

SUMMARY
The primary purpose of this investigation was to determine whether ApoE−/− mice, when subjected to chronic stress, exhibit lesions characteristic of human vulnerable plaque and, if so, to determine the time course of such changes. We found that the lesions were remarkably similar to human vulnerable plaque, and that the time course of lesion progression raised interesting insights into the process of plaque development. Lard-fed mixed-background ApoE−/− mice exposed to chronic stress develop lesions with large necrotic core, thin fibrous cap and a high degree of inflammation. Neovascularization and intraplaque hemorrhage are observed in over 80% of stressed animals at 20 weeks of age. Previously described models report a prevalence of only 13% for neovascularization observed at a much later time point, between 36 and 60 weeks of age. Thus, our new stress-induced model of advanced atherosclerotic plaque provides an improvement over what is currently available. This model offers a tool to further investigate progression of plaque phenotype to a more vulnerable phenotype in humans. Our findings also suggest a possible use of this stress-induced model to determine whether therapeutic interventions have effects not only on plaque burden, but also, and importantly, on plaque vulnerability.
INTRODUCTION
Human vulnerable plaque is characterized and distinguished from stable plaque by the presence of a large lipid core, thin fibrous cap, inflammatory cells and neovascularization. These vulnerable lesions are prone to rupture, thereby resulting in thrombus formation, vessel occlusion and often death (Arbustini et al., 1999; Davies, 2000; Falk, 1983; Virmani et al., 2000; Virmani et al., 2005). A deeper understanding of the cellular and molecular mechanisms contributing to plaque instability would have enormous clinical impact. However, progress has been hindered by the lack of an acceptable animal model of the human vulnerable plaque (Falk et al., 2007; Jackson, 2007; Jackson et al., 2007; Schwartz et al., 2007).
Previous attempts to develop animal models with human ‘vulnerable plaque-like’ phenotypes have focused on the C57BL ApoE−/− mouse or on larger animal models such as hypercholesterolemic rabbits (Aikawa et al., 1998; Pakala et al., 2003) and pigs (Gössl et al., 2007), as well as pigs with both hypercholesterolemia and induced diabetes (Mohler et al., 2008). Although each model has one or more elements recognized as being present in human vulnerable plaque, it is the general consensus that existing animal models do not adequately represent human vulnerable plaque (Finn et al., 2010). Even though it is likely that no animal model will be found that exactly reproduces the complex morphology and biology of human lesions that are prone to rupture, given the importance of this problem, continued efforts to develop models that resemble human lesions as closely as possible seem warranted.
In humans, stress is perceived as an increasingly prevalent feature of daily life and has been shown to increase atherosclerotic plaque development and the incidence of acute myocardial infarction (Malzburg, 1937; Yan et al., 2003), although little is known about the mechanisms linking stress to acute myocardial infarction. The melding of these two interests, vulnerable plaque and stress, helped define the direction we took in developing an experimental model that closely mimics vulnerable plaque in humans.
In this regard, there is considerable variability in human responses to stress. Genetic differences in the hypothalamic-pituitary-adrenocortical axis and the resulting differences in the response to stress might partially explain this variability (Binder et al., 2004; van Rossum et al., 2006). Thus, McCutcheon et al. reported that mice on a mixed background of C57BL and Sv129 are more affected by stress than animals on a C57BL background, as measured by plasma corticosterone levels following stress (McCutcheon et al., 2008). Johnson et al. also described ‘plaque rupture’ in the brachiocephalic artery of the lard-fed ApoE−/− mouse on a mixed background of C57BL and Sv129 (Johnson et al., 2005; Williams et al., 2002), although these findings remain controversial (Falk et al., 2007; Schwartz et al., 2007).
Due to inherent differences that make such mixed-background mice more susceptible to the effects of stress, we thought it would be productive to further characterize the lard-fed, mixed background ApoE−/− mouse model. Specifically, we hypothesized that mixed genetic background mice (C57BL and Sv129), exposed to chronic stress, are more prone to develop atherosclerotic lesions that closely mimic human vulnerable plaque-like lesions.
TRANSLATIONAL IMPACT.
Clinical issue
Although atherosclerosis has a major impact on health, particularly in developed nations, the atherosclerotic lesion burden does not kill; it is plaque rupture that kills. Thus, acute myocardial infarction, unstable angina and stroke are most commonly precipitated by plaque rupture. Attention is now being directed, therefore, to the possibility that the processes leading to plaque vulnerability, including genetic factors, are different from the processes leading to atherosclerosis. Progress towards a deeper understanding of the cellular and molecular mechanisms contributing to plaque instability, which would have enormous clinical impact, has been hindered to date by the lack of an acceptable animal model of the human vulnerable plaque. In humans, stress is an increasingly prevalent feature of daily life that increases both atherosclerotic plaque development and the incidence of acute myocardial infarction.
Results
In this study, the authors modeled vulnerable plaque-like lesions by exposing ApoE−/− mice (a mouse model of atherogenesis) to chronic stress. Mice on a mixed background of C57BL and Sv129 are more affected by stress than animals on a C57BL background, as measured by plasma corticosterone levels following stress. Therefore, the authors investigated atherogenesis and plaque rupture in ApoE−/− mixed-background mice (C57BL and Sv129) exposed to chronic stress. Lard-fed mixed-background ApoE−/− mice exposed to chronic stress developed lesions with a large necrotic core, thin fibrous cap and a high degree of inflammation. Neovascularization and intraplaque hemorrhage were observed in over 80% of stressed animals at 20 weeks of age. By contrast, previously described models reported a neovascularization prevalence of only 13%, which was not observed until between 36 and 60 weeks of age. These findings suggest that this new stress-induced model of advanced atherosclerotic plaque provides an improvement over currently available models.
Implications and future directions
The new model of stress-induced advanced atherosclerotic plaque described in this study provides a tool for the investigation of the mechanisms involved in the progression of plaques to a more vulnerable phenotype in humans. It might also prove useful in determining whether therapeutic interventions have effects on plaque vulnerability as well as on plaque burden. Finally, future studies using this model that examine the pathways through which stress modulates lesion phenotype, including the role of epigenetics, could provide therapeutic targets for advancing clinical care.
RESULTS
Lesion scoring; mixed background versus C57
Cross-sections of the brachiocephalic artery were assessed by a blinded observer and scored according to the criteria described and illustrated in Fig. 1. At 16-17 weeks of age, stressed ApoE−/− mice on a mixed C57BL/6J, Sv129 genetic background had significantly more class V lesions than stressed ApoE−/− mice on the C57BL/6J background (40% versus 0%; P=0.04, Fig. 2). These findings led us to focus our detailed studies on stress-induced effects on lesion phenotype in the mixed genetic background mice.
Fig. 1.
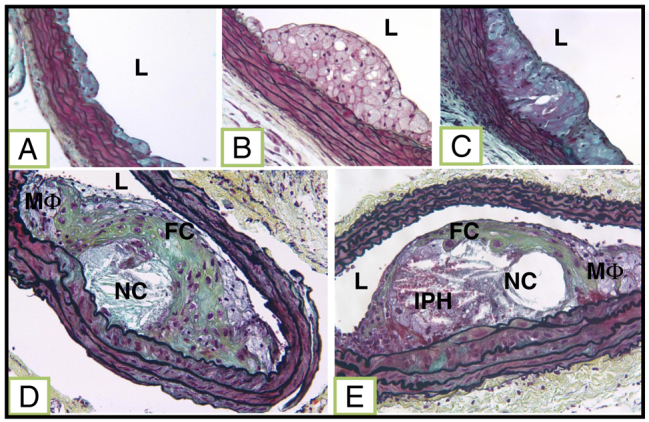
Lesion classification. (A) Class I: single layer of foam cells. (B) Class II: multiple layers of foam cells. (C) Class III: multiple layers of foam cells with free cholesterol and rare SMCs. (D) Class IV: well-formed necrotic core (NC) and cholesterol clefts with overlying fibrous cap and some degree of macrophage (MΦ) invasion. (E) Class V (complex atheroma) all the class IV features plus neovascularization and/or intraplaque hemorrhage (IPH). L indicates lumen.
Fig. 2.
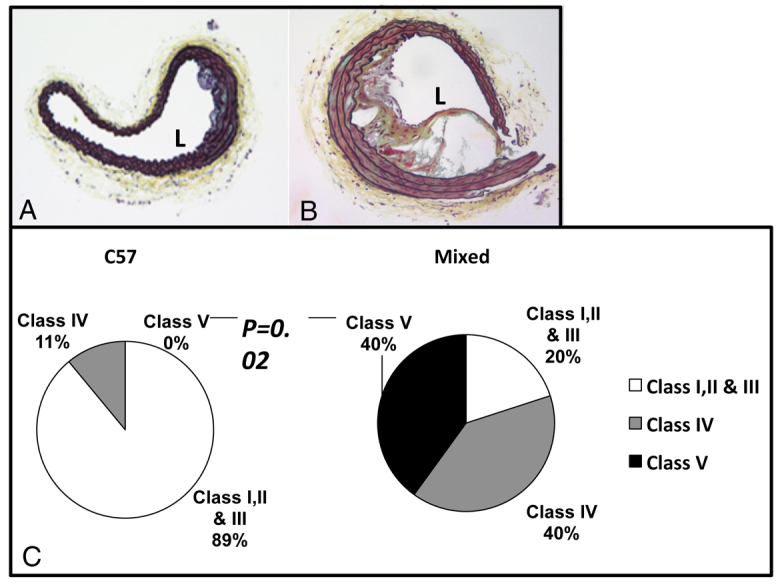
Comparison of brachiocephalic lesions in 16-week-old stressed mice of different backgrounds. (A,B) Brachiocephalic lesions of 16-week-old stressed C57 (A) and stressed mixed-background (B) mice. (C) Pie chart shows distribution of different lesion grades between C57 and mixed-background mice.
Neuropeptide Y immunoreactivity
Blood samples from 20-week-old mice were divided into platelet-rich (PRP) and platelet-poor (PPP) plasma fractions, and neuropeptide Y immunoreactivity (NPY-ir) was measured using an ELISA. NPY contained in the platelets (as measured in PRP) was elevated in stressed mice (16.44±1.26 ng/ml versus 8.66±0.87 ng/ml, P=0.001), but NPY present in the plasma (PPP) was not different between stressed and non-stressed mice (5.58±0.38 ng/ml versus 5.3±0.46 ng/ml; P not significant, n=9 per group, Fig. 3A).
Fig. 3.
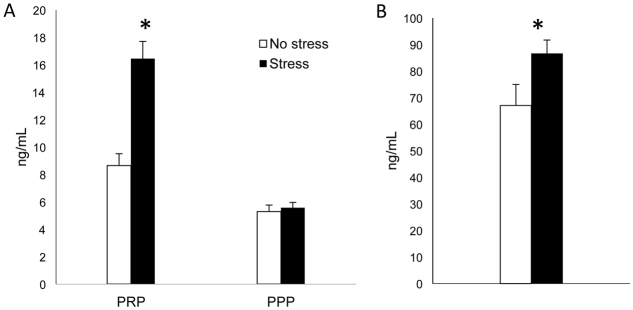
NPY and corticosterone levels in 20-week-old stressed and unstressed mice. (A) NPY measurements in PRP and PPP collected from mixed-background ApoE−/− mice at 20 weeks of age. NPY levels are significantly greater in the PRP from stressed mice (n=9) compared with non-stressed mice (n=9; *P=0.001). (B) Urine corticosterone measurements at 20 weeks of age. Stressed mice (n=12) have higher levels of corticosterone than unstressed animals (n=6; *P=0.04).
Corticosterone measurements
Urine was collected at 20 weeks of age and corticosterone levels were measured using an ELISA. Corticosterone levels were higher in the urine of stressed mice compared with non-stressed mice (86.71±5.0 ng/ml versus 67.05±7.9 ng/ml, P=0.04, n=12 and 6, respectively; Fig. 3B).
Percentage stenosis
Percentage stenosis was measured in the brachiocephalic arteries at 16, 20 and 40 weeks of age. Although there was no effect of stress, the percentage stenosis significantly increased with age (P<0.0001; Fig. 4A). All of the progression appeared to occur within the first 20 weeks, with no further progression evident after 20 weeks.
Fig. 4.

Percentage stenosis, ratio of lipid core to lesion area, fibrous cap thickness and percentage of class IV and class V lesions in stressed and unstressed mice of different ages. (A) Percentage stenosis increases with age from 16 to 20 weeks (P<0.01). At 16 weeks, stressed animals had greater percentage stenosis than unstressed animals (*P=0.02). (B) Ratio of lipid core to lesion area increases with age (P<0.01). (C) Fibrous cap thickness decreases with age (P=0.02). There is a trend for thinner fibrous cap in stressed animals (P=0.05). At 16 weeks, only one non-stressed mouse had a fibrous cap. (D) Percentage of class IV and V lesions increases with stress in 16-week-old mice (*P=0.02) and with age from 16 to 20 weeks (**P=0.01). Complex atheroma (class V) increases significantly with stress at the age of 20 weeks (#, P=0.03); however, there are significantly less complex atheroma (class V) in the 40-week-old mice compared with 20-week-old mice (##, P<0.01). All stressed animals at 20 weeks (100%) had class IV or V lesions.
Necrotic core
The necrotic core area was measured and the percentage of necrotic core to lesion area was determined at all time points. Stress did not significantly effect this parameter, although the necrotic core percentage of the lesion area increased with age (P<0.0001; Fig. 4B).
Fibrous cap thickness
The fibrous cap thickness was measured at all time points. Overall, fibrous cap thickness decreased with age (P=0.02). Interestingly, across all ages, the stressed mice tended to have thinner fibrous caps than unstressed mice (11.3 μm versus 18.5 μm, respectively, P=0.05; Fig. 4C).
Lesion classification
Lesions were assessed on a scale of 0-5 as described in Materials and Methods. Fig. 1 and Fig. 4D show that the percentage of class IV and V lesions increases with stress in 16-week-old mice (P=0.02), and with age from 16 to 20 weeks (P=0.01). Interestingly, the percentage of animals having complex atheroma (class V) increased with stress at younger ages (i.e. at 20 weeks of age: non-stressed, 20% versus stressed, 83%; P=0.03). Of note, there were significantly fewer complex atheroma (class V) in the 40-week-old mice compared to the 20-week-old animals (P<0.01).
Intraplaque hemorrhage and neovascularization
Stressed mice aged 20 weeks had significantly more lesions with intraplaque hemorrhage or neovascularization, as measured by Ter119 staining and/or the presence of red blood cell-containing vessels within the lesions, compared with non-stressed mice (83% versus 20%; P=0.03, Fig. 5). Lesions from 40-week-old mice did not show Ter119 staining in either group (data not shown). Additional examples of 20-week lesions are shown in Fig. 6.
Fig. 5.

Neovascularization, intraplaque hemorrhage and inflammation in brachiocephalic lesions of a 20-week-old mixed-background ApoE−/− stressed mouse. (A) Longitudinal section of a 20-week-old brachiocephalic lesion shows neovascularization (arrows). (B) Same lesion as in A with higher magnification; CD 31 staining confirms the presence of neovascularization (arrow). (C) Cross-section of a lesion with Movat staining shows RBCs (arrow) in the plaque. (D) Movat staining of a different cut of the same lesion with higher magnification shows the presence of intraplaque hemorrhage in the shoulder area. (E)Ter119 staining of the same lesion confirms the presence of RBCs (arrow). (F) CD 31 staining of the same lesion shows presence of endothelial cells (arrow) in the plaque. (G,H) Mac3 staining of 20-week-old mixed-background ApoE−/− non-stressed (G) and stressed mice (H). (I) Bar graphs show that 20-week-old mixed-background ApoE−/− stressed mice have more neovascularization (NV) and/or intraplaque hemorrhage (IPH) than non-stressed mice. (J) Bar graph shows greater degree of macrophage content in lesions from 20-week-old mixed-background ApoE−/− stressed animals compared with non-stressed mice of the same age.
Fig. 6.
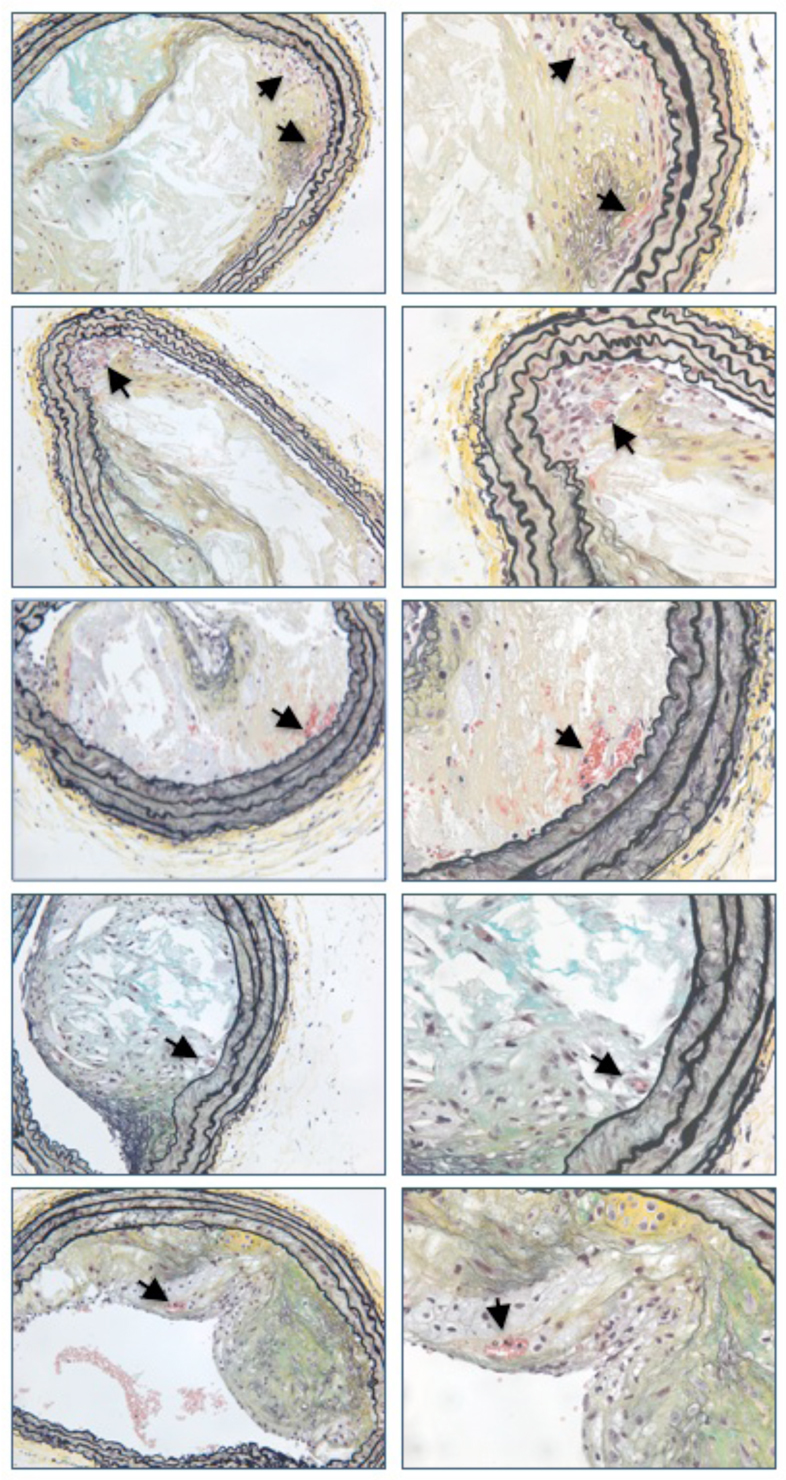
Examples of intraplaque hemorrhage and neovascularization in five different 20-week-old mixed-background ApoE−/− mice. Images on the left are taken at 20×. Images on the right are taken at 40× and correspond to the lesion depicted immediately to the left. Arrows indicate intraplaque hemorrhage and/or neovascularization.
Inflammation
There was greater macrophage infiltration (as measured by Mac3 staining) in the lesions of stressed mice compared with unstressed mice at 20 weeks of age (12.58% versus 4.99%; P=0.02, Fig. 5). Neither stressed nor unstressed mice at 40 weeks of age had significant Mac3 staining in their lesions (data not shown).
Lesion NPY
At 20 weeks of age, the lesions from the stressed mice had an increased percentage area of NPY immunostaining compared with the lesions from unstressed mice (P=0.03; Fig. 7).
Fig. 7.
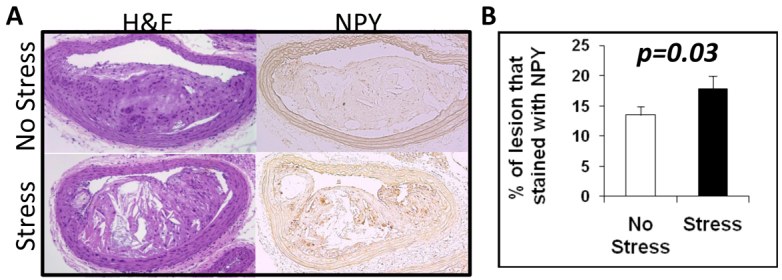
Impact of stress on lesion NPY in 20-week-old mixed-background ApoE−/− mice. (A) H&E and NPY immunostaining of a brachiocephalic lesion from non-stressed and stressed mice. (B) Bar graph shows that brachiocephalic lesions from stressed mice have more NPY-immunopositivity compared with non-stressed mice (P=0.03).
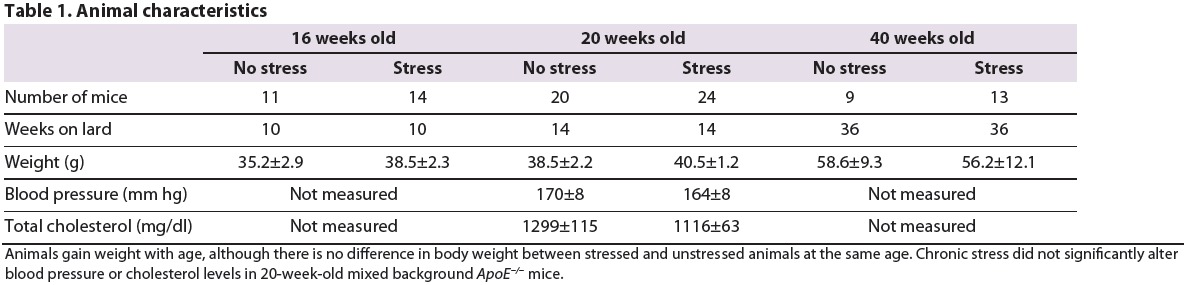
Blood pressure and cholesterol measurements
At 20 weeks of age, there was no difference in blood pressure or total cholesterol levels between stressed and non-stressed mice. (Table 1).
Table 1.
Animal characteristics
DISCUSSION
Many publications have described various animal models of atherosclerotic lesions that have included several elements of human vulnerable plaque such as necrotic core (Mazzolai et al., 2004), inflammation (Li et al., 2010; Mazzolai et al., 2004; Roncal et al., 2010), intraplaque hemorrhage (Rosenfeld et al., 2000) and fibrous cap thickness (Li et al., 2010; Mazzolai et al., 2004). Some investigators have described the occurrence of plaque rupture (Johnson et al., 2005; Johnson and Jackson, 2001; Rosenfeld et al., 2000; Williams et al., 2002), including ‘buried fibrous caps’ thought to reflect prior episodes of rupture. Others have published models using cuffs placed around the carotid artery to produce a ‘vulnerable’ lesion (Cheng et al., 2006; Sasaki et al., 2006). Cheng et al. reported in one such study a 28% incidence of intraplaque hemorrhage (as measured by the presence of erythrocytes and by Prussian blue reaction with iron deposits) (Cheng et al., 2006). Studies by Rosenfeld et al. detailed the presence of intraplaque hemorrhage in the innominate artery of ApoE−/− mice, aged 42-60 weeks (Rosenfeld et al., 2000). However, the authors emphasized that the lesions present in these mice lacked microvessels, a crucial component of human vulnerable plaque. Thus, although each of these models contain one or more elements of human vulnerable plaque, it appears to be the general consensus that, to date, the overall plaque phenotype of ‘vulnerable plaque’ animal models lacks crucial comparability to human vulnerable plaque and that the physiological relevance of such models to human disease remains unclear (Falk et al., 2007; Finn et al., 2010; Schwartz et al., 2007).
In humans, stress is one of the major risk factors for the development of atherosclerosis. An epidemiologic study by Yusuf et al. of nearly 30,000 participants showed that psychosocial stress is among one of the top modifiable risk factors for cardiovascular disease (Yusuf et al., 2001). Previously, Gu et al. found that chronic mild stress in ApoE−/− mice resulted in greater lesion area and, importantly, increased aortic expression of specific genes in the TLR4 signaling pathway, including TLR4, MyD88, NF- κB, MCP-1, IL-1β, TNF-α and sICAM-1 (Gu et al., 2009), the gene products of which could induce a greater inflammatory response.
We therefore included stress in our model in order to determine not only whether stress resulted in a lesion phenotype that more closely mimics human disease but, as importantly (because it is widely believed that myocardial infarction and sudden cardiac death can be triggered by periods of intense stress) (Malzburg, 1937; Yan et al., 2003), whether the psycho-physical type of stress we employed modulates changes from a stable to a more complex plaque phenotype. In deciding which mouse strain to use in our studies, we took into account the inherent differences between mouse strains in their responses to stress. In this regard, we were intrigued by the study by McCutcheon et al. that showed that mice on a mixed background of C57 and SV129 are more affected by stress (McCutcheon et al., 2008). Hence, with the thought that stress might induce more of a ‘vulnerable-like’ phenotype, we decided to utilize this mixed strain.
In our study, we used the cold stress model mainly because it is known to activate the sympathetic nervous system and is linked to the development of experimental vascular occlusion (Li et al., 2005). This model imposes not only a physical stress, but the accompanying feelings of restraint and helplessness also probably mimic psychosocial stress. Previous studies by our group have demonstrated that this type of stress can trigger the same neurohormonal changes as social defeat stress (Kuo et al., 2007). Elevated platelet NPY and corticosterone confirm that our mice were experiencing stress (Fig. 3).
Our findings bear out our hypothesis that the mixed strain mouse model develops, in a time-related manner, progressively more severe lesions in the brachiocephalic artery and that chronic stress increases many of the vulnerable plaque-like components found within these lesions. In an attempt to categorize and therefore more easily discuss the lesions, we classified the plaques in relation to progressive phenotypic severity from class I to class V (Fig. 1), the definitions of which are detailed in the Materials and Methods. The higher grade lesions are classes IV and V. Class IV (referred to as a fibrous cap atheroma) contains a well-formed necrotic core with overlying fibrous cap containing smooth muscle cells (SMCs), with occasional clusters of macrophages. Class V (referred to as a ‘complex’ atheroma) has a well-formed necrotic core, macrophage infiltration, and fibrous cap, plus various degrees of intraplaque neovascularization or hemorrhage.
Using this classification, we first found that the mixed strain mice subjected to chronic stress had more severe lesions than the stressed ApoE−/− C57 mouse (Fig. 2). This finding validates our hypothesis that the mixed-background mouse is more prone to develop stress-induced atherosclerotic plaque. We next found that the 20-week-old mixed strain mice exhibited frequent neovascularisation or plaque hemorrhage as well as macrophage infiltration, findings that were exacerbated by chronic stress (Fig. 5). In addition, we observed that the severity of the lesions changed with time. Thus, the percentage necrotic core area and percentage number of lesions with intraplaque hemorrhage or neovascularization all increased with time between 16 to 20 weeks. However, the more severe class IV-V lesions seemed to peak at 20 weeks, reaching a plateau so that no further progression was seen after 20 weeks (Fig. 4D).
Importantly, chronic cold stress imposed 4 weeks prior to sacrifice accelerates lesion progression in the mice sacrificed at 16 and 20 weeks of age. In particular, the incidence of neovascularization and intraplaque hemorrhage was 83% in the 20-week-old chronically stressed mice, compared with only 20% in the 20-week-old non-stressed animals. There was also a trend for thinner fibrous caps in the stressed mice compared with the non-stressed animals (Fig. 4C).
By 40 weeks of age, brachiocephalic lesions of the mixed-background mice revealed sparse cellularity and did not develop more complex lesions when exposed to chronic stress (Fig. 4). These findings suggest that there is a window of time during which the lesions are susceptible to the effects of stress (between 16 and 20 weeks of age), whereas if stress is initiated after the lesions are already well-established, or after a certain age, stress-induced phenotype modulation does not occur.
A significant contributor to necrotic core enlargement in human lesions is thought to be intraplaque hemorrhage occurring from a leaky fragile network of intraplaque vasa vasorum (Kolodgie et al., 2003). Until now, such neovascularization has not been definitively reported in the brachiocephalic artery of existing mouse models. In the plaques of 20-week-old stressed mixed strain mice we have not only demonstrated intraplaque hemorrhage using a staining technique that uses an antibody specific for red blood cell (RBC) membranes (Ter119), but also observed RBC-filled microvessels that are lined by CD31+ cells (Fig. 4). Moulton et al. described the presence of CD31+ structures in advanced aortic sinus lesions in much older ApoE−/− mice (36-60 weeks) (Moulton et al., 1999; Moulton et al., 2003), which they interpreted as evidence of neovascularization. However, their interpretation has been questioned (Rosenfeld et al., 2000). Furthermore, the described neovascularization was only found in 13% of lesions (Moulton et al., 1999). In our study, we found over an 80% prevalence of neovascularization or intraplaque hemorrhage in the stressed animals at a much earlier time point. Because CD31 immunopositivity is not entirely specific for endothelial cells, our independent finding of RBC-containing vessels and the demonstration of intraplaque hemorrhage suggest that, in our model, leaky and/or fragile neovessels contribute to intraplaque hemorrhage and lesion progression. We recognize, however, that we cannot entirely discount intraplaque hemorrhage occurring consequent to cap disruption.
The primary combined purpose of this investigation was to determine whether our mixed strain model, when subjected to chronic stress, exhibits lesions characteristic of human vulnerable plaque and, if so, to determine the time course of such changes. We did find that the lesions that developed were remarkably similar in many ways to human vulnerable plaque, and that the time course of the appearance of these lesions raised interesting insights into plaque development.
Because the complex atherosclerotic lesions we observed were associated with an increase in several markers of stress, including NPY, we hypothesize that this neuropeptide might play a role in linking stress with atherosclerotic plaque complexity. NPY, a major sympathetic co-transmitter, is abundant in the brain and heart (Zukowska-Grojec and Wahlestedt, 1993). Plasma NPY levels increase during stress such as cold exposure and treadmill exercise (Kaijser et al., 1994; Zukowska-Grojec et al., 1996), and also in myocardial ischemia, congestive heart failure and hypertension (Zukowska-Grojec and Wahlestedt, 1993). NPY has certain activities that could predispose to lesion vulnerability. Thus, NPY augments neointimal development in response to vascular injury (Li et al., 2005; Li et al., 2003) and can increase angiogenesis (Lee et al., 2003; Pons et al., 2003; Zukowska-Grojec et al., 1998a), a commonly accepted component of vulnerable plaques in patients.
We found that chronic stress leads to an increase in platelet-rich plasma NPY (Fig. 3A). Based upon this observation, and the finding that chronic stress can enhance atherosclerotic lesion complexity, we thought it worthwhile to quantify the NPY burden in the lesions. Immunohistochemical staining demonstrated that NPY is in fact elevated in the lesions following chronic stress exposure (Fig. 7). Most of the angiogenic activity mediated by NPY, which might lead to plaque neovascularization and progression, is through the Y2 receptor (Lee et al., 2003; Pons et al., 2003; Zukowska-Grojec et al., 1998b; Zukowska-Grojec and Wahlestedt, 1993). Future studies designed to elucidate the role of the Y2 receptor in lesion progression and phenotype are warranted.
It is important to discuss a limitation in our study. We have defined class V as lesions containing either neovascularization or hemorrhage. At 40 weeks of age, we do not see evidence of class V lesions, i.e. no signs of either neovascularization or hemorrhage, either in the non-stressed or stressed mice. By contrast, 80% of the stressed 20-week-old mice had class V lesions. It therefore could be that older mice do not respond to stress by forming more complex lesions, i.e. lesion maturation or age itself convey some resistance to stress-induced complex lesion development. Alternatively, because ∼20% of the non-stressed 20-week-old mice also had class V lesions, it is possible that the 40-week-old mice did have class V lesions earlier (i.e. at 20 weeks) and that the lesions regressed over time. Our findings do not enable us to select between these two possibilities.
In conclusion, ApoE−/− mice exposed to chronic stress can develop lesions with large necrotic core, thin fibrous cap, a high degree of inflammation and intraplaque hemorrhage or neovascularization. Based on our preliminary results, we speculate that stress might induce some of its deleterious effects on the atherosclerotic plaque through NPY signaling pathways.
This model of stress-induced lesion development offers a tool to further investigate progression of plaque phenotype to a more vulnerable phenotype in humans. Our findings also suggest a possible use of this stress-induced model to determine whether therapeutic interventions have effects not only on plaque burden, but also, and importantly, on plaque vulnerability. Future studies to examine the pathways through which stress modulates lesion phenotype, including the role of NPY signaling, could provide therapeutic targets for advancing clinical care.
MATERIALS AND METHODS
Animal model
C57BL/6J ApoE−/− mice were purchased from the Jackson Laboratory and placed on a lard-containing diet (Harlan Teklad, TD 05282) comprising 21% lard and 0.15% added cholesterol at 6 weeks of age. Mixed-background ApoE−/− mice were generated as follows: C57BL/6J ApoE−/− and Sv129 mice were obtained from the Jackson Laboratory and crossed. Female offspring were then crossed with male C57BL/6J ApoE−/− mice, and pups were genotyped to identify homozygous ApoE−/− animals. Genetic screening was performed to determine the percentage of C57BL and Sv129 background in a random sampling of 10 mice (Taconic). All samples were between 77 and 91% C57BL genotype. Mice were weaned by gender and housed with littermates at no more than four mice per cage. At 6 weeks of age, male pups were placed on the lard-containing diet described above (Harlan Teklad, TD 05282). At 12, 16 and 36 weeks of age, animals were divided into stressed and non-stressed groups by cage. Mice in the stressed groups were subjected to 4 weeks of chronic stress by placing them in a cage containing 1 cm of iced water for 1 hour per day, 5 days per week. Mice assigned to stressed groups were stressed together with their cage mates and returned to their home cage following the stress session. All mice were housed in the same room and exposed to the same light dark cycle and diet. At 16, 20 and 40 weeks, animals were heavily sedated (using ketamine 80 mg/kg plus xylazine 10 mg/kg subcutaneously) and exsanguinated while under anesthesia. Animals were perfused with 4% PFA and the brachiocephalic arteries collected. C57BL/6J ApoE−/− were only studied at 16 weeks of age. All protocols involving animals were approved by the Institutional Animal Care and Use Committee at MedStar Health Research Institute.
Lesion scoring
Sections were selected by identifying the slide with the largest and most complex lesions. Two slides prior and two slides after the identified slide were also selected for analysis. All lesions were scored by a blinded observer using the following scoring system: class I, single layer of macrophage foam cells; class II, multiple layers of macrophage foam cells; class III, multiple layers of macrophage foam cells with free cholesterol, necrotic core and rare smooth muscle cells; class IV, well-formed necrotic core with overlying fibrous cap containing smooth muscle cells (SMCs), clusters of macrophage might also be present; and class V, large necrotic core and fibrous cap plus various degrees of intraplaque neovascularization or hemorrhage and macrophage infiltration; we refer to this lesion as a complex atheroma.
Stress markers
NPY-ir levels were measured using a commercially available ELISA kit (Bachem). Blood was collected from the inferior vena cava using a syringe containing 0.1 ml of sodium citrate, then transferred to EDTA-containing tubes and placed on ice. Samples were centrifuged at 380 g for 5 minutes at 4°C and platelet-rich plasma (PRP) isolated. To isolate platelet-poor plasma (PPP), the remaining blood was further centrifuged at 10,000 g for 2 minutes. Peptides were extracted from the PPP and PRP using a C18 Sep-column, according to the manufacturer's instructions (Bachem). NPY-ir was then measured in extracts from PPP and PRP by ELISA.
Corticosterone was measured in urine samples using a commercially available ELISA kit, according to the manufacturer's instructions (Kamiya BioMed). All urine samples were obtained at noon, prior to the stress session one time between days 26-28 (for stressed mice). Each mouse was placed in an empty cage for 1 hour and urine was collected from the empty cages.
Histology
Cross-sections (8 μm) of the brachiocephalic artery were stained with hematoxylin and eosin (H&E) and Movat's pentachrome. Immunohistochemical staining for Ter119, which stains for erythroid cells (eBioscience); CD31, which stains for vascular endothelium (Abcam); Mac 3, which stains for macrophages (BD Biosciences); and NPY (which is released from sympathetic nerves and found in platelets, and many other cell types) (Sigma) was performed and lesions were digitally photographed. Lesion area, percentage stenosis, necrotic core area and ratio of necrotic core to lesion area were determined in a blinded fashion in five sections per animal. Vessel stenosis was measured using NIH ImageJ software. The total lumen area was measured by tracing around the inner vessel wall. The total lesion area was measured by tracing around the lesion. The total lesion area was divided by the total lumen area to determine percentage stenosis.
We defined necrotic core as the acellular areas inside the plaque. These areas can contain cell debris, cholesterol and/or proteoglycan. The lesion area was first measured by a blinded observer using Image ProPlus software. The same observer selected the areas of interest within the lesion that fit our necrotic core definition. The ratio was obtained by dividing the total necrotic area by the lesion area. Three adjacent sections that had the largest lesion area were selected for each mouse and the average was reported.
A section was considered to have neovascularization if red blood cell-containing vessels were visible in the lesion. CD31 staining provided further confirmation of the presence of vessels. Intraplaque hemorrhage was identified by the presence of Ter119 staining. Macrophages were identified by Mac 3 staining. Image ProPlus was used to quantify the amount of Mac3 and NPY staining present in the lesions by a blinded observer, using pixel selection.
Fibrous cap was identified by examination and identification of endothelial and smooth muscle cells in Movats Pentachrome stained slides. Fibrous cap thickness was measured at the thinnest segment.
Blood pressure measurement
Animals were anesthetized with isoflurane and warmed to a tail temperature of 35±1°C. Blood pressure in the tail was measured non-invasively (AD Instruments). A Moor FLPI device was used to detect the flow immediately distal to the blood pressure cuff. Systolic blood pressure was recorded at the point that blood flow started to rise with cuff deflation. All the measurements were done between days 26 and 28. Five consecutive readings were recorded 1 minute apart. The average of five readings was reported per mouse.
Cholesterol measurements
Total cholesterol was measured in the blood using a commercially available kit (Pointe Scientific).
Statistics
Data were analyzed by STATA software (Statacorp, College Station, Texas). All descriptive data are expressed as mean ± s.e.m. Differences in means were analyzed by Student's t-test or one-way ANOVA with Bonferroni correction. Before and after results were analyzed by paired t-test. Ratio data were compared by Chi-square test. Probability values of less than 0.05 were considered significant.
Acknowledgments
FUNDING: The majority of this work was supported by internal funds, with partial support from The National Institutes of Health, National Heart, Lung and Blood Institute [grant number 2R37HL055310-12].
Footnotes
COMPETING INTERESTS: The authors declare that they have no competing or financial interests.
AUTHOR CONTRIBUTIONS: A.H.N., N.A., J.U.T., J.A.A., X.-Z.P., R.M.L.-S., S.S., L.O.A., L.L., K.A. and M.S.B. performed the experiments. A.H.N., N.A., F.K., R.V., Z.Z., S.E.E. and M.S.B. analyzed the data. A.H.N., N.A., S.E.E. and M.S.B. prepared the manuscript.
References
- Aikawa M., Rabkin E., Okada Y., Voglic S. J., Clinton S. K., Brinckerhoff C. E., Sukhova G. K., Libby P. (1998). Lipid lowering by diet reduces matrix metalloproteinase activity and increases collagen content of rabbit atheroma: a potential mechanism of lesion stabilization. Circulation 97, 2433-2444 [DOI] [PubMed] [Google Scholar]
- Arbustini E., Dal Bello B., Morbini P., Burke A. P., Bocciarelli M., Specchia G., Virmani R. (1999). Plaque erosion is a major substrate for coronary thrombosis in acute myocardial infarction. Heart 82, 269-272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binder E. B., Salyakina D., Lichtner P., Wochnik G. M., Ising M., Pütz B., Papiol S., Seaman S., Lucae S., Kohli M. A., et al. (2004). Polymorphisms in FKBP5 are associated with increased recurrence of depressive episodes and rapid response to antidepressant treatment. Nat. Genet. 36, 1319-1325 [DOI] [PubMed] [Google Scholar]
- Cheng C., Tempel D., van Haperen R., van der Baan A., Grosveld F., Daemen M. J., Krams R., de Crom R. (2006). Atherosclerotic lesion size and vulnerability are determined by patterns of fluid shear stress. Circulation 113, 2744-2753 [DOI] [PubMed] [Google Scholar]
- Davies M. J. (2000). The pathophysiology of acute coronary syndromes. Heart 83, 361-366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk E. (1983). Plaque rupture with severe pre-existing stenosis precipitating coronary thrombosis. Characteristics of coronary atherosclerotic plaques underlying fatal occlusive thrombi. Br. Heart J. 50, 127-134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falk E., Schwartz S. M., Galis Z. S., Rosenfeld M. E. (2007). Putative murine models of plaque rupture. Arterioscler. Thromb. Vasc. Biol. 27, 969-972 [DOI] [PubMed] [Google Scholar]
- Finn A. V., Nakano M., Narula J., Kolodgie F. D., Virmani R. (2010). Concept of vulnerable/unstable plaque. Arterioscler. Thromb. Vasc. Biol. 30, 1282-1292 [DOI] [PubMed] [Google Scholar]
- Gössl M., Versari D., Mannheim D., Ritman E. L., Lerman L. O., Lerman A. (2007). Increased spatial vasa vasorum density in the proximal LAD in hypercholesterolemia – implications for vulnerable plaque-development. Atherosclerosis 192, 246-252 [DOI] [PubMed] [Google Scholar]
- Gu H., Tang C., Peng K., Sun H., Yang Y. (2009). Effects of chronic mild stress on the development of atherosclerosis and expression of toll-like receptor 4 signaling pathway in adolescent apolipoprotein E knockout mice. J. Biomed. Biotechnol. 2009, 613879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson C. L. (2007). Defining and defending murine models of plaque rupture. Arterioscler. Thromb. Vasc. Biol. 27, 973-977 [DOI] [PubMed] [Google Scholar]
- Jackson C. L., Bennett M. R., Biessen E. A., Johnson J. L., Krams R. (2007). Assessment of unstable atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol. 27, 714-720 [DOI] [PubMed] [Google Scholar]
- Johnson J. L., Jackson C. L. (2001). Atherosclerotic plaque rupture in the apolipoprotein E knockout mouse. Atherosclerosis 154, 399-406 [DOI] [PubMed] [Google Scholar]
- Johnson J., Carson K., Williams H., Karanam S., Newby A., Angelini G., George S., Jackson C. (2005). Plaque rupture after short periods of fat feeding in the apolipoprotein E-knockout mouse: model characterization and effects of pravastatin treatment. Circulation 111, 1422-1430 [DOI] [PubMed] [Google Scholar]
- Kaijser L., Pernow J., Berglund B., Grubbström J., Lundberg J. M. (1994). Neuropeptide Y release from human heart is enhanced during prolonged exercise in hypoxia. J. Appl. Physiol. 76, 1346-1349 [DOI] [PubMed] [Google Scholar]
- Kolodgie F. D., Gold H. K., Burke A. P., Fowler D. R., Kruth H. S., Weber D. K., Farb A., Guerrero L. J., Hayase M., Kutys R., et al. (2003). Intraplaque hemorrhage and progression of coronary atheroma. N. Engl. J. Med. 349, 2316-2325 [DOI] [PubMed] [Google Scholar]
- Kuo L. E., Kitlinska J. B., Tilan J. U., Li L., Baker S. B., Johnson M. D., Lee E. W., Burnett M. S., Fricke S. T., Kvetnansky R., et al. (2007). Neuropeptide Y acts directly in the periphery on fat tissue and mediates stress-induced obesity and metabolic syndrome. Nat. Med. 13, 803-811 [DOI] [PubMed] [Google Scholar]
- Lee E. W., Michalkiewicz M., Kitlinska J., Kalezic I., Switalska H., Yoo P., Sangkharat A., Ji H., Li L., Michalkiewicz T., et al. (2003). Neuropeptide Y induces ischemic angiogenesis and restores function of ischemic skeletal muscles. J. Clin. Invest. 111, 1853-1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Lee E. W., Ji H., Zukowska Z. (2003). Neuropeptide Y-induced acceleration of postangioplasty occlusion of rat carotid artery. Arterioscler. Thromb. Vasc. Biol. 23, 1204-1210 [DOI] [PubMed] [Google Scholar]
- Li L., Jönsson-Rylander A. C., Abe K., Zukowska Z. (2005). Chronic stress induces rapid occlusion of angioplasty-injured rat carotid artery by activating neuropeptide Y and its Y1 receptors. Arterioscler. Thromb. Vasc. Biol. 25, 2075-2080 [DOI] [PubMed] [Google Scholar]
- Li L., Cai X. J., Feng M., Rong Y. Y., Zhang Y., Zhang M. (2010). Effect of adiponectin overexpression on stability of preexisting plaques by inducing prolyl-4-hydroxylase expression. Circ. J. 74, 552-559 [DOI] [PubMed] [Google Scholar]
- Malzburg B. (1937). Mortality among patients with involution melancholia. Am. J. Psychiatry 93, 1231-1238 [Google Scholar]
- Mazzolai L., Duchosal M. A., Korber M., Bouzourene K., Aubert J. F., Hao H., Vallet V., Brunner H. R., Nussberger J., Gabbiani G., et al. (2004). Endogenous angiotensin II induces atherosclerotic plaque vulnerability and elicits a Th1 response in ApoE-/- mice. Hypertension 44, 277-282 [DOI] [PubMed] [Google Scholar]
- McCutcheon J. E., Fisher A. S., Guzdar E., Wood S. A., Lightman S. L., Hunt S. P. (2008). Genetic background influences the behavioural and molecular consequences of neurokinin-1 receptor knockout. Eur. J. Neurosci. 27, 683-690 [DOI] [PubMed] [Google Scholar]
- Mohler E. R., 3rd, Sarov-Blat L., Shi Y., Hamamdzic D., Zalewski A., Macphee C., Llano R., Pelchovitz D., Mainigi S. K., Osman H., et al. (2008). Site-specific atherogenic gene expression correlates with subsequent variable lesion development in coronary and peripheral vasculature. Arterioscler. Thromb. Vasc. Biol. 28, 850-855 [DOI] [PubMed] [Google Scholar]
- Moulton K. S., Heller E., Konerding M. A., Flynn E., Palinski W., Folkman J. (1999). Angiogenesis inhibitors endostatin or TNP-470 reduce intimal neovascularization and plaque growth in apolipoprotein E-deficient mice. Circulation 99, 1726-1732 [DOI] [PubMed] [Google Scholar]
- Moulton K. S., Vakili K., Zurakowski D., Soliman M., Butterfield C., Sylvin E., Lo K. M., Gillies S., Javaherian K., Folkman J. (2003). Inhibition of plaque neovascularization reduces macrophage accumulation and progression of advanced atherosclerosis. Proc. Natl. Acad. Sci. USA 100, 4736-4741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakala R., Leborgne L., Cheneau E., Chan R. C., Yazdi H., Fournadjiev J., Weber D., Hellinga D., Kolodgie F., Virmani R., et al. (2003). Radiation-induced atherosclerotic plaque progression in a hypercholesterolemic rabbit: a prospective vulnerable plaque model?. Cardiovasc. Radiat. Med. 4, 146-151 [DOI] [PubMed] [Google Scholar]
- Pons J., Kitlinska J., Ji H., Lee E. W., Zukowska Z. (2003). Mitogenic actions of neuropeptide Y in vascular smooth muscle cells: synergetic interactions with the beta-adrenergic system. Can. J. Physiol. Pharmacol. 81, 177-185 [DOI] [PubMed] [Google Scholar]
- Roncal C., Buysschaert I., Gerdes N., Georgiadou M., Ovchinnikova O., Fischer C., Stassen J. M., Moons L., Collen D., De Bock K., et al. (2010). Short-term delivery of anti-PlGF antibody delays progression of atherosclerotic plaques to vulnerable lesions. Cardiovasc. Res. 86, 29-36 [DOI] [PubMed] [Google Scholar]
- Rosenfeld M. E., Polinsky P., Virmani R., Kauser K., Rubanyi G., Schwartz S. M. (2000). Advanced atherosclerotic lesions in the innominate artery of the ApoE knockout mouse. Arterioscler. Thromb. Vasc. Biol. 20, 2587-2592 [DOI] [PubMed] [Google Scholar]
- Sasaki T., Kuzuya M., Nakamura K., Cheng X. W., Shibata T., Sato K., Iguchi A. (2006). A simple method of plaque rupture induction in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 26, 1304-1309 [DOI] [PubMed] [Google Scholar]
- Schwartz S. M., Galis Z. S., Rosenfeld M. E., Falk E. (2007). Plaque rupture in humans and mice. Arterioscler. Thromb. Vasc. Biol. 27, 705-713 [DOI] [PubMed] [Google Scholar]
- van Rossum E. F., Binder E. B., Majer M., Koper J. W., Ising M., Modell S., Salyakina D., Lamberts S. W., Holsboer F. (2006). Polymorphisms of the glucocorticoid receptor gene and major depression. Biol. Psychiatry 59, 681-688 [DOI] [PubMed] [Google Scholar]
- Virmani R., Kolodgie F. D., Burke A. P., Farb A., Schwartz S. M. (2000). Lessons from sudden coronary death: a comprehensive morphological classification scheme for atherosclerotic lesions. Arterioscler. Thromb. Vasc. Biol. 20, 1262-1275 [DOI] [PubMed] [Google Scholar]
- Virmani R., Kolodgie F. D., Burke A. P., Finn A. V., Gold H. K., Tulenko T. N., Wrenn S. P., Narula J. (2005). Atherosclerotic plaque progression and vulnerability to rupture: angiogenesis as a source of intraplaque hemorrhage. Arterioscler. Thromb. Vasc. Biol. 25, 2054-2061 [DOI] [PubMed] [Google Scholar]
- Williams H., Johnson J. L., Carson K. G., Jackson C. L. (2002). Characteristics of intact and ruptured atherosclerotic plaques in brachiocephalic arteries of apolipoprotein E knockout mice. Arterioscler. Thromb. Vasc. Biol. 22, 788-792 [DOI] [PubMed] [Google Scholar]
- Yan L. L., Liu K., Matthews K. A., Daviglus M. L., Ferguson T. F., Kiefe C. I. (2003). Psychosocial factors and risk of hypertension: the Coronary Artery Risk Development in Young Adults (CARDIA) study. JAMA 290, 2138-2148 [DOI] [PubMed] [Google Scholar]
- Yusuf S., Reddy S., Ounpuu S., Anand S. (2001). Global burden of cardiovascular diseases: Part II: variations in cardiovascular disease by specific ethnic groups and geographic regions and prevention strategies. Circulation 104, 2855-2864 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z., Wahlestedt C. (1993). Origins and actions of Neuropeptide Y in the cardiovascular circulation. Totawa, NJ: Human Press [Google Scholar]
- Zukowska-Grojec Z., Dayao E. K., Karwatowska-Prokopczuk E., Hauser G. J., Doods H. N. (1996). Stress-induced mesenteric vasoconstriction in rats is mediated by neuropeptide Y Y1 receptors. Am. J. Physiol. 270, H796-H800 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z., Karwatowska-Prokopczuk E., Fisher T. A., Ji H. (1998a).Mechanisms of vascular growth-promoting effects of neuropeptide Y: role of its inducible receptors. Regul. Pept. 75-76, 231-238 [DOI] [PubMed] [Google Scholar]
- Zukowska-Grojec Z., Karwatowska-Prokopczuk E., Rose W., Rone J., Movafagh S., Ji H., Yeh Y., Chen W. T., Kleinman H. K., Grouzmann E., et al. (1998b). Neuropeptide Y:a novel angiogenic factor from the sympathetic nerves and endothelium. Circ. Res. 83, 187-195 [DOI] [PubMed] [Google Scholar]