

Abstract
Angiogenic therapy, which involves the use of an exogenous stimulus to promote blood vessel growth, is an attractive approach for the treatment of ischemic diseases. It has been shown in animal models that the stimulation of blood vessel growth leads to the growth of the whole vascular tree, improvement of ischemic tissue perfusion and improved muscle aerobic energy metabolism. However, very few positive results have been gained from Phase 2 and 3 clinical angiogenesis trials. Many reasons have been given for the failures of clinical trials, including poor transgene expression (in gene-therapy trials) and instability of the vessels induced by therapy. In this Review, we discuss the selection of preclinical models as one of the main reasons why clinical translation has been unsuccessful thus far. This issue has received little attention, but could have had dramatic implications on the expectations of clinical trials. We highlight crucial differences between human patients and animal models with regards to blood flow and pressure, as well as issues concerning the chronic nature of ischemic diseases in humans. We use these as examples to demonstrate why the results from preclinical trials might have overestimated the efficacy of angiogenic therapies developed to date. We also suggest ways in which currently available animal models of ischemic disease could be improved to better mimic human disease conditions, and offer advice on how to work with existing models to avoid overestimating the efficacy of new angiogenic therapies.
Introduction
Ischemic diseases: the clinical picture
Ischemic diseases make up a group of cardiovascular diseases that result from inadequate oxygenation of tissues such as the heart (causing coronary artery disease), brain (cerebrovascular disease) and peripheral muscles (peripheral arterial disease). The principal pathophysiological process causing ischemic diseases is atherosclerosis. Atherosclerosis is a progressive disease that usually affects large arteries, in which the accumulation of lipids, inflammatory cells and fibrous material in the inner arterial wall culminates in the formation of stenotic and occlusive lesions (Lusis, 2000). Humans with significant stenosis of the arterial lumen experience reversible ischemic pain during exercise, which manifests as stable angina pectoris or intermittent claudication. Progression of the stenosis is associated with ischemic pain at rest, which manifests as unstable angina pectoris or critical chronic limb ischemia. Acute and life-threatening cardiovascular events, such as myocardial infarction and stroke, can occur upon rupture of an atherosclerotic lesion and subsequent atherothrombotic events.
Conventional treatment strategies
Treatment of ischemic diseases is strongly based on prevention of disease progression. Primary prevention involves addressing lifestylerelated issues such as smoking, a sedentary lifestyle, poor diet and being overweight. Primary and secondary prevention involves pharmacological management of cardiovascular risk factors such as hyperglycemia, hyperlipidemia and hypertension (Norgren et al., 2007). Although statins have been reported to decrease atherosclerotic plaque size and improve the function of the vascular endothelium (Davignon, 2004; Ylä-Herttuala et al., 2011), no pharmacological treatment exists to treat the ischemic tissue. In patients with intermittent ischemic symptoms, anticoagulants, vasodilators and exercise training can be used to relieve symptoms. Revascularization procedures, such as percutaneous transluminal angioplasty, intravascular catheter-mediated thrombolysis, thrombendarterectomy or bypass surgery are performed in patients with critical symptoms to improve blood circulation (Gibbons et al., 2003; Antman et al., 2004; Norgren et al., 2007). However, many patients cannot be treated with conventional revascularization strategies because of a poor overall health status or underlying comorbidities. Moreover, a substantial portion of patients undergoing revascularization procedures does not benefit from the treatments or experiences restenosis, resulting in poor prognosis and diminished quality of life. These challenges have encouraged the search for novel therapeutic alternatives to treat ischemic diseases.
Concept of angiogenic therapy
The essential role of angiogenesis in the growth and recovery of tissues gave birth to the concept of angiogenic therapy – i.e. promoting blood vessel growth as a potential therapeutic approach for the treatment of ischemic diseases (Folkman, 1971). The aim of angiogenic therapy in ischemic diseases is to stimulate blood vessel growth in areas of poor vascularization in an attempt to increase blood supply, and to support tissue function and recovery. Blood vessel growth can be induced by three means: angiogenesis, arteriogenesis and vasculogenesis (Fig. 1). Angiogenesis is defined as the process of capillary vessel growth, and can occur by: (1) proliferation of pre-existing vascular endothelial cells into new capillary sprouts (sprouting angiogenesis); (2) enlargement of preexisting capillaries; or (3) by intussusception or bridging of preexisting vessels into smaller daughter vessels (splitting angiogenesis) (Fig. 1) (Risau, 1997; Rissanen et al., 2005; Makanya et al., 2009; Carmeliet and Jain, 2011). Arteriogenesis is defined as the process of growth of collateral arteries, whereby pre-existing arterioles enlarge and remodel into large vessels that bypass the arterial occlusion (Simons, 2005; Schaper, 2009). Arteriogenesis is initiated by blood-pressure- and shear-stress-mediated mechanisms, whereas angiogenesis is triggered by hypoxia, resulting in the production of vascular growth factors in the ischemic tissue. In ischemic diseases, both angiogenesis and arteriogenesis take place endogenously. Similarly, during angiogenic therapies, stimulation of both angiogenesis and arteriogenesis might be beneficial (Rissanen et al., 2005). Whereas angiogenesis and arteriogenesis are well known to take place in adult tissues, by definition vasculogenesis (i.e. the de novo formation of the vascular plexus from endothelial precursor cells) is usually considered to take place only during early development. However, the incorporation of vascular stem or progenitor cells into vessel structures (i.e. postnatal vasculogenesis) has gained attention in some studies of angiogenic cell therapies. Thus, the relevance of vasculogenesis in adult tissues remains unclear (Asahara et al., 1997; Rafii and Lyden, 2003; Balsam et al., 2004; Ziegelhoeffer et al., 2004; Fang and Salven, 2011).
Fig. 1.
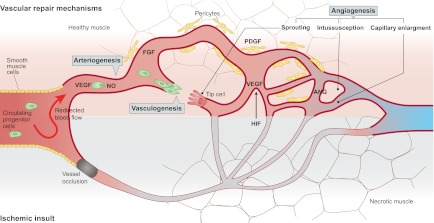
Post-ischemic vascular repair mechanisms and the growth factors involved. The top half of the diagram shows angiogenic vascular repair processes that can take place after an ischemic insult (arterial occlusion), which is displayed in the bottom half of the diagram. Upon an arterial occlusion, arteriogenesis (i.e. collateral growth) is induced by the redirection of blood flow, causing increased shear stress and subsequent cytokine production in the vascular endothelium. Factors such as VEGF and NO are responsible for the enlargement and growth of the collaterals, whereas factors such as PDGF and FGF mediate the stabilization of the vessels by recruiting pericytes. The hypoxic tissue distal to the occlusion (bottom half of the diagram) expresses transcription factors such as HIF, which enables the production of angiogenic proteins such as VEGF and ANG, which are involved in the modulation of the distal vasculature to make connections to the opening collaterals (angiogenesis). Angiogenesis can include sprouting, intussusception or capillary enlargement (see text for details). Postnatal vasculogenesis might also contribute to post-ischemic vascular repair via the incorporation of circulating endothelial progenitor cells into the forming vascular structures.
Methods for inducing blood vessel growth
Stimulation of vascular growth can be achieved by exogenous administration of pro-angiogenic agents such as members of the vascular endothelial growth factor family (notably VEGF-A165 and the mature form of VEGF-D) (Ferrara, 2004; Ylä-Herttuala et al., 2007), members of the fibroblast growth factor family (notably FGF- 2 and FGF-4) (Ylä-Herttuala and Alitalo, 2003; Murakami and Simons, 2008), members of the platelet-derived growth factor family (notably PDGF-BB) (Fredriksson et al., 2004), angiopoetins (notably ANG-1) (Davis et al., 1996), hepatocyte growth factor (HGF) (Aoki et al., 2000; Rissanen and Ylä-Herttuala, 2007), insulin-like growth factors (IGFs) (Delafontaine et al., 2004), members of the hypoxiainducible factor family (notably HIF-1α) (Pajusola et al., 2005; Patel et al., 2005) and nitric oxide synthase (NOS) (Brevetti et al., 2003b; Cooney et al., 2006), among others (Fig. 1).
To deliver the angiogenic agents, three main approaches have been tested in preclinical settings: protein, gene and cell therapies. In protein therapy, recombinant proteins are used directly to induce therapeutic effects (Ruel and Sellke, 2003). However, a major limitation of this approach is the very short half-life of exogenous proteins in target tissues, resulting in only transient therapeutic effects (Annex and Simons, 2005). In contrast, gene therapy uses non-viral or viral vectors to carry a gene construct encoding a therapeutic protein into target tissues, where it is abundantly expressed by the target cells (Ylä-Herttuala and Alitalo, 2003). The idea of cell therapy in its present form is that transplanted cells function as protein factories with the capability of producing multiple endogenous growth factors, meaning that the transplanted cells will induce vascular growth mainly in a paracrine manner, rather than directly replacing damaged cells (Menasché, 2010). However, at least at the preclinical level, the efficacy of cell therapies has not yet reached that of gene therapy to promote vascular growth.
Therapeutic potential of angiogenic factors
The potential of angiogenic therapies to revascularize tissues has been extensively studied in animal models of myocardial and peripheral ischemia (summarized in Table 1). Using these models, preclinical studies using several individual angiogenic factors, including VEGFs, FGFs, HIF-1α and HGF, have showed significant improvements in clinically relevant end points such as increased regional perfusion, improved exercise tolerance and tissue energy metabolism, improved myocardial function, and protection against ischemic damage. Although most of these factors have also been tested in clinical trials, the promising preclinical potential has thus far not been translated into clinical success.
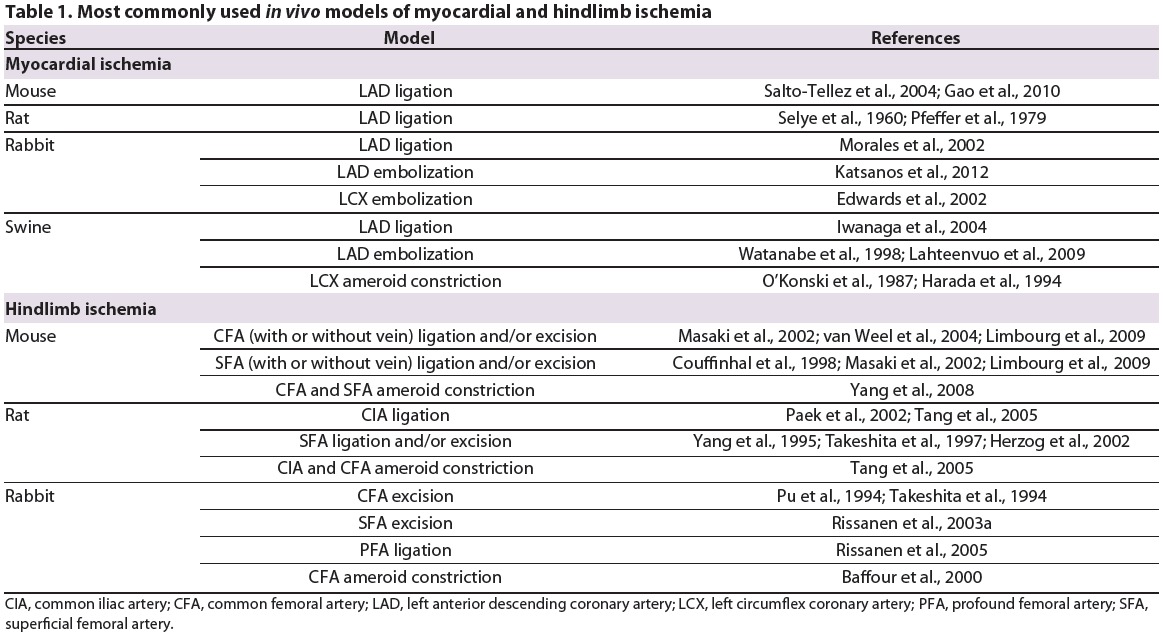
Table 1.
Most commonly used in vivo models of myocardial and hindlimb ischemia.
Testing candidate therapies in models of myocardial ischemia
Animal models have been used to demonstrate the potential of angiogenic therapies for myocardial ischemia. In experimental settings, myocardial ischemia is most commonly induced by ligation of the anterior descending branch of the left coronary artery. The occlusion can be achieved by introducing a cathetermediated embolization coil or a surgical ligature to block the coronary flow, or by surgically placing ameroid constrictors around the coronary vessels to resemble more slowly progressing occlusion (Watanabe et al., 1998; Hughes et al., 2003; Madeddu et al., 2006; Abarbanell et al., 2010; Gao et al., 2010) (Table 1). In a porcine model of myocardial ischemia, intramyocardial injection with a plasmid encoding VEGF caused neoangiogenesis followed by improved regional myocardial perfusion and function (Choi et al., 2006). In another study, plasmid- or adenovirus-mediated VEGF-A gene transfer induced significant post-ischemia neovascularization and improved left ventricular function in a rat model of myocardial infarction (Hao et al., 2007). More recently, adenovirus-mediated overexpression of VEGF-B in a porcine model of myocardial ischemia was shown to induce myocardiumspecific vascular growth, and to improve myocardial perfusion and ejection fraction (Lähteenvuo et al., 2009). Adenoviral gene transfer of FGF-2 was shown to enhance arteriogenesis and echocardiographic parameters of left ventricular function in a porcine model of chronic ischemia (Horvath et al., 2002). In addition, intracoronary injection of adenoviral FGF-4 resulted in improved myocardial perfusion and increased regional function in a porcine model of stress-induced myocardial infarction (Gao et al., 2004).
Testing candidate therapies in models of peripheral ischemia
Angiogenic factors have also shown promise in experimental models of peripheral ischemia. Experimental limb ischemia is typically induced by ligation and/or excision of the common femoral artery or common iliac artery, with or without accompanying ligation of potential collateral sources (Pu et al., 1994; Couffinhal et al., 1998; Waters et al., 2004; Madeddu et al., 2006; Limbourg et al., 2009) (Table 1). In ischemic rabbit hind limbs, plasmid or adenoviral gene transfer of VEGF-A demonstrated neoangiogenesis, resulting in increased skeletal muscle perfusion (Takeshita et al., 1996; Korpisalo et al., 2008a). Additionally, adenoviral VEGF-A was shown to induce the growth of the whole vascular tree, including the growth of collateral arteries (Rissanen et al., 2005). Furthermore, adenoviral gene transfer of either placental growth factor (PIGF; a member of the VEGF family) or VEGF-A improved local perfusion, aerobic energy metabolism and exercise tolerance in ischemic rabbit hind limbs (Gowdak et al., 2000; Korpisalo et al., 2008b). Repeated (but not single) intramuscular injections of plasmid encoding VEGF-A were found to increase microvasculature, resulting in effective protection against ischemic muscle damage (Olea et al., 2009). Furthermore, adenovirus-mediated overexpression of FGF-4 was reported to induce therapeutic angiogenesis and arteriogenesis, increasing local muscle perfusion (Rissanen et al., 2003a). Finally, proteins such as the transcription factor HIF-1α and HGF, which can simultaneously stimulate the expression of multiple growth factors involved in post-ischemic vascular response and tissue recovery, have been reported to achieve more physiological angiogenic responses than individual growth factors alone (Taniyama et al., 2001; Patel et al., 2005; Pyun et al., 2010; Li et al., 2011). Notably, tissue edema has been acknowledged as a side effect of angiogenic treatments (Weis and Cheresh, 2005); growth factors such as the mature form of VEGF-D, capable of stimulating the growth of lymphatic vessels, might help to relieve this side effect (Rissanen et al., 2003b).
Results from clinical trials
Despite these promising results from preclinical studies, clinical trials testing angiogenic therapies have thus far failed to demonstrate clear-cut evidence of therapeutic efficacy: improvements in primary outcome measures such as improved exercise performance, decreased rates of amputation, or decreased mortality have not been observed (Tongers et al., 2008; Belch et al., 2011; Hedman et al., 2011; Mitsos et al., 2012). In addition, evidence of biological activity of the drugs (such as increased angiogenic factor levels in plasma or improved regional perfusion at injection sites) has rarely been reported (Baumgartner et al., 1998; Hedman et al., 2003; Gupta et al., 2009; Muona et al., 2012). However, an acceptable safety profile and improvement in secondary or supportive end points such as symptomatic improvement has been achieved (Baumgartner et al., 1998; Hedman et al., 2003; Gupta et al., 2009; Muona et al., 2012).
Several explanations have been provided to account for the lack of efficacy in clinical trials. Concerning the angiogenic drug, problems including inadequate therapeutic doses, insufficient duration of exposure, compromised delivery and poor vector transduction efficiency have been discussed (Rissanen and Ylä- Herttuala, 2007; Gupta et al., 2009; Hedman et al., 2011; Zachary and Morgan, 2011; Mitsos et al., 2012). In addition, potential patient-related issues proposed to underlie inefficacy include defects in the response to angiogenic stimuli due to existing comorbidities, the use of other medications, circulating angiogenic inhibitors, lack of target receptor expression in target tissues, lack of viable muscle tissue required for a therapeutic response, and growth factor resistance in a chronically ischemic environment (Simons and Ware, 2003; Ylä-Herttuala and Alitalo, 2003; Rissanen and Ylä-Herttuala, 2007; Gupta et al., 2009). However, one of the major shortcomings in this area – and one that has not yet been rigorously acknowledged – is the lack of animal models that accurately recapitulate key features of the human disease. This deficiency has probably contributed to the inadequate and overoptimistic conclusions drawn from preclinical data thus far, and to the poor outcome of drug development. Improved preclinical models, and improved preclinical study design, will be essential prerequisites to move forward in developing treatments for ischemic diseases. We address these challenges in the remainder of this article.
Challenges in modeling ischemic diseases
The ideal animal model of cardiovascular ischemic disease does not currently exist. At one extreme, it can be claimed that a truly accurate model is not possible, given that the clinical disease is so heterogeneous and each patient is unique. In addition to arterial occlusions, patients with ischemic diseases have various comorbidities (Table 2) that can affect the development and severity of atherosclerotic plaques, and the responsiveness of muscle tissue to the subsequent ischemic injury. Thus far, none of the existing animal models of ischemic disease can fully reproduce the comorbidities of chronic ischemia that occur in humans, or are too complicated to be used in preclinical development. Importantly, using simple models to test a new therapy can lead to overly optimistic results of that therapy's potential to treat a complex chronic condition. In the following sections, we discuss the main problems with currently used ischemia models and suggest how models could be improved to better resemble human ischemic diseases.
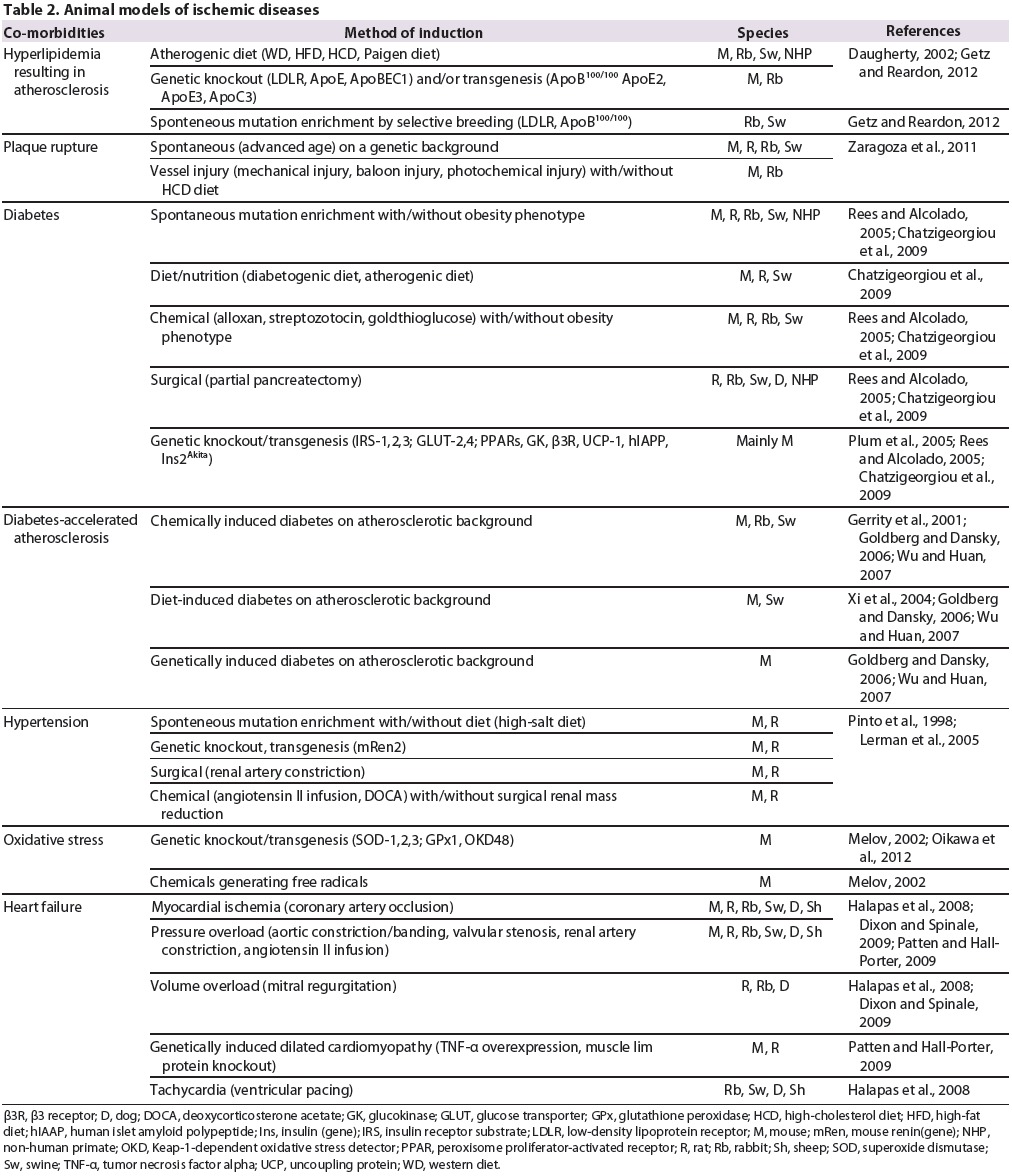
Table 2.
Animal models of ischemic diseases.
Acute ligation models
The occlusion pathophysiology and tissue recovery that occur after an acute arterial ligation are very different in animal models than in human chronic ischemic diseases. An experimental acute vessel occlusion (Fig. 2A) results in an immediate vascular response that generates a powerful, endogenous stimulus for collateral growth (i.e. arteriogenesis) in otherwise healthy animals. This type of acute ischemia induced in young, healthy animals mainly reflects the situation in a limited subgroup of patients (such as young patients with traumatic injuries or arterial emboli), who require immediate medical interventions and are not typically enrolled in angiogenic therapy clinical trials. Under physiological conditions, an acute arterial occlusion causes pre-existing collaterals to function as possible anastomoses to bypass the occlusion (both in animal models and in humans). The occlusion generates a pressure gradient between the proximal and distal ends of the occluded vessel, resulting in a redirection of blood flow towards the collaterals (Fig. 2A). Subsequently, the increased flow generates a dramatic rise in collateral artery wall shear stress, which again alters endothelial gene expression (Pipp et al., 2004). As a primary response, nitric oxide (NO)-induced vessel vasodilatation and flow-mediated arterial remodeling take place, decreasing vascular resistance by increasing the collateral vessel diameter (Heil et al., 2006). Indeed, multiple lines of evidence in the literature illustrate the remarkably fast recovery of perfusion after sudden occlusion of a major artery in animal models of experimental ischemia, suggesting the existence of a strong natural compensatory collateral response (Hershey et al., 2001; Scholz et al., 2002; Buschmann et al., 2003; Tang et al., 2005; Yang et al., 2008; Ziegler et al., 2010). The strong collateralization in animal models provides inflow to ischemic muscles in a way that does not occur in human patients; this could have a major impact on the results of angiogenic studies.
Fig. 2.

Differences in collateral growth and tissue recovery in animal models versus humans with arterial occlusive disease. (A) After an acute arterial ligation (shown here in a mouse), there is a strong pressure gradient between the proximal and distal sides of the occlusion (orange line). This redirects the blood flow into adjacent arterioles and causes a strong shear-stress-mediated opening of collateral channels, which restore blood flow into the hypoxic areas. In the hypoxic tissues, ischemic tissue damage (necrosis) occur if the blood flow is not restored within the first hours after the occlusion. Necrosis induces acute inflammation, and recruited inflammatory cells produce angiogenic cytokines such as VEGF. The hypoxia itself activates factors such as HIF that stimulate the production of VEGF among other factors, and angiogenesis. Distal angiogenesis, along with the growth of collaterals, contributes to the tissue recovery by the formation of connections between the collaterals and the distal capillaries. (B) Upon a gradual occlusion, for example by using an ameroid constrictor, the slow development of the occlusion produces an accommodation window for the collaterals to open. The pressure gradient is never as high as after acute ligation because the blood flow is gradually redirected to the adjacent arterioles, which have time to open to balance the pressure change caused by the gradual occlusion with the constrictor. There is no acute tissue damage, because the collaterals start to compensate for the lack of blood flow before it completely stops. Therefore, there is also less inflammation. The amount of hypoxia and tissue damage varies according to the availability of collaterals and causes comparable distal angiogenic responses. If collaterals are effectively recruited, there is very little tissue hypoxia and therefore little endogenous growth factor upregulation, but also no tissue damage requiring recovery. (C) In human ischemic diseases, atherosclerotic plaques develop all around the arterial tree, causing blood pressure to decrease gradually following each plaque. Thus, a single occlusion in the periphery causes a very small pressure gradient between the proximal and distal sides of the occlusion, as the blood pressure is low already above the occlusion. Accordingly, there is very little blood flow to redirect towards the collaterals and minimal shear stress to open the collaterals, which results in insufficient collateralization. Distal tissue suffers from gradually developing chronic hypoxia that manifests by the replacement of muscle fibers with less-energy-consuming fibrotic tissue and chronic inflammation. The tissue might be less prone to respond to angiogenic stimuli owing to endothelial dysfunction and other factors. The insufficient collaterals and angiogenic signaling limit tissue recovery, eventually leading to total necrosis and loss of tissue function if not effectively treated. Artery names are shown in panel A.
Furthermore, an acute arterial occlusion leading to distal tissue hypoxia causes the activation of an acute inflammatory-angiogenicmyogenic response against ischemic tissue injury (Fig. 2A) (Scholz et al., 2003; Silvestre et al., 2008). The outcome of this response involves: the recruitment and activation of inflammatory cells; VEGF-induced activation, proliferation and migration of endothelial cells; and activation of satellite cells in the periphery of the ischemia-injured myofibers. These events all support the formation of new blood vessels – i.e. angiogenesis and regeneration of the ischemic muscle. Thus, endogenous arteriogenesis and angiogenesis have a profound contribution to the recovery of blood flow and muscle function in animal models with acutely induced ischemic injuries. In contrast, these endogenous recovery mechanisms do not function sufficiently in patients with chronic ischemic symptoms and co-morbidities (Fig. 2C). Thus, acute ligation models should not be used to model chronic ischemia. If they must be used, the results should be interpreted with extreme caution because both vascular and muscular responses to the ischemic insult are very different in these models compared with humans with ischemic diseases. These differences could dramatically affect the effects of angiogenic therapies.
Gradually developing occlusions
Gradually tightening constrictors such as ameroids are often used to model chronic ischemia. Such systems are thought to better recapitulate human ischemic disease pathophysiology and atherosclerotic plaque progression than acute ligations (Tio et al., 1999; Baffour et al., 2000; Tang et al., 2005; Yang et al., 2008). However, a gradually developing occlusion does not cause chronic ischemia in otherwise healthy animals. Instead, the gradual development of the occlusion results in an accommodation window during which the muscle has time to adapt to hypoxia [i.e. to grow collaterals (Fig. 2B). Hence, a smaller amount of necrosis, acute inflammatory responses and endogenous angiogenic stimuli take place than with acute arterial occlusion (Fig. 2B). Thus, ischemia (defined by ischemic tissue injury) might not in fact take place. Instead, gradually developing occlusions might just involve transient tissue hypoxia (Fig. 2B) and thus model a milder form of ischemia. This might be more suitable than an acute ischemia model in certain settings, e.g. for studying hypoxia-sensitive tissues such as the myocardium or brain. In humans with chronic ischemia, the lack of vast necrosis, acute inflammation and endogenous angiogenic stimuli are similar to what is observed in gradual occlusion models. However, the crucial difference between the experimental models and patients is that the patients, owing to their co-morbidities, do not have sufficient growth of collaterals to counteract the evolution of the plaques, despite this accommodation window (Fig. 2C). Thus, the patients have chronic tissue damage whereby necrotic tissue is replaced by fibrotic tissue, and acute inflammation is replaced by chronic inflammation. Furthermore, these patients not only have decreased endogenous angiogenic stimuli, but also diminished angiogenic signaling because there is very little viable tissue left to mediate the effects (Fig. 2C).
Another reason for the insufficient collateralization in patients with ischemic diseases might be stenotic lesions throughout the major feeding arteries: in the legs, the carotid arteries, the aorta and the coronary vessels, among others (Fig. 2C). This results in a gradually decreasing pressure gradient along the arterial tree away from the heart [demonstrated as a decreased ankle-brachial blood pressure index (ABI) in peripheral arterial disease], which might hinder the opening of collateral vessels owing to the lack of shear stress and decreased flow when an arterial occlusion takes place (Fig. 2C). Thus, to better mimic the human ischemic disease, it might be essential to control collateralization in animal models, instead of controlling the speed of occlusion.
Atherosclerotic models
A plain ischemia model is never an ischemic disease model. Unfortunately, most animals do not naturally develop atherosclerotic plaques nor an atherosclerotic ischemic disease. There are several genetic and/or diet-induced models of atherosclerosis and related co-morbidities [involving rodents, rabbits, pigs and even non-human primates (Table 2)], but they are often not suitable for modeling ischemia. For example, the Watanabe heritable hyperlipidemic (WHHL) rabbit, which is genetically prone to hyperlipidemia, develops sporadic, spontaneous atherosclerotic plaques; however, plaque development is slow, and the sporadic nature of the plaques makes it difficult to use this animal as an ischemia model (Watanabe, 1980; Shiomi and Ito, 2009). IGF-II/LDLR−/−ApoB100 mice are hyperlipidemic with type 2 diabetes and exhibit early-onset severe atherosclerotic plaques (Heinonen et al., 2007). However, even with 80-100% stenosis in coronary vessels, the cardiac outcome is normal in these mice owing to adaptive remodeling and myocardial hibernation (Heinonen et al., 2011). Thus, it seems that genetic modifications of two important ischemic risk factors (hyperlipidemia and diabetes) is not enough to mimic human ischemic disease because of compensatory mechanisms that occur in young, otherwise healthy animals. Combining more co-morbidities in a single model might reduce the compensatory responses and create a more human-like disease progression. However, even then, the potential of animals to spontaneously recover from ischemia needs to be well validated, while keeping in mind that patients do not recover in the same way.
Models for endothelial dysfunction and oxidative stress
Impairment in the functional properties of the endothelium (i.e. endothelial dysfunction) has been proposed as another potential cause for the lack of natural regenerative responses in humans suffering from ischemic disease (Kinnaird et al., 2008; Boodhwani and Sellke, 2009; Sun et al., 2009). Endothelial dysfunction seems to be related especially to oxidative stress and the reduction of endothelial NO production and signaling (McQuaid and Keenan, 1997; Boger, 2004; Sun et al., 2009; Vita and Hamburg, 2010). Endothelial dysfunction is also intensified in the presence of different co-morbidities such as diabetes, hyperlipidemia, chronic inflammation or the lack of the nutrifying blood flow (Brevetti et al., 2003a; Münzel et al., 2008; Vita and Hamburg, 2010). Although endothelial dysfunction might be present in many of the current animal models for ischemic diseases, the specific role of oxidative stress on therapeutic outcomes has rarely been explored owing to the confounding effects of different co-morbidities. For example, hyperglycemia in diabetes causes oxidative stress, but might also have direct effects on endothelial dysfunction. Specific knockout models for oxidative stress have been developed (Table 2) and might offer a useful tool for the future study of the specific effects of oxidative stress on angiogenic therapies.
The impact of aging on ischemic disease
Most animal studies have failed to acknowledge the impact of aging on endogenous repair mechanisms and exogenous therapeutic outcomes. Aging is associated with diminished post-ischemic recovery both in humans and animals (Faber et al., 2011; Lähteenvuo and Rosenzweig, 2012). The muscle composition in elderly patients is affected not only by age-related changes, but also by diet, physical activity and hormonal control. This can result in replacement of the muscle tissue with fat and connective tissue, age-related decline of muscle fibers, reduced muscle performance or progressive deficits in metabolic capacity (Rissanen et al., 2002). Although physical activity has been proven to positively influence almost all aspects of cardiovascular diseases, elderly patients are not always able to exercise owing to disease-related pain, tiredness or weakness. In contrast, animals, by evolution, have been programmed to move to ensure survival. Using aged animals would help to better mimic the human disease. However, in animals, the detection of significant atherosclerotic stenoses, before they become life threatening but still cause ischemic symptoms, will require substantial numbers of animals, careful monitoring and further developments in non-invasive imaging techniques.
Predicting clinical potential using currently available animal models
Until the ideal preclinical model is developed, we need to work with our less-than-perfect animal models. Because it will be difficult to create a single animal model that can completely mimic the human ischemic disease, we need to acknowledge that results from a single model will probably not predict how a treatment will work in the clinic. To better predict the potential of new angiogenic therapies in clinical trials, we might thus need to use several different disease models to evaluate the effects of different comorbidities on the therapeutic efficacy in a step-by-step manner, using clinically relevant end points.
Experimental setups
There are some obvious differences between animals and humans with ischemic disease, including physiology, body size and the distance over which collateral arteries need to grow. Currently, these are features that must be controlled by the requirements of clinical trial applications, at least to some extent, by using larger animal models to support data from small rodents before a therapy can be tested in clinics. However, in terms of cardiovascular ischemic diseases, there is no requirement to use disease models – only ischemia models – when applying for permission to test a novel therapeutic agent in clinical trials. In light of the problems with the currently available ischemia models that we have discussed here, there is a clear need for updating the requirements, because the models do not accurately predict how the treatment will work in patients. A step-wise analysis of the effects of different co-morbidities, such as diabetes, hypercholesterolemia, oxidative stress and aging, on the efficacy of angiogenic therapies needs to be examined. Proven efficacy in heterogeneous animal models (instead of only in a very specific homogenous animal population) will increase the probability that a therapy will work in the clinic.
Heterogeneity of preclinical models will probably increase variation in the results, but this should not be seen as a disadvantage. Rather, investigators should study the reasons underlying variation in response to angiogenic therapy (e.g. if the duration or the level of hyperglycemia affects angiogenic responses in diabetic animals). Identifying a limiting factor or co-morbidity that results in a poor treatment response can also help in the selection of patients for clinical trials. Importantly, publishing not only positive findings, but also neutral and especially negative findings, might offer crucial information that could be used to determine why the clinical trials carried out thus far have mostly failed.
Preclinical end points
There are currently also major differences between the end points used in preclinical animal studies and clinical trials. In animal models, demonstrating histological evidence of angiogenesis and healing of perfusion after an acute arterial occlusion is common practice. In contrast, in clinical studies the end points in addition to evidence of improved perfusion include healing of chronic ischemic ulcers, decreased amputation rates, decreased mortality, improved quality of life and improved walking distance. Of course, the lack of true chronic ischemic disease models means that end points such as decreased amputation rate or decreased mortality cannot be studied, because results obtained with non-diseased animals would probably lead to overly optimistic predictions. However, the functionality of the newly formed angiogenic vessels and the benefit of the angiogenic therapy on ischemic muscle survival should be regularly studied in animal models. Besides histological evidence of vascular growth, there should be requirements to demonstrate improved blood flow in the immediate area of the angiogenic therapy as well as to demonstrate therapeutic benefits on muscle function or energy metabolism.
Conclusions
In this Review, we have discussed the selection of preclinical models as one of the main reasons for the failures in clinical angiogenic trials. It is an obvious issue, that has received little attention. However, this has probably had, and will continue to have, a great impact on expectations from the clinical trials, if not acknowledged properly. As we have discussed, a simple ischemia model is never a disease model. For example, the induction of ischemia using acute ligations or gradually occluding constrictors in animal models might have produced an overly optimistic view of the potential of angiogenic therapies in humans, in whom chronic arterial disease blood-pressure- and shear-stress-related recovery mechanisms are quite different than in the animal models. In addition, diseaserelated co-morbidities that are present in humans can strongly affect the results of angiogenic therapies in the clinics. These comorbidities (Table 2) should be more carefully taken into account in preclinical testing of angiogenic therapies so as to better predict the clinical effects of the therapy. To ensure the future success of angiogenic therapies in the clinic, it is essential that more attention is focused on developing animal models that better mimic human ischemic disease.
Acknowledgments
FUNDING: This work was supported by grants from the Academy of Finland, the Sigrid Juselius Foundation and the Kuopio University Hospital.
Footnotes
These authors contributed equally to this work
COMPETING INTERESTS: The authors have no competing financial interests to declare.
REFERENCES
- Abarbanell A. M., Herrmann J. L., Weil B. R., Wang Y., Tan J., Moberly S. P., Fiege J. W., Meldrum D. R. (2010). Animal models of myocardial and vascular injury. J. Surg. Res. 162, 239-249 [DOI] [PubMed] [Google Scholar]
- Annex B. H., Simons M. (2005). Growth factor-induced therapeutic angiogenesis in the heart: protein therapy. Cardiovasc. Res. 65, 649-655 [DOI] [PubMed] [Google Scholar]
- Antman E. M., Anbe D. T., Armstrong P. W., Bates E. R., Green L. A., Hand M., Hochman J. S., Krumholz H. M., Kushner F. G., Lamas G. A., et al. (2004). ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction – executive summary. A report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Writing Committee to revise the 1999 guidelines for the management of patients with acute myocardial infarction). J. Am. Coll. Cardiol. 44, 671-719 [DOI] [PubMed] [Google Scholar]
- Aoki M., Morishita R., Taniyama Y., Kida I., Moriguchi A., Matsumoto K., Nakamura T., Kaneda Y., Higaki J., Ogihara T. (2000). Angiogenesis induced by hepatocyte growth factor in non-infarcted myocardium and infarcted myocardium: up-regulation of essential transcription factor for angiogenesis, ets. Gene Ther. 7, 417-427 [DOI] [PubMed] [Google Scholar]
- Asahara T., Murohara T., Sullivan A., Silver M., van der Zee R., Li T., Witzenbichler B., Schatteman G., Isner J. M. (1997). Isolation of putative progenitor endothelial cells for angiogenesis. Science 275, 964-966 [DOI] [PubMed] [Google Scholar]
- Baffour R., Garb J. L., Kaufman J., Berman J., Rhee S. W., Norris M. A., Friedmann P. (2000). Angiogenic therapy for the chronically ischemic lower limb in a rabbit model. J. Surg. Res. 93, 219-229 [DOI] [PubMed] [Google Scholar]
- Balsam L. B., Wagers A. J., Christensen J. L., Kofidis T., Weissman I. L., Robbins R. C. (2004). Haematopoietic stem cells adopt mature haematopoietic fates in ischaemic myocardium. Nature 428, 668-673 [DOI] [PubMed] [Google Scholar]
- Baumgartner I., Pieczek A., Manor O., Blair R., Kearney M., Walsh K., Isner J. M. (1998). Constitutive expression of phVEGF165 after intramuscular gene transfer promotes collateral vessel development in patients with critical limb ischemia. Circulation 97, 1114-1123 [DOI] [PubMed] [Google Scholar]
- Belch J., Hiatt W. R., Baumgartner I., Driver I. V., Nikol S., Norgren L., Van Belle E. (2011). Effect of fibroblast growth factor NV1FGF on amputation and death: a randomised placebo-controlled trial of gene therapy in critical limb ischaemia. Lancet 377, 1929-1937 [DOI] [PubMed] [Google Scholar]
- Boger R. H. (2004). Asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, explains the “L-arginine paradox” and acts as a novel cardiovascular risk factor. J. Nutr. 134, 2842S-2847S; discussion 2853S. [DOI] [PubMed] [Google Scholar]
- Boodhwani M., Sellke F. W. (2009). Therapeutic angiogenesis in diabetes and hypercholesterolemia: influence of oxidative stress. Antioxid. Redox Signal. 11, 1945-1959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brevetti G., Silvestro A., Di Giacomo S., Bucur R., Di Donato A., Schiano V., Scopacasa F. (2003a). Endothelial dysfunction in peripheral arterial disease is related to increase in plasma markers of inflammation and severity of peripheral circulatory impairment but not to classic risk factors and atherosclerotic burden. J. Vasc. Surg. 38, 374-379 [DOI] [PubMed] [Google Scholar]
- Brevetti L. S., Chang D. S., Tang G. L., Sarkar R., Messina L. M. (2003b). Overexpression of endothelial nitric oxide synthase increases skeletal muscle blood flow and oxygenation in severe rat hind limb ischemia. J. Vasc. Surg. 38, 820-826 [DOI] [PubMed] [Google Scholar]
- Buschmann I. R., Voskuil M., van Royen N., Hoefer I. E., Scheffler K., Grundmann S., Hennig J., Schaper W., Bode C., Piek J. J. (2003). Invasive and non-invasive evaluation of spontaneous arteriogenesis in a novel porcine model for peripheral arterial obstructive disease. Atherosclerosis 167, 33-43 [DOI] [PubMed] [Google Scholar]
- Carmeliet P., Jain R. K. (2011). Molecular mechanisms and clinical applications of angiogenesis. Nature 473, 298-307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzigeorgiou A., Halapas A., Kalafatakis K., Kamper E. (2009). The use of animal models in the study of diabetes mellitus. In Vivo 23, 245-258 [PubMed] [Google Scholar]
- Choi J. S., Kim K. B., Han W., Kim D. S., Park J. S., Lee J. J., Lee D. S. (2006). Efficacy of therapeutic angiogenesis by intramyocardial injection of pCK-VEGF165 in pigs. Ann. Thorac. Surg. 82, 679-686 [DOI] [PubMed] [Google Scholar]
- Cooney R., Hynes S. O., Duffy A. M., Sharif F., O'Brien T. (2006). Adenoviralmediated gene transfer of nitric oxide synthase isoforms and vascular cell proliferation. J. Vasc. Res. 43, 462-472 [DOI] [PubMed] [Google Scholar]
- Couffinhal T., Silver M., Zheng L. P., Kearney M., Witzenbichler B., Isner J. M. (1998). Mouse model of angiogenesis. Am. J. Pathol. 152, 1667-1679 [PMC free article] [PubMed] [Google Scholar]
- Daugherty A. (2002). Mouse models of atherosclerosis. Am. J. Med. Sci. 323, 3-10 [DOI] [PubMed] [Google Scholar]
- Davignon J. (2004). Beneficial cardiovascular pleiotropic effects of statins. Circulation 109 Suppl. 1, III39-III43 [DOI] [PubMed] [Google Scholar]
- Davis S., Aldrich T. H., Jones P. F., Acheson A., Compton D. L., Jain V., Ryan T. E., Bruno J., Radziejewski C., Maisonpierre P. C., et al. (1996). Isolation of angiopoietin-1, a ligand for the TIE2 receptor, by secretion-trap expression cloning. Cell 87, 1161-1169 [DOI] [PubMed] [Google Scholar]
- Delafontaine P., Song Y. H., Li Y. (2004). Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels. Arterioscler. Thromb. Vasc. Biol. 24, 435-444 [DOI] [PubMed] [Google Scholar]
- Dixon J. A., Spinale F. G. (2009). Large animal models of heart failure: a critical link in the translation of basic science to clinical practice. Circ. Heart Fail. 2, 262-271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards R., Yousef Z., Rakhit R., Wright M., Rosenthal E., Redwood S., Marber M. (2002). A model of closed chest regional myocardial infarction in the rabbit: a clinically relevant in vivo assay system of post-infarction remodelling. Basic Res. Cardiol. 97, 374-383 [DOI] [PubMed] [Google Scholar]
- Faber J. E., Zhang H., Lassance-Soares R. M., Prabhakar P., Najafi A. H., Burnett M. S., Epstein S. E. (2011). Aging causes collateral rarefaction and increased severity of ischemic injury in multiple tissues. Arterioscler. Thromb. Vasc. Biol. 31, 1748-1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S., Salven P. (2011). Stem cells in tumor angiogenesis. J. Mol. Cell. Cardiol. 50, 290-295 [DOI] [PubMed] [Google Scholar]
- Ferrara N. (2004). Vascular endothelial growth factor: basic science and clinical progress. Endocr. Rev. 25, 581-611 [DOI] [PubMed] [Google Scholar]
- Folkman J. (1971). Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 285, 1182-1186 [DOI] [PubMed] [Google Scholar]
- Fredriksson L., Li H., Eriksson U. (2004). The PDGF family: four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 15, 197-204 [DOI] [PubMed] [Google Scholar]
- Gao M. H., Lai N. C., McKirnan M. D., Roth D. A., Rubanyi G. M., Dalton N., Roth D. M., Hammond H. K. (2004). Increased regional function and perfusion after intracoronary delivery of adenovirus encoding fibroblast growth factor 4: report of preclinical data. Hum. Gene Ther. 15, 574-587 [DOI] [PubMed] [Google Scholar]
- Gao E., Lei Y. H., Shang X., Huang Z. M., Zuo L., Boucher M., Fan Q., Chuprun J. K., Ma X. L., Koch W. J. (2010). A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 107, 1445-1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrity R. G., Natarajan R., Nadler J. L., Kimsey T. (2001). Diabetes-induced accelerated atherosclerosis in swine. Diabetes 50, 1654-1665 [DOI] [PubMed] [Google Scholar]
- Getz G. S., Reardon C. A. (2012). Animal models of atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 32, 1104-1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbons R. J., Abrams J., Chatterjee K., Daley J., Deedwania P. C., Douglas J. S., Ferguson T. B., Jr, Fihn S. D., Fraker T. D., Jr, Gardin J. M., et al. (2003). ACC/AHA 2002 guideline update for the management of patients with chronic stable angina – summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee on the Management of Patients With Chronic Stable Angina). Circulation 107, 149-158 [DOI] [PubMed] [Google Scholar]
- Goldberg I. J., Dansky H. M. (2006). Diabetic vascular disease: an experimental objective. Arterioscler. Thromb. Vasc. Biol. 26, 1693-1701 [DOI] [PubMed] [Google Scholar]
- Gowdak L. H., Poliakova L., Wang X., Kovesdi I., Fishbein K. W., Zacheo A., Palumbo R., Straino S., Emanueli C., Marrocco-Trischitta M., et al. (2000). Adenovirus-mediated VEGF (121) gene transfer stimulates angiogenesis in normoperfused skeletal muscle and preserves tissue perfusion after induction of ischemia. Circulation 102, 565-571 [DOI] [PubMed] [Google Scholar]
- Gupta R., Tongers J., Losordo D. W. (2009). Human studies of angiogenic gene therapy. Circ. Res. 105, 724-736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halapas A., Papalois A., Stauropoulou A., Philippou A., Pissimissis N., Chatzigeorgiou A., Kamper E., Koutsilieris M. (2008). In vivo models for heart failure research. In Vivo 22, 767-780 [PubMed] [Google Scholar]
- Hao X., Månsson-Broberg A., Grinnemo K. H., Siddiqui A. J., Dellgren G., Brodin L. A., Sylvén C. (2007). Myocardial angiogenesis after plasmid or adenoviral VEGF-A(165) gene transfer in rat myocardial infarction model. Cardiovasc. Res. 73, 481-487 [DOI] [PubMed] [Google Scholar]
- Harada K., Grossman W., Friedman M., Edelman E. R., Prasad P. V., Keighley C. S., Manning W. J., Sellke F. W., Simons M. (1994). Basic fibroblast growth factor improves myocardial function in chronically ischemic porcine hearts. J. Clin. Invest. 94, 623-630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedman M., Hartikainen J., Syvänne M., Stjernvall J., Hedman A., Kivelä A., Vanninen E., Mussalo H., Kauppila E., Simula S., et al. (2003). Safety and feasibility of catheter-based local intracoronary vascular endothelial growth factor gene transfer in the prevention of postangioplasty and in-stent restenosis and in the treatment of chronic myocardial ischemia: phase II results of the Kuopio Angiogenesis Trial (KAT). Circulation 107, 2677-2683 [DOI] [PubMed] [Google Scholar]
- Hedman M., Hartikainen J., Ylä-Herttuala S. (2011). Progress and prospects: hurdles to cardiovascular gene therapy clinical trials. Gene Ther. 18, 743-749 [DOI] [PubMed] [Google Scholar]
- Heil M., Eitenmüller I., Schmitz-Rixen T., Schaper W. (2006). Arteriogenesis versus angiogenesis: similarities and differences. J. Cell. Mol. Med. 10, 45-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinonen S. E., Leppänen P., Kholová I., Lumivuori H., Häkkinen S. K., Bosch F., Laakso M., Ylä-Herttuala S. (2007). Increased atherosclerotic lesion calcification in a novel mouse model combining insulin resistance, hyperglycemia, and hypercholesterolemia. Circ. Res. 101, 1058-1067 [DOI] [PubMed] [Google Scholar]
- Heinonen S. E., Merentie M., Hedman M., Mäkinen P. I., Loponen E., Kholová I., Bosch F., Laakso M., Ylä-Herttuala S. (2011). Left ventricular dysfunction with reduced functional cardiac reserve in diabetic and non-diabetic LDL-receptor deficient apolipoprotein B100-only mice. Cardiovasc. Diabetol. 10, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershey J. C., Baskin E. P., Glass J. D., Hartman H. A., Gilberto D. B., Rogers I. T., Cook J. J. (2001). Revascularization in the rabbit hindlimb: dissociation between capillary sprouting and arteriogenesis. Cardiovasc. Res. 49, 618-625 [DOI] [PubMed] [Google Scholar]
- Herzog S., Sager H., Khmelevski E., Deylig A., Ito W. D. (2002). Collateral arteries grow from preexisting anastomoses in the rat hindlimb. Am. J. Physiol. Heart Circ. Physiol. 283, H2012-H2020 [DOI] [PubMed] [Google Scholar]
- Horvath K. A., Doukas J., Lu C. Y., Belkind N., Greene R., Pierce G. F., Fullerton D. A. (2002). Myocardial functional recovery after fibroblast growth factor 2 gene therapy as assessed by echocardiography and magnetic resonance imaging. Ann. Thorac. Surg. 74, 481-487 [DOI] [PubMed] [Google Scholar]
- Hughes G. C., Post M. J., Simons M., Annex B. H. (2003). Translational physiology: porcine models of human coronary artery disease: implications for preclinical trials of therapeutic angiogenesis. J. Appl. Physiol. 94, 1689-1701 [DOI] [PubMed] [Google Scholar]
- Iwanaga K., Takano H., Ohtsuka M., Hasegawa H., Zou Y., Qin Y., Odaka K., Hiroshima K., Tadokoro H., Komuro I. (2004). Effects of G-CSF on cardiac remodeling after acute myocardial infarction in swine. Biochem. Biophys. Res. Commun. 325, 1353-1359 [DOI] [PubMed] [Google Scholar]
- Katsanos K., Mitsos S., Koletsis E., Bravou V., Karnabatidis D., Kolonitsiou F., Diamantopoulos A., Dougenis D., Siablis D. (2012). Transauricular embolization of the rabbit coronary artery for experimental myocardial infarction: comparison of a minimally invasive closed-chest model with open-chest surgery. J. Cardiothorac. Surg. 7, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinnaird T., Stabile E., Zbinden S., Burnett M. S., Epstein S. E. (2008). Cardiovascular risk factors impair native collateral development and may impair efficacy of therapeutic interventions. Cardiovasc. Res. 78, 257-264 [DOI] [PubMed] [Google Scholar]
- Korpisalo P., Karvinen H., Rissanen T. T., Kilpijoki J., Marjomäki V., Baluk P., McDonald D. M., Cao Y., Eriksson U., Alitalo K., et al. (2008a). Vascular endothelial growth factor-A and platelet-derived growth factor-B combination gene therapy prolongs angiogenic effects via recruitment of interstitial mononuclear cells and paracrine effects rather than improved pericyte coverage of angiogenic vessels. Circ. Res. 103, 1092-1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korpisalo P., Rissanen T. T., Bengtsson T., Liimatainen T., Laidinen S., Karvinen H., Markkanen J. E., Gröhn O. H., Ylä-Herttuala S. (2008b). Therapeutic angiogenesis with placental growth factor improves exercise tolerance of ischaemic rabbit hindlimbs. Cardiovasc. Res. 80, 263-270 [DOI] [PubMed] [Google Scholar]
- Lähteenvuo J., Rosenzweig A. (2012). Effects of aging on angiogenesis. Circ. Res. 110, 1252-1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lähteenvuo J. E., Lähteenvuo M. T., Kivelä A., Rosenlew C., Falkevall A., Klar J., Heikura T., Rissanen T. T., Vähäkangas E., Korpisalo P., et al. (2009). Vascular endothelial growth factor-B induces myocardium-specific angiogenesis and arteriogenesis via vascular endothelial growth factor receptor-1- and neuropilin receptor-1-dependent mechanisms. Circulation 119, 845-856 [DOI] [PubMed] [Google Scholar]
- Lerman L. O., Chade A. R., Sica V., Napoli C. (2005). Animal models of hypertension: an overview. J. Lab. Clin. Med. 146, 160-173 [DOI] [PubMed] [Google Scholar]
- Li M., Liu C., Bin J., Wang Y., Chen J., Xiu J., Pei J., Lai Y., Chen D., Fan C., et al. (2011). Mutant hypoxia inducible factor-1α improves angiogenesis and tissue perfusion in ischemic rabbit skeletal muscle. Microvasc. Res. 81, 26-33 [DOI] [PubMed] [Google Scholar]
- Limbourg A., Korff T., Napp L. C., Schaper W., Drexler H., Limbourg F. P. (2009). Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind-limb ischemia. Nat. Protoc. 4, 1737-1748 [DOI] [PubMed] [Google Scholar]
- Lusis A. J. (2000). Atherosclerosis. Nature 407, 233-241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeddu P., Emanueli C., Spillmann F., Meloni M., Bouby N., Richer C., Alhenc Gelas F., Van Weel V., Eefting D., Quax P. H., et al. (2006). Murine models of myocardial and limb ischemia: diagnostic end-points and relevance to clinical problems. Vascul. Pharmacol. 45, 281-301 [DOI] [PubMed] [Google Scholar]
- Makanya A. N., Hlushchuk R., Djonov V. G. (2009). Intussusceptive angiogenesis and its role in vascular morphogenesis, patterning, and remodeling. Angiogenesis 12, 113-123 [DOI] [PubMed] [Google Scholar]
- Masaki I., Yonemitsu Y., Yamashita A., Sata S., Tanii M., Komori K., Nakagawa K., Hou X., Nagai Y., Hasegawa M., et al. (2002). Angiogenic gene therapy for experimental critical limb ischemia: acceleration of limb loss by overexpression of vascular endothelial growth factor 165 but not of fibroblast growth factor-2. Circ. Res. 90, 966-973 [DOI] [PubMed] [Google Scholar]
- McQuaid K. E., Keenan A. K. (1997). Endothelial barrier dysfunction and oxidative stress: roles for nitric oxide? Exp. Physiol. 82, 369-376 [DOI] [PubMed] [Google Scholar]
- Melov S. (2002). Animal models of oxidative stress, aging, and therapeutic antioxidant interventions. Int. J. Biochem. Cell Biol. 34, 1395-1400 [DOI] [PubMed] [Google Scholar]
- Menasché P. (2010). Cell therapy for peripheral arterial disease. Curr. Opin. Mol. Ther. 12, 538-545 [PubMed] [Google Scholar]
- Mitsos S., Katsanos K., Koletsis E., Kagadis G. C., Anastasiou N., Diamantopoulos A., Karnabatidis D., Dougenis D. (2012). Therapeutic angiogenesis for myocardial ischemia revisited: basic biological concepts and focus on latest clinical trials. Angiogenesis 15, 1-22 [DOI] [PubMed] [Google Scholar]
- Morales C., González G. E., Rodríguez M., Bertolasi C. A., Gelpi R. J. (2002). Histopathologic time course of myocardial infarct in rabbit hearts. Cardiovasc. Pathol. 11, 339-345 [DOI] [PubMed] [Google Scholar]
- Münzel T., Sinning C., Post F., Warnholtz A., Schulz E. (2008). Pathophysiology, diagnosis and prognostic implications of endothelial dysfunction. Ann. Med. 40, 180-196 [DOI] [PubMed] [Google Scholar]
- Muona K., Mäkinen K., Hedman M., Manninen H., Ylä-Herttuala S. (2012). 10-year safety follow-up in patients with local VEGF gene transfer to ischemic lower limb. Gene Ther. 19, 392-395 [DOI] [PubMed] [Google Scholar]
- Murakami M., Simons M. (2008). Fibroblast growth factor regulation of neovascularization. Curr. Opin. Hematol. 15, 215-220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norgren L., Hiatt W. R., Dormandy J. A., Nehler M. R., Harris K. A., Fowkes F. G. R. (2007). Inter-society consensus for the management of peripheral arterial disease (TASC II). Eur. J. Vasc. Endovasc. Surg. 33 Suppl. 1, S1-S75 [DOI] [PubMed] [Google Scholar]
- O'Konski M. S., White F. C., Longhurst J., Roth D., Bloor C. M. (1987). Ameroid constriction of the proximal left circumflex coronary artery in swine. A model of limited coronary collateral circulation. Am. J. Cardiovasc. Pathol. 1, 69-77 [PubMed] [Google Scholar]
- Oikawa D., Akai R., Tokuda M., Iwawaki T. (2012). A transgenic mouse model for monitoring oxidative stress. Sci. Rep. 2, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olea F. D., Vera Janavel G., Cuniberti L., Yannarelli G., Cabeza Meckert P., Cors J., Valdivieso L., Lev G., Mendiz O., Bercovich A., et al. (2009). Repeated, but not single, VEGF gene transfer affords protection against ischemic muscle lesions in rabbits with hindlimb ischemia. Gene Ther. 16, 716-723 [DOI] [PubMed] [Google Scholar]
- Paek R., Chang D. S., Brevetti L. S., Rollins M. D., Brady S., Ursell P. C., Hunt T. K., Sarkar R., Messina L. M. (2002). Correlation of a simple direct measurement of muscle pO(2) to a clinical ischemia index and histology in a rat model of chronic severe hindlimb ischemia. J. Vasc. Surg. 36, 172-179 [DOI] [PubMed] [Google Scholar]
- Pajusola K., Künnapuu J., Vuorikoski S., Soronen J., André H., Pereira T., Korpisalo P., Ylä-Herttuala S., Poellinger L., Alitalo K. (2005). Stabilized HIF- 1alpha is superior to VEGF for angiogenesis in skeletal muscle via adeno-associated virus gene transfer. FASEB J. 19, 1365-1367 [DOI] [PubMed] [Google Scholar]
- Patel T. H., Kimura H., Weiss C. R., Semenza G. L., Hofmann L. V. (2005). Constitutively active HIF-1alpha improves perfusion and arterial remodeling in an endovascular model of limb ischemia. Cardiovasc. Res. 68, 144-154 [DOI] [PubMed] [Google Scholar]
- Patten R. D., Hall-Porter M. R. (2009). Small animal models of heart failure: development of novel therapies, past and present. Circ. Heart Fail. 2, 138-144 [DOI] [PubMed] [Google Scholar]
- Pfeffer M. A., Pfeffer J. M., Fishbein M. C., Fletcher P. J., Spadaro J., Kloner R. A., Braunwald E. (1979). Myocardial infarct size and ventricular function in rats. Circ. Res. 44, 503-512 [DOI] [PubMed] [Google Scholar]
- Pinto Y. M., Paul M., Ganten D. (1998). Lessons from rat models of hypertension: from Goldblatt to genetic engineering. Cardiovasc. Res. 39, 77-88 [DOI] [PubMed] [Google Scholar]
- Pipp F., Boehm S., Cai W. J., Adili F., Ziegler B., Karanovic G., Ritter R., Balzer J., Scheler C., Schaper W., et al. (2004). Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expression in the pig hind limb. Arterioscler. Thromb. Vasc. Biol. 24, 1664-1668 [DOI] [PubMed] [Google Scholar]
- Plum L., Wunderlich F. T., Baudler S., Krone W., Brüning J. C. (2005). Transgenic and knockout mice in diabetes research: novel insights into pathophysiology, limitations, and perspectives. Physiology (Bethesda) 20, 152-161 [DOI] [PubMed] [Google Scholar]
- Pu L. Q., Jackson S., Lachapelle K. J., Arekat Z., Graham A. M., Lisbona R., Brassard R., Carpenter S., Symes J. F. (1994). A persistent hindlimb ischemia model in the rabbit. J. Invest. Surg. 7, 49-60 [DOI] [PubMed] [Google Scholar]
- Pyun W. B., Hahn W., Kim D. S., Yoo W. S., Lee S. D., Won J. H., Rho B. S., Park Z. Y., Kim J. M., Kim S. (2010). Naked DNA expressing two isoforms of hepatocyte growth factor induces collateral artery augmentation in a rabbit model of limb ischemia. Gene Ther. 17, 1442-1452 [DOI] [PubMed] [Google Scholar]
- Rafii S., Lyden D. (2003). Therapeutic stem and progenitor cell transplantation for organ vascularization and regeneration. Nat. Med. 9, 702-712 [DOI] [PubMed] [Google Scholar]
- Rees D. A., Alcolado J. C. (2005). Animal models of diabetes mellitus. Diabet. Med. 22, 359-370 [DOI] [PubMed] [Google Scholar]
- Risau W. (1997). Mechanisms of angiogenesis. Nature 386, 671-674 [DOI] [PubMed] [Google Scholar]
- Rissanen T. T., Ylä-Herttuala S. (2007). Current status of cardiovascular gene therapy. Mol. Ther. 15, 1233-1247 [DOI] [PubMed] [Google Scholar]
- Rissanen T. T., Vajanto I., Hiltunen M. O., Rutanen J., Kettunen M. I., Niemi M., Leppänen P., Turunen M. P., Markkanen J. E., Arve K., et al. (2002). Expression of vascular endothelial growth factor and vascular endothelial growth factor receptor-2 (KDR/Flk-1) in ischemic skeletal muscle and its regeneration. Am. J. Pathol. 160, 1393-1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rissanen T. T., Markkanen J. E., Arve K., Rutanen J., Kettunen M. I., Vajanto I., Jauhiainen S., Cashion L., Gruchala M., Närvänen O., et al. (2003a). Fibroblast growth factor 4 induces vascular permeability, angiogenesis and arteriogenesis in a rabbit hindlimb ischemia model. FASEB J. 17, 100-102 [DOI] [PubMed] [Google Scholar]
- Rissanen T. T., Markkanen J. E., Gruchala M., Heikura T., Puranen A., Kettunen M. I., Kholová I., Kauppinen R. A., Achen M. G., Stacker S. A., et al. (2003b). VEGF-D is the strongest angiogenic and lymphangiogenic effector among VEGFs delivered into skeletal muscle via adenoviruses. Circ. Res. 92, 1098-1106 [DOI] [PubMed] [Google Scholar]
- Rissanen T. T., Korpisalo P., Markkanen J. E., Liimatainen T., Ordén M. R., Kholová I., de Goede A., Heikura T., Gröhn O. H., Ylä-Herttuala S. (2005). Blood flow remodels growing vasculature during vascular endothelial growth factor gene therapy and determines between capillary arterialization and sprouting angiogenesis. Circulation 112, 3937-3946 [DOI] [PubMed] [Google Scholar]
- Ruel M., Sellke F. W. (2003). Angiogenic protein therapy. Semin. Thorac. Cardiovasc. Surg. 15, 222-235 [DOI] [PubMed] [Google Scholar]
- Salto-Tellez M., Yung Lim S., El-Oakley R. M., Tang T. P., ALmsherqi Z. A., Lim S. K. (2004). Myocardial infarction in the C57BL/6J mouse: a quantifiable and highly reproducible experimental model. Cardiovasc. Pathol. 13, 91-97 [DOI] [PubMed] [Google Scholar]
- Schaper W. (2009). Collateral circulation: past and present. Basic Res. Cardiol. 104, 5-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholz D., Ziegelhoeffer T., Helisch A., Wagner S., Friedrich C., Podzuweit T., Schaper W. (2002). Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J. Mol. Cell. Cardiol. 34, 775-787 [DOI] [PubMed] [Google Scholar]
- Scholz D., Thomas S., Sass S., Podzuweit T. (2003). Angiogenesis and myogenesis as two facets of inflammatory post-ischemic tissue regeneration. Mol. Cell. Biochem. 246, 57-67 [PubMed] [Google Scholar]
- Selye H., Bajusz E., Grasso S., Mendell P. (1960). Simple techniques for the surgical occlusion of coronary vessels in the rat. Angiology 11, 398-407 [DOI] [PubMed] [Google Scholar]
- Shiomi M., Ito T. (2009). The Watanabe heritable hyperlipidemic (WHHL) rabbit, its characteristics and history of development: a tribute to the late Dr. Yoshio Watanabe. Atherosclerosis 207, 1-7 [DOI] [PubMed] [Google Scholar]
- Silvestre J. S., Mallat Z., Tedgui A., Lévy B. I. (2008). Post-ischaemic neovascularization and inflammation. Cardiovasc. Res. 78, 242-249 [DOI] [PubMed] [Google Scholar]
- Simons M. (2005). Angiogenesis: where do we stand now? Circulation 111, 1556-1566 [DOI] [PubMed] [Google Scholar]
- Simons M., Ware J. A. (2003). Therapeutic angiogenesis in cardiovascular disease. Nat. Rev. Drug Discov. 2, 863-872 [DOI] [PubMed] [Google Scholar]
- Sun L., Bai Y., Du G. (2009). Endothelial dysfunction – an obstacle of therapeutic angiogenesis. Ageing Res. Rev. 8, 306-313 [DOI] [PubMed] [Google Scholar]
- Takeshita S., Pu L. Q., Stein L. A., Sniderman A. D., Bunting S., Ferrara N., Isner J. M., Symes J. F. (1994). Intramuscular administration of vascular endothelial growth factor induces dose-dependent collateral artery augmentation in a rabbit model of chronic limb ischemia. Circulation 90, II228-II234 [PubMed] [Google Scholar]
- Takeshita S., Weir L., Chen D., Zheng L. P., Riessen R., Bauters C., Symes J. F., Ferrara N., Isner J. M. (1996). Therapeutic angiogenesis following arterial gene transfer of vascular endothelial growth factor in a rabbit model of hindlimb ischemia. Biochem. Biophys. Res. Commun. 227, 628-635 [DOI] [PubMed] [Google Scholar]
- Takeshita S., Isshiki T., Mori H., Tanaka E., Eto K., Miyazawa Y., Tanaka A., Shinozaki Y., Hyodo K., Ando M., et al. (1997). Use of synchrotron radiation microangiography to assess development of small collateral arteries in a rat model of hindlimb ischemia. Circulation 95, 805-808 [DOI] [PubMed] [Google Scholar]
- Tang G. L., Chang D. S., Sarkar R., Wang R., Messina L. M. (2005). The effect of gradual or acute arterial occlusion on skeletal muscle blood flow, arteriogenesis, and inflammation in rat hindlimb ischemia. J. Vasc. Surg. 41, 312-320 [DOI] [PubMed] [Google Scholar]
- Taniyama Y., Morishita R., Aoki M., Nakagami H., Yamamoto K., Yamazaki K., Matsumoto K., Nakamura T., Kaneda Y., Ogihara T. (2001). Therapeutic angiogenesis induced by human hepatocyte growth factor gene in rat and rabbit hindlimb ischemia models: preclinical study for treatment of peripheral arterial disease. Gene Ther. 8, 181-189 [DOI] [PubMed] [Google Scholar]
- Tio R. A., Tkebuchava T., Scheuermann T. H., Lebherz C., Magner M., Kearny M., Esakof D. D., Isner J. M., Symes J. F. (1999). Intramyocardial gene therapy with naked DNA encoding vascular endothelial growth factor improves collateral flow to ischemic myocardium. Hum. Gene Ther. 10, 2953-2960 [DOI] [PubMed] [Google Scholar]
- Tongers J., Roncalli J. G., Losordo D. W. (2008). Therapeutic angiogenesis for critical limb ischemia: microvascular therapies coming of age. Circulation 118, 9-16 [DOI] [PubMed] [Google Scholar]
- van Weel V., Deckers M. M., Grimbergen J. M., van Leuven K. J., Lardenoye J. H., Schlingemann R. O., van Nieuw Amerongen G. P., van Bockel J. H., van Hinsbergh V. W., Quax P. H. (2004). Vascular endothelial growth factor overexpression in ischemic skeletal muscle enhances myoglobin expression in vivo. Circ. Res. 95, 58-66 [DOI] [PubMed] [Google Scholar]
- Vita J. A., Hamburg N. M. (2010). Does endothelial dysfunction contribute to the clinical status of patients with peripheral arterial disease? Can. J. Cardiol. 26, 45A-50A [DOI] [PubMed] [Google Scholar]
- Watanabe Y. (1980). Serial inbreeding of rabbits with hereditary hyperlipidemia (WHHL-rabbit). Atherosclerosis 36, 261-268 [DOI] [PubMed] [Google Scholar]
- Watanabe E., Smith D. M., Sun J., Smart F. W., Delcarpio J. B., Roberts T. B., Van Meter C. H., Jr, Claycomb W. C. (1998). Effect of basic fibroblast growth factor on angiogenesis in the infarcted porcine heart. Basic Res. Cardiol. 93, 30-37 [DOI] [PubMed] [Google Scholar]
- Waters R. E., Terjung R. L., Peters K. G., Annex B. H. (2004). Preclinical models of human peripheral arterial occlusive disease: implications for investigation of therapeutic agents. J. Appl. Physiol. 97, 773-780 [DOI] [PubMed] [Google Scholar]
- Weis S. M., Cheresh D. A. (2005). Pathophysiological consequences of VEGFinduced vascular permeability. Nature 437, 497-504 [DOI] [PubMed] [Google Scholar]
- Wu K. K., Huan Y. (2007). Diabetic atherosclerosis mouse models. Atherosclerosis 191, 241-249 [DOI] [PubMed] [Google Scholar]
- Xi S., Yin W., Wang Z., Kusunoki M., Lian X., Koike T., Fan J., Zhang Q. (2004). A minipig model of high-fat/high-sucrose diet-induced diabetes and atherosclerosis. Int. J. Exp. Pathol. 85, 223-231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. T., Ogilvie R. W., Terjung R. L. (1995). Heparin increases exerciseinduced collateral blood flow in rats with femoral artery ligation. Circ. Res. 76, 448-456 [DOI] [PubMed] [Google Scholar]
- Yang Y., Tang G., Yan J., Park B., Hoffman A., Tie G., Wang R., Messina L. M. (2008). Cellular and molecular mechanism regulating blood flow recovery in acute versus gradual femoral artery occlusion are distinct in the mouse. J. Vasc. Surg. 48, 1546-1558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylä-Herttuala S., Alitalo K. (2003). Gene transfer as a tool to induce therapeutic vascular growth. Nat. Med. 9, 694-701 [DOI] [PubMed] [Google Scholar]
- Ylä-Herttuala S., Rissanen T. T., Vajanto I., Hartikainen J. (2007). Vascular endothelial growth factors: biology and current status of clinical applications in cardiovascular medicine. J. Am. Coll. Cardiol. 49, 1015-1026 [DOI] [PubMed] [Google Scholar]
- Ylä-Herttuala S., Bentzon J. F., Daemen M., Falk E., Garcia-Garcia H. M., Herrmann J., Hoefer I., Jukema J. W., Krams R., Kwak B. R., et al. (2011). Stabilisation of atherosclerotic plaques. Position paper of the European Society of Cardiology (ESC) Working Group on atherosclerosis and vascular biology. Thromb. Haemost. 106, 1-19 [DOI] [PubMed] [Google Scholar]
- Zachary I., Morgan R. D. (2011). Therapeutic angiogenesis for cardiovascular disease: biological context, challenges, prospects. Heart 97, 181-189 [DOI] [PubMed] [Google Scholar]
- Zaragoza C., Gomez-Guerrero C., Martin-Ventura J. L., Blanco-Colio L., Lavin B., Mallavia B., Tarin C., Mas S., Ortiz A., Egido J. (2011). Animal models of cardiovascular diseases. J. Biomed. Biotechnol. 2011, 497841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelhoeffer T., Fernandez B., Kostin S., Heil M., Voswinckel R., Helisch A., Schaper W. (2004). Bone marrow-derived cells do not incorporate into the adult growing vasculature. Circ. Res. 94, 230-238 [DOI] [PubMed] [Google Scholar]
- Ziegler M. A., Distasi M. R., Bills R. G., Miller S. J., Alloosh M., Murphy M. P., George Akingba A., Sturek M., Dalsing M. C., Unthank J. L. (2010). Marvels, mysteries, and misconceptions of vascular compensation to peripheral artery occlusion. Microcirculation 17, 3-20 [DOI] [PMC free article] [PubMed] [Google Scholar]