

Abstract
The potency of clinical-grade T cells can be improved by combining gene therapy with immunotherapy to engineer a biologic product with the potential for superior (i) recognition of tumor-associated antigens (TAAs), (ii) persistence after infusion, (iii) potential for migration to tumor sites, and (iv) ability to recycle effector functions within the tumor microenvironment. Most approaches to genetic manipulation of T cells engineered for human application have used retrovirus and lentivirus for the stable expression of CAR1-3. This approach, although compliant with current good manufacturing practice (GMP), can be expensive as it relies on the manufacture and release of clinical-grade recombinant virus from a limited number of production facilities. The electro-transfer of nonviral plasmids is an appealing alternative to transduction since DNA species can be produced to clinical grade at approximately 1/10th the cost of recombinant GMP-grade virus. To improve the efficiency of integration we adapted Sleeping Beauty (SB) transposon and transposase for human application4-8. Our SB system uses two DNA plasmids that consist of a transposon coding for a gene of interest (e.g. 2nd generation CD19-specific CAR transgene, designated CD19RCD28) and a transposase (e.g. SB11) which inserts the transgene into TA dinucleotide repeats9-11. To generate clinically-sufficient numbers of genetically modified T cells we use K562-derived artificial antigen presenting cells (aAPC) (clone #4) modified to express a TAA (e.g. CD19) as well as the T cell costimulatory molecules CD86, CD137L, a membrane-bound version of interleukin (IL)-15 (peptide fused to modified IgG4 Fc region) and CD64 (Fc-γ receptor 1) for the loading of monoclonal antibodies (mAb)12. In this report, we demonstrate the procedures that can be undertaken in compliance with cGMP to generate CD19-specific CAR+ T cells suitable for human application. This was achieved by the synchronous electro-transfer of two DNA plasmids, a SB transposon (CD19RCD28) and a SB transposase (SB11) followed by retrieval of stable integrants by the every-7-day additions (stimulation cycle) of γ-irradiated aAPC (clone #4) in the presence of soluble recombinant human IL-2 and IL-2113. Typically 4 cycles (28 days of continuous culture) are undertaken to generate clinically-appealing numbers of T cells that stably express the CAR. This methodology to manufacturing clinical-grade CD19-specific T cells can be applied to T cells derived from peripheral blood (PB) or umbilical cord blood (UCB). Furthermore, this approach can be harnessed to generate T cells to diverse tumor types by pairing the specificity of the introduced CAR with expression of the TAA, recognized by the CAR, on the aAPC.
Keywords: Immunology, Issue 72, Cellular Biology, Medicine, Molecular Biology, Cancer Biology, Biomedical Engineering, Hematology, Biochemistry, Genetics, T-Lymphocytes, Antigen-Presenting Cells, Leukemia, Lymphoid, Lymphoma, Antigens, CD19, Immunotherapy, Adoptive, Electroporation, Genetic Engineering, Gene Therapy, Sleeping Beauty, CD19, T cells, Chimeric Antigen Receptor, Artificial Antigen Presenting Cells, Clinical Trial, Peripheral Blood, Umbilical Cord Blood, Cryopreservation, Electroporation
Protocol
Day 0 or Before
1. Isolation of Mononuclear Cells (MNC) from PB and UCB
Dilute PB with equal volume, and UCB with four volumes of PBS-EDTA.
Slowly layer diluted blood (25 ml) onto Ficoll (12 ml) in a 50 ml centrifuge tube(s) and centrifuge at 400 x g for 30 - 40 min (no brake).
Collect and transfer the mononuclear cell fraction (interface) using a transfer pipette to a fresh 50 ml centrifuge tube.
Bring up the volume to 50 ml with PBS-EDTA and centrifuge at 450 x g for 10 min.
Aspirate the supernatant, gently re-suspend the cell pellet(s) in 50 ml of Complete Culture Media (CCM) and, centrifuge at 400 x g for 10 min.
Gently re-suspend and pool the cell pellets in CCM and perform a cell count using Trypan blue exclusion method (Cellometer, PBMC program).
MNC can now be used for electroporation (Nucleofection) or cryopreserved for future use.
2. Preparation of T Cells for Electroporation on Day 0
If using cryopreserved MNC, quickly thaw sufficient cells for a full scale electroporation (2 x 108 adding ~20% to account for cell loss during centrifugation and 2 hr incubation) in 37 °C water bath. If using freshly isolated MNC proceed to step 2.3.
Gently re-suspend and transfer cells to an appropriately sized centrifuge tube(s) contained pre-warmed Complete Phenol-Free RPMI culture media (PF-RPMI) and centrifuge at 200 x g for 10 min (no brake), aspirate supernatant.
Re-suspend the MNC in PF-RPMI, perform a cell count (Cellometer) and transfer cells to an appropriately sized cell culture vessel(s) at a concentration of 106 cells/ml.
Incubate in a humidified 37 °C/5% CO2 incubator for 2 hr ± 30 min.
Transfer the MNC to sterile centrifuge tube(s), spin at 200 x g for 5 min (no brakes), aspirate the supernatant and gently re-suspend and combine the cell pellet(s) in PF-RPMI.
Perform a cell count (Cellometer) and calculate the volume of the cell suspension required (2 x 108 MNC).
Transfer the calculated volume to a sterile 50 ml centrifuge tube and spin at 200 x g for 10 min (no brake).
Aspirate the supernatant so that no residual media remains and gently re-suspend by tapping side of tube.
3. Electroporation (Nucleofection) of MNC (Full Scale Process Using 10 Cuvettes) on Day 0
Pre-incubate a sterile 12-well plate with 10 wells containing 4 ml of warm PF-RPMI in a humidified 37 °C/5% CO2 incubator.
Prepare and pre-warm the Lonza Nucleofector Solution Human T cell kit (reconstituted per manufacturer's instructions, www.lonza.com) to ambient temperature in a Biosafety Cabinet (BSC).
Prepare Nucleofector solution/DNA master mix by adding 100 μl of supplemented Nucleofector solution, 15 μg of transposon (supercoiled DNA plasmid designated as CD19RCD28/pSBSO) and 5 μg of transposase (supercoiled DNA plasmid designated as pCMV-SB11) per reaction/cuvette.
Disperse the cell pellet (from step 2.8) by gently tapping the side of the centrifuge tube and re-suspend in Nucleofection solution/DNA master mix (Final cell concentration: 2 x 107 cells/100 μl).
Carefully transfer 100 μl of the cell suspension (from step 3.4) to each of ten (10) Lonza Nucleofection cuvettes, being careful to avoid bubbles.
Tap the cuvette once, and electroporate using program U-014 (for unstimulated T cells).
Transfer the cuvettes and the 12-well plate (Step 3.1) to the BSC.
Harvest the electroporated cells from each cuvette using an Amaxa fine tip transfer pipette, by adding ~500 μl of the pre-warmed culture medium from the corresponding well (12-well plate prepared in Step 3.1) and return the plate to a humidified 37 °C/5% CO2 incubator for 2 hr ± 30 min.
Following the 2-hr incubation, harvest and transfer the cells from all wells to a sterile centrifuge tube.
Wash cells by centrifugation at 140 x g for 8 min, ambient temperature, no brake and aspirate and discard the supernatant so that no residual medium covers the cell pellet.
Disperse the cell pellet by gently tapping the side of the centrifuge tube and gently re-suspend in CCM to achieve a single cell suspension.
Perform cell count and adjust cell concentration to 106 cells/ml in CCM.
Transfer cell suspension to cell culture flask(s) and place in incubator overnight.
Process steps 2.1 to 3.8 were repeated for control: EGFP-transfected cells (5 x 106 cells/cuvette with 5 μg Amaxa control EGFP supercoiled plasmid, pmaxGFP).
Day 1 of 1st and Subsequent Stimulation Cycles
4. Analysis of CAR Expression by Flow Cytometry on Day 1
Harvest the electroporated cells and perform a cell count using Trypan blue exclusion method (Hemocytometer).
Stain cells (1 to 2 x 106) with antibody specific for CD3, CD4, CD8, and human IgG Fcγ (as a measurement of CAR expression).
- Acquire cells on FACS Calibur and analyze the data using FCS Express software to calculate expression of CAR.
- Calculate CAR+ cells in culture by the formula: (No. of Total viable cells) x (% CAR+ cells) = No. of CAR+ cells
5. Preparation of aAPC (clone #4) on Day 1. aAPC (clone #4) were Derived from K562 Cells (Parental Line Obtained from American Type Culture Collection) to Co-express Desired T Cell Co-stimulatory Molecule
Thaw an aliquot of frozen 100 Gy irradiated aAPC in a 37 °C water bath.
Cells are washed twice by centrifugation at 400 x g, 10 min in CCM and counted using Cellometer (Trypan blue exclusion).
Calculate number of viable aAPC required for stimulation: (No. of CAR+ cells) x 2 = No. of irradiated aAPC required
6. aAPC-mediated Stimulation of CAR+ T Cells on Day 1 Beginning of 1st and Subsequent Stimulation Cycles
- Mix electroporated cells (expressing CAR) and γ-irradiated aAPC (clone #4) in a sterile container at a ratio of 1:2 (CAR+ cell: viable aAPC) in CCM.
- Note the aAPC ratio is adjusted for the expression of CAR based on flow cytometry the day after electroporation.
Add IL-21 (30 ng/ml) to the cell suspension.
Aliquot in T-75 cm2 flask(s) and/or Vue Life Culture bags at a concentration of 106 cells/ml and return to the incubator.
Days 3, 5
7. Continued Culture of CAR+ T Cells
Perform a half-media change, replenish IL-21, and maintain T cells at a concentration of 106 cells/ml.
Day 7
8. End of First aAPC-mediated Stimulation Cycle
Harvest cells, count and stain for CD3, CD4, CD8, and Fcγ (CAR) and proceed to step 10.1.
9. Depletion of CD56+ Cells (Usually between 7 and 14 days after electroporation)
Perform a CD56 depletion using paramagnetic beads if CD56+CD3neg lymphocytes ≥ 10%.
Stimulation Cycles #2, #3, & #4 Corresponding to Days 8 → 14, Days 15 → 21, & Days 22 → 28
10. Recursive Addition of aAPC to Propagate T Cells to Clinically-sufficient Numbers
Repeat the stimulation process (4 times) as described in steps 4.3-8.1.
Add IL-2 (50U/ml) to the cultures beginning on Days 7, 14 and 21, & then at each media change (three times a week, on a Monday-Wednesday-Friday schedule).
- Cryopreserve (archive) excess T cells as needed.
- T cells frozen using controlled rate freezer.
Day 28
11. End of Last aAPC-mediated Stimulation Cycle: Harvest T Cells
Cryopreserve T cells for release testing and infusion.
Representative Results
We report that electro-transfer of DNA plasmids and propagation of T cells on γ-irradiated aAPC can be used to generate clinically-appealing numbers of T cells derived from PB and UCB for human applications. These genetically modified T cells express an introduced CAR that recognizes the TAA CD19, independent of major histocompatibility complex. The SB-derived DNA plasmids to express the (i) transposon, a 2nd generation CAR (CD19RCD28) that signals through CD28 and CD3-ε14, and (ii) transposase, SB1115, have been previously described13,16,17. The plasmids used in the current study were produced commercially by Waisman Clinical Biomanufacturing Facility (Madison, WI). The aAPC (clone #4), derived from K562 cells (parental line obtained from American Type Culture Collection), co-express desired T cell co-stimulatory molecules (each introduced molecule at 390% on cell surface of aAPC), as previously described12. Here we show that CD19-specific T cells could be generated from mononuclear cells (MNC) derived from PB or UCB using SB transposition to introduce the CAR followed by addition of aAPC to numerically expand the T cells in a CAR-dependent manner (Figures 1, 4)13,18. Ten cuvettes (2x107 MNC/cuvette) are electroporated for each recipient using 15 μg of DNA plasmid (CD19RCD28/pSBSO) coding for transposon (CAR) and 5 μg of DNA plasmid (pCMV-SB11) coding for transposase (SB11). The number of cuvettes can be reduced if MNC are limiting or scaled back for laboratory work. The day of electroporation is defined as "Day 0" of Stimulation cycle #1. As controls for flow cytometry and culture conditions, autologous T cells are mock electroporated (without DNA plasmid) and numerically expanded on γ-irradiated aAPC (clone #4) that had been pre-loaded with OKT3 to cross-link CD3 to sustain T cell proliferation. We routinely assess the efficiency of electrotransfer and viability of the T cells the day after electroporation (Figure 2B). The expression of EGFP from control DNA plasmid (designated pmaxGFP) and CAR at this initial time point reflects protein expression from the integrated and episomal plasmid. Typically, the day after electroporation we measure EGFP expression at ~60% and CAR expression at ~40% (Figure 2A) with T cell viability between 40-50%. Recursive additions of γ-irradiated aAPC in the presence of soluble recombinant human IL-2 and IL-21 retrieve T cells stably expressing CAR (CD19RCD28). CD3negCD56+ NK cells are depleted from the culture using CD56-specific paramagnetic beads if the percentage of these NK cells is ≥10% and especially if the percentage of CAR expressed on the T cells is low. This depletion prevents the rapid overgrowth of NK cells which interferes with the ability of aAPC to sustain the proliferation of CAR+ T cells. On occasion, depletion of NK cells from CAR+ T cells is undertaken during the last two stimulation cycles, but this introduces a loss of desired cells due to co-expression of CD56 on some CAR+ T cells. The T cells were grown in a functionally closed system using Vue Life culture bags past Day 14. A subset of the genetically modified and propagated T cells are typically cryopreserved at Day 14 or Day 21 (end of Stimulation cycles #2 or #3) of co-culture on aAPC to serve as a source of archived material for future analyses and to be thawed if unanticipated problems subsequently occur during the manufacturing process. T cells are typically harvested on or about Day 28 of culture (Figure 3) that routinely express >90% CAR and are >80% viable (Figure 2C, D). We have previously shown that, after four weeks of co-culture on aAPC the average fold-expansion of CD3+ T cells is 19,800±11,313 with CAR+ expression being 90%±7.5 13. These T cells are cryopreserved and undergo in-process and release testing that informs on the safety and therapeutic potential of the manufactured product. Release testing is undertaken in compliance with clinical laboratory improvement amendments (CLIA) to generate a certificate of analysis prior to infusion into recipients on clinical trials.
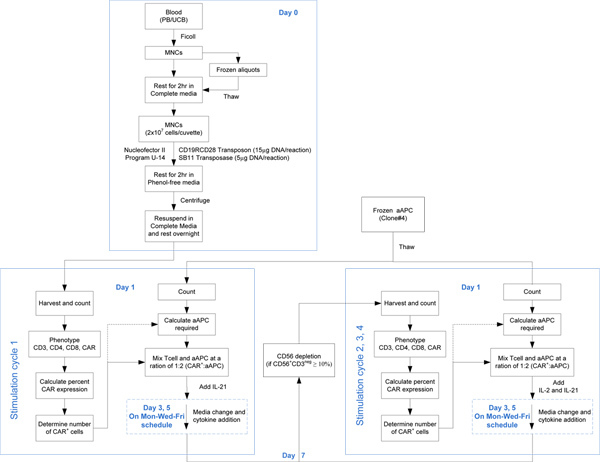 Figure 1. Steps outlining the process to electroporate and propagate CAR+ T cells from PB and UCB. Click here to view larger figure.
Figure 1. Steps outlining the process to electroporate and propagate CAR+ T cells from PB and UCB. Click here to view larger figure.
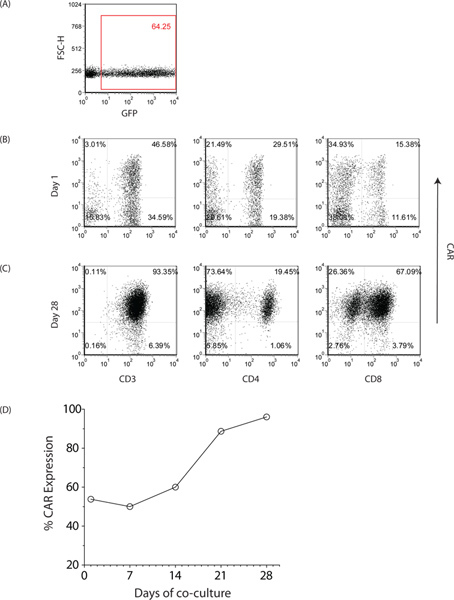 Figure 2. Characterization of genetically modified T cells from PB. (A) Expression of EGFP at Day 0 of first stimulation cycle to assess the efficiency of gene transfer. Expression of CD19-specific CAR (CD19RCD28) as assessed by flow cytometry on CD3+, CD8+ and CD4+ T cells at (B) approximately 24 hr after electroporation and (C) 28 days after co-culture on aAPC. Similar expression of CAR was observed with UCB-derived T cells. (D) Kinetics of CAR expression. Click here to view larger figure.
Figure 2. Characterization of genetically modified T cells from PB. (A) Expression of EGFP at Day 0 of first stimulation cycle to assess the efficiency of gene transfer. Expression of CD19-specific CAR (CD19RCD28) as assessed by flow cytometry on CD3+, CD8+ and CD4+ T cells at (B) approximately 24 hr after electroporation and (C) 28 days after co-culture on aAPC. Similar expression of CAR was observed with UCB-derived T cells. (D) Kinetics of CAR expression. Click here to view larger figure.
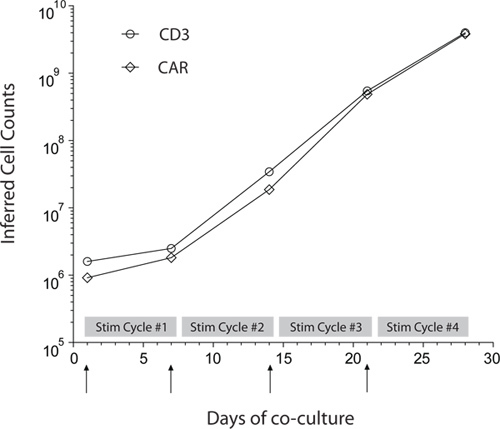 Figure 3. Propagation of PB-derived CAR+ T cells. Rate of numeric expansion of CD3+ and CAR+ T cells derived from PB by repeated co-culture on γ-irradiated aAPC in presence of recombinant human soluble IL-2 and IL-21. Upward arrows indicate the additions of γ-irradiated aAPC that mark the beginning of each Stimulation cycle. UCB-derived CAR+ T cells exhibit similar rates of numeric expansion.
Figure 3. Propagation of PB-derived CAR+ T cells. Rate of numeric expansion of CD3+ and CAR+ T cells derived from PB by repeated co-culture on γ-irradiated aAPC in presence of recombinant human soluble IL-2 and IL-21. Upward arrows indicate the additions of γ-irradiated aAPC that mark the beginning of each Stimulation cycle. UCB-derived CAR+ T cells exhibit similar rates of numeric expansion.
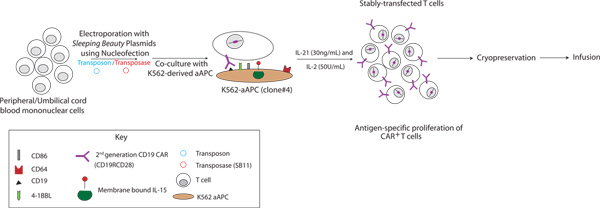 Figure 4. Schematic of the manufacturing process using SB and aAPC systems to genetically modify and propagate CAR+ T cells derived from PB and UCB. CD19-specific CAR+ T cells were generated by electro-transfer of SB-derived supercoiled DNA plasmids and subsequent co-culture on K562-derived aAPC (clone #4) in the presence of recombinant human soluble IL-2 and IL-21. Click here to view larger figure.
Figure 4. Schematic of the manufacturing process using SB and aAPC systems to genetically modify and propagate CAR+ T cells derived from PB and UCB. CD19-specific CAR+ T cells were generated by electro-transfer of SB-derived supercoiled DNA plasmids and subsequent co-culture on K562-derived aAPC (clone #4) in the presence of recombinant human soluble IL-2 and IL-21. Click here to view larger figure.
| T-cell source | Transposon* | Transposase | T-cell infusion | IRB # | NIH-OBA # | IND # |
| Autologous (patient-derived)** | CD19RCD28 | SB11 | After autologous hematopoietic stem-cell transplantation | 2007-0635 | 0804-922 | 14193 |
| Allogeneic (donor-derived) | CD19RCD28 | SB11 | After allogeneic hematopoietic stem-cell transplantation | 2009-0525 | 0910-1003 | 14577 |
| Allogeneic (donor-derived) | CD19RCD28 | SB11 | After allogeneic umbilical cord blood transplantation | 2010-0835 | 1001-1022 | 14739 |
Table 1. Clinical trials under the auspices of the FDA at MDACC to infuse CD19-specific CAR+ T-cells propagated on aAPC. *T-cells are rendered specific for CD19 through enforced expression of SB transposon coding for a 2nd generation CAR, designated CD19RCD28, that signals through CD28 and CD3-z. **Trial described in reference # 7
Discussion
Transposon and transposase systems, such as from piggyBac12,19 and SB18,20-22, are non-viral approaches to gene therapy that are an alternative to viral-mediated transduction of clinical grade CAR+ T cells. The SB was chosen as the gene transfer system based on its potential for human gene therapy1,6,23. We developed the dual technologies of SB transposition (to introduce a CAR) and recursive addition of γ-irradiated aAPC (to retrieve genetically modified T cells stably expressing a CAR) to serve as platform technologies in the manufacture of TAA-specific T cells in compliance with cGMP for Phase I/II trials (Figure 4). After 28 days (four 7-day stimulation cycles) of co-culture on γ-irradiated aAPC, we are typically able to generate at least ~1010 genetically modified T cells suitable for human applications. As needed, additional stimulation cycles can be undertaken to generate larger numbers of genetically modified T cells. Furthermore, if less CAR+ T cells are needed, the approach to electroporation and propagation can be scaled back employing fewer cuvettes and carrying forward just a sub-set of the numerically expanded T cells for subsequent rounds of proliferation on aAPC at the beginning of each Stimulation cycle. The majority of the electroporated and propagated T cells harvested for infusion stably express the CAR. The outgrowth of CD4+ and CD8+ T cells expressing our 2nd generation CAR include cells with a memory/naive phenotype and exhibit three hallmarks of re-directed specificity. Firstly, the genetically modified T cells specifically lyse CD19+ targets, secondly, produce IFN-γ in response to CD19+ stimulator cells and thirdly, proliferate in response to CD19+ feeder cells, all in a CAR-dependent manner13,18. Our approach to integrate a CAR transgene by electro-transfer of non-viral DNA plasmids from the SB system can be undertaken in quiescent primary T cells derived from PB and UCB. We and others have genetically modified K562 cells to serve as aAPC to efficiently propagate clinically-sufficient numbers of T cells for infusion24,25. The aAPC and tissue culture environment (e.g. the addition of IL-21) have been have been modified to generate patient- and donor-derived CD19-specific T cells for infusion after hematopoietic stem-cell transplantation (Table 1)13,18. We can produce CAR+ T cells from PB simply obtained by venipuncture which avoids the cost, discomfort, and inconvenience of obtaining MNC from PB by apheresis. The ability to derive large numbers of CAR+ T cells from small numbers of MNC is particularly appealing for infusing T cells after allogeneic UCB transplantation. The small size and anonymity of the neonatal donor precludes re-accessing this individual at a later time point and only limited numbers of harvested MNC are available as starting material for T cell manufacture to avoid interfering with hematopoiesis. Further advances to the manufacturing process are currently underway to include a high throughput electroporation device coupled with a fully closed WAVE bioreactor to minimize handling. In aggregate, the SB and aAPC are appealing platforms to generate CD19-specific CAR+ T cells that can be adapted to generate large numbers of genetically modified T cells that can recognize alternative cell-surface TAAs in compliance with cGMP.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors would like to thank Dr. Carl June (University of Pennsylvania) for help generating and providing aAPC clone #4 and Dr. Perry Hackett (University of Minnesota) for help with the SB system.
Grant support: Cancer Center Core Grant (CA16672); RO1 (CA124782, CA120956, CA141303); R33 (CA116127); P01 (CA148600); SPORE (CA136411); Albert J Ward Foundation; Burroughs Wellcome Fund; Gillson Longenbaugh Foundation; Cancer Prevention and Research Institute of Texas; CLL Global Research Foundation; Department of Defense; Estate of Noelan L. Bibler; Harry T. Mangurian, Jr., Fund for Leukemia Immunotherapy; Institute of Personalized Cancer Therapy; Leukemia and Lymphoma Society; Lymphoma Research Foundation; MDACC's Sister Institution Network Fund; Miller Foundation; Mr. Herb Simons; Mr. and Mrs. Joe H. Scales; Mr. Thomas Scott; National Foundation for Cancer Research; Pediatric Cancer Research Foundation; Production Assistance for Cellular Therapies (PACT); William Lawrence and Blanche Hughes Children's Foundation.
References
- Jena B, Dotti G, Cooper LJ. Redirecting T-cell specificity by introducing a tumor-specific chimeric antigen receptor. Blood. 2010;116:1035–1044. doi: 10.1182/blood-2010-01-043737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ertl HC, Zaia J, Rosenberg SA, et al. Considerations for the clinical application of chimeric antigen receptor T cells: observations from a recombinant DNA Advisory Committee Symposium held. Cancer Res. 2010;71:3175–3181. doi: 10.1158/0008-5472.CAN-10-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn DB, Dotti G, Brentjens R, et al. CARs on track in the clinic. Mol. Ther. 2011;19:432–438. doi: 10.1038/mt.2011.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronovich EL, McIvor RS, Hackett PB. The Sleeping Beauty transposon system: a non-viral vector for gene therapy. Hum. Mol Genet. 2011;20:R14–R20. doi: 10.1093/hmg/ddr140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett PB, Largaespada DA, Cooper LJ. A transposon and transposase system for human application. Mol. Ther. 2010;18:674–683. doi: 10.1038/mt.2010.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izsvak Z, Hackett PB, Cooper LJ, Ivics Z. Translating Sleeping Beauty transposition into cellular therapies: victories and challenges. Bioessays. 2010;32:756–767. doi: 10.1002/bies.201000027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebriaei P, Huls H, Bipulendu J, et al. Infusing CD19-directed T cells to augment disease control in patients undergoing autologous hematopoietic stem-cell transplantation for advanced B-lymphoid malignancies. Hum. Gene Ther. 2012;23(5):444–450. doi: 10.1089/hum.2011.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams DA. Sleeping beauty vector system moves toward human trials in the United States. Mol. Ther. 2008;16:1515–1516. doi: 10.1038/mt.2008.169. [DOI] [PubMed] [Google Scholar]
- Geurts AM, Hackett CS, Bell JB, et al. Structure-based prediction of insertion-site preferences of transposons into chromosomes. Nucleic Acids Res. 2006;34:2803–2811. doi: 10.1093/nar/gkl301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivics Z, Hackett PB, Plasterk RH, Izsvak Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell. 1997;91:501–510. doi: 10.1016/s0092-8674(00)80436-5. [DOI] [PubMed] [Google Scholar]
- Izsvak Z, Ivics Z. Sleeping beauty transposition: biology and applications for molecular therapy. Mol Ther. 2004;9:147–156. doi: 10.1016/j.ymthe.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Manuri PV, Wilson MH, Maiti SN, et al. piggyBac transposon/transposase system to generate CD19-specific T cells for the treatment of B-lineage malignancies. Hum. Gene Ther. 2010;21:427–437. doi: 10.1089/hum.2009.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Figliola MJ, Dawson MJ, et al. Reprogramming CD19-specific T cells with IL-21 signaling can improve adoptive immunotherapy of B-lineage malignancies. Cancer Res. 2011;71:3516–3527. doi: 10.1158/0008-5472.CAN-10-3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowolik CM, Topp MS, Gonzalez S, et al. CD28 costimulation provided through a CD19-specific chimeric antigen receptor enhances in vivo persistence and antitumor efficacy of adoptively transferred T cells. Cancer Res. 2006;66:10995–11004. doi: 10.1158/0008-5472.CAN-06-0160. [DOI] [PubMed] [Google Scholar]
- Jin Z, Maiti S, Huls H, et al. The hyperactive Sleeping Beauty transposase SB100X improves the genetic modification of T cells to express a chimeric antigen receptor. Gene Ther. 2011;18:849–856. doi: 10.1038/gt.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies JK, Singh H, Huls H, et al. Combining CD19 redirection and alloanergization to generate tumor-specific human T cells for allogeneic cell therapy of B-cell malignancies. Cancer Res. 2010;70:3915–3924. doi: 10.1158/0008-5472.CAN-09-3845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kebriaei P, Kelly SS, Manuri P, et al. CARs: driving T-cell specificity to enhance anti-tumor immunity. Front. Biosci. (Schol. Ed) 2012;4:520–531. doi: 10.2741/282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Manuri PR, Olivares S, et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008;68:2961–2971. doi: 10.1158/0008-5472.CAN-07-5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakazawa Y, Huye LE, Salsman VS, et al. PiggyBac-mediated cancer immunotherapy using EBV-specific cytotoxic T-cells expressing HER2-specific chimeric antigen receptor. Mol. Ther. 2011;19:2133–2143. doi: 10.1038/mt.2011.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang G, Yu L, Cooper LJ, Hollomon M, Huls H, Kleinerman ES. Genetically modified T cells targeting interleukin-11 receptor alpha-chain kill human osteosarcoma cells and induce the regression of established osteosarcoma lung metastases. Cancer Res. 2012;72:271–281. doi: 10.1158/0008-5472.CAN-11-2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Guo H, Kang J, et al. Sleeping Beauty transposon-mediated engineering of human primary T cells for therapy of CD19+ lymphoid malignancies. Mol. Ther. 2008;16:580–589. doi: 10.1038/sj.mt.6300404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Wilber A, McIvor RS, Zhou X. DNA transposons for modification of human primary T lymphocytes. Methods Mol. Biol. 2009;506:115–126. doi: 10.1007/978-1-59745-409-4_9. [DOI] [PubMed] [Google Scholar]
- Hackett PB, Jr, Aronovich EL, Hunter D, et al. Efficacy and safety of Sleeping Beauty transposon-mediated gene transfer in preclinical animal studies. Curr. Gene Ther. 2011;11:341–349. doi: 10.2174/156652311797415827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suhoski MM, Golovina TN, Aqui NA, et al. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol. Ther. 2007;15:981–988. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numbenjapon T, Serrano LM, Singh H, et al. Characterization of an artificial antigen-presenting cell to propagate cytolytic CD19-specific T cells. Leukemia. 2006;20:1889–1892. doi: 10.1038/sj.leu.2404329. [DOI] [PubMed] [Google Scholar]