi: 10.1177/107110070502600708. [DOI] [PubMed] [Google Scholar]
- 42.Burns J., Ouvrier R., Estilow T., Shy R., Laura M., Pallant J.F., Lek M., Muntoni F., Reilly M.M., Pareyson D., et al. Validation of the Charcot-Marie-Tooth disease pediatric scale as an outcome measure of disability. Ann. Neurol. 2012;71:642–652. doi: 10.1002/ana.23572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lathrop G.M., Lalouel J.M., Julier C., Ott J. Strategies for multilocus linkage analysis in humans. Proc. Natl Acad. Sci. USA. 1984;81:3443–3446. doi: 10.1073/pnas.81.11.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cottingham R.W., Jr., Idury R.M., Schaffer A.A. Faster sequential genetic linkage computations. Am. J. Hum. Genet. 1993;53:252–263. [PMC free article] [PubMed] [Google Scholar]
- 45.Kong X., Murphy K., Raj T., He C., White P.S., Matise T.C. A combined linkage-physical map of the human genome. Am. J. Hum. Genet. 2004;75:1143–1148. doi: 10.1086/426405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li H., Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brewer M., Changi F., Antonellis A., Fischbeck K., Polly P., Nicholson G., Kennerson M. Evidence of a founder haplotype refines the X-linked Charcot-Marie-Tooth (CMTX3) locus to a 2.5 Mb region. Neurogenetics. 2008;9:191–195. doi: 10.1007/s10048-008-0126-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wynn R.M., Kato M., Chuang J.L., Tso S.C., Li J., Chuang D.T. Pyruvate dehydrogenase kinase-4 structures reveal a metastable open conformation fostering robust core-free basal activity. J. Biol. Chem. 2008;283:25305–25315. doi: 10.1074/jbc.M802249200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.O'Brien M. for the Guarantors of Brain. Aids to the Examination of the Peripheral Nervous System. Saunders Elsevier, London; 2010. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.