

Abstract
The exploration of brain epigenomes, which consist of various types of DNA methylation and covalent histone modifications, is providing new and unprecedented insights into the mechanisms of normal neural development, neurological disease and aging. Traditionally, chromatin defects in brain were considered static lesions of early development that occurred in the context of rare genetic syndromes but it is now clear that mutations and maladaptations of the epigenetic machinery cover a much wider continuum, including adult-onset neurodegenerative disease. Here, we describe how recent advances in neuroepigenetics have contributed to an improved mechanistic understanding of developmental and degenerative brain disorders, as well as how they could influence the development of future therapies for these conditions.
Introduction
In the living cell, the functional definition of the human genome goes far beyond its linear sequence of 6 (or when haploid, 3) billion basepairs. The ‘epi-(greek for ‘over’, ‘above’)genome’, with its rich cache of highly regulated, structural modifications of DNA cytosine and histone residues and variants, defines the moldings and three-dimensional structure of the genomic material inside the cell nucleus, thereby providing a molecular bridge between genes and the environment. The epigenome is also responsible for orchestrating the myriads of transcriptional units, condensed chromatin clusters and many other features that distinguish between various cell types and development- or disease-states that share the same genome within the same subject (see also Box 1, 2 and Fig. 1).
Box 1: The organization and composition of the epigenome.
The basic unit of chromatin is the nucleosome, which consists of 146 bp of genomic DNA wrapped around an octamer composed of the core histones, H2A, H2B, H3 and H4. Chromatin fibers are defined as arrays of these nucleosomes, which are connected by linker DNA and linker histones (Figure 1). The combined set of DNA and histone modifications and histone variants provide the major building blocks for the epigenome117,118.
DNA methylation
Two related but functionally very different types of DNA modifications, methylation (m) and hydroxymethylation (hm) of cytosine carbon 5 (C5) mostly in CpG dinucleotides, occur primarily within CpG-enriched islands (which areoften defined by a GC percentage >50% across a minimum of 200 bp)119. The mC5 and hmC5 markings show a strikingly different distribution, with hmC5 mostly confined to the 5’ ends of genes, at amounts that overall correlate with gene expression activity120,121. In contrast, only a minute portion (<3%) of mC5 locates to CpG islands at the 5’ end of genes, where it is thought to function as a repressive mark, while the remaining 97% of mC5 is found in intra- and intergenic sequences and within DNA repeats122.
Post-translational histone modifications (PTMs)
According to recent studies, the number of amino acid residue-specific PTMs in a typical vertebrate cell may be as high as 130123. PTMs include mono (me1), di (me2)- and tri (me3) methylation, acetylation and crotonylation, polyADP-ribosylation and small protein (for example, ubiquitin and SUMO) modification of specific lysine residues, as well as arginine methylation and citrullination, serine phosphorylation, tyrosine hydroxylation, and several others123–125. Various combinations and interrelations of these site- and residue-specific PTMs show close association with the functional architecture of chromatin, and specific epigenetic signatures have been identified for proximal promoters and gene bodies at sites of actual or potential transcription, for enhancer and other regulatory sequences and for condensed and silenced chromatin126.
Histone variants
Apart from the core histones H2A,H2B,H3 and H4, metazoan genomes encode a number of histone variants that provide another layer of epigenetic regulation. Some of the well known variants include H3.3, H2A.Z and H2A.X which, in contrast to the canonical histones, are subject to replication-independent expression and assembly127, and have strong effects on nucleosome stability and compaction128.
Linker histones
These histones, such as H1, are crucial for the three-dimensional architecture of chromatin and the ‘zigzag’ arrangement of nucleosomes by regulating linker DNA folding, and the amounts of linker DNA strongly correlate with nucleosome repeat length 129 (Figure 1). Importantly, the amounts of H1 in neurons are much lower than in most other cell types129, which explains the observation that the average nucleosome repeat length in rat neurons is 40bp shorter than in glia (162 versus 201 bp)130. These neuron-specific features could have implications for disease vulnerability, because the Rett syndrome protein MeCP2 competes with H1 for linker DNA binding sites131 and, furthermore, H1 levels are markedly increased in Mecp2 deficient mouse brains36.
Box 2: Readers, writers and erasers of chromatin marks.
It is still under debate whether the epigenome’s constituents play a causal part in establishing functional chromatin states. For example, the presence of ‘open chromatin’-associated histone modifications and variants at a specific locus in the genome is thought to reflect a process driven by transcriptional regulation and dynamic repositioning of nucleosomes. From this perspective, at least some DNA and histone modifications function merely as a ‘cog’ in the chromatin remodeling machinery but are not necessarily key drivers132. Whatever the importance of DNA and histone modifications in driving the chromatin remodeling process, an increasing number of molecules that attach (‘writers’), or that erase DNA or histone modifications, or that bind (‘readers’) to a specific epigenetically modified site, have emerged as key players in the pathophysiology and potential treatment of neurological disease. Most, or virtually all, epigenetic markings that have been studied to date in brain are subject to a bidirectional and potentially highly dynamic regulation in the context of neuronal activity and various other paradigms133,134.
The underlying molecular machineries that contribute to the epigenome are often complex; for example, three DNA methyltransferases (DNMT1, DNMT3a and DNMT3b) establish and maintain DNA methylation marks. The actions of these enzymes are counterbalanced by active demethylation pathways involving mC5 hydroxylation and oxidation via ten-eleven translocation (TET) dioxygenases, or activation-induced deaminase (AID) and APOBEC-mediated deamination of mC5 or hmC5, followed by base excision repair-mediated replacement with (unmethylated) cytosine135,136. Other systems show a surprising degree of diversity, or perhaps redundancy, at the genetic level. For example, the various families of histone methyltransferases and demethylases together could easily account for > 100 genes in a mammalian genome137,138. Proteins that bind to a specific epigenetic mark are defined by their characteristic reader module; well studied examples include the methyl CpG binding domain (MBD) for mC5-DNA, the bromodomain for lysine acetylation, and the ‘chromo’, ‘Tudor’, ‘MBT’, ‘WD40repeat’ and ‘PHD finger’ domains that target methylated lysines or arginines in a residuespecific manner125. Conversely, specific methyl-lysine marks could become the target of 50–100 reader proteins. For the ‘open chromatin’ mark histone H3-trimethyl-lysine 4 (H3K4me3), the reader proteins include many components of the RNA polymerase II-associated transcriptional initiation complex, while other marks such as H3K9me3 are primarily targeted by transcriptional repressors and regulators of chromatin condensation139.
Figure 1. The epigenome and chromatin organization.
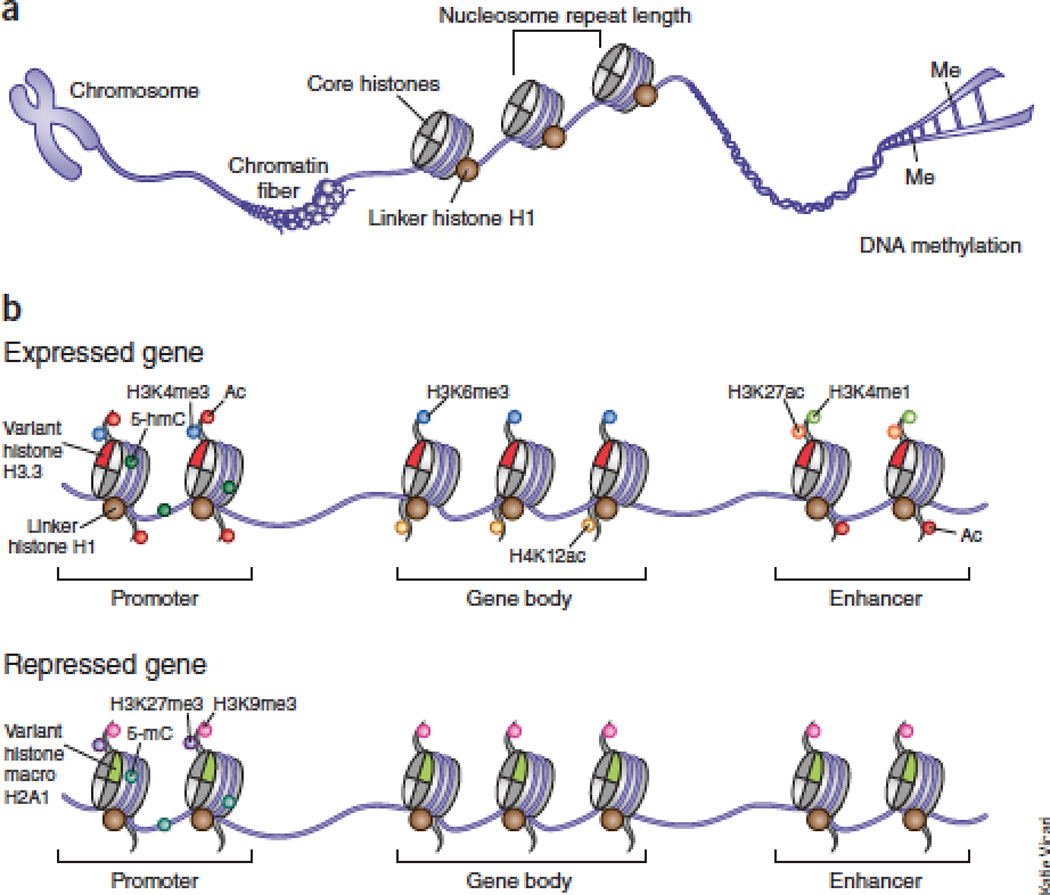
The top part shows that chromosomes are organized into domains of loose (euchromatin) or highly condensed (heterochromatin) chromatin and other loosely defined higher order structures (such as ‘globules’), some of which are tethered to the nuclear membrane. The bottom part shows 11 nm ‘beads-on-a-string’ chromatin fiber comprised of nucleosomal arrays connected by linker DNA, and linker histones as major regulators of nucleosomal repeat length. The distribution of DNA methylation and a small subset of > 100 posttranslational histone markings, linker histones and core histone variants represents differential regulation at (left) active promoters and gene bodies as opposed to enhancers, or (right) silenced and repressed chromatin, as indicated.
In this review, we describe how the field of epigenetics is dramatically reshaping the current thinking about neurological and neurodegenerative diseases. Remarkably, only a few years ago, this field’s primary focus was on a single mark, DNA methylation, in the context of cell division and early development. At first glance, these topics seemed to bear little relevance to the postnatal and adult brain that has a large proportion of postmitotic and highly differentiated cells, as in humans, the majority of neurons develop, differentiate and permanently exit from the cell cycle many weeks prior to birth. However, three recent major developments—each of which will be highlighted in this review—have the potential importance of epigenetics in brain development and disease to be reconsidered. First, human studies have indicated that the epigenetic landscape remains ‘plastic’ throughout all periods of brain development and aging, and ongoing dynamic regulation occurs even in neurons and other postmitotic constituents of the brain1–4. Second, disordered chromatin organization and function have been ascribed key pathogenic roles not only in several neurodevelopmental syndromes of early childhood, but also in a subset of adult onset hereditary neurodegenerative disorders5,6. Third, a rapidly expanding repertoire of chromatin modifying drugs has shown an unexpected therapeutic potential for a wide range of degenerative and functional disorders of the nervous system 7–11. These three lines of discoveries have now coalesced, thereby igniting an enormous interest in the chromatin-associated mechanisms of neurological disease and providing the foundations for a new discipline, ‘neuroepigenetics’12.
Epigenetics and the mature brain
While the focus of this review is on epigenetic changes in various neurological diseases, the first issue to consider is whether the normal course of maturation and aging is associated with changes in the brain’s epigenome (which is defined by the combined set of DNA methylation markings and histone modifications and variants, see also Box 1). On the one hand, this hypothesis that aging is associated with brain epigenetic changes is attractive, given that there are widespread age-related changes in gene expression in the cerebral cortex, including the downregulation of many neuronal genes13,14. However, in contrast to the accumulation of somatic mutations and other structural brain DNA changes that affect promoter function during aging (which are likely to be irreversible)15, most or perhaps all epigenetic markings studied to date (Box 1) are now thought to be reversible, and there is no a priori reason why there would be the unidirectional accumulation of a specific epigenetic mark in aging brain chromatin. Nonetheless, an increasing body of literature indicates that a substantial reorganization of the brain epigenome occurs during postnatal development and aging. Human cerebral cortex, for example, shows complex and gene-specific changes in the amounts of methylcytosine (mC5; cytosines are methylated at the carbon 5 position), and there is a steady rise in mC5 at many promoters that continues into old age in conjunction with subtle changes (mostly a decline) in expression of transcripts originating from these promoters 2,3. Such age-related epigenetic drifts could impact vulnerability to neurodegenerative disease. For example, in mouse cerebellum the levels of the mC5 derivative, hydroxymethyl-cytosine (5hmC; see also Box 1), are subject to a 10-fold increase from postnatal week 1 to adulthood16. Notably, among the genes that are affected by increasing 5hmC amounts at their promoters during cerebellar maturation, pathways for aging-related neurodegenerative diseases and angiogenesis were overrepresented and included at least 15 genes linked to hereditary forms of spinocerebellar ataxia, a neurological syndrome defined by severe motor dysfunction with the degeneration of cerebellar Purkinje neurons and other systems16. Also, of relevance, ten-eleven translocation (TET) proteins are responsible for converting mC5 to hmC5, and the active domains of these proteins belong to the same dioxygenase superfamily as hypoxia-inducible factor (HIF), an oxygen sensor that has been ascribed with a key role in angiogenesis and oxidative stress responses16. Perhaps TET proteins also function as oxygen sensors, thereby linking the epigenetic status of brain cells to (mal)adaptive processes related to impaired tissue perfusion and oxygen supply.
As well as changes in DNA methylation during development and aging, the epigenetic landscapes of histone post-translational modifications (PTMs) are also subject to dynamic changes. An age-dependent regulation of histone methylation markings that can differentiate between open and repressive chromatin has been documented for the human prefrontal and cerebellar cortex17, and hundreds of loci undergo substantial chromatin remodeling in cortical neurons during the transition from infancy to advanced age4. Furthermore, the brains from mice that are prone to accelerated senescence (the SAMP8 line) and have learning and memory deficits show age-related drifts in histone PTMs. These epigenetic drifts are defined by a loss of the markings associated with active gene expression, such as histone H4 lysine 20 monomethyl (H4-K20me1) and H3-K36me3 (Figure 1 and Box 1), in conjunction with a robust rise in the repressive mark, H3-K27me318. Likewise, in the hippocampus of 16 month old wild-type mice, genomic regions associated with actively expressed genes show a robust decline in acetylated H4-K1219, a histone PTM that like H3-K36me3 is linked to the transcriptional elongation process20. In addition, histone deacetylase inhibitor-induced upregulation of H4-K12ac dramatically improves hippocampal-dependent learning and memory in aged mice19. It is possible that age-related drifts in brain epigenomes negatively affect neuronal 10,15 and oligodendroglial21 transcriptomes, thereby contributing to a decline in the signaling capacity of nerve cells, defects in axon myelination and other molecular defects that have been linked to cognitive disorders of the adult brain with22 or without neurodegeneration14. Thus, together, these findings, leave little doubt that brain epigenomes are indeed subject to dynamic changes throughout all periods of maturation and aging, and this may have important implications for the neurobiology of disease.
MONOGENETIC NEUROLOGICAL DISEASE ASSOCIATED WITH EPIGENETIC CHANGES
Chromatin remodeling and the proper assignment of epigenetic marks on the genome are of fundamental importance for brain ontogenesis. These processes are also key control points in the stepwise transition from pluripotency to neural precursors to terminally differentiated neurons and glia23, and are involved in developmental events such as neuronal migration and connectivity formation24. Perhaps unsurprisingly, to date more than a dozen neurological syndromes have been linked to single gene mutations in DNA methyltransferase and histone modifying enzymes, or their ‘reader’ proteins (Table 1). Interestingly, however, this list includes not only embryonic defects and multi-organ syndromes, but also specific neurological disorders that shown an onset of symptoms in early childhood (for example, Rett syndrome), or neurodegeneration and regression starting after adolescence (some cases with Kleefstra syndrome) or as late as the third or fourth decade of life (for example, hereditary sensory and autonomic neuropathy with early onset dementia, type 1, HSAN1) (Table 1). Using the molecular defects in these rare monogenic causes of intellectual disability and adult onset dementia as a starting point, the unraveling of the underlying pathophysiology of these diseases could then open up hitherto unexplored therapeutic avenues both for these specific conditions and, eventually, perhaps for a broader spectrum of neurological disease.
Table 1.
Monogenetic brain disorders associated with DNA methylation and histone modification defects
| Gene (OMIN*) | Function | Syndrome(s) | OMIM # (disease) |
|---|---|---|---|
| ATRX (Xq21.1) *300032 | Replication-independent nucleosome remodeling and histone H3.3 incorporation | Alpha-thalassaemia, X-linked with mental retardation (ATRX), autism40 | #301040 |
|
CREBBP (16p13.3) *600140 EP300 (22q13.2) *602700 |
Transcriptional co-activator, histone acetyl-transferase | Rubinstein-Taybi syndrome (RSTS) 1 and 2105 | RSTS1/#180849 RSTS2/#613684 |
| DNMT1 (19p13.2) *126375 | DNA methyltransferase. Disease mutations are associated with hypomethylated repeats and promoters | Hereditary hensory and autonomic neuropathy type 1 with adult-onset dementia (HSAN1E)5, autosomal dominant cerebellar ataxia, deafness and narcolepsy (ADCA-DN)6. | #614116 |
| DNMT3B (20q11.21) *602900 | DNA methyltransferase. Disease mutations are associated with hypomethylation of pericentric repeats | Immunodeficiency, centromere instability, facial anomalies (ICF1) mental retardation syndrome28,106 | #242860 |
| ZBTB24 (6q21) *614064 | Transcriptional repressor and regulator of DNA methylation at pericentric repeats | Immunodeficiency, centromere Instability, facial anomalies (ICF2) mental retardation syndrome28,29 | #614069 |
| KDM5C (Xp11.22)/JARID1C *314690 | Histone H3-lysine 4 demethylase | X-linked mental retardation107, autism108 | #300534 |
| KMT1D (9q34.3) (also:EHMT1) *607001 | Histone H3-lysine 9 methyltransferase | Kleefstra (mental retardation) syndrome109, schizophrenia110, non-specific psychiatric phenotypes and neurodegenerative disease in post-adolescence period111 | #610253 |
| KMT3B (5q35.2-q35.3) (also NSD1) *606681 | Histone H3-lysine 36 and H4-lysine 20 methyltransferase | Sotos (mental retardation) syndrome112 | #117550 |
| PHF8 (Xp11.22) *300560 | Histone H3-lysine 9 demethylase and transcriptional activator | X-linked mental retardation withoutcleft lip and/or palate (Siderius-Hamel)113,114 | #300263 |
| RSK2 (Xp22.12) *300075 | Serine/threonine kinase (of both histones and non-histone proteins) | Coffin-Lowry X-linked mental retardation syndrome115 | #303600 |
| MECP2 (Xq28) *300005 | Methyl CpG binding protein | Rett and other neurodevelopmental syndromes, autism116 | #312750 |
Mutations affecting DNA methylation and neurological disease
Hypomorphic (partial loss-of-function) mutations in the DNA methyltransferase DNMT3B are responsible for a multiorgan syndrome - Immunodeficiency, Centromere Instability, Facial anomalies (ICF 1) - which includes mental retardation and defective brain development25,26. Cultured cells from subjects diagnosed with ICF show imbalances in repressive (mC5 and H3K27me3) and facilitative (H3K4me3 and H3K9ac) markings at pericentric (the ‘middle’ portions of the chromosome that surround the centromere, or the contact point for mitotic spindle attachment during cell division) and DNA repeats26 and at hundreds of promoters throughout the genome. This is thought to contribute to the dysregulated expression of genes important for brain development, immune defense and many other key functions27. A near identical syndrome, ICF 2, which is also defined by DNA hypomethylation defects but at a different set of pericentric repeat sequences (Fig. 3), is caused by mutations in Zinc Finger and BTB domain containing 24 (ZBTB24), which encodes a putative transcriptional repressor28,29.
Mutations and structural variants in the X-linked gene MECP2, which encodes a methyl-CpG-binding protein, have been linked to Rett syndrome (RTT), a disorder of early childhood with an incidence of 1 in 10,000 that is associated with developmental and cognitive regression and a broad range of neurological symptoms29,30. Although males with RTT typically succumb in the perinatal period, females survive as they are somatic mosaics for the MECP2 gene due to X-inactivation, and often appear grossly normal at birth before typically developing symptoms before the age of 2 years. Since the initial discovery of MECP2 mutations as a cause of RTT30, both loss-of-function mutations and copy number increases of MECP2 have been linked to various neurodevelopmental diseases, including individuals diagnosed with childhood onset schizophrenia and autism31. Studies in Mecp2 mutant mouse lines have provided insights into an unexpectedly complex molecular pathophysiology and linked specific neurological RTT phenotypes to transcriptional regulation at specific promoter sequences (Box 3). However, as for ICF syndrome, the chromatin pathology in RTT goes far beyond dysregulated promoter activity and is likely to involve pericentromeric repeats and other heterochromatic domains. For example, many RTT cases show decreased clustering of the chromocenters (which represent clusters of highly condensed chromatin) inside the nucleus and Mecp2 deficient neurons do not show the expected chromocenter expansions after induction of neuronal activity32,33. Neuronal chromatin, which contains much higher amounts of Mecp2 compared to glia34,35, shows a global disorganization in Mecp2 deficient brains, with supranormal amounts of the linker histone H1, hyperacetylation of nucleosome core histones, and de-repression of transcriptional activity at pericentromeric repeats, retrotransposons and other normally silent DNA repeats36. Alterations in the amounts of Mecp2, which could compete with H1 for occupancy of linker DNA36, could also affect nucleosome repeat length and the general architecture of chromatin fibers (Figure 1 and Box 1).
Box 3: The complex pathophysiology of MeCP2 [Au: this box title will probably be too long, so I’ve suggested shortening it].
Among the growing list of genes associated with a brain-specific chromatin disorder, the largest share of the literature relates to the Rett syndrome-associated gene MECP2. The following discussion on emerging views of MECP2-associated transcriptional (dys)regulation exemplifies the challenges and complexities in the endeavor to identify and pinpoint the molecular pathology even in a monogenetic disease.
Studies in both humans and micehave shown that phenotypic consequences arise when the amounts of functional MeCP2 protein drop below or rise above its normal amounts in brain140. Recent comparisons of the transcriptomes from gain- and loss-of-function Mecp2 mutant mice to those from wildtype mice have provided some insights into the complexities of MeCP2-mediated transcriptional regulation at specific promoter sequences and have alsoimplicated specific transcriptional changes in some of the emotional and behavioral changes that have been observed across the clinical spectrum of MECP2 mutations. For example, in basal forebrain including the amygdala, more than 32 genes with an established role in the regulation of anxiety and social behaviors, including the neuropeptide corticotropin-releasing hormone (Crh) and the G-protein coupled mu-opioid receptor (Oprm1), were highly sensitive to Mecp2 gene dosage141,142. In particular, elevated levels of anxiety in mice that overexpress Mecp2 are thought to be directly related to increased MeCP2-binding and upregulated transcription at the Crh and Oprm1 genes141. Conversely, decreased anxiety in some Mecp2 loss of function mutants has been linked to decreased expression of Crh and other gene expression changes142.
These results, taken together, seem counterintuitive to the originally described role of MeCP2 as a transcriptional repressor and, adding to the complexity, neuronal activity induces MeCP2 phosphorylation at multiple serine sites, thereby altering the protein’s affinity to its target gene promoters143. In vivo, ablation of a phospho-serine site downstream of MeCP2’s transcriptional repression domain (S421) enhanced binding of MeCP2 to the brain-derived neurotrophic factor (Bdnf) promoter, resulting in increased neurotrophin expression and improved hippocampal learning and plasticity144(see figure below). Thus, a highly complicated, multi-layered process is involved in fine-tuning of MeCP2 expression, which in turn controls many genes that have a key regulatory role in higher order behaviors.
Figure for Box 3 Fine-tuning of MeCP2 function and higher order behavior. (top) A schematic presentation of MeCP2 protein is shown, including methyl-CpG binding (MBD) and transcriptional repressor (TRD) domains, and neuronal activity-sensitive phosphorylation sites. (bottom). In adult mouse forebrain, promoter-bound MeCP2 upregulates the expression of key regulatory factors that controll emotional and affective states, learning and memory. In vivo, phosphorylation of MeCP2-serine 421 decreases promoter binding and gene expression, with corresponding changes in behavioral states. These observations were established by work in the amygdala, hypothalamus and hippocampus of Mecp2 mutant and wildtype mice (in schematic mouse brain section, amygdala, blue, hippocampus, green, hypothalamus, purple).
![Box 3: The complex pathophysiology of MeCP2 [Au: this box title will probably be too long, so I’ve suggested shortening it]](https://cdn.ncbi.nlm.nih.gov/pmc/blobs/7dde/3596876/5b92e973401b/nihms444352f3.jpg)
It was recently discovered that monogenetic brain disorders associated with defective DNA methylation are not limited to neurodevelopmental diseases but also include some cases who have been diagnosed with HSAN15, a rare neurodegenerative condition characterized by various neuropathies and early onset dementia in the third or fourth decade of life. The mutations found in this condition are positioned in the coding sequence of the DNA methyltransferase 1 (DNMT1) gene, specifically within the targeting sequence domain important for the nuclear localization and preferential enrichment of this enzyme at pericentric and other repeat DNA, as well as for its activity and stability5. Indeed, peripheral blood cells from individuals with HSAN1 showed hypomethylated at DNA repeat sequences and subtle methylation imbalances (mostly hypo-) at hundreds of gene promoters5. Furthermore, additional mutations in close proximity to the targeting sequence domain of DNMT1 are found in some kindreds with autosomal dominant cerebellar ataxia, deafness and narcolepsy (ADCA-DN), which is also associated with early onset dementia6. At first glance, it is surprising that these DNMT1 loss-of-function mutations selectively affect postmitotic neurons. DNMT1 functions as part of the DNA replication machinery during the S phase of the cell cycle, and tracks the replication fork to remethylate the newly synthesized DNA strands37, which are mechanisms considered relevant only for dividing cells. However, DNMT1 may also play a part in maintaining the amount of DNA methylation in quiescent and even postmitotic cells, because this protein remains enriched at the pericentric repeats and other condensed portions of the genome after S-phase is completed38. This cell cycle-independent DNMT1 function is dependent on it having an intact targeting sequence domain38, and therefore one could speculate that the deleterious DNMT1 mutations in the affected individuals with HSAN1 result in faulty chromatin remodeling at repeat sequences of differentiated cells, including neurons. Of note, mutant mice that are mosaic for a prenatal Dnmt1 gene deletion in neural precursors lose virtually all of their brain cells with DNA hypomethylation within one month after birth39, and it will be interesting to find out whether similar phenotypes would emerge in animals in which Dnmt1 mutations are limited to the portions of the targeting sequence domain that are mutated in HSAN1 pedigrees.
It is remarkable that each of these three neurological conditions involving dysregulated DNA methylation—ICF, RETT and HSAN1—is associated with widespread chromatin defects across the genome of differentiated CNS cells, including pericentric and other repeat-rich domains that until now have been barely explored in the neurosciences. It will be extremely interesting to follow future developments in this area, which are likely to shed more light on the role of epigenetic regulation of these repeat DNAs in healthy and diseased brains.
Heritable brain disorders with histone defects
Loss-of-function mutations in the ATRX (α-thalassemia, mental retardation, X-linked) gene, which encodes a multifunctional chromatin regulator, are responsible for various X-linked mental retardation syndromes40. ATRX protein expression progressively increases during brain development and, in mice, Atrx ablation results in excessive apoptosis and defective migration of young neurons40,41. Another Atrx mutant mouse line, which hasa deletion in the Atrx zinc-finger motif, is a genetic model for similar human cases who are afflicted with a milder form of X-linked mental retardation42. This mouse model shows impaired learning behaviors, elongated, structurally abnormal dendritic spines (a classical neuropathological abnormality found in various neurodevelopmental conditions43) and defective synaptic signaling in the frontal cortex 42. However, the molecular mechanisms that link ATRX mutations to these neurological phenotypes remain unclear. One possible mechanism involves H3.3, a histone H3 variant that was originally associated with active transcription but was subsequently linked to ribosomal44, telomeric and pericentric repeats45–47. ATRX, together with its binding partner and histone chaperone, the death domain associated protein DAXX, is essential for replication-independent H3.3 incorporation into nucleosomes 45–47. It has been suggested that ATRX preferentially targets G-rich tandem repeats which tend to form four-stranded aberrant DNA structures (called G-quadruplexes) and refolds them back into doublestranded DNA and a regular nucleosomal organization44.
Whether any of these mechanisms are indeed relevant in the context of neurological disease remains to be investigated. However, notably there are at least there monogenetic disorders that show prominent epigenetic dysregulation of repeat DNA, including the aforementioned ICF1 and ICF2 syndromes and ATRX (Figure 3). Further work will be required to clarify whether the observed chromatin defects at the sites of repeat DNA are indeed a major factor for the disordered neurodevelopment common to these conditions. Furthermore, numerous deleterious mutations in histone modifying enzymes have been identified as monogenetic cause for neurodevelopmental or neurodegenerative disease (Table 1).
DISORDERED CHROMATIN IN NEURODEGENERATIVE DISEASE
Historically, studies on chromatin structure and function, and epigenetically driven concepts in general received little attention by researchers exploring the pathophysiology of slowly progressing neurodegenerative disorders such as Alzheimer’s, Parkinson’s or Huntington’s disease. Two independent lines of discovery have now moved epigenetics towards the center stage in these fields. First, initial reports of the therapeutic benefits of histone deacetylase inhibitor drugs in the nervous system utilized preclinical models for Huntington’s disease, a hereditary condition caused by the excessive expansion of a CAG repeat in the huntingtin gene48,49. Second, the genetic findings described earlier in the review link rare types of adult onset dementia associated with hereditary neuropathy5 or cerebellar ataxia6 to deleterious mutations in proteins that requlate DNA or histone methylation. These findings may imply that other, more common types of adult-onset neurodegenerative disease could also be related to the defective regulation of brain chromatin.
Indeed, ‘synucleinpathies’ such as Parkinson’s disease and dementia with Lewy bodies (DLB) are associated with loss of DNMT1 protein from the cell nuclei in brains from patients with these conditions and brains from transgenic mice that overexpress synuclein50. This in turn leads to deregulation of DNA methylation at the promoters of several disease-associated genes, including that of alpha-synuclein itself 50. There is additional, indirect, evidence that the delicate balance in the amounts of DNMT1 and other DNA methyltransferases, including DNMT3a, inside the nuclei of brain cells plays an important part in neuronal health and function. For example, the in vivo induction of motor neuron apoptosis by toxic drugs or peripheral nerve lesions is associated with a pre-apoptotic rise in the amounts of DNMT1 and DNMT3a in the nuclei of motor neurons51. Furthermore, the amounts of methyl-cytosine and the expression of DNMT are reportedly increased in the pyramidal neurons in the cerebral cortex of subjects diagnosed with the sporadic motor neuron disease amyotrophic lateral sclerosis,(ALS)51. Adding further complexity, there is evidence that DNMT1 and DNMT3a are not only targeted to the nucleus but are also abundant in mitochondria, including those that are localized in distal neuronal processes and synapses51. Whether or not these DNA methyltransferase enzymes regulate mitochondrial functions in brain cells remains to be investigated, but preliminary evidence suggests that mitochondrial DNA, like nuclear DNA, is subject to the methylation and hydroxymethylation of cytosine residues52.
Likewise, histone modifying enzymes could play a part in neurodegenerative disease. Because much of the evidence so far has been correlative, the neuroepigenetic field eagerly awaits further work that can confirm (or refute) that histone modifying enzumes have a key role in the etiology or progress of neurodegenerative disorders. For example, the pathological sequestration of transcription factors vital for neuronal health, such as the cAMP response binding protein CREB and its partner, the histone acetyl-transferase CREB-binding protein48,53 (CBP, and mutations in the gene encoding CBP are responsible for Rubinstein-Taybi syndrome, Table 1), has also been associated with the beta amyloid plaques in brains from individuals with Alzheimer’s disease54 and the polyglutamine aggregates and nuclear inclusions in Huntington’s chorea55. Furthermore, a hyperactivity of transglutaminase TG2 in Huntington’s disease leads to excess nuclear amounts of lysine ε-amino - glutamine γ-carboxamide bonds, which are resistant to proteolysis56. This results in an abnormal configuration of nuclear actin-cofilin complexes57 and potentially abnormal histone polyamination, which leads to defective expression of a transcription factor with an essential role in mitochondrial biogenesis, peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1-α)58. Furthermore, excessive histone H3K9 methylation59 and increased amounts of macroH2A1 (a variant histone broadly associated with repressive chromatin remodeling) expression60 have been observed in blood and brain tissues from individuals with Huntington’s disease, including the striatum and frontal cortex that are heavily affected by the underlying neurodegenerative process in this disease. Such changes, in conjunction with additional defects in nucleosomal structure and function, may contribute to the decreased expression of the neurotrophic factor BDNF, dopamine receptors, MAP kinase signaling components and other transcriptional changes that have been observed in the striatum from people with Huntington’s disease61. Therefore, the hypothesis that histone modifying enzymes are involved in the pathophysiology of neurodegenerative diseases is clearly an interesting one that warrants further investigation.
Epigenetic targets in the treatment of neurological disease
Therapeutic targeting of histone acetylation
There is great enthusiasm in academia and industry to develop new epigenetic therapies, and this has been further boosted by the recent U.S. Food and Drug Administration approvals for the potent histone deacetylase inhibitors (HDACi) suberoylanilide hydroxamic acid (SAHA, trade name vorinostat) and romidepsin (trade name Istodax) in the treatment of hematologic malignancies. Indeed, a large body of preclinical work has suggested that HDACi might have therapeutic potential in a surprisingly wide range of neurological conditions. These include acute brain injury and stroke paradigms, various neurodegenerative conditions such as Parkinson’s, Alzheimer’s, triplet Repeat disease (including Huntington’s and spinocerebellar ataxias),motor neuron disease8–11, as well as depression and other psychiatric illnesses62–64(Table 2). However, it is still unclear whether HDACi would indeed benefit the patients affected by any of these conditions.
Table 2.
Chromatin modifying drugs as potential therapies for neurological disease
| Drug type and mechanism |
Representative compound(s) |
CNS effects in preclinical models | FDA approval for non-neurological condition |
|---|---|---|---|
| Sequesters DNMT at DNA replication fork | 5-aza-cytidine, zebularine (nucleoside analogues) | Disrupted hippocampal learning and also reward behavior87–90 in wild-type mice and rats. Conferred stroke protection in mice 93,94 |
Yes |
| DNMT active site inhibitor | RG108 | Prevented cell death in a mouse model of motor neuron disease model 51. Impaired hippocampal learning92 in rats. |
|
| Class I/II HDAC inhibitor | phenylbutyrate, SAHA, TSA | Had a broad neuroprotective profile in acute and chronic injury and neurodegeneration models, including ischemia, Huntington’s and other polyglutamine andtriplet diseases, and a Parkinson’s disease model (MPTP toxicity)8–11 Improved hippocampus-dependent learning and cognition and exerted antidepressant-like effects 62,63 |
Yes |
| Class III HDAC inhibitor | nicotinamide AGK2 | Reduced amounts of phospho-tau and improved cognition in hippocampal learning tasks in a mouse model of Alzheimer’s disease73 Reduced α-synuclein toxicity and dopaminergic cell death in a Drosophila model of Parkinson’s disease 74 | |
| Histone methyltransferase Glp/G9a inhibitor | BIX-01294 | Enhanced reward behavior in mice after exposure to a stimulant drug (cocaine)85 | |
| p300 histone acetyl transferase inhibitor | C646 | Reduced amounts of tau l and neurotoxicity in cultured rat neurons and cells from people with Alzheimer’s disease71. | |
| Topoisomerase I inhibitor | Topotecan, irinotecan | Reversed imprinting-mediated silencing of Ube3a(the Angelman’s syndrome gene) in mouse brain 96 | Yes |
| Topoisomerase II inhibitor | Etoposide, dexrazoxane | Reversed imprinting-mediated silencing of Ube3a in mouse brain96 | Yes |
Moreover, like their counterparts - the histone acetyltransferases (HATs) -both the class I/II HDACs (commonly defined by their zinc-containing catalytic domain) and the (nicotinamide NAD+ dependent) class III HDACs, or sirtuins, regulate lysine acetylation of non-histone proteins65,66. Indeed, non-histone targets of sirtuin 1 are thought to mediate the molecule’s neuroprotective and cognitive phenotypes67,68. Such promiscuity of HDACs for their target proteins adds further complexity to the interpretation of their therapeutic potential that has emerged in models of neurological disease. Consider the example of fronto-temporal dementia (FTD), a genetically heterogeneous neurodegenerative condition that primarily affects the frontal lobes and is associated with at least three different types of inclusion bodies in brain tissue 69. Haploinsufficiency for PROGRANULIN (GRN) underlies one type of FTD that is defined by brain inclusions containing TAR-DNA binding protein 43 (TDP-43) 69. Importantly, GRN expression is robustly upregulated by the HDAC SAHA, and it has been suggested that SAHA could become a promising treatment for FTD70. In contrast, FTDP-17, or fronto-temporal dementia with parkinsonism, is defined by the abnormal accumulation of the microtubule binding protein tau in the brain due to excessive tau acetylation71. Thus, blocking tau acetylation by inhibiting the HAT CBP and promoting tau deacetylation via activation of the HDAC SIRT1 has been proposed as a new therapeutic avenue to treat FTDP-17 and other ‘taupathies’ such as Alzheimer’s disease71. Notably, upregulation of SIRT1 also suppresses β-amyloid production in the brains of mice modeling Alzheimer’s disease72. Interestingly, however, the pan-sirtuin inhibitor nicotinamide also has therapeutic benefit in an Alzheimer’s model consisting of transgenic mice that overexpress tau73. Although nicotinamide is a fairly broadly acting drug that has many effects beyond sirtuin inhibition, a subset of SIRT2-specific inhibitors elicits therapeutic benefits in some models of Parkinson’s disease74. Thus, depending on the type of neurodegenerative disease and the specific HDACs and HATs involved, modulation of HDAC and HAT activity could have very different and potentially opposing clinical implications. Furthermore, HDACi are generally considered to promote neuronal growth and differentiation, but there is also evidence that they could have potentially detrimental effects on the orderly maturation of astro- and oligodendrocytes75–77. Therefore, caution is warranted when evaluating the potential benefits of these drugs in multiple sclerosis76, white matter ischemia9 and other neurological conditions that involve the damage or injury of the brain’s non-neuronal cellular constituents. In addition, the activation of normally epigenetically silenced retrotransposons could be another potential HDACi side effect78 (Box 4).
Box 4: Epigenetic control of the brain’s ‘jumping genes’.
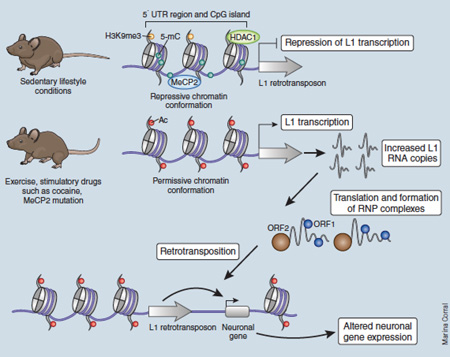
Retrotransposons, or ‘jumping genes’, are commonly defined as mobile elements that can proliferate in the germline of plants and mammals, thus contributing to the potential for massive genome expansion during the course of evolution145. Approximately 40–50% of the human genome is derived from mobile elements and, more importantly, several classes of retrotransposons, which duplicate via RNA intermediates and then insert into a different site of the genome, remain active even in present day humans146. Some of the best known examples of retrotransposons include LINE1 (L1), Alu and SVA elements146, and it likely that new insertions by these elements contributes to genome plasticity in individual brain cells147. In particular, the hippocampus and striatum are defined by much higher rates of de novo retrotransposition compared to other somatic tissues such as blood, heart and liver147,148. It has been estimated that each hippocampal cell could harbor up to 800 new insertions, indicating a hitherto unexpected degree of genomic plasticity148. Interestingly, both L1 and Alu-based insertion events into human brain genomes disproportionally affect actively expressed genes, including many that are pivotal for synaptic transmission such as those encodingthe dopamine receptor DRD3, the amino acid transporters SLC6A5, SLC6A6 and SLC6A9 and the RAI1 transcription factor that has been previously implicated in schizophrenia and the 17p11.2 (Smith Magenis) deletion syndrome147. Therefore, retrotransposon activity could contribute to neuronal diversity149 and even drug- or experience-dependent plasticity150,151 (Box Figure).
Insertional mutagenesis could also have potentially detrimental consequences, for example, theaberrant activation or repression of neighboring genes, exon shuffling and gene deletions 149. Importantly, epigenetic regulators are vital for a cell’s ability to suppress retrotransposon activity and L1, Alu and other elements that utilize the L1 replication machinery are epigenetically silenced via repressive DNA methylation and histone modifications152. Therefore, when repressive chromatin remodeling at sites of mobile elements is ‘exchanged’ with transcriptional activation complexes such as when the WNT3a/β-catenin complex binds to the L1 promoter153,154, excessive amounts of insertional mutagenesis could cause cell damage. For example, loss of the repressive DNA methyl-reader protein MECP2 in neural cultures or in the brains of mice with MECP2 mutation results in a modest excess of L1 activity36,155 in conjunction with changes in global chromatin states (Box Figure). Increased L1 retrotransposition was also recently detected in cells from people with ataxia telangiectasia, an autosomal recessive neurodegenerative condition caused by mutations of the DNA damage sensor and serine/threonine kinase ATM78. Loss of ATM results in increased reverse transcriptase efficiency, thereby fostering new L1 insertions in the genome78. It is conceivable that Rett syndrome and ataxia telangiectasia are merely the ‘tip of the iceberg’ and that numerous other neurological conditions with links to transposon dysregulation may emerge in the near future.
Figure for Box 4: Epigenetic regulation of retrotransposon activity in the nervous system. A schematic presentation of the LINE-1 (L1) retrotransposon is shown, including the CpG island upstream of two open reading frames (ORFs) for transcriptional control. Epigenetic ‘brakes’ preventing excess L1 transcription in the brain include DNA methylation and repressive chromatin remodeling, involving the actions of the histone deacetylase HDAC1 and the methyl-CpG-binding protein MECP2. Physiological activities are associated with mild relaxation of L1 repression, while stimulant drugs-of-abuse or loss of MECP2 could substantially upregulate the number of L1 RNA copies. The L1 RNAs are then translated, assembled into complexes for active retrotranspositions, thus resulting in genome toxicity through excess amounts of de novo genome integrations
In addition to HAT and HDAC activators or inhibitors, other types of drugs that interfere with histone acetylation pathways have recently emerged. For example, the bromodomains of the BET (bromodomain and extraterminal) protein family, which are classical ‘readers’ of acetylated chromatin and facilitate transcriptional activation79, can be targeted by highly specific small-molecule inhibitors that have been shown to correct transcriptional dysregulation in hematopoetic and solid malignancies80,81, inflammation and other medical conditions previously shown to be HDACi-sensitive82. Because multiple BET reader proteins, including Brd (bromodomain-containing proteins) 1–3 show robust and widespread expression in the mature mouse brain (www.brain-map.org)83, it will be interesting to explore bromodomain inhibitors in the context of the some of the neurological disease models discussed above. As for the HAT and HDAC drugs, the specificity and side effect profiles of bromodomain inhibitors will also requires additional investigation.
Therapeutic targeting of histone methylation
The clinical potential of drugs that can interfere with the regulation of histone methylation is also a promising but as yet largely unexplored field. There are probably up to 100 histone methyltransferases (KMTs) and demethylases (KDMs) encoded in the human genome, and many of these enzymes are defined by functional domains outside of the catalytic site that are thought to contribute to target specificity and genomic occupancy patterns7. Like for the HDACi, a subset of histone methyltransferase inhibitors are in clinical trials for cancer treatment, and are likely to be explored in the context of neurological disease in the not too distant future7. An interesting candidate is the small molecule BIX-01294, which is an inhibitor for the histone H3K9-specific methyltransferases G9a/Glp84. This drug de-represses neuronal gene expression84 and, when administered directly into the ventral striatum (a key structure in the brain’s addiction circuitry), strongly enhances the development of reward behaviors in mice exposed to the stimulant drug cocaine85. It remains to be determined if BIX-01294 elicits learning and memory-enhancing effects outside of the stimulant addiction paradigm. However, it is interesting to note that the drug’s mechanism of action could, at least in part, involve the inhibition of G9a/Glp-mediated repressive chromatin remodeling at the promoters of Bdnf, Cdk5, Arc and other genes that function as key regulators for spine density and synaptic connectivity in the brain85.
Therapeutic targeting of DNA methylation
Several DNA methylation inhibitors, including the cytidine analogues 5-azacytidine (5-Aza-CR), zebularine and nucleoside analogs that sequester DNMT enzymes after being incorporated into DNA86, have been well characterized and are approved or are in preclinical and clinical trials for the treatment of cancer86. When administered directly into brain tissue of mice and rats, these and other types of DNMT inhibitors disrupt synaptic plasticity and hippocampal learning and memory, and are powerful modulators of reward and addiction behaviors87–90. However, nucleoside analogs are thought to act primarily at sites of DNA synthesis and replication during the cell cycle, and thus at first glance, they seem to be of little relevance for postmitotic neurons and glia. Notably, though drugs such as N-Phthalyl-L-tryptophan/RG 108, which interfere with sites at which DNMT is active independently of DNA replication, still elicit a robust impairment of hippocampal learning and memory91,92. The potential of drugs interfering with DNMT activity may go beyond these examples of synaptic and behavioral plasticity. For example, treatment with DNMT inhibitors can confer stroke protection after mild ischemia in mice and, furthermore, haploinsufficiency for the Dnmt1 gene in mice is associated with smaller infarction volumes after acute ischemia and stroke 93,94. Furthermore, a recent study reported a strong, anti-apoptotic effect of RG 108, which inhibits the active site of DNMT, in an injury-based mouse model of ALS51.
Topoisomerase inhibitors as potential neurotherapeutics
Topoisomerases (topos) are DNA cleaving enzymes that are important in the processes of replication and recombination, transcription and chromatin remodeling95. A recent study using an unbiased high-content screening approach in mouse primary cortical neurons discovered that a diverse group of molecules that function as topoisomerase I or II inhibitors can unlock the expression of the normally epigenetically silenced paternal allele of the gene encoding ubiquitin protein ligase E3A (Ube3a). These topo inhibitors [mediate their effect by reducing the expression of the imprinted Ube3a antisense RNA (Ube3a-ATS)96. The expression of this antisense RNA is normally repressed on the maternal chromosome in conjunction with the allele-specific DNA methylation of an imprinting center (which isa DNA or chromatin structure that carries epigenetic information about parental origin)97. Like hundreds of other loci defined by parent-of-origin-specific gene expression, Ube3a was considered epigenetically stable throughout life98. This hypothesis now needs to be revised, however, given that even a single intrathecal infusion of the FDA-approved topoisomerase inhibitor topotecan was sufficient to relieve silencing of the paternal Ube3a (sense) transcript in lumbar spinal neurons for an extended period of at least 3 months96. The most obvious explanation for topotecan’s mechanism of action - altered DNA methylation of the Ube3a imprinting center - has been ruled out, thus the underlying mechanism(s) remain a mystery.
The reactivation of paternal UBE3A expression via topoisomerase inhibition could provide a starting point to investigate potential therapies for the neurodevelopmental disorder Angelman syndrome, which is caused by loss of function mutations and deletions at the maternal UBE3A locus99–101, and for which there are currently no effective treatments. Although it is presently not known whether topoisomerase-mediated reversal of imprinting-related gene expression is specific to the UBE3A locus, this issue could be addressed by, for example, fusing topoisomerase enzymes to customized motifs for sequence-specific binding at UBE3A. For example, chi zinc finger nucleases or transcription activator-like (TAL) effectors of plant pathogenic bacteria have been fused to the FokI restriction enzyme, allowingthe induction of ‘custom-made’ DNA strand breaks ‘ at specific and even at unique loci in the genome102,103. It is certainly worth exploring whether similar strategies can be used tocreate a chimeric protein comprised of a topoisomerase enzyme fused to a custom-made zinc finger or TAL effector sequence that can specifically target the UBE3A imprinting center.
Outlook
Neuroepigenetics, as a field, is evolving at a rapid pace. We are beginning to understand more about the neural mechanisms that mediate the reversible and bidirectional regulation of DNA methylation and various histone modifications. Furthermore, as discussed in this review, the epigenetic dysregulation of gene expression and chromatin architecture could play a prominent part in the pathophysiology of various neurological disorders. However, in many of these disease conditions the mutant epigenetic regulator seemingly operates in a highly complex and multifunctional manner at a large number of genomic loci. For example, Rett, ICF1 and 2 and HSAN1 syndromes are a heterogeneous group of neurodevelopmental and neurodegenerative disorders caused by mutations in DNA methyl-reader and methyl-writer proteins, and each disease exhibits disordered DNA methylation at numerous single copy genes as well as in specific repeat DNA sequences in subdomains of constitutive heterochromatin. It will be important to clarify, for each disorder, which genomic loci affected are important for disease manifestation, progression and the potential of eventual cure, and which are mere bystanders and irrelevant to the disease process.
For the numerous types of chromatin modifying drugs that have shown some promise in preclinical studies, it will be important to pinpoint their key mechanism of action. For example, are the beneficial effects of HDAC inhibitors in various acute and chronic neurodegenerative and cognitive disorders due to their broad effects on histone modifications or, instead, due to changes in the acetylation of non-histone proteins (such as the presynaptic molecule and regulator of vesicle release Bruchpilot104) or a combination thereof? Whatever the underlying mechanism of action, we predict that, as in the fields of oncology and general medicine, which are currently pursuing hundreds of clinical trials with epigenetic drug targets, the treatment options for neurodevelopmental and neurodegenerative diseases will soon be enriched by an array of chromatin compounds that are emerging from preclinical and translational research (Table 2).
Figure 2. Example monogenetic brain disorders with a heterochromatin defect.
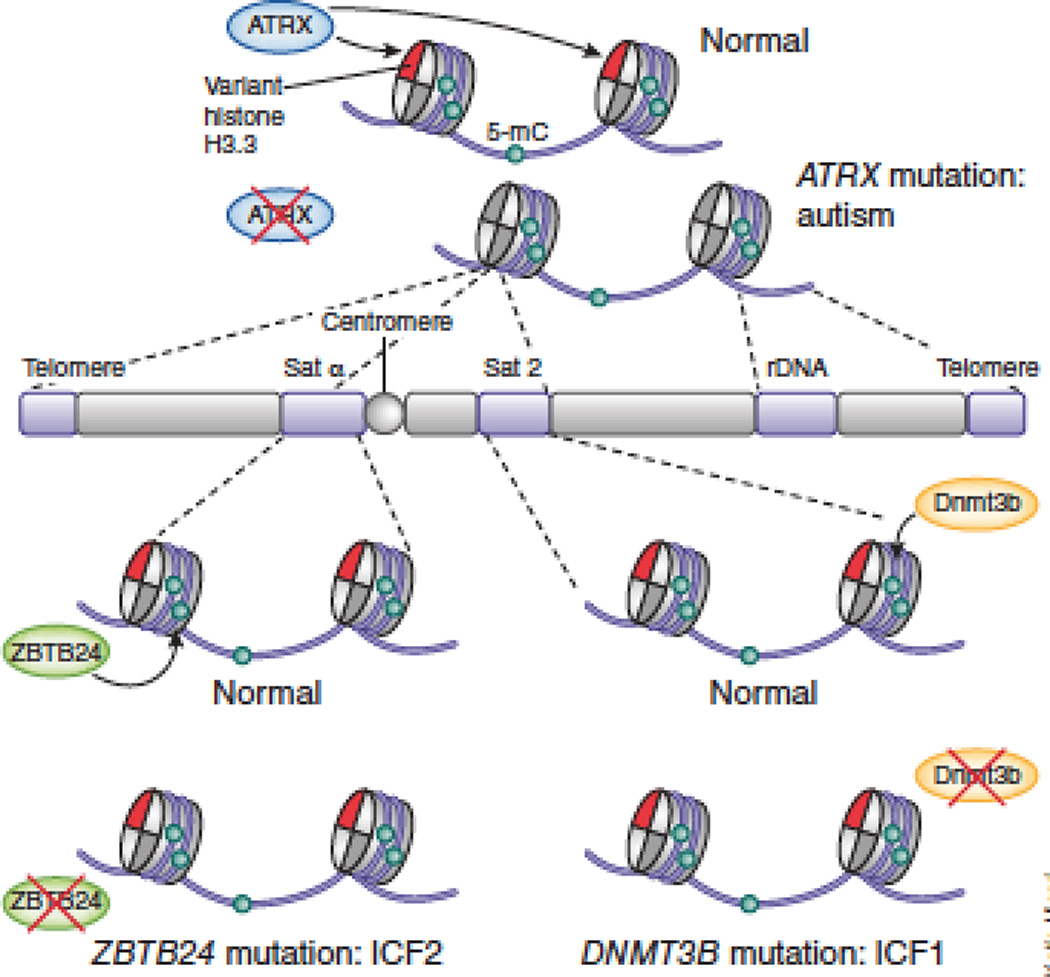
Three examples of monogenetic brain disorders associated with defects in heterochromatin are shown. Mutations in genes encoding the histone demethylase DNMT3B (associated with the disorder ICF1) or the transcriptional repressor ZBTB24 (associated with the disorder ICF2) result in hypo(DNA mC5) methylation of different types of pericentric satellite repeats28,106. The multifunctional chromatin regulator ATRX controls the incorporation of the variant histone H3.3 not only just into nucleosomes surrounding the transcription start sites of active genes but also at various repeat sequences, as indicated. Loss of function mutations in ATRX have been associated with various X-linked mental retardation syndromes.
Acknowledgement
Work in the authors laboratory is supported by funds from the National Institutes of Health (NINDS, NIMH, NIDA), DARPA and Autism Speaks.
REFERENCES
- 1.Numata S, et al. DNA methylation signatures in development and aging of the human prefrontal cortex. Am J Hum Genet. 2012;90:260–272. doi: 10.1016/j.ajhg.2011.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Siegmund KD, et al. DNA methylation in the human cerebral cortex is dynamically regulated throughout the life span and involves differentiated neurons. PLoS One. 2007;2:e895. doi: 10.1371/journal.pone.0000895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hernandez DG, et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum Mol Genet. 2011;20:1164–1172. doi: 10.1093/hmg/ddq561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheung I, et al. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci U S A. 2010;107:8824–8829. doi: 10.1073/pnas.1001702107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klein CJ, et al. Mutations in DNMT1 cause hereditary sensory neuropathy with dementia and hearing loss. Nat Genet. 2011;43:595–600. doi: 10.1038/ng.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Winkelmann J, et al. Mutations in DNMT1 cause autosomal dominant cerebellar ataxia, deafness and narcolepsy. Hum Mol Genet. 2012 doi: 10.1093/hmg/dds035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peter CJ, Akbarian S. Balancing histone methylation activities in psychiatric disorders. Trends Mol Med. 2011;17:372–379. doi: 10.1016/j.molmed.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chuang DM, Leng Y, Marinova Z, Kim HJ, Chiu CT. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009;32:591–601. doi: 10.1016/j.tins.2009.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baltan S, Murphy SP, Danilov CA, Bachleda A, Morrison RS. Histone deacetylase inhibitors preserve white matter structure and function during ischemia by conserving ATP and reducing excitotoxicity. J Neurosci. 2011;31:3990–3999. doi: 10.1523/JNEUROSCI.5379-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer A, Sananbenesi F, Mungenast A, Tsai LH. Targeting the correct HDAC(s) to treat cognitive disorders. Trends Pharmacol Sci. 2010;31:605–617. doi: 10.1016/j.tips.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 11.Tsou AY, Friedman LS, Wilson RB, Lynch DR. Pharmacotherapy for Friedreich ataxia. CNS Drugs. 2009;23:213–223. doi: 10.2165/00023210-200923030-00003. [DOI] [PubMed] [Google Scholar]
- 12.Day JJ, Sweatt JD. DNA methylation and memory formation. Nat Neurosci. 2010;13:1319–1323. doi: 10.1038/nn.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Erraji-Benchekroun L, et al. Molecular aging in human prefrontal cortex is selective and continuous throughout adult life. Biol Psychiatry. 2005;57:549–558. doi: 10.1016/j.biopsych.2004.10.034. [DOI] [PubMed] [Google Scholar]
- 14.Tang B, et al. Normal human aging and early-stage schizophrenia share common molecular profiles. Aging Cell. 2009;8:339–342. doi: 10.1111/j.1474-9726.2009.00468.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lu T, et al. Gene regulation and DNA damage in the ageing human brain. Nature. 2004;429:883–891. doi: 10.1038/nature02661. [DOI] [PubMed] [Google Scholar]
- 16.Szulwach KE, et al. 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat Neurosci. 2011;14:1607–1616. doi: 10.1038/nn.2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stadler F, et al. Histone methylation at gene promoters is associated with developmental regulation and region-specific expression of ionotropic and metabotropic glutamate receptors in human brain. J Neurochem. 2005;94:324–336. doi: 10.1111/j.1471-4159.2005.03190.x. [DOI] [PubMed] [Google Scholar]
- 18.Wang CM, Tsai SN, Yew TW, Kwan YW, Ngai SM. Identification of histone methylation multiplicities patterns in the brain of senescence-accelerated prone mouse 8. Biogerontology. 2010;11:87–102. doi: 10.1007/s10522-009-9231-5. [DOI] [PubMed] [Google Scholar]
- 19.Peleg S, et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science. 2010;328:753–756. doi: 10.1126/science.1186088. [DOI] [PubMed] [Google Scholar]
- 20.Hargreaves DC, Horng T, Medzhitov R. Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 2009;138:129–145. doi: 10.1016/j.cell.2009.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Copray S, Huynh JL, Sher F, Casaccia-Bonnefil P, Boddeke E. Epigenetic mechanisms facilitating oligodendrocyte development, maturation, and aging. Glia. 2009;57:1579–1587. doi: 10.1002/glia.20881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yankner BA, Lu T, Loerch P. The aging brain. Annu Rev Pathol. 2008;3:41–66. doi: 10.1146/annurev.pathmechdis.2.010506.092044. [DOI] [PubMed] [Google Scholar]
- 23.Ho L, Crabtree GR. Chromatin remodelling during development. Nature. 2010;463:474–484. doi: 10.1038/nature08911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fuentes P, Canovas J, Berndt FA, Noctor SC, Kukuljan M. CoREST/LSD1 Control the Development of Pyramidal Cortical Neurons. Cereb Cortex. 2011 doi: 10.1093/cercor/bhr218. [DOI] [PubMed] [Google Scholar]
- 25.Hansen RS, et al. The DNMT3B DNA methyltransferase gene is mutated in the ICF immunodeficiency syndrome. Proc Natl Acad Sci U S A. 1999;96:14412–14417. doi: 10.1073/pnas.96.25.14412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- 27.Jin B, et al. DNA methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet. 2008;17:690–709. doi: 10.1093/hmg/ddm341. [DOI] [PubMed] [Google Scholar]
- 28.de Greef JC, et al. Mutations in ZBTB24 are associated with immunodeficiency, centromeric instability, and facial anomalies syndrome type 2. Am J Hum Genet. 2011;88:796–804. doi: 10.1016/j.ajhg.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chouery E, et al. A novel deletion in ZBTB24 in a Lebanese family with Immunodeficiency, Centromeric Instability, and Facial Anomalies Syndrome Type 2. Clin Genet. 2011 doi: 10.1111/j.1399-0004.2011.01783.x. [DOI] [PubMed] [Google Scholar]
- 30.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 31.Ramocki MB, et al. Autism and other neuropsychiatric symptoms are prevalent in individuals with MeCP2 duplication syndrome. Ann Neurol. 2009;66:771–782. doi: 10.1002/ana.21715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Agarwal N, et al. MeCP2 Rett mutations affect large scale chromatin organization. Hum Mol Genet. 2011 doi: 10.1093/hmg/ddr346. [DOI] [PubMed] [Google Scholar]
- 33.Singleton MK, et al. MeCP2 is required for global heterochromatic and nucleolar changes during activity-dependent neuronal maturation. Neurobiol Dis. 2011;43:190–200. doi: 10.1016/j.nbd.2011.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Akbarian S, et al. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. 2001;8:784–791. doi: 10.1006/nbdi.2001.0420. [DOI] [PubMed] [Google Scholar]
- 35.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 36.Skene PJ, et al. Neuronal MeCP2 is expressed at near histone-octamer levels and globally alters the chromatin state. Mol Cell. 2010;37:457–468. doi: 10.1016/j.molcel.2010.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865–873. doi: 10.1016/0092-8674(92)90561-p. [DOI] [PubMed] [Google Scholar]
- 38.Easwaran HP, Schermelleh L, Leonhardt H, Cardoso MC. Replication-independent chromatin loading of Dnmt1 during G2 and M phases. EMBO Rep. 2004;5:1181–1186. doi: 10.1038/sj.embor.7400295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan G, et al. DNA hypomethylation perturbs the function and survival of CNS neurons in postnatal animals. J Neurosci. 2001;21:788–797. doi: 10.1523/JNEUROSCI.21-03-00788.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Berube NG, et al. The chromatin-remodeling protein ATRX is critical for neuronal survival during corticogenesis. J Clin Invest. 2005;115:258–267. doi: 10.1172/JCI22329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seah C, et al. Neuronal death resulting from targeted disruption of the Snf2 protein ATRX is mediated by p53. J Neurosci. 2008;28:12570–12580. doi: 10.1523/JNEUROSCI.4048-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shioda N, et al. Aberrant calcium/calmodulin-dependent protein kinase II (CaMKII) activity is associated with abnormal dendritic spine morphology in the ATRX mutant mouse brain. J Neurosci. 2011;31:346–358. doi: 10.1523/JNEUROSCI.4816-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kaufmann WE, Moser HW. Dendritic anomalies in disorders associated with mental retardation. Cereb Cortex. 2000;10:981–991. doi: 10.1093/cercor/10.10.981. [DOI] [PubMed] [Google Scholar]
- 44.Law MJ, et al. ATR-X syndrome protein targets tandem repeats and influences allele-specific expression in a size-dependent manner. Cell. 2010;143:367–378. doi: 10.1016/j.cell.2010.09.023. [DOI] [PubMed] [Google Scholar]
- 45.Lewis PW, Elsaesser SJ, Noh KM, Stadler SC, Allis CD. Daxx is an H3.3-specific histone chaperone and cooperates with ATRX in replication-independent chromatin assembly at telomeres. Proc Natl Acad Sci U S A. 2010;107:14075–14080. doi: 10.1073/pnas.1008850107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eustermann S, et al. Combinatorial readout of histone H3 modifications specifies localization of ATRX to heterochromatin. Nat Struct Mol Biol. 2011;18:777–782. doi: 10.1038/nsmb.2070. [DOI] [PubMed] [Google Scholar]
- 47.Baumann C, Viveiros MM, De La Fuente R. Loss of maternal ATRX results in centromere instability and aneuploidy in the mammalian oocyte and pre-implantation embryo. PLoS Genet. 2010;6 doi: 10.1371/journal.pgen.1001137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Steffan JS, et al. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- 49.Ferrante RJ, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington's disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Desplats P, et al. Alpha-synuclein sequesters Dnmt1 from the nucleus: a novel mechanism for epigenetic alterations in Lewy body diseases. J Biol Chem. 2011;286:9031–9037. doi: 10.1074/jbc.C110.212589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chestnut BA, et al. Epigenetic Regulation of Motor Neuron Cell Death through DNA Methylation. J Neurosci. 2011;31:16619–16636. doi: 10.1523/JNEUROSCI.1639-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Shock LS, Thakkar PV, Peterson EJ, Moran RG, Taylor SM. DNA methyltransferase 1, cytosine methylation, and cytosine hydroxymethylation in mammalian mitochondria. Proc Natl Acad Sci U S A. 2011;108:3630–3635. doi: 10.1073/pnas.1012311108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nucifora FC, Jr, et al. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- 54.Caccamo A, Maldonado MA, Bokov AF, Majumder S, Oddo S. CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2010;107:22687–22692. doi: 10.1073/pnas.1012851108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wilburn B, et al. An antisense CAG repeat transcript at JPH3 locus mediates expanded polyglutamine protein toxicity in Huntington's disease-like 2 mice. Neuron. 2011;70:427–440. doi: 10.1016/j.neuron.2011.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kazemi-Esfarjani P, La Spada AR. Deja vu with a twist: transglutaminases in bioenergetics and transcriptional dysfunction in Huntington's disease. EMBO Mol Med. 2010;2:335–337. doi: 10.1002/emmm.201000092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Munsie L, et al. Mutant huntingtin causes defective actin remodeling during stress: defining a new role for transglutaminase 2 in neurodegenerative disease. Hum Mol Genet. 2011;20:1937–1951. doi: 10.1093/hmg/ddr075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McConoughey SJ, et al. Inhibition of transglutaminase 2 mitigates transcriptional dysregulation in models of Huntington disease. EMBO Mol Med. 2010;2:349–370. doi: 10.1002/emmm.201000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ryu H, et al. ESET/SETDB1 gene expression and histone H3 (K9) trimethylation in Huntington's disease. Proc Natl Acad Sci U S A. 2006;103:19176–19181. doi: 10.1073/pnas.0606373103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu Y, et al. Transcriptional modulator H2A histone family, member Y (H2AFY) marks Huntington disease activity in man and mouse. Proc Natl Acad Sci U S A. 2011;108:17141–17146. doi: 10.1073/pnas.1104409108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stack EC, et al. Modulation of nucleosome dynamics in Huntington's disease. Hum Mol Genet. 2007;16:1164–1175. doi: 10.1093/hmg/ddm064. [DOI] [PubMed] [Google Scholar]
- 62.Morris MJ, Karra AS, Monteggia LM. Histone deacetylases govern cellular mechanisms underlying behavioral and synaptic plasticity in the developing and adult brain. Behav Pharmacol. 2010;21:409–419. doi: 10.1097/FBP.0b013e32833c20c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Covington HE, 3rd, et al. Antidepressant actions of histone deacetylase inhibitors. J Neurosci. 2009;29:11451–11460. doi: 10.1523/JNEUROSCI.1758-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Schroeder FA, Lin CL, Crusio WE, Akbarian S. Antidepressant-like effects of the histone deacetylase inhibitor, sodium butyrate, in the mouse. Biol Psychiatry. 2007;62:55–64. doi: 10.1016/j.biopsych.2006.06.036. [DOI] [PubMed] [Google Scholar]
- 65.Zhao W, et al. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dokmanovic M, Marks PA. Prospects: histone deacetylase inhibitors. J Cell Biochem. 2005;96:293–304. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
- 67.Jeong H, et al. Sirt1 mediates neuroprotection from mutant huntingtin by activation of the TORC1 and CREB transcriptional pathway. Nat Med. 2011 doi: 10.1038/nm.2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Libert S, et al. SIRT1 Activates MAO-A in the Brain to Mediate Anxiety and Exploratory Drive. Cell. 2011;147:1459–1472. doi: 10.1016/j.cell.2011.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rohrer JD, et al. Clinical and neuroanatomical signatures of tissue pathology in frontotemporal lobar degeneration. Brain. 2011;134:2565–2581. doi: 10.1093/brain/awr198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cenik B, et al. Suberoylanilide hydroxamic acid (vorinostat) up-regulates progranulin transcription: rational therapeutic approach to frontotemporal dementia. J Biol Chem. 2011;286:16101–16108. doi: 10.1074/jbc.M110.193433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Min SW, et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron. 2010;67:953–966. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell. 2010;142:320–332. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 73.Green KN, et al. Nicotinamide restores cognition in Alzheimer's disease transgenic mice via a mechanism involving sirtuin inhibition and selective reduction of Thr231-phosphotau. J Neurosci. 2008;28:11500–11510. doi: 10.1523/JNEUROSCI.3203-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Outeiro TF, et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317:516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
- 75.Hsieh J, Nakashima K, Kuwabara T, Mejia E, Gage FH. Histone deacetylase inhibition-mediated neuronal differentiation of multipotent adult neural progenitor cells. Proc Natl Acad Sci U S A. 2004;101:16659–16664. doi: 10.1073/pnas.0407643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pedre X, et al. Changed histone acetylation patterns in normal-appearing white matter and early multiple sclerosis lesions. J Neurosci. 2011;31:3435–3445. doi: 10.1523/JNEUROSCI.4507-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu J, Casaccia P. Epigenetic regulation of oligodendrocyte identity. Trends Neurosci. 2010;33:193–201. doi: 10.1016/j.tins.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Coufal NG, et al. Ataxia telangiectasia mutated (ATM) modulates long interspersed element-1 (L1) retrotransposition in human neural stem cells. Proc Natl Acad Sci U S A. 2011;108:20382–20387. doi: 10.1073/pnas.1100273108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rahman S, et al. The Brd4 extraterminal domain confers transcription activation independent of pTEFb by recruiting multiple proteins, including NSD3. Mol Cell Biol. 2011;31:2641–2652. doi: 10.1128/MCB.01341-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Delmore JE, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–917. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Filippakopoulos P, et al. Selective inhibition of BET bromodomains. Nature. 2010;468:1067–1073. doi: 10.1038/nature09504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nicodeme E, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010;468:1119–1123. doi: 10.1038/nature09589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lein ES, et al. Genome-wide atlas of gene expression in the adult mouse brain. Nature. 2007;445:168–176. doi: 10.1038/nature05453. [DOI] [PubMed] [Google Scholar]
- 84.Kubicek S, et al. Reversal of H3K9me2 by a small-molecule inhibitor for the G9a histone methyltransferase. Mol Cell. 2007;25:473–481. doi: 10.1016/j.molcel.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 85.Maze I, et al. Essential role of the histone methyltransferase G9a in cocaine-induced plasticity. Science. 2010;327:213–216. doi: 10.1126/science.1179438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kelly TK, De Carvalho DD, Jones PA. Epigenetic modifications as therapeutic targets. Nat Biotechnol. 2010;28:1069–1078. doi: 10.1038/nbt.1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Levenson JM, et al. Evidence that DNA (cytosine-5) methyltransferase regulates synaptic plasticity in the hippocampus. J Biol Chem. 2006;281:15763–15773. doi: 10.1074/jbc.M511767200. [DOI] [PubMed] [Google Scholar]
- 88.Han J, et al. Effect of 5-aza-2-deoxycytidine microinjecting into hippocampus and prelimbic cortex on acquisition and retrieval of cocaine-induced place preference in C57BL/6 mice. Eur J Pharmacol. 2010;642:93–98. doi: 10.1016/j.ejphar.2010.05.050. [DOI] [PubMed] [Google Scholar]
- 89.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53:857–869. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 90.LaPlant Q, et al. Dnmt3a regulates emotional behavior and spine plasticity in the nucleus accumbens. Nat Neurosci. 2010;13:1137–1143. doi: 10.1038/nn.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lubin FD, Roth TL, Sweatt JD. Epigenetic regulation of BDNF gene transcription in the consolidation of fear memory. J Neurosci. 2008;28:10576–10586. doi: 10.1523/JNEUROSCI.1786-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Miller CA, et al. Cortical DNA methylation maintains remote memory. Nat Neurosci. 2010;13:664–666. doi: 10.1038/nn.2560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Endres M, Fan G, Meisel A, Dirnagl U, Jaenisch R. Effects of cerebral ischemia in mice lacking DNA methyltransferase 1 in post-mitotic neurons. Neuroreport. 2001;12:3763–3766. doi: 10.1097/00001756-200112040-00032. [DOI] [PubMed] [Google Scholar]
- 94.Endres M, et al. DNA methyltransferase contributes to delayed ischemic brain injury. J Neurosci. 2000;20:3175–3181. doi: 10.1523/JNEUROSCI.20-09-03175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Salerno S, et al. Recent advances in the development of dual topoisomerase I and II inhibitors as anticancer drugs. Curr Med Chem. 2010;17:4270–4290. doi: 10.2174/092986710793361252. [DOI] [PubMed] [Google Scholar]
- 96.Huang HS, et al. Topoisomerase inhibitors unsilence the dormant allele of Ube3a in neurons. Nature. 2011 doi: 10.1038/nature10726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bressler J, et al. The SNRPN promoter is not required for genomic imprinting of the Prader-Willi/Angelman domain in mice. Nat Genet. 2001;28:232–240. doi: 10.1038/90067. [DOI] [PubMed] [Google Scholar]
- 98.Reik W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature. 2007;447:425–432. doi: 10.1038/nature05918. [DOI] [PubMed] [Google Scholar]
- 99.Kishino T, Lalande M, Wagstaff J. UBE3A/E6-AP mutations cause Angelman syndrome. Nat Genet. 1997;15:70–73. doi: 10.1038/ng0197-70. [DOI] [PubMed] [Google Scholar]
- 100.Matsuura T, et al. De novo truncating mutations in E6-AP ubiquitin-protein ligase gene (UBE3A) in Angelman syndrome. Nat Genet. 1997;15:74–77. doi: 10.1038/ng0197-74. [DOI] [PubMed] [Google Scholar]
- 101.Sutcliffe JS, et al. The E6-Ap ubiquitin-protein ligase (UBE3A) gene is localized within a narrowed Angelman syndrome critical region. Genome Res. 1997;7:368–377. doi: 10.1101/gr.7.4.368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Porteus MH, Baltimore D. Chimeric nucleases stimulate gene targeting in human cells. Science. 2003;300:763. doi: 10.1126/science.1078395. [DOI] [PubMed] [Google Scholar]
- 103.Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333:1843–1846. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 104.Miskiewicz K, et al. ELP3 controls active zone morphology by acetylating the ELKS family member Bruchpilot. Neuron. 2011;72:776–788. doi: 10.1016/j.neuron.2011.10.010. [DOI] [PubMed] [Google Scholar]
- 105.Roelfsema JH, Peters DJ. Rubinstein-Taybi syndrome: clinical and molecular overview. Expert Rev Mol Med. 2007;9:1–16. doi: 10.1017/S1462399407000415. [DOI] [PubMed] [Google Scholar]
- 106.Ehrlich M, et al. ICF, an immunodeficiency syndrome: DNA methyltransferase 3B involvement, chromosome anomalies, and gene dysregulation. Autoimmunity. 2008;41:253–271. doi: 10.1080/08916930802024202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Michelson DJ, et al. Evidence report: Genetic and metabolic testing on children with global developmental delay: report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology. 2011;77:1629–1635. doi: 10.1212/WNL.0b013e3182345896. [DOI] [PubMed] [Google Scholar]
- 108.Adegbola A, Gao H, Sommer S, Browning M. A novel mutation in JARID1C/SMCX in a patient with autism spectrum disorder (ASD) Am J Med Genet A. 2008;146A:505–511. doi: 10.1002/ajmg.a.32142. [DOI] [PubMed] [Google Scholar]
- 109.Kleefstra T, et al. Further clinical and molecular delineation of the 9q subtelomeric deletion syndrome supports a major contribution of EHMT1 haploinsufficiency to the core phenotype. J Med Genet. 2009;46:598–606. doi: 10.1136/jmg.2008.062950. [DOI] [PubMed] [Google Scholar]
- 110.Kirov G, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Verhoeven WM, Egger JI, Vermeulen K, van de Warrenburg BP, Kleefstra T. Kleefstra syndrome in three adult patients: further delineation of the behavioral and neurological phenotype shows aspects of a neurodegenerative course. Am J Med Genet A. 2011;155A:2409–2415. doi: 10.1002/ajmg.a.34186. [DOI] [PubMed] [Google Scholar]
- 112.Berdasco M, et al. Epigenetic inactivation of the Sotos overgrowth syndrome gene histone methyltransferase NSD1 in human neuroblastoma and glioma. Proc Natl Acad Sci U S A. 2009;106:21830–21835. doi: 10.1073/pnas.0906831106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kleine-Kohlbrecher D, et al. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol Cell. 2010;38:165–178. doi: 10.1016/j.molcel.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Fortschegger K, et al. PHF8 targets histone methylation and RNA polymerase II to activate transcription. Mol Cell Biol. 2010;30:3286–3298. doi: 10.1128/MCB.01520-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pereira PM, Schneider A, Pannetier S, Heron D, Hanauer A. Coffin-Lowry syndrome. Eur J Hum Genet. 2010;18:627–633. doi: 10.1038/ejhg.2009.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Percy AK. Rett syndrome: exploring the autism link. Arch Neurol. 2011;68:985–989. doi: 10.1001/archneurol.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nat Med. 2011;17:330–339. doi: 10.1038/nm.2305. [DOI] [PubMed] [Google Scholar]
- 118.Li G, Reinberg D. Chromatin higher-order structures and gene regulation. Curr Opin Genet Dev. 2011;21:175–186. doi: 10.1016/j.gde.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929–930. doi: 10.1126/science.1169786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jin SG, Wu X, Li AX, Pfeifer GP. Genomic mapping of 5-hydroxymethylcytosine in the human brain. Nucleic Acids Res. 2011;39:5015–5024. doi: 10.1093/nar/gkr120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Song CX, et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68–72. doi: 10.1038/nbt.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maunakea AK, et al. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature. 2010;466:253–257. doi: 10.1038/nature09165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Tan M, et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell. 2011;146:1016–1028. doi: 10.1016/j.cell.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 125.Taverna SD, Li H, Ruthenburg AJ, Allis CD, Patel DJ. How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol. 2007;14:1025–1040. doi: 10.1038/nsmb1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12:7–18. doi: 10.1038/nrg2905. [DOI] [PubMed] [Google Scholar]
- 127.Woodcock CL. Chromatin architecture. Curr Opin Struct Biol. 2006;16:213–220. doi: 10.1016/j.sbi.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 128.Jin C, Felsenfeld G. Nucleosome stability mediated by histone variants H3.3 and H2A.Z. Genes Dev. 2007;21:1519–1529. doi: 10.1101/gad.1547707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Woodcock CL, Skoultchi AI, Fan Y. Role of linker histone in chromatin structure and function: H1 stoichiometry and nucleosome repeat length. Chromosome Res. 2006;14:17–25. doi: 10.1007/s10577-005-1024-3. [DOI] [PubMed] [Google Scholar]
- 130.Pearson EC, Bates DL, Prospero TD, Thomas JO. Neuronal nuclei and glial nuclei from mammalian cerebral cortex. Nucleosome repeat lengths, DNA contents and H1 contents. Eur J Biochem. 1984;144:353–360. doi: 10.1111/j.1432-1033.1984.tb08471.x. [DOI] [PubMed] [Google Scholar]
- 131.Ghosh RP, Horowitz-Scherer RA, Nikitina T, Shlyakhtenko LS, Woodcock CL. MeCP2 binds cooperatively to its substrate and competes with histone H1 for chromatin binding sites. Mol Cell Biol. 2010;30:4656–4670. doi: 10.1128/MCB.00379-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Henikoff S, Shilatifard A. Histone modification: cause or cog? Trends Genet. 2011 doi: 10.1016/j.tig.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 133.Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Day JJ, Sweatt JD. Epigenetic mechanisms in cognition. Neuron. 2011;70:813–829. doi: 10.1016/j.neuron.2011.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Guo JU, Su Y, Zhong C, Ming GL, Song H. Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell. 2011;145:423–434. doi: 10.1016/j.cell.2011.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bhutani N, Burns DM, Blau HM. DNA demethylation dynamics. Cell. 2011;146:866–872. doi: 10.1016/j.cell.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Copeland RA, Solomon ME, Richon VM. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 2009;8:724–732. doi: 10.1038/nrd2974. [DOI] [PubMed] [Google Scholar]
- 138.Rotili D, Mai A. Targeting Histone Demethylases: A New Avenue for the Fight against Cancer. Genes & Cancer. 2011 doi: 10.1177/1947601911417976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Vermeulen M, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 140.Chao HT, Zoghbi HY. MeCP2: only 100% will do. Nat Neurosci. 2012;15:176–177. doi: 10.1038/nn.3027. [DOI] [PubMed] [Google Scholar]
- 141.Samaco RC, et al. Crh and Oprm1 mediate anxiety-related behavior and social approach in a mouse model of MECP2 duplication syndrome. Nat Genet. 2012;44:206–211. doi: 10.1038/ng.1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Goffin D, et al. Rett syndrome mutation MeCP2 T158A disrupts DNA binding, protein stability and ERP responses. Nat Neurosci. 2011;15:274–283. doi: 10.1038/nn.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Tao J, et al. Phosphorylation of MeCP2 at Serine 80 regulates its chromatin association and neurological function. Proc Natl Acad Sci U S A. 2009;106:4882–4887. doi: 10.1073/pnas.0811648106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Li H, Zhong X, Chau KF, Williams EC, Chang Q. Loss of activity-induced phosphorylation of MeCP2 enhances synaptogenesis, LTP and spatial memory. Nat Neurosci. 2011;14:1001–1008. doi: 10.1038/nn.2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Kazazian HH., Jr Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- 146.Cordaux R, Batzer MA. The impact of retrotransposons on human genome evolution. Nat Rev Genet. 2009;10:691–703. doi: 10.1038/nrg2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Baillie JK, et al. Somatic retrotransposition alters the genetic landscape of the human brain. Nature. 2011;479:534–537. doi: 10.1038/nature10531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Coufal NG, et al. L1 retrotransposition in human neural progenitor cells. Nature. 2009;460:1127–1131. doi: 10.1038/nature08248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Singer T, McConnell MJ, Marchetto MC, Coufal NG, Gage FH. LINE-1 retrotransposons: mediators of somatic variation in neuronal genomes? Trends Neurosci. 2010;33:345–354. doi: 10.1016/j.tins.2010.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Muotri AR, Zhao C, Marchetto MC, Gage FH. Environmental influence on L1 retrotransposons in the adult hippocampus. Hippocampus. 2009;19:1002–1007. doi: 10.1002/hipo.20564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Maze I, et al. Cocaine dynamically regulates heterochromatin and repetitive element unsilencing in nucleus accumbens. Proc Natl Acad Sci U S A. 2011;108:3035–3040. doi: 10.1073/pnas.1015483108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- 153.Muotri AR, et al. Somatic mosaicism in neuronal precursor cells mediated by L1 retrotransposition. Nature. 2005;435:903–910. doi: 10.1038/nature03663. [DOI] [PubMed] [Google Scholar]
- 154.Kuwabara T, et al. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat Neurosci. 2009;12:1097–1105. doi: 10.1038/nn.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Muotri AR, et al. L1 retrotransposition in neurons is modulated by MeCP2. Nature. 2010;468:443–446. doi: 10.1038/nature09544. [DOI] [PMC free article] [PubMed] [Google Scholar]