

Abstract
Sex steroid hormones estradiol and progesterone play an important role in vascular adaptations during pregnancy. However, little is known about the role of androgens. Plasma testosterone (T) levels are elevated in preeclampsia, mothers with polycystic ovary, and pregnant African-American women, who have endothelial dysfunction and develop gestational hypertension. We tested whether elevated T alters vascular adaptations during pregnancy and if these alterations depend upon endothelium-derived factors such as prostacyclin, endothelium-derived hyperpolarizing factor (EDHF), and nitric oxide (NO). Pregnant Sprague Dawley rats were injected with vehicle (n=12) or T propionate [0.5 mg/Kg/day from gestation day (GD) 15–19;n=12] to increase plasma T levels 2-fold, similar to that observed in preeclampsia. Telemetric blood pressures and endothelium-dependent vascular reactivity were assessed with wire-myograph system. Phospho-eNOS and total-eNOS were examined in mesenteric arteries (MA). Mean arterial pressures were significantly higher starting from GD19 until delivery in T-treated dams. Endothelium-dependent relaxation responses to acetylcholine were significantly lower in MA of T-treated dams (pD2 (-log EC50)=7.05±0.06; Emax=89.4±1.89) compared to controls (pD2=7.38±0.04; Emax=99.9±0.97). Further assessment of endothelial factors showed NO-mediated relaxations were blunted in T-treated MA (Emax=45.4±5.48) compared to controls (Emax=76.49±5.06); however, prostacyclin- and EDHF-mediated relaxations were unaffected. Relaxation to sodium nitroprusside was unaffected with T-treatment. Phosphorylations of eNOS at Ser1177 were decreased and at Thr495 were increased in T-treated MA without changes in total-eNOS levels. In conclusion, elevated maternal T, at concentrations relevant to abnormal clinical conditions, cause hypertension associated with blunting of NO-mediated vasodilation. Testosterone may induce the increased vascular resistance associated with pregnancy-induced hypertension.
Keywords: blood pressure, eNOS phosphorylation, hypertension, vasodilation, mesenteric arteries, EDHF, prostacyclin
INTRODUCTION
Pregnancy is characterized by major cardiovascular adaptations, including marked decreases in systemic vascular resistance and mean arterial pressure (MAP) and an increase in maternal cardiac output and total blood volume.1,2 Studies in rats suggest that the enhanced endothelial vasodilatory actions allow peripheral vessels to accommodate these increases in blood flow and volume.3 Consistently, maternal vascular adaptations are accompanied by enhanced release of 3 major endothelium-derived vasodilatory factors including nitric oxide (NO),3-5 prostacyclin (PGI2),6-9 and endothelium-derived hyperpolarizing factor (EDHF).10,11 Further, there are concomitant pregnancy-induced increases in mRNA and protein expression of endothelial NO synthase (eNOS),4,5,12,13 prostaglandin H synthase (PGHS),7,14 and EDHF activity.11 Altered vascular adaptations during pregnancy are directly related to several maternal/fetal vascular pathologies, such as increased systemic vascular resistance, hypertension, proteinuria, poor placental growth, decreased nutrient transport, and low birth weight, which are characteristics of preeclampsia.15
Most studies have investigated the beneficial role of sex steroids hormones, especially estradiol and progesterone, and cardiovascular function during pregnancy.2 However little is known about the role of androgens. The relationship between testosterone and maternal cardiovascular function deserves special consideration because plasma levels of testosterone are elevated approximately 2- to 3-fold in pathological pregnancies such as in preeclampsia,16-20 and androgen levels in preeclamptic women positively correlate with higher average systolic blood pressure.20 Also, pregnant hyperandrogenemia women with polycystic ovary syndrome (PCOS)21 and pregnant African-American women have high maternal and cord blood testosterone levels22-24 and are at increased risk for developing preeclampsia.25,26 Previous studies have focused on the effects of elevated maternal testosterone on cardiovascular consequences in the offspring,27-29 but, surprisingly, there is a paucity of literature about the effect of testosterone on the maternal cardiovascular system during pregnancy.
Most investigators have studied the effect of elevated testosterone in cardiovascular function of males and nonpregnant females.30-32 Elevated testosterone has been shown to decrease endothelial function through its influence on the production and/or function of NO27,33-35, PGI2,36 and EDHF37 and affect vascular reactivity38,39 and systemic blood pressure.30-32 Thus, it is possible that the functions of endothelium in decreasing the vascular adaptations associated with pregnancy are altered in response to elevated testosterone. Therefore, we hypothesized that elevated testosterone may decrease endothelium-mediated cardiovascular adaptations of pregnancy. We tested this hypothesis by injecting testosterone into pregnant rats, mimicking the 2-fold elevation in plasma testosterone levels observed in preeclamptic women,27,28,40 to investigate 1) whether the systemic arterial pressure is enhanced in testosterone–treated compared with control pregnant rats; 2) whether endothelium-dependent vascular relaxation, particularly in the resistance mesenteric arteries, is inhibited in testosterone–treated compared with control pregnant rats; and 3) whether the testosterone-induced changes in vascular relaxation involve alterations in the endothelium-dependent PGI2-, EDHF-, or NO-mediated pathways.
METHODS
Animals and treatment
All experimental procedures were in accordance with National Institutes of Health guidelines (NIH Publication No. 85–23, revised 1996) with approval by the Animal Care and Use Committee at the University of Texas Medical Branch at Galveston. Timed pregnant Sprague-Dawley rats (day 4 or day 12 of gestation; copulation plug on day 1; Charles River, Wilmington, MA) were used in the experiment. After acclimatization, on gestational day (GD) 13, dams were randomly divided into 2 groups. Dams in the treatment group were subcutaneously injected with testosterone propionate (0.5 mg/Kg/day, n=12) for 5 days from GD 15–19. The control group received vehicle (sesame oil, n=12). This dose and duration of exposure is commonly used to mimic plasma testosterone levels observed in preeclamptic women.27,28,40 Blood samples were taken from the femoral vein 2 hours after testosterone injection on GD19 and analyzed for testosterone levels (Enzo Life Sciences, Farmingdale, NY) as reported previously.27,28 We injected rats with testosterone instead of dihydrotestosterone because it is the levels of testosterone but not dihydrotestosterone that are increased and correlate with pregnancies complications.16-20 Moreover, this model is not associated with changes in levels of maternal estradiol, progesterone, or corticosterone.40 Detailed methods for telemetric measurement of blood pressure, vascular reactivity studies, determination of eNOS expression and phosphorylation and plasma nitrate/nitrite (NOx) analysis are described in supplementary section.
Statistical analysis
All data are presented as mean±SEM. Responses to acetylcholine (ACh) are expressed as percent relaxation of the initial PE contraction. Vasodilator concentration response curves were fitted to a log-logistic sigmoid relationship, and pD2 values (negative logarithm of the molar concentration of vasodilator that produced 50% of the maximal response) and Emax (maximal relaxation effect) were calculated (GraphPad Prism, La Jolla, CA). Repeated measures ANOVA (treatment and time as factors) with a Bonferroni post hoc were used for comparisons of blood pressures and dose response curves between control and treatment groups. Percent maximal relaxation, pD2, NOx levels, eNOS mRNA expression and protein levels were compared between control and treatment groups using unpaired Student’s t test. Statistical significance was defined as P < 0.05. The letter n represents number of rats.
RESULTS
The length of gestation and mean litter size were not significantly affected by testosterone treatment (n=8 litters in each group). Fetal weights (control: 2.62±0.06g; testosterone-treated: 1.99±0.08 g), placental weights on GD 20 (control: 0.54±0.08 g; testosterone-treated: 0.43±0.13 g), and birth weight of pups (control: 6.30±0.19 g; testosterone-treated: 5.75±0.19 g) were significantly reduced (P<0.05; n=8 litters in each group) in the testosterone-treated group compared with controls, consistent with our previous reports.27,28,40 The plasma testosterone levels on GD 19 (2 h after injection) from testosterone-treated dams were 2.1±0.17 ng/mL compared to 1.0±0.11 ng/mL in vehicle-treated control dams.
Mean arterial pressure and heart rate measurements
Rats are nocturnal animals and continuous monitoring of blood pressure by telemetry revealed a characteristic circadian pattern with higher arterial pressure and heart rate values during the dark cycle (active phase) compared to the light cycle (Fig. 1). In control animals (n= 7), MAP was steady up to GD 17/18 and then progressively decreased as pregnancy advanced, reaching a nadir on GD21 and then increasing up to delivery on GD22. In testosterone-treated rats (n=7), MAP was similar to control rats during the early phase of treatment; however, the MAP did not show the expected decrease that occurred in control pregnant rats. Pregnant rats on testosterone had significantly higher MAP starting from GD19 until delivery on GD22 compared to the respective time point in controls (Fig. 1A; n=7 rats in each group; P<0.05). Changes in both systolic and diastolic blood pressures were similar to that of MAP; therefore, data are not shown. No differences in heart rate were observed between controls and testosterone-treated dams (Fig. 1B; n=7 rats in each group).
Figure 1.

Mean arterial pressure (MAP) and heart rate in control and testosterone-treated pregnant rats. MAP (A) and heart rate (B) were continuously monitored via telemetry catheters in femoral artery from gestational day (GD) 14 until delivery in control and testosterone-treated (0.5 mg/kg/day, s/c from GD 15–19) pregnant rats. MAP and heart rate values are presented in 12-h intervals showing circadian variation; nighttime periods are shaded. Data points represent the mean±SEM of measurements in 7 rats in each group. *P≤0.05 vs control.
Mesenteric vasodilator function
Testosterone treatment of pregnant rats did not alter phenylephrine (PE)-induced contractile responses but significantly decreased vessel sensitivity to ACh-induced vasodilation. The responses for ACh were significantly reduced in testosterone-treated pregnant rats (pD2: 7.05± 0.06; n=9; P <0.05) compared with controls (pD2: 7.38±0.04; n=9) (Fig. 2 and Table 1). The maximal responses to ACh were also significantly decreased in testosterone-treated pregnant rats (Emax:89.4±1.89%; n=9) compared with controls (Emax:99.9±0.97%; n=9).
Figure 2.
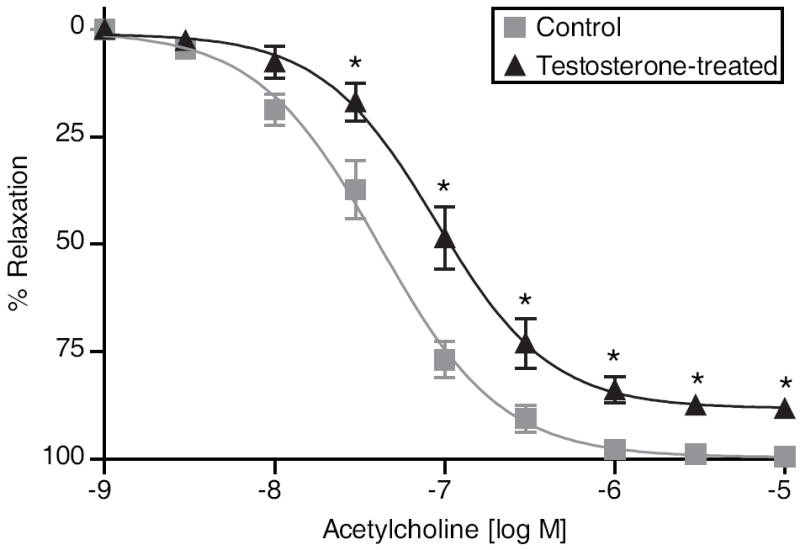
Endothelium-dependent relaxation in mesenteric arterial rings. A submaximal phenylephrine contraction (EC80) was elicited, acetylcholine (ACh) was added, and the percent relaxation of phenylephrine contraction was measured. Data points represent the mean±SEM of measurements in 18 to 24 vascular rings from 9 rats of each group. *P≤0.05 vs control.
Table 1.
The Emax and pD2 of concentration response curves induced by ACh in PE-precontracted resistance mesenteric arteries of control and testosterone-treated pregnant rats.
| Vasodilatory component | pD2 (-log M) | Emax (%) | ||
|---|---|---|---|---|
| Control | Testosterone-treated | Control | Testosterone-treated | |
| Total dilation | 7.38±0.04 | 7.05±0.06* | 99.9±0.97 | 89.4±1.89* |
| PGI2-mediated† | ND‡ | ND | 13.04±4.62 | 10.45±3.99 |
| EDHF-mediated# | 6.45±0.23 | 6.26±0.30 | 77.68±5.75 | 79.08±5.32 |
| NO-mediated¶ | 4.85±2.38 | ND | 76.49±5.06 | 42.26±5.95* |
Data represent the mean±SEM from 7 rats in each group.
P ≤ 0.05 vs corresponding measurements in control.
The PGI2-mediated vasodilation was studied after inhibition of eNOS and EDHF pathways, leaving PGI2 as the only intact pathway.
ND- not determined.
The EDHF-mediated vasodilation was studied after inhibition of eNOS (L-NAME,10-4 mol/l) and PGI2, leaving EDHF as the only intact pathway.
The NO-mediated vasodilation was studied after inhibition of PGI2 (indomethacin,10-5 mol/l) and EDHF (apamin and charybdotoxin, 10-7 mol/l each) pathways, leaving NO as the only intact pathway.
To address the involvement of products generated by PGHS, EDHF, and eNOS activities in testosterone-impaired mesenteric endothelial vasodilation, we examined ACh-induced relaxation in the absence or presence of specific inhibitors. Inhibition of eNOS and EDHF pathways, leaving PGI2 as the only intact pathway, showed minimal relaxation to ACh, and there was no difference between control (n=7) and testosterone-treated pregnant rats (n=8) (Fig. 3A and Table 1). Inhibition of PGI2 and eNOS pathways, leaving EDHF as the only intact pathway, showed substantial relaxation to ACh; however, there were no significant differences between control (n=7) and testosterone-treated rats (n=8) (Fig. 3B and Table 1). However, inhibition of PGI2 and EDHF pathways, leaving NO as the only intact pathway, showed significant relaxation to ACh, and this relaxation response was significantly lower in the mesenteric arteries of testosterone-treated pregnant rats (Emax: 42.26±5.95%; n=9) compared with control pregnant rats (Emax: 76.49±5.06%; n=9) (Fig 3C and Table 1). Blockade of all 3 pathways with inhibitors completely abolished ACh-induced relaxation (data not shown). Overall, these data imply that testosterone treatment does not affect PGI2 or EDHF components of relaxation, but only inhibits the NO component of relaxation responses to ACh. Sodium nitroprusside caused concentration-dependent relaxation of PE contraction that was not different in mesenteric arteries of control (n=6) and testosterone-treated (n=6) pregnant rats (Fig. 4).
Figure 3.

PGI2-, EDHF-, and NO-mediated endothelium-dependent relaxation in mesenteric arterial rings. Submaximal phenylephrine contraction (EC80) was elicited, acetylcholine (ACh) was added to arterial rings in presence of selective inhibitors as described in methods and Table 1, and then the percentage of relaxation to phenylephrine contraction was measured to determine (A) PGI2-, (B) EDHF-, and (C) NO-mediated mesenteric arterial relaxation. Data (mean±SEM) represent measurements from 14 to 24 vascular rings from 7–9 rats per treatment group. *P≤0.05 vs control.
Figure 4.

Endothelium-independent relaxation in mesenteric arteries. Submaximal phenylephrine contraction (EC80) was elicited in endothelium-denuded vascular rings, increasing concentrations of sodium nitroprusside (SNP) were added, and then the percentage of relaxation to phenylephrine contraction was measured. Data points represent mean±SEM of measurements in 10 to 12 mesenteric arterial rings from 6 rats of each group.
eNOS expression and phosphorylation
eNOS mRNA levels were significantly reduced (P =0.0251; n=7 in each group) in mesenteric arteries of testosterone-treated pregnant rats compared with control pregnant rats (Fig. 5A). However, immunoblot analysis indicated that there were no significant differences in protein levels of total eNOS in mesenteric arteries between control and testosterone-treated pregnant rats (Fig. 5B; n=5 in each group). Examination of phosphorylation status of eNOS, as an indicator activity state, demonstrated site-specific changes (Fig. 6A). When expressed as a ratio of total eNOS, phosphorylation at Ser1177 was significantly lower (3.3-fold) in the mesenteric arteries of testosterone-treated pregnant rats compared with controls (Fig. 6B; P = 0.0021; n=5 in each group). Phosphorylation at Ser635 was unchanged with testosterone treatment (Fig. 6C). In contrast, phosphorylation at Thr495 was significantly higher (1.3-fold) in testosterone-treated compared with control rats (Fig. 6D; P =0.0117; n=5 in each group). Human umbilical vein in culture exposed to testosterone also resulted in similar alterations in eNOS phosphorylation (see supplementary section).
Figure 5.

Endothelial nitric oxide synthase (eNOS) expression. (A) Quantitative RT-PCR analysis of eNOS expression in mesenteric arteries isolated from testosterone-treated and control pregnant rats. (B) Total eNOS protein expression in mesenteric arteries of testosterone-treated and control pregnant rats. Representative Western blots for eNOS and actin are shown at top; blot density obtained from densitometric scanning of eNOS normalized to actin is shown at bottom. Values are given as means±SEM of 5 to 7 rats in each group. *P≤0.05 vs control.
Figure 6.

Phosphorylation of eNOS in mesenteric arteries isolated from control and testosterone-treated pregnant rats. Tissue lysates were immunoblotted with antibodies recognizing Ser1177-, Ser635,- or Thr495-phosphorylated eNOS, and blots were reprobed with anti-eNOS antibody. (A) Representative Western blots of phospho-eNOS and total eNOS. The level of (B) Ser1177, (C) Ser635 or (D) Thr495 phosphorylation of eNOS was quantified by scanning densitometry and normalized to total eNOS. Values are means±SE; n = 5 for each group. *P≤0.05 vs control.
Changes in plasma NOx levels
The level of plasma NOx at GD20 was significantly lower in testosterone-treated dams compared with controls (control=4.0± 0.71 μmol/l and testosterone-treated=2.6±0.37 μmol/l; P =0.002, n=12 in each group).
DISCUSSION
To our knowledge, this is the first study evaluating the effect of elevated testosterone levels on maternal vascular adaptations in pregnant animals together with a detailed investigation of the underlying mechanisms. Major findings are that elevated testosterone levels lead to 1) increases in mean arterial pressure in pregnant rats, 2) inhibition of endothelium-dependent mesenteric arterial relaxation, and 3) decreases in vascular mesenteric relaxations that are mediated by alterations in the endothelium-dependent NO-pathway, but not EDHF- or PGI2-mediated pathways. Moreover, testosterone-induced reductions in endothelial NO activity are associated with decreased phosphorylation of excitatory eNOS at Ser1177 and increased phosphorylation of inhibitory eNOS at Thr495. Importantly, these testosterone effects in pregnant rats are observed at an in vivo concentration similar to that observed in pathological pregnancies, such as in preeclampsia.
The arterial pressure in normal pregnant rats is stable until GD17 or 18 and then gradually decreases until GD21. Similarly, in pregnant women, arterial pressure is stable during early part of first trimester and then gradually decreases reaching a nadir during the second trimester.2,41 Interestingly, elevated testosterone prevented the decrease in blood pressure observed in normal pregnancy such that the testosterone-treated pregnant rats have higher blood pressure compared to control pregnant rats. The lack of a pregnancy-related fall in blood pressure indicates a failure in normal cardiovascular adaptations and is considered to be a cardinal feature of preeclampsia.42 This failure suggests the mechanisms controlling blood pressure during pregnancy are perturbed by elevated testosterone. This effect on arterial pressure increase is without any changes in heart rate, indicating that testosterone does not affect the sympathetic activity. In the search for the possible mechanisms involved in the testosterone-induced increases in systemic arterial pressure, we found that elevated testosterone caused inhibition of ACh-induced relaxation of resistance mesenteric arteries, suggesting that elevated testosterone inhibits an endothelium-dependent relaxation pathway.
In the systemic circulation, the principal endothelium-dependent vasodilators are NO, PGI2, and EDHF. Studies have shown that PGI2 plays an important role in mediating vascular relaxation in uterine arteries;43 however, its contribution in the mesenteric arterial relaxation, and in general to systemic blood pressure, is minimal, which is consistent with our findings and previous data illustrating that infusion of indomethacin into pregnant rats does not induce hypertension.44 Although a previous study reported that testosterone inhibits PGI2 production in cultured aortic vascular smooth muscle cells in vitro,36 we find that the PGI2-mediated relaxation was not affected by the presence of elevated testosterone. This observation is also supported by similar levels of PGHS mRNA in the mesenteric arteries of testosterone-treated and control rats (data not shown). The current study shows that EDHF contributes substantially to mesenteric arterial relaxation during pregnancy, supporting earlier findings,43 but the presence of elevated testosterone does not alter EDHF-mediated relaxation. However, in cerebral arteries of male rats, testosterone inhibited EDHF-mediated relaxation.37 The differences between our study and that of Gonzales et al37 may be due to the sex, pregnancy status, or vascular bed examined. Indeed, sex and, in particular, pregnancy status can play a major role in many aspects of vascular function.
The most striking finding of our study is that NO-mediated mesenteric vasodilation was significantly decreased in testosterone-treated pregnant rats compared with controls, suggesting that elevated testosterone selectively blunts endothelial NO function. It has been suggested that during pregnancy there is a relative predominance of NO-dependent vasodilation45 that may increase its susceptibility to the effect of elevated testosterone. The decreased NO-mediated arterial relaxation in testosterone-treated pregnant rats is not due to decreased vascular smooth muscle sensitivity to NO, as relaxation of mesenteric rings to sodium nitroprusside, an exogenous NO donor, was not different between control and testosterone-treated rats. This suggests that the decreased relaxation in the testosterone-treated-rats is more likely due to changes in the synthesis/release of NO. The current study demonstrates a significant decrease in basal eNOS mRNA expression, but a similar difference was not noted at the level of the protein. Similar differences in eNOS mRNA but not protein expression levels were reported in pigs and sheep.33,46,47
Testosterone-mediated decreases in the mesenteric vascular function via the NO pathway were accompanied by concomitant decreases in the eNOS phosphorylation activity state. Immunoblotting demonstrated that the excitatory Ser1177 eNOS was decreased while no differences were observed at the excitatory Ser635 levels. Ser1177 eNOS is the most widely investigated excitatory phosphorylation site in the systemic vasculature and is regulated by numerous kinases including AKT and AMPK and our studies suggest that testosterone specially affects the excitatory Ser1177 site. Our studies also show that testosterone increases phosphorylation at the inhibitory Thr495 eNOS. Thr495 eNOS, an inhibitory site in the Ca+2-calmodulin binding region of eNOS is reported to be significantly phosphorylated in the caveolar subcellular domain of endothelial cells and its dephosphorylation leads to increased enzyme activity.48 Expressing as a ratio of excitatory Ser1177 to inhibitory Thr495 eNOS would better suggest eNOS activity state. Testosterone-treatment significantly decreased the Ser1177/Thr495 ratio (0.97 ±0.06, n=5, P<0.05) in mesenteric vessels compared to controls (3.6 ±0.7, n=5). However, the ratio of Ser635/Thr495 ratio in mesenteric arteries of testosterone treated rats (2.6 ±0.2) was not significantly different compared to controls (3.0 ±0.1). Thus, these findings suggest that chronic testosterone administration during gestation not only leads to decreased activity state of eNOS (as measured by decreased Ser1177 phosphorylation), but also produces decreased availability of eNOS (due to increased Thr495 phosphorylation) that is essential for eNOS activation. Endothelial cells in culture treated with testosterone also produced similar alterations in eNOS phosphorylation (see supplementary figure). Consistent with the decreased eNOS activity state, the levels of plasma NOx were significantly lower in testosterone-treated dams compared with controls. Also, in vitro treatment of human umbilical vein endothelial cells with testosterone decreased NOx production (see supplementary figure). Future studies will be designated to examine testosterone-induced subcellular partitioning of eNOS and the signaling mechanisms, including the role of kinases that regulates the multisite phosphorylation state of eNOS.
Although this study demonstrates a role for NOS system, testosterone may also lead to increases in systemic blood pressure through other mechanisms including increase in renal tubular sodium and water reabsorption, activation of specific vasoconstrictor systems including the rennin- angiotensin,30,49,50 endothelin-151 and thromboxane,38,52 as well as increasing oxidative stress.53 Blood pressure increase in testosterone-treated animals may also be due to increase in sympathetic or decrease in the parasympathetic function. Numerous studies provide evidence that dysfunction in endothelial NO production is an important factor for the induction of gestational hypertension. For example, in pregnant rats, subcutaneous infusions of L-NAME from day 17 to 22 of gestation resulted in sustained hypertension, proteinuria, and intrauterine growth retardation.54,55 Similarly, pregnant baboons showed an increase in MAP in middle and later pregnancy, but not early pregnancy, after administration of L-NAME.56 Many factors are proposed to play a role for impairing endothelial NO production during pregnancy; our study for the first time demonstrates a potential role for elevated testosterone in causing endothelial dysfunction that may lead to hypertensive diseases of pregnancy. Also, it is possible that the decreased endothelial NO function may consequently decrease production and activity of other factors that cause pregnancy-associated vascular adaptations such as vascular endothelial growth factor57,58 and placental growth factor.59 Ultimately, placental blood flow and transfer of nutrients may be adversely affected, and this may contribute to abnormal fetal growth and development.40 In the present study, placental weights and pups born to testosterone-treated dams were significantly smaller than those born to control dams.
In conclusion, elevated testosterone is associated with increased arterial pressure and selectively inhibits the endothelium-dependent NO-mediated vascular relaxation pathway in resistance vessels of pregnant rats. These results have important implications in determining the underlying factors for the adverse effects of testosterone on fetal growth and development.
PERSPECTIVES
Several studies show that the plasma levels of testosterone are 2-fold higher in preeclamptic pregnancies compared to normal pregnancies. Additionally, elevated testosterone during pregnancy is associated with abnormal fetal growth and development leading to adult-life diseases, yet the mechanisms underlying the detrimental effects of testosterone remains unknown. In the present study, clinically relevant concentrations of testosterone produced increases in arterial pressure with significant effects, particularly on the vasculature of pregnant rats. The endothelium-dependent relaxation pathway involving the NO activity in endothelial cells is inhibited in systemic vessels of pregnant rats with elevated testosterone. The results suggest a role for testosterone as a possible mediator of increased vascular resistance and elevated blood pressure during pregnancy. Therefore, some of the vascular effects observed during preeclampsia may indeed be androgen-mediated. The ability of elevated testosterone to influence cardiovascular function during pregnancy may contribute to some of the negative effects of testosterone on fetal growth and development. Understanding testosterone’s influences on the cardiovascular system could lead to new therapeutic approaches to antagonize some hypertensive effects during pregnancy. Furthermore, these results provide a novel approach to understanding the underlying factors that contribute to the pathogenesis of fetal origins of adult diseases.
NOVELTY AND SIGNIFICANCE.
What Is New?
In contrast to the well-studied beneficial roles of estrogen and progesterone in maternal cardiovascular adaptations to pregnancy, this study shows that elevated maternal plasma testosterone at levels similar to those observed in preeclampsia, PCOS mothers, and pregnant African-American women leads to increases in mean arterial pressure and inhibition of endothelium-dependent mesenteric arterial relaxation in pregnant rats.
Testosterone-mediated decreases in vascular mesenteric relaxation are mediated by alterations in the endothelium-dependent nitric oxide—but not EDHF and prostacyclin—components.
Testosterone-induced reductions in endothelial nitric oxide activity are associated with decreased phosphorylation of excitatory eNOS at Ser1177 and increased phosphorylation of inhibitory eNOS at Thr495.
What Is Relevant?
Many pregnancy pathologies, like preeclampsia, that are associated with systemic hypertension and endothelial dysfunctions also have increased testosterone levels. Our studies imply that the cardiovascular dysfunctions observed in hypertensive disorders of pregnancy may indeed be androgen-mediated.
Novel compounds that may inhibit excessive androgen action might be used to reduce the severity of hypertension and endothelial dysfunction in preeclamptic patients.
Elevated maternal testosterone levels affect fetal growth and program offspring to develop cardiovascular dysfunctions later in life. These effects of testosterone on maternal cardiovascular function may provide a novel approach to understanding the pathogenesis of fetal origins of adult diseases.
Summary
This article is the first to show that exogenous administration of testosterone to pregnant rats to increase plasma testosterone levels by 2-fold similar to that observed in abnormal clinical conditions like preeclampsia, affect cardiovascular adaptations to pregnancy. Elevated testosterone impairs maternal vascular adaptations by causing hypertension and, specifically, blunting NO-mediated vasodilation in resistance mesenteric arteries.
Acknowledgments
SOURCES OF FUNDING
Financial Support from the National Institute of Health (NIH) through grants HD069750, HL58144, and HL102866 is greatly appreciated.
Footnotes
CONFLICT OF INTEREST
None
Reference List
- 1.Thornburg KL, Jacobson SL, Giraud GD, Morton MJ. Hemodynamic changes in pregnancy. Semin Perinatol. 2000;24:11–14. doi: 10.1016/s0146-0005(00)80047-6. [DOI] [PubMed] [Google Scholar]
- 2.Magness RR. Maternal cardiovascular and other physiologic responses to the endocrinology of pregnancy. In: Bazer FW, editor. The Endocrinology of Pregnancy. Totowa, NJ: Humana Press Inc.; 1998. pp. 507–539. [Google Scholar]
- 3.Conrad KP, Joffe GM, Kruszyna H, Kruszyna R, Rochelle LG, Smith RP, Chavez JE, Mosher MD. Identification of increased nitric oxide biosynthesis during pregnancy in rats. FASEB J. 1993;7:566–571. [PubMed] [Google Scholar]
- 4.Williams DJ, Vallance PJ, Neild GH, Spencer JA, Imms FJ. Nitric oxide-mediated vasodilation in human pregnancy. Am J Physiol. 1997;272:H748–H752. doi: 10.1152/ajpheart.1997.272.2.H748. [DOI] [PubMed] [Google Scholar]
- 5.Sladek SM, Magness RR, Conrad KP. Nitric oxide and pregnancy. Am J Physiol. 1997;272:R441–R463. doi: 10.1152/ajpregu.1997.272.2.R441. [DOI] [PubMed] [Google Scholar]
- 6.Kawano M, Mori N. Prostacyclin producing activity of human umbilical, placental and uterine vessels. Prostaglandins. 1983;26:645–662. doi: 10.1016/0090-6980(83)90201-0. [DOI] [PubMed] [Google Scholar]
- 7.Magness RR, Shideman CR, Habermehl DA, Sullivan JA, Bird IM. Endothelial vasodilator production by uterine and systemic arteries. V. Effects of ovariectomy, the ovarian cycle, and pregnancy on prostacyclin synthase expression. Prostaglandins Other Lipid Mediat. 2000;60:103–118. doi: 10.1016/s0090-6980(99)00055-6. [DOI] [PubMed] [Google Scholar]
- 8.Magness RR, Rosenfeld CR, Hassan A, Shaul PW. Endothelial vasodilator production by uterine and systemic arteries. I. Effects of ANG II on PGI2 and NO in pregnancy. Am J Physiol. 1996;270:H1914–H1923. doi: 10.1152/ajpheart.1996.270.6.H1914. [DOI] [PubMed] [Google Scholar]
- 9.Magness RR, Mitchell MD, Rosenfeld CR. Uteroplacental production of eicosanoids in ovine pregnancy. Prostaglandins. 1990;39:75–88. doi: 10.1016/0090-6980(90)90096-e. [DOI] [PubMed] [Google Scholar]
- 10.Gillham JC, Kenny LC, Baker PN. An overview of endothelium-derived hyperpolarising factor (EDHF) in normal and compromised pregnancies. Eur J Obstet Gynecol Reprod Biol. 2003;109:2–7. doi: 10.1016/s0301-2115(03)00044-7. [DOI] [PubMed] [Google Scholar]
- 11.Gokina NI, Kuzina OY, Vance AM. Augmented EDHF signaling in rat uteroplacental vasculature during late pregnancy. Am J Physiol Heart Circ Physiol. 2010;299:H1642–H1652. doi: 10.1152/ajpheart.00227.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nelson SH, Steinsland OS, Wang Y, Yallampalli C, Dong YL, Sanchez JM. Increased nitric oxide synthase activity and expression in the human uterine artery during pregnancy. Circ Res. 2000;87:406–411. doi: 10.1161/01.res.87.5.406. [DOI] [PubMed] [Google Scholar]
- 13.Magness RR, Sullivan JA, Li Y, Phernetton TM, Bird IM. Endothelial vasodilator production by uterine and systemic arteries. VI. Ovarian and pregnancy effects on eNOS and NO(x) Am J Physiol Heart Circ Physiol. 2001;280:H1692–H1698. doi: 10.1152/ajpheart.2001.280.4.H1692. [DOI] [PubMed] [Google Scholar]
- 14.Bird IM, Sullivan JA, Di T, Cale JM, Zhang L, Zheng J, Magness RR. Pregnancy-dependent changes in cell signaling underlie changes in differential control of vasodilator production in uterine artery endothelial cells. Endocrinology. 2000;141:1107–1117. doi: 10.1210/endo.141.3.7367. [DOI] [PubMed] [Google Scholar]
- 15.Powe CE, Levine RJ, Karumanchi SA. Preeclampsia, a disease of the maternal endothelium: the role of antiangiogenic factors and implications for later cardiovascular disease. Circulation. 2011;123:2856–2869. doi: 10.1161/CIRCULATIONAHA.109.853127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Salamalekis E, Bakas P, Vitoratos N, Eleptheriadis M, Creatsas G. Androgen levels in the third trimester of pregnancy in patients with preeclampsia. Eur J Obstet Gynecol Reprod Biol. 2006;126:16–19. doi: 10.1016/j.ejogrb.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 17.Acromite MT, Mantzoros CS, Leach RE, Hurwitz J, Dorey LG. Androgens in preeclampsia. Am J Obstet Gynecol. 1999;180:60–63. doi: 10.1016/s0002-9378(99)70150-x. [DOI] [PubMed] [Google Scholar]
- 18.Ghorashi V, Sheikhvatan M. The relationship between serum concentration of free testosterone and pre-eclampsia. Endokrynol Pol. 2008;59:390–392. [PubMed] [Google Scholar]
- 19.Carlsen SM, Romundstad P, Jacobsen G. Early second-trimester maternal hyperandrogenemia and subsequent preeclampsia: a prospective study. Acta Obstet Gynecol Scand. 2005;84:117–121. doi: 10.1111/j.0001-6349.2005.00493.x. [DOI] [PubMed] [Google Scholar]
- 20.Troisi R, Potischman N, Roberts JM, Ness R, Crombleholme W, Lykins D, Siiteri P, Hoover RN. Maternal serum oestrogen and androgen concentrations in preeclamptic and uncomplicated pregnancies. Int J Epidemiol. 2003;32:455–460. doi: 10.1093/ije/dyg094. [DOI] [PubMed] [Google Scholar]
- 21.Sir-Petermann T, Maliqueo M, Angel B, Lara HE, Perez-Bravo F, Recabarren SE. Maternal serum androgens in pregnant women with polycystic ovarian syndrome: possible implications in prenatal androgenization. Hum Reprod. 2002;17:2573–2579. doi: 10.1093/humrep/17.10.2573. [DOI] [PubMed] [Google Scholar]
- 22.Rohrmann S, Sutcliffe CG, Bienstock JL, Monsegue D, Akereyeni F, Bradwin G, Rifai N, Pollak MN, Agurs-Collins T, Platz EA. Racial variation in sex steroid hormones and the insulin-like growth factor axis in umbilical cord blood of male neonates. Cancer Epidemiol Biomarkers Prev. 2009;18:1484–1491. doi: 10.1158/1055-9965.EPI-08-0817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Agurs-Collins T, Rohrmann S, Sutcliffe C, Bienstock JL, Monsegue D, Akereyeni F, Bradwin G, Rifai N, Pollak MN, Platz EA. Racial variation in umbilical cord blood sex steroid hormones and the insulin-like growth factor axis in African-American and white female neonates. Cancer Causes Control. 2012;23:445–454. doi: 10.1007/s10552-011-9893-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Potischman N, Troisi R, Thadhani R, Hoover RN, Dodd K, Davis WW, Sluss PM, Hsieh CC, Ballard-Barbash R. Pregnancy hormone concentrations across ethnic groups: implications for later cancer risk. Cancer Epidemiol Biomarkers Prev. 2005;14:1514–1520. doi: 10.1158/1055-9965.EPI-04-0869. [DOI] [PubMed] [Google Scholar]
- 25.Iavazzo C, Vitoratos N. Polycystic ovarian syndrome and pregnancy outcome. Arch Gynecol Obstet. 2010;282:235–239. doi: 10.1007/s00404-010-1495-0. [DOI] [PubMed] [Google Scholar]
- 26.Samadi AR, Mayberry RM, Reed JW. Preeclampsia associated with chronic hypertension among African-American and White women. Ethn Dis. 2001;11:192–200. [PubMed] [Google Scholar]
- 27.Sathishkumar K, Elkins R, Yallampalli U, Balakrishnan M, Yallampalli C. Fetal programming of adult hypertension in female rat offspring exposed to androgens in utero. Early Hum Dev. 2011;87:407–414. doi: 10.1016/j.earlhumdev.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chinnathambi V, Balakrishnan M, Yallampalli C, Sathishkumar K. Prenatal testosterone exposure leads to hypertension that is gonadal hormone-dependent in adult rat male and female offspring. Biol Reprod. 2012;137:1–7. doi: 10.1095/biolreprod.111.097550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King AJ, Olivier NB, Mohankumar PS, Lee JS, Padmanabhan V, Fink GD. Hypertension caused by prenatal testosterone excess in female sheep. Am J Physiol Endocrinol Metab. 2007;292:E1837–E1841. doi: 10.1152/ajpendo.00668.2006. [DOI] [PubMed] [Google Scholar]
- 30.Yanes LL, Sartori-Valinotti JC, Iliescu R, Romero DG, Racusen LC, Zhang H, Reckelhoff JF. Testosterone-dependent hypertension and upregulation of intrarenal angiotensinogen in Dahl salt-sensitive rats. Am J Physiol Renal Physiol. 2009;296:F771–F779. doi: 10.1152/ajprenal.90389.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Reckelhoff JF, Zhang H, Granger JP. Testosterone exacerbates hypertension and reduces pressure-natriuresis in male spontaneously hypertensive rats. Hypertension. 1998;31:435–439. doi: 10.1161/01.hyp.31.1.435. [DOI] [PubMed] [Google Scholar]
- 32.Chen MJ, Yang WS, Yang JH, Chen CL, Ho HN, Yang YS. Relationship between androgen levels and blood pressure in young women with polycystic ovary syndrome. Hypertension. 2007;49:1442–1447. doi: 10.1161/HYPERTENSIONAHA.106.083972. [DOI] [PubMed] [Google Scholar]
- 33.Chatrath R, Ronningen KL, Severson SR, LaBreche P, Jayachandran M, Bracamonte MP, Miller VM. Endothelium-dependent responses in coronary arteries are changed with puberty in male pigs. Am J Physiol Heart Circ Physiol. 2003;285:H1168–H1176. doi: 10.1152/ajpheart.00029.2003. [DOI] [PubMed] [Google Scholar]
- 34.Park KM, Kim JI, Ahn Y, Bonventre AJ, Bonventre JV. Testosterone is responsible for enhanced susceptibility of males to ischemic renal injury. J Biol Chem. 2004;279:52282–52292. doi: 10.1074/jbc.M407629200. [DOI] [PubMed] [Google Scholar]
- 35.Makinen JI, Perheentupa A, Irjala K, Pollanen P, Makinen J, Huhtaniemi I, Raitakari OT. Endogenous testosterone and brachial artery endothelial function in middle-aged men with symptoms of late-onset hypogonadism. Aging Male. 2011;14:237–242. doi: 10.3109/13685538.2011.593655. [DOI] [PubMed] [Google Scholar]
- 36.Nakao J, Change WC, Murota SI, Orimo H. Testosterone inhibits prostacyclin production by rat aortic smooth muscle cells in culture. Atherosclerosis. 1981;39:203–209. doi: 10.1016/0021-9150(81)90070-8. [DOI] [PubMed] [Google Scholar]
- 37.Gonzales RJ, Krause DN, Duckles SP. Testosterone suppresses endothelium-dependent dilation of rat middle cerebral arteries. Am J Physiol Heart Circ Physiol. 2004;286:H552–H560. doi: 10.1152/ajpheart.00663.2003. [DOI] [PubMed] [Google Scholar]
- 38.Gonzales RJ, Ghaffari AA, Duckles SP, Krause DN. Testosterone treatment increases thromboxane function in rat cerebral arteries. Am J Physiol Heart Circ Physiol. 2005;289:H578–H585. doi: 10.1152/ajpheart.00958.2004. [DOI] [PubMed] [Google Scholar]
- 39.Ojeda NB, Royals TP, Black JT, Dasinger JH, Johnson JM, Alexander BT. Enhanced sensitivity to acute angiotensin II is testosterone dependent in adult male growth-restricted offspring. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1421–R1427. doi: 10.1152/ajpregu.00096.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sathishkumar K, Elkins R, Chinnathambi V, Gao H, Hankins GD, Yallampalli C. Prenatal testosterone-induced fetal growth restriction is associated with down-regulation of rat placental amino acid transport. Reprod Biol Endocrinol. 2011;9:110. doi: 10.1186/1477-7827-9-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bosio PM, McKenna PJ, Conroy R, O’Herlihy C. Maternal central hemodynamics in hypertensive disorders of pregnancy. Obstet Gynecol. 1999;94:978–984. doi: 10.1016/s0029-7844(99)00430-5. [DOI] [PubMed] [Google Scholar]
- 42.Ishikuro M, Obara T, Metoki H, Ohkubo T, Yaegashi N, Kuriyama S, Imai Y. Blood pressure changes during pregnancy. Hypertens Res. 2012;35:563–561. doi: 10.1038/hr.2012.33. [DOI] [PubMed] [Google Scholar]
- 43.Cooke CL, Davidge ST. Pregnancy-induced alterations of vascular function in mouse mesenteric and uterine arteries. Biol Reprod. 2003;68:1072–1077. doi: 10.1095/biolreprod.102.009886. [DOI] [PubMed] [Google Scholar]
- 44.Conrad KP, Colpoys MC. Evidence against the hypothesis that prostaglandins are the vasodepressor agents of pregnancy. Serial studies in chronically instrumented, conscious rats. J Clin Invest. 1986;77:236–245. doi: 10.1172/JCI112282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cook JL, Zhang Y, Davidge ST. Vascular function in alcohol-treated pregnant and nonpregnant mice. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1449–R1455. doi: 10.1152/ajpregu.2001.281.5.R1449. [DOI] [PubMed] [Google Scholar]
- 46.Wood CE, Chen GF, Keller-Wood M. Expression of nitric oxide synthase isoforms is reduced in late-gestation ovine fetal brainstem. Am J Physiol Regul Integr Comp Physiol. 2005;289:R613–R619. doi: 10.1152/ajpregu.00722.2004. [DOI] [PubMed] [Google Scholar]
- 47.Jayachandran M, Mukherjee R, Steinkamp T, LaBreche P, Bracamonte MP, Okano H, Owen WG, Miller VM. Differential effects of 17beta-estradiol, conjugated equine estrogen, and raloxifene on mRNA expression, aggregation, and secretion in platelets. Am J Physiol Heart Circ Physiol. 2005;288:H2355–H2362. doi: 10.1152/ajpheart.01108.2004. [DOI] [PubMed] [Google Scholar]
- 48.Ramadoss J, Liao WX, Morschauser TJ, Lopez GE, Patankar MS, Chen DB, Magness RR. Endothelial caveolar hub regulation of adenosine triphosphate-induced endothelial nitric oxide synthase subcellular partitioning and domain-specific phosphorylation. Hypertension. 2012;59:1052–1059. doi: 10.1161/HYPERTENSIONAHA.111.189498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fortepiani LA, Yanes L, Zhang H, Racusen LC, Reckelhoff JF. Role of androgens in mediating renal injury in aging SHR. Hypertension. 2003;42:952–955. doi: 10.1161/01.HYP.0000099241.53121.7F. [DOI] [PubMed] [Google Scholar]
- 50.Henriques TA, Huang J, D’Souza SS, Daugherty A, Cassis LA. Orchidectomy, but not ovariectomy, regulates angiotensin II-induced vascular diseases in apolipoprotein E-deficient mice. Endocrinology. 2004;145:3866–3872. doi: 10.1210/en.2003-1615. [DOI] [PubMed] [Google Scholar]
- 51.Pearson LJ, Yandle TG, Nicholls MG, Evans JJ. Regulation of endothelin-1 release from human endothelial cells by sex steroids and angiotensin-II. Peptides. 2008;29:1057–1061. doi: 10.1016/j.peptides.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 52.Matsuda K, Ruff A, Morinelli TA, Mathur RS, Halushka PV. Testosterone increases thromboxane A2 receptor density and responsiveness in rat aortas and platelets. Am J Physiol. 1994;267:H887–H893. doi: 10.1152/ajpheart.1994.267.3.H887. [DOI] [PubMed] [Google Scholar]
- 53.Reckelhoff JF, Yanes LL, Iliescu R, Fortepiani LA, Granger JP. Testosterone supplementation in aging men and women: possible impact on cardiovascular-renal disease. Am J Physiol Renal Physiol. 2005;289:F941–F948. doi: 10.1152/ajprenal.00034.2005. [DOI] [PubMed] [Google Scholar]
- 54.Molnar M, Suto T, Toth T, Hertelendy F. Prolonged blockade of nitric oxide synthesis in gravid rats produces sustained hypertension, proteinuria, thrombocytopenia, and intrauterine growth retardation. Am J Obstet Gynecol. 1994;170:1458–1466. doi: 10.1016/s0002-9378(94)70179-2. [DOI] [PubMed] [Google Scholar]
- 55.Yallampalli C, Garfield RE. Inhibition of nitric oxide synthesis in rats during pregnancy produces signs similar to those of preeclampsia. Am J Obstet Gynecol. 1993;169:1316–1320. doi: 10.1016/0002-9378(93)90299-x. [DOI] [PubMed] [Google Scholar]
- 56.Hennessy A, Gillin AG, Duggin GG, Horvath JS, Tiller DJ. Low-dose nitro-L-arginine administration in baboon (Papio hamadryas) pregnancy. Clin Exp Pharmacol Physiol. 1999;26:849–852. doi: 10.1046/j.1440-1681.1999.03158.x. [DOI] [PubMed] [Google Scholar]
- 57.Ahmed A, Perkins J. Angiogenesis and intrauterine growth restriction. Baillieres Best Pract Res Clin Obstet Gynaecol. 2000;14:981–998. doi: 10.1053/beog.2000.0139. [DOI] [PubMed] [Google Scholar]
- 58.Reynolds LP, Redmer DA. Angiogenesis in the placenta. Biol Reprod. 2001;64:1033–1040. doi: 10.1095/biolreprod64.4.1033. [DOI] [PubMed] [Google Scholar]
- 59.Groesch KA, Torry RJ, Wilber AC, Abrams R, Bieniarz A, Guilbert LJ, Torry DS. Nitric oxide generation affects pro- and anti-angiogenic growth factor expression in primary human trophoblast. Placenta. 2011;32:926–931. doi: 10.1016/j.placenta.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]