

SUMMARY
Atrial fibrillation (AF) is the most common cardiac arrhythmia and carries a significant risk of stroke and heart failure. The molecular etiologies of AF are poorly understood, leaving patients with limited therapeutic options. AF has been recognized as an inherited disease in almost 30% of patient cases. However, few genetic loci have been identified and the mechanisms linking genetic variants to AF susceptibility remain unclear. By sequencing 193 probands with lone AF, we identified a Q76E variant within the coding sequence of the bone morphogenetic protein (BMP) antagonist gremlin-2 (GREM2) that increases its inhibitory activity. Functional modeling in zebrafish revealed that, through regulation of BMP signaling, GREM2 is required for cardiac laterality and atrial differentiation during embryonic development. GREM2 overactivity results in slower cardiac contraction rates in zebrafish, and induction of previously identified AF candidate genes encoding connexin-40, sarcolipin and atrial natriuretic peptide in differentiated mouse embryonic stem cells. By live heart imaging in zebrafish overexpressing wild-type or variant GREM2, we found abnormal contraction velocity specifically in atrial cardiomyocytes. These results implicate, for the first time, regulators of BMP signaling in human AF, providing mechanistic insights into the pathogenesis of the disease and identifying potential new therapeutic targets.
INTRODUCTION
Atrial fibrillation (AF) carries a significant risk of stroke and heart failure and is associated with substantial morbidity and mortality (Feinberg et al., 1995; Stewart et al., 2002). Up to 30% of patients with AF have a family history of the condition, suggesting that the disease has a broad genetic basis (Darbar, 2008; Lubitz et al., 2010; Miyasaka et al., 2006). Cases of ‘lone’ AF, defined by the presence of sustained arrhythmia in the absence of structural heart disease or other identifiable causes in patients younger than 65 years of age, further underscore the contribution of genetic variation to the development of AF (Parvez and Darbar, 2010). Recent studies have identified both common and rare genetic variants contributing to AF susceptibility. Positional cloning and candidate gene approaches have implicated mutations in genes encoding ion channels, gap junctions and signaling molecules in isolated cases and small kindreds (Abraham et al., 2010; Gollob et al., 2006; Hodgson-Zingman et al., 2008). Genome-wide association studies (GWAS) have also recognized AF susceptibility loci (Ellinor et al., 2012) on chromosomes 4q25 near PITX2 (Gudbjartsson et al., 2007; Ritchie et al., 2012), 1q21 in KCNN3 (Ellinor et al., 2010) and 16q22 in ZFHX3 (Gudbjartsson et al., 2009). Even so, most cases of lone AF remain of unknown etiology, are poorly penetrant, and segregate in isolated cases or small families, rendering the identification of causative genes and the design of new therapeutic strategies particularly challenging (Darbar et al., 2012). Moreover, there is a paucity of functional modeling of known variants that could be used to draw putative molecular and cellular pathways contributing to AF symptoms.
In many cases of AF, electrical signals initiate in ectopic atrial locations, often close to the muscle sleeves of the pulmonary veins (Haïssaguerre et al., 1998; Levin et al., 2009). Pulmonary veins and pulmonary myocardium develop from pharyngeal mesoderm, a process that depends on transcription factor PITX2 (Liu et al., 2002; Mommersteeg et al., 2007). Recent evidence suggests that aberrant activation of embryonic mechanisms of atrial and pulmonary myocardium development can lead to AF (Mommersteeg et al., 2009) and genetic studies have linked PITX2 to AF patients (Gudbjartsson et al., 2007; Ritchie et al., 2012). Heterozygote Pitx2 knockout mice, which have only 40% lower Pitx2 expression than wild types, are also prone to arrhythmias, indicating that even modest changes in Pitx2 protein levels might promote AF (Kirchhof et al., 2011).
During development, Pitx2 expression is regulated by BMP signaling (Furtado et al., 2008; Monteiro et al., 2008; Schlange et al., 2002). It was previously shown that the secreted BMP antagonist Grem2 (Pearce et al., 1999; Sudo et al., 2004) is highly expressed in pharyngeal mesoderm (Müller et al., 2006), similarly to PITX2, suggesting that GREM2 might modulate BMP signaling, thus acting upstream of PITX2. To test this possibility, and evaluate potential involvement of GREM2 in AF, we re-sequenced the GREM2 coding region in families of the Vanderbilt AF Registry (Darbar et al., 2007). We discovered a GREM2 variant in two independent probands with familial AF with a higher frequency than found in the general population. To assess whether the variant contributes to AF, we investigated the role of GREM2 in cardiac development and assayed the arrhythmogenic potential of the GREM2 variant in animal models in vivo and cultured cells in vitro. Our results show that during embryonic development in zebrafish Grem2 is required for proper pitx2 expression, cardiac laterality and atrial differentiation. GREM2 Q76E overactivity results in slower cardiac contraction rates and induction of previously identified AF candidate genes. These findings show that aberrant GREM2 activity probably contributes to AF, providing mechanistic insights into the pathogenesis of the disease.
TRANSLATIONAL IMPACT.
Clinical issue
Atrial fibrillation (AF) is the most prominent heart arrhythmia and carries a significant risk of stroke and heart failure. It affects 2-5 million people in the United States, including young individuals (primarily in an idiopathic form) and older individuals (usually as a complication of various cardiovascular diseases). In most cases of AF, electrical signals begin in aberrant trigger areas, often close to the pulmonary veins. As the molecular causes of AF are not well understood, current treatments are mostly empiric. In addition, treatments can carry significant risks, are frequently ineffective or provide only temporary symptom relief.
Results
By sequencing the gene encoding the bone morphogenetic protein (BMP) antagonist Gremlin-2 (GREM2) in a cohort of idiopathic AF patients, the authors discovered a Q76E variant that increases the capacity of GREM2 to inhibit BMP signaling. The authors went on to analyze this human mutation in a zebrafish model, and assessed its function in live hearts. They made the novel finding that GREM2 is a crucial regulator of the cardiac rhythm gene network that acts upstream of Pitx2 (an important AF-promoting transcription factor). Additional in vivo experiments showed that GREM2 hyperactivity specifically slows conduction velocity in the atrium, without affecting ventricular contraction.
Implications and future directions
These results provide important novel mechanistic insights into the pathogenesis of AF. In addition, they provide a new platform for drug development. A major challenge in modern medicine is to understand how the patchwork of genetic variation in each human patient eventually leads to disease. This work provides an experimental template to evaluate the pathologic contribution of genetic variants in AF, and might contribute to finding new AF treatments.
RESULTS
Presence of a GREM2 Q76E variant in two independent probands with lone AF
To identify new variants contributing to AF, we sequenced a cohort consisting of 193 probands with lone AF for GREM2 and identified a single c.226C>G variant in two independent probands (Darbar et al, 2007; Table 1). C.226C>G is a rare variant (rs142343894) with a significantly higher frequency in the AF cohort than found in the general population represented by 1560 subjects in the 1000 Genomes Project (0.5% compared to 0.03%, P=0.034) (Clarke et al, 2012; Mills et al, 2011) (Fig. 1A; supplementary material Table S1). C.226C>G is a missense mutation within exon 2 of the GREM2 gene that results in substitution of glutamine 76 for glutamic acid (Q76E). BMP antagonists such as GREM2 share the same overall protein structure as BMP ligands. Q76 is highly conserved across species and adjacent to C73, the first of the group of six cysteine residues that form the cystine knot motif required for protein folding, dimerization and interaction with BMP receptors (Avsian-Kretchmer and Hsueh, 2004) (Fig. 1B,C).
Table 1.
Clinical characteristics of AF patients
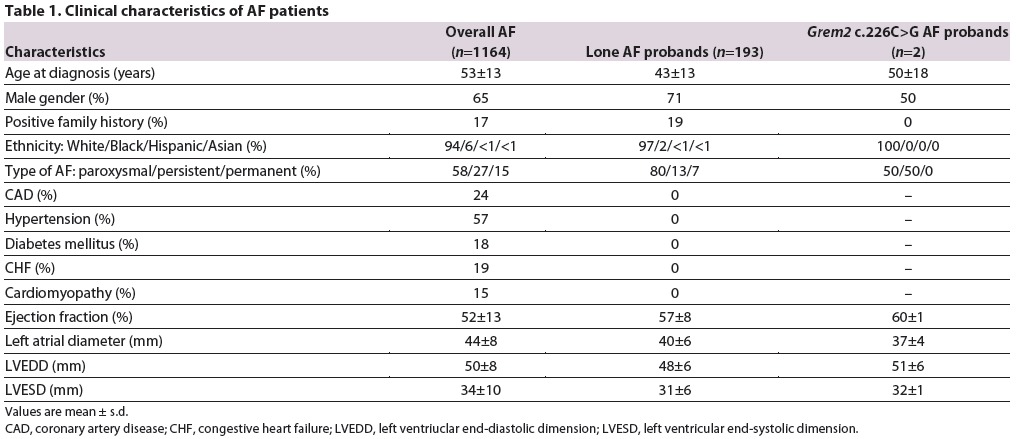
Fig. 1.
Identification of GREM2 variant in probands with lone AF. (A) Pedigrees of two families with AF and probands heterozygous for the GREM2 c.226C>G variant allele that segregates in one of the families. Symbols indicate phenotypes as follows: solid, positive for AF; open, unaffected at >65 years of age. Due to the variable expressivity of AF and late onset of symptoms, patients age <65 years are considered to be of undetermined phenotype and are indicated by gray shading (A-III, B-III). Chromatographs are of proband carrying the Q76E mutation in GREM2 and wild-type control. (B) Protein model of GREM2 indicates the position of the amino acid substitution (red). The location of the six cysteines of the cystine knot are marked in green. Two additional cysteines C' and C” linking adjacent loops are indicated, as well as the unpaired Cx in the heel of the cysteine knot. The Q76E substitution is immediately adjacent to the conserved first cysteine (C73) of the cystine-knot motif at the base of the first of the two loops. (C) Sequence alignment shows that the Q76E substitution is in a highly conserved glutamine (Q) residue across species. (D) Relative expression levels of ID2 in HEK293 cells transfected with decreasing amounts of plasmids expressing wild-type GREM2 or GREM2-Q76E and treated with BMP4 recombinant protein. GREM2-Q76E decreased ID2 expression at lower levels than wild-type GREM2, suggesting that the variant was more potent at antagonizing BMP than wild-type GREM2. ***P<0.0001 versus no BMP4; ##P<0.01, ###P<0.0001 versus wild-type GREM2.
The Q76E variant increases the inhibitory activity of GREM2
To determine whether the p.Q76E variant alters the ability of GREM2 to antagonize BMP signaling, we transfected expression plasmids carrying wild-type GREM2 or GREM2-Q76E into human embryonic kidney cells. We then exposed cells to recombinant BMP4 and assessed induction of ID2, a prototypical BMP signaling gene target. At maximum expression levels, both wild-type and variant GREM2 efficiently inhibited BMP signaling; however, by titrating the amount of transfected plasmids, we found that at lower concentrations the Q76E variant was at least twofold more potent at antagonizing BMP than wild-type GREM2 (Fig. 1D). These data suggest that the Q76E substitution generates a hypermorphic allele.
Grem2 is required for asymmetric cardiac development and atrial differentiation in zebrafish
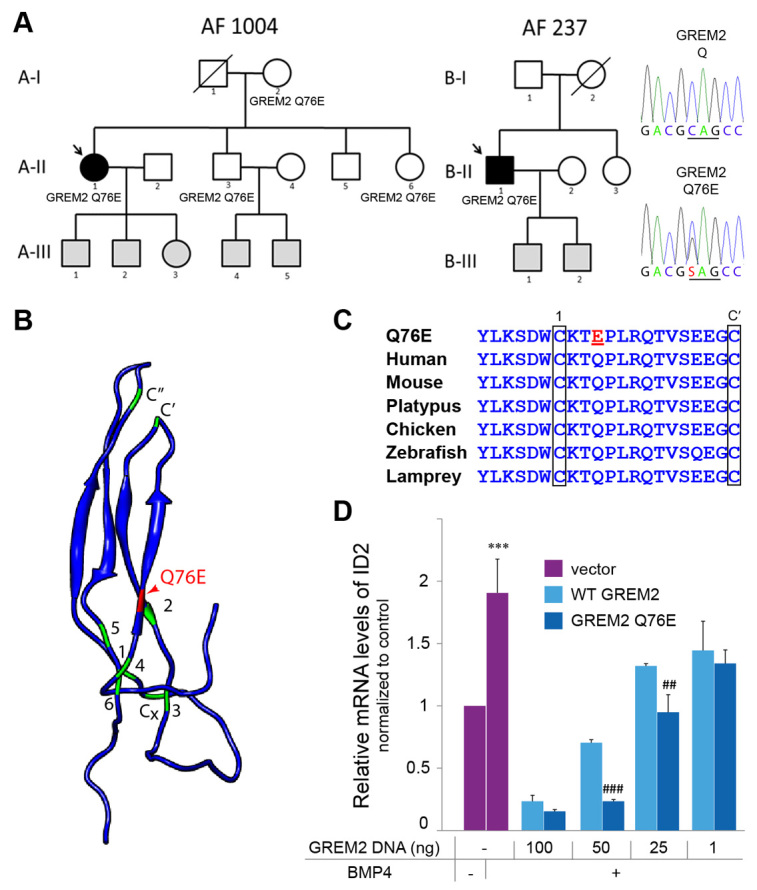
Recent studies have implicated GREM2 in placode neurogenesis and craniofacial patterning (Kriebitz et al, 2009; Zuniga et al, 2011). However, its role in cardiac development and cardiac function is unknown. To address this issue, we turned to the zebrafish model that offers excellent genetic and embryological tools for studying cardiac structure and function in vivo. By examining the expression of grem2 and cardiac-specific genes, we found that grem2 was present in the ventral portion of the pharyngeal arch mesoderm, immediately adjacent to the cardiac field, at the time that cardiac progenitor cells migrate from the midline to the left side of the embryo (Fig. 2). Because BMPs are known to modulate proliferation and differentiation of cardiac progenitor cells (Chocron et al, 2007; Schultheiss et al, 1997), the juxtaposition of the Grem2-positive and cardiac territories raised the possibility that Grem2 shapes the gradient of BMP activity across the migratory path of developing cardiomyocytes and thus affects their fate.
Fig. 2.
Expression ofgrem2in pharyngeal arch primordia during cardiac tube migration. (A,B) Whole mount images of grem2 (purple)- and cmlc2 (red)-stained embryos in dorsal (A) and lateral (B) views, anterior to the left. grem2 is expressed in the first two pharyngeal arches (blue arrows point to the left first arch, black arrows to the left second arch) as the cardiac progenitors begin to assemble in the midline. During cardiac jogging to the left, the heart tube (yellow arrowhead) is positioned next to grem2-expressing pharyngeal mesoderm and subsequently passes ventrally to the grem2-expression domain in the left first pharyngeal arch. (C) Cross-sections of zebrafish embryos double-labeled for grem2 and cmlc2. h, hours post fertilization; A, anterior; P, posterior; nt, neural tube; y, yolk.
To test this possibility, we employed loss-of-function approaches in zebrafish using morpholino antisense oligonucleotides (described in supplementary material Fig. S1). We found that depletion of grem2 led to randomization of the cardiac axis, jogging and looping defects, as well as varied shape of the heart tube (Fig. 3A). Replacement experiments with zebrafish or mouse mRNAs rescued the morpholino defects, suggesting that the phenotype was specific to loss of Grem2 function (supplementary material Figs S2, S3). In zebrafish grem2 morphants, expression of both atrial- and ventricular-specific myosin heavy chains (amhc and vmhc) was reduced, with amhc being more affected. The deficits in cardiac gene expression at 19 hours post-fertilization (hpf) was consistent with a smaller atrium in older embryos (48 hpf), as evaluated by antibody staining of ventricular and atrial tissues (Fig. 3B). In addition, expression of pitx2, which controls cardiac laterality (Essner et al., 2000; Ryan et al., 1998), was bilaterally upregulated when cardiac progenitors occupy the concentric territory adjacent to the midline, whereas the expression of the nodal antagonist lefty2, which is asymmetrically expressed on the left side when the cardiac tube has jogged to the left (Bisgrove et al., 1999), was absent (Fig. 3B). Grem2 does not appear to be required for asymmetric gene activity during early developmental stages, as illustrated by the normal left side expression of the nodal gene spaw in posterior mesoderm at 16 hpf (supplementary material Fig. S4). These data indicate that Grem2 depletion results in abnormal expression of pitx2 and lefty2, leading to cardiac laterality defects. In addition, atrial development appears to be particularly sensitive to Grem2 depletion.
Fig. 3.

Loss of Grem2 leads to cardiac jogging and looping defects, aberrant expression ofpitx2and abnormal development of cardiac chambers. (A) In situ hybridization analysis using cmlc2 riboprobe shows that in wild-type (WT) embryos the heart jogs leftward, whereas cardiac morphogenesis is randomized in grem2 morphants (grem2 MO). The cardiac tube, consisting of a single atrium (A, white arrowheads) and a single ventricle (V), is thinner, shorter and fails to loop in grem2 morphants. Left/Right (L/R) axis orientation is indicated. (B) In situ hybridization analysis using vmhc and amhc riboprobes shows that loss of Grem2 leads to lower expression levels of both vmhc and amhc, with amhc being essentially absent at 19 hpf (arrowheads). Pitx2 expression (black arrowheads) at 19 hpf is enhanced (white arrowheads), whereas lefty2 expression at 24 hpf is abolished (black arrowheads). Quantification of expression levels in wild-type and morphant embryos is shown below the corresponding images (expression is relative to wild type, which was set as arbitrary value 1). Immunofluorescence analysis using MF20 (red, labels both ventricular and atrial cardiomyocytes) and S46 (green, labels only atrial cardiomyocytes, which appear yellow) antibodies shows abnormal development of both chambers, with the atrium being more deformed and reduced in size than the ventricle. Arrowheads mark the position of the atrio-ventricular boundary. Scale bars: 50 μm. *P<0.05.
Grem2 regulates BMP signaling activity in cardiac development
Previous work has shown that precise levels of BMP signaling are necessary for cardiac morphogenesis (Chocron et al., 2007). To test whether Grem2 regulates BMP signaling during zebrafish cardiac development, we analyzed phosphorylated Smad1/5/8 (pSmad) proteins, the main intracellular mediators of activated BMP signaling. We found strong pSmad protein upregulation in the somitic boundaries (Fig. 4A) and the heart of grem2 morphants compared with wild-type controls (Fig. 4B), suggesting that Grem2 plays a key role in modulating BMP signaling during cardiogenesis. To test whether attenuation of the high BMP signaling levels in grem2 morphants could reverse the cardiac deficits, we exposed grem2-depleted embryos to dorsomorphin (DM), a chemical inhibitor of BMP signaling (Hao et al., 2008) and analyzed cardiac development using cmlc2 and amhc riboprobes. Incubation of grem2 morphants with DM restored the expression domains of cardiac genes to levels comparable with wild-type embryos, particularly in the atrium (Fig. 4C), whereas DM had no effect on wild-type, DMSO-treated control embryos at concentrations used in these experiments. Furthermore, the differentiation of the atrial bulb itself was restored in 65% of the DM-treated morphants, as assessed by the size of the heart area and the appearance of the morphological characteristics of the atrial bulb (Fig. 4C,D). These data show that aberrantly high BMP signaling in grem2 morphants is responsible, at least in part, for the observed cardiac defects and that Grem2 is a crucial regulator of BMP signaling during cardiac development and essential for atrial patterning and differentiation.
Fig. 4.

Loss of Grem2 leads to increased BMP signaling and cardiac defects that can be reversed by the BMP inhibitor dorsomorphin. (A) Whole mount immunohistochemistry using antibodies recognizing the phosphorylated forms of Smads1/5/8 shows stronger pSmad protein staining in grem2 morphants than in controls. Arrowhead marks somite boundaries. (B) Frontal close up views of the heart after pSmad1/5/8 antibody staining at 48 hpf shows sharply increased nuclear staining in cardiomyocytes of Grem2-depleted embryos. The heart is outlined by dotted lines; arrowheads mark pSmad-positive nuclei. (C) Wild-type (WT) embryos and grem2 morphants were incubated with dorsomorphin or its vehicle DMSO between 16 and 48 hpf and stained at 48 hpf with cmlc2 and amhc probes to visualize the ventricle (V) and atrium (A). Dorsomorphin treatment restored atrial patterning and differentiation (arrowheads point to the atrioventricular boundary in wild-type hearts; dotted lines demarcate ventricle-atrium boundary in grem2 morphants). (D) Quantification of heart size (Heart Area) and atrial bulb restoration (Presence of Atrial Bulb) in DMSO-treated controls (WT), DMSO-treated grem2 morphants (MO) and grem2 morphants treated with dorsomorphin (MO+DM). Dorsomorphin treatment restores atrial development. Error bars represent s.d. P-values and number of embryos analyzed (n) are indicated. b, brain; e, eye; m, mouth; y, yolk.
Grem2 overexpression expands atrial cardiomyogenesis
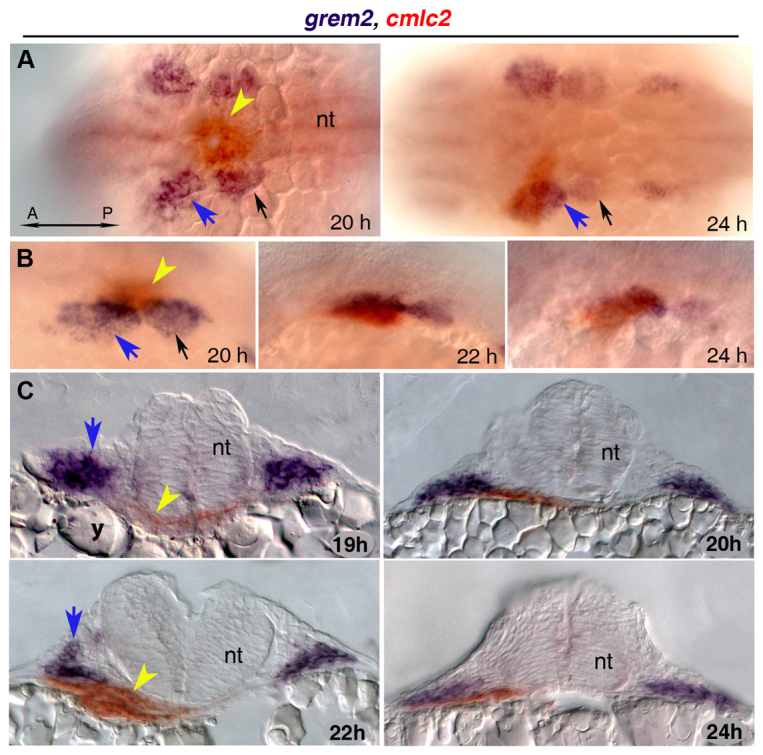
To establish whether Grem2 is sufficient to induce atrial formation in developing embryos, we overexpressed Grem2 by injecting grem2 mRNA into one-cell-stage cmlc2-egfp transgenic zebrafish embryos and evaluated cardiac gene expression. Resultant embryos showed cardiac development not only in its native location, but also contractile activity in ectopic areas in the posterior trunk at 48 hpf (Fig. 5A). At earlier stages (13 hpf), we observed supernumerary expression domains of gata-5, a transcription factor that promotes cardiac specification in mesodermal progenitor cells (Reiter et al., 1999) (Fig. 5B). These results indicate that Grem2 is sufficient to induce ectopic cardiomyogenesis in mesodermal progenitor cells. To determine the molecular characteristics of the ectopic cardiac territories, we stained grem2-overexpressing embryos with amhc and vmhc riboprobes (Fig. 5C). The expanded ectopic myocardium expressed exclusively atrial mhc (amhc), whereas ventricular mhc (vmhc) expression in the native hearts was significantly reduced as compared with wild type, indicating that high Grem2 activity promotes atrial development and differentiation.
Fig. 5.
Overexpression of Grem2 induces ectopic atrial myocardium. (A) Lateral view of live wild-type (WT) and grem2 mRNA-injected (15 pg) cmlc2-egfp transgenic embryos (grem2 OE) at 48 hpf shows ectopic cardiac tissue (arrowhead). Anterior-posterior (A/P) and dorsal-ventral (D/V) axes are indicated. (B) In situ hybridization shows ectopic sites expressing gata-5 along the paraxial mesoderm (arrowheads). (C) WT zebrafish embryos were injected with grem2 mRNA at the 1-4 cell stage; non-injected embryos served as controls. At 24 hpf, Grem2 overexpression (OE) led to enlarged or ectopic (arrowhead) amhc expression in 100% of injected embryos, whereas vmhc expression was reduced (arrow) or absent in 54.4%. These results remained consistent at 48 hpf, with 96% of embryos displaying ectopic amhc expression (arrowhead), and 52.2% displaying reduced vmhc expression (arrow). Insets display enlarged frontal images. Normal atrium (A) expressing amhc and ventricle (V) expressing vmhc are marked; dorsal (24 hpf) and lateral (48 hpf) views; anterior is to the left in all panels.
GREM2 Q76E variant is arrhythmogenic
The role of Grem2 in zebrafish atrial development, as well as the association between human GREM2-Q76E and AF, led us to hypothesize that, in addition to its role in cardiac morphogenesis, GREM2 is an upstream regulatory factor of the genetic network controlling cardiac rhythm. To test this possibility, we compared the expression levels of previously identified AF candidate genes (Antzelevitch et al, 2007; Firouzi et al, 2004; Gollob et al, 2006; Gudbjartsson et al., 2007; Hodgson-Zingman et al, 2008; Shanmugam et al., 2011) in differentiated mouse embryonic stem cells stably transfected with empty vector (control), human wild-type GREM2 and variant GREM2-Q76E expressing plasmids (Fig. 6A). The results show that the expression levels of AF candidate genes were significantly increased by wild-type and variant GREM2, supporting the hypothesis that GREM2 regulates genes essential for maintaining cardiac rhythm.
Fig. 6.

GREM2 activity regulates cardiac rhythm. (A) Stable mouse embryonic stem cell lines, generated with empty vector (control) or vectors expressing wild-type GREM2 or GREM2-Q76E, were allowed to form embryoid bodies. RNA was then isolated at differentiation day 8 and analyzed by qRT-PCR. The results showed that overexpression of GREM2 and GREM2-Q76E leads to upregulation of genes encoding calcium channel, voltage-dependent, L type, alpha 1C subunit (Cacna1c), natriuretic atrial peptide (Nppa), potassium voltage-gated channel subfamily E member 2 (Kcne2), connexin-40 (Gja5) and sarcolipin (Sln). (B) Quantification of cardiac contraction rates in vivo in Tg(cmlc2:EGFP) zebrafish embryos overexpressing human GREM2 and GREM2-Q76E variant. Both wild-type GREM2 and GREM2-Q76E slowed contraction rates, but the effect was more pronounced in GREM2-Q76E-injected embryos. (C) Digital cardiography of Tg(cmlc2:EGFP) zebrafish at 48 hpf showed regular ventricular rhythm and discordant atrial rhythm in wild-type GREM2 and variant GREM2-Q76E injected embryos (arrows). (D) Tracking of two points in the proximal (1) and distal (2) atrium over time with quantification of time spent by each point in systole showed that the time spent in systole was not disrupted in the proximal point, but was significantly increased in the distal point in both wild-type (WT) and variant-injected embryos, suggesting a possible deterioration of the contraction wave as it traveled across the atrium. *P<0.05, **P<0.001, ***P<0.0001 versus control; #P<0.01, ##P<0.001 versus wild-type GREM2.
GREM2 levels are crucial for normal cardiac rhythm
To determine the functional impact of the GREM2 and GREM2-Q76E on cardiac rhythm, we assessed contraction rates in zebrafish embryos injected with human wild-type GREM2 or GREM2-Q76E mRNAs. We found that contraction rates were significantly slower in embryos overexpressing either form of GREM2 compared with non-injected controls, but the effect was more pronounced in variant than wild-type GREM2-injected embryos (Fig. 6B).
To evaluate whether the observed abnormal contraction rate defect reflects irregular rhythm, abnormal contraction signal propagation, or uncoupling of atrial from ventricular contractions, we developed a method of optical digital cardiography (DCG) for in vivo quantification of heart function in zebrafish. We found that the sequential rhythms of atrium and ventricle were synchronized and thus not perturbed by GREM2 or GREM2-Q76E overexpression (Fig. 6C; supplementary material Movies 1-3). However, quantification of the DCG output showed that although ventricular rhythm remained regular, the atrial rhythm was consistently abnormal in GREM2- and GREM2-Q76E-injected embryos (Fig. 6C; supplementary material Movies 1-3). We concentrated our analysis on morphologically normal hearts; however, it is noteworthy that the severity of the arrhythmias increased in morphologically abnormal hearts (not shown). Overall, these data support the hypothesis that GREM2 regulates cardiac rhythm in the atrium, and establish that higher than normal GREM2 activity, either by overexpression or gain-of-function mutations such as the Q76E variant, have arrhythmogenic potential.
To probe the mechanism of irregular atrial contraction, we investigated whether contraction signal propagation was disrupted by assessing spatiotemporal conduction velocity on two distant test points, upstream (point 1) and downstream (point 2) along the contraction wave of the atrium (Fig. 6D). We found that upstream contraction velocities were similar in all three conditions (control, wild type, variant), whereas downstream contraction velocities were slowed in hearts overexpressing GREM2 or variant GREM2-Q76E as compared with controls. Thus, it appears that high GREM2 activity distorts atrial contraction velocity and wavefront propagation.
DISCUSSION
Advanced genetics approaches and sequencing tools have identified a broad spectrum of genetic variation in the coding region of human genes. The current challenge of modern medicine is to determine the contribution of individual pathogenic variants to complex diseases such as familial AF. Our analysis of the GREM2 variant Q76E and the cardiac imaging approaches developed here provide a template for functional modeling of AF-associated variants in zebrafish and differentiating embryonic stem cells. Moreover, because GWAS studies have linked the GREM2 genetic locus to coronary vascular disease (Wellcome Trust Case Control Consortium, 2007), the characterization of the role of GREM2 in cardiac development and function might have broader implications for understanding the contribution of BMP signaling and its specific modulators to cardiovascular health.
Specifically, our results show that Grem2, a BMP antagonist that is required for pharyngeal mesoderm patterning, is essential for zebrafish cardiac development. Loss of Grem2 leads to overexpression of pitx2 and downregulation of lefty2, two crucial regulators of asymmetric cardiac development (Bisgrove et al., 1999; Essner et al., 2000), randomizing jogging and looping. Moreover, Grem2 knockdown increases the levels of activated phosphorylated Smad and BMP signaling within cardiac cells, interfering with both ventricular and atrial differentiation. Atrial differentiation in particular is compromised at high levels of BMP signaling, but favored at low levels.
Our in vitro and in vivo data also support the notion that the GREM2 Q76E mutation generates a hypermorphic allele and a pathogenic genetic variant that increases the risk of AF. Although the allele shows poor penetrance, as often with AF loci, functional analyses in zebrafish revealed that higher than normal GREM2 levels expand atrial differentiation, but reduce cardiac contraction rates and disturb contraction propagation in atrial cardiomyocytes. Because the GREM2 Q76E variant appears to be a stronger BMP inhibitor than wild-type GREM2, it probably has no measurable effect at areas of high GREM2 expression, but might interfere with BMP signaling at border zones of GREM2 and BMP activity. In this setting, overactive GREM2 could promote ectopic differentiation of mesenchymal cells around pulmonary veins to sleeves of atrial muscle, and thus establish foci of ectopic arrhythmogenic activity. Alternatively, GREM2, by inhibiting BMP signaling, might function upstream to directly or indirectly downregulate PITX2c expression (Furtado et al., 2008). Thus, increased GREM2 activity could lower PITX2c levels, altering the expression of atrial genes and raising the risk of AF.
Treatment of AF poses significant challenges, with a clear need for new therapeutic targets. The functional modeling of the human GREM2 mutation suggests that defective BMP signaling leads to AF, providing mechanistic insights into the pathogenesis of the disease and a host of novel therapeutic targets. To the best of our knowledge, GREM2 is the first morphogen implicated in cardiac laterality that also regulates cardiac contraction. This raises the intriguing possibility that the pathways establishing asymmetric cardiac development also control the genetic networks of cardiac rhythm, although the precise molecular mechanisms will need to be further investigated in future studies. These findings open new avenues for exploring the causes of AF, ultimately leading to better diagnostic and therapeutic strategies.
MATERIALS AND METHODS
Gene nomenclature
Gene names in the text are according to nomenclature guidelines, with human genes in capitals (e.g. GREM2), mouse first letter capital (Grem2) and zebrafish in small letters (grem2). Gene names are in italic and protein names in roman font.
Study subjects and sample collection
The study protocol was approved by the Vanderbilt University Institutional Review Board and participants took part after informed written consent was obtained. Patients ≥18 years of age with ECG-confirmed AF were enrolled in the Vanderbilt AF Registry, which comprises clinical and genetic databases (Darbar et al, 2007). A detailed medical and drug history was obtained for all patients at enrollment. Clinical characteristics of study subjects are shown in Table 1.
Whole blood was collected for genomic DNA extraction and analysis. The GREM2 coding and flanking regions were amplified by polymerase chain reaction (PCR) using primers 5'-ACCAGATCAAGGAGGTGCTG-3' and 5'-AGAAGTGCTT-GCTGCTGAGG-3', and analyzed using the Reveal Discovery System to identify aberrant conformers, which were then directly sequenced (Abraham et al, 2010). Two unrelated lone AF subjects with GREM2 variants were identified, namely a woman with persistent AF since age 62, and a man with AF since age 37. Both reported vagal triggers, with the former reporting post-prandial and nocturnal onset and the latter reporting nocturnal/ obstructive sleep apnea-related onset. As control cohort, subsections of BAM files from 1560 samples containing chromosome 1:240656549-240656551 were downloaded from 1000 Genome Project data (Exome alignment dated 2011-11-14) (Clarke et al, 2012; Mills et al, 2011) and SNPs were called using SAMtools (Li et al, 2009). The minor allele frequency (MAF) of the GREM2 Q76E (rs142343894) is 0.03% in the 1000 Genomes (1/3120) compared with 0.5% (2/193) in the Vanderbilt Registry of AF probands. Allele frequencies between 1000 Genome Project data and our re-sequencing data were compared using two-tailed Fisher's exact test.
Zebrafish maintenance
Zebrafish, Danio rerio, were maintained as described (Müller et al., 2006). Zebrafish embryos were obtained from crosses between AB and TL lines or the Tg(cmlc2:EGFP)twu34 line.
In situ hybridization and antibody staining
Whole-mount in situ hybridization was performed as described (Müller et al., 2006; Wang et al, 2011). The hybridization mix for the double staining with grem2 and cmlc2 contained two digoxigenin-labeled RNA antisense probes for grem2 (250 ng of grem2 coding region probe and 500 ng of grem2 3'-UTR probe per 250 μl of hybridization mix) and 250 ng of fluorescein-labeled cmlc2 probe. For all other markers, we used 250 ng of digoxigenin-labeled RNA antisense probe per 250 μl hybridization mix.
Whole-mount antibody staining was performed as described (Melville et al, 2011; Sarmah et al, 2010). Mouse monoclonal antibodies MF20 against sarcomeric myosin heavy chain and S46 against atrial myosin heavy chain were obtained from the Developmental Studies Hybridoma Bank, diluted 1:10 in blocking solution, and applied to embryos overnight at 4°C. After five washes in PBS-Triton, embryos were incubated overnight at 4°C with secondary antibodies conjugated to AlexaFluor 488 and AlexaFluor 555 from Molecular Probes, diluted 1:400 in blocking solution. After six washes in PBT-DMSO, samples were transferred to 1:1 glycerol-PBT and stored at 4°C. Samples were incubated overnight at 4°C with anti-pSmad1/5/8 antibody (diluted 1:400 in blocking buffer; Cell Signaling Technology) then washed and incubated with pre-adsorbed alkaline phosphatase-conjugated goat anti-rabbit IgG (diluted 1:1000 in blocking buffer; Jackson ImmunoResearch) for 2 hours at room temperature.
To quantify changes in gene expression levels, signal intensity at the region of interest was measured in the green channel using NIH ImageJ. The background was subtracted and the signal normalized to non-injected, wild-type controls. Statistics were performed using the two-tailed Student's t-test, with P<0.05 being considered significant.
Loss- and gain-of-function experiments and dorsomorphin treatment
We used the following morpholino (MO) antisense oligonucleotides (Gene Tools) to block the splicing (MO1) and translation (MO2) of the zebrafish grem2 transcript: MO1 5'-ACTGCTCATC-CTGGAACACAGAGAG-3' and MO2 5'-ACACAGCGCCAC-CTTACTGCTCATC-3' (supplementary material Fig. S1). Morpholinos were reconstituted in water. Embryos at 1- to 4-cell stage were injected with 1 nl of morpholino solution into the yolk, close to the blastomeres, with an air injector from WPI (PV820 Pneumatic PicoPump). A concentration of 0.6 mM for MO1 or 0.4 mM for MO2 produced a fully penetrant and expressive phenotype in more than 85% of the injected embryos (morphants) without unspecific toxic effects.
Morphant embryos and non-injected controls were incubated in 2 μM dorsomorphin (Sigma-Aldrich) (Hao et al., 2008) dissolved in DMSO, or in DMSO alone, from 16 hpf until fixation at 48 hpf.
We injected 1 nl zebrafish grem2 mRNA at 10-15 pg/nl into the yolk. Human wild-type and variant GREM2 mRNAs were injected at various concentrations ranging from 2 to 5 pg/nl. Grem2 overexpression caused the expected defect of lack of BMP signaling during early embryogenesis, i.e. dorsalization of embryos. Only embryos with mild dorsalized phenotypes were processed for molecular characterization at later stages in order to visualize cardiac defects. For all shown experiments, 2.5 pg/nl of human GREM2 mRNA was used because we found it to be the highest concentration that resulted in normal cardiac morphology but abnormal rhythm.
Cardiac morphology and contractility in live embryos
For optical digital cardiography (DCG), live zebrafish Tg(cmlc2:EGFP)twu34 embryos at 48 hpf were embedded into 1.5% agarose and continuous images of the whole heart were taken on a Axio Imager Z1 microscope with a EC Plan-Neofluar 5×/0.15 M27 lens for 30 seconds using Axiovision software (Zeiss). Files were then imported into ImageJ and converted to binary using the ‘threshold’ command. The atrium or ventricle was selected and the two-dimensional surface area was calculated for each frame using the ‘analyze particles’ command. The output values were collected and plotted in Microsoft Excel. Analysis was performed on four embryo hearts for each condition. To determine whether signal propagation was uniform across the atrium, we selected two points on the atrium: one near the location where the contraction wave begins (point 1), and one far from both the initiation site and the valve that the signal moves towards (point 2). Point 2 was at a 45° angle to the line drawn through the valve.
In order to track points 1 and 2, the two points were marked with a single pixel and the move of the pixel was measured in ∼100 successive frames using ImageJ to report the coordinates of each selected point. The coordinates were then plotted in Microsoft Excel, assigning as 0 the coordinate at the most expanded location (i.e. furthest distance from the valve) during diastole. The coordinates were then converted to distance values from location 0 and the distances plotted over time as a line chart. Thus, peak values in the middle chart of Fig. 6D represent point locations at the end of systole.
In Grem2 overexpressing hearts, peaks corresponding to distal point 2 appeared broader than the peaks of proximal point 1, indicating that point 2 spent a longer time in systole than point 1 in overexpressing embryos. To quantify the average time that points 1 and 2 spent in systole, we calculated the size of peak areas above the mid-contraction point (Fig. 6D, right panel).
Cloning of human wild-type and variant GREM2
The GREM2 wild-type coding sequence was amplified from human genomic DNA using forward primer 5'-CTAGCGAATTCA-TGTTCTGGAAGCTTTCCCTGTCCTTGTTC-3' and reverse primer 5'-CTAGCGGATCCTCACTGCTTGTCCGAGTCGCT -CAGGTTCAC-3'. Cloning sites EcoRI and BamHI are underlined. The resulting amplicon was digested with EcoRI and BamHI and cloned into pcDNA3.1/Myc-His(-)A (Invitrogen). The Q76E mutation was introduced with primers 5'-AGTGACTG-GTGCAAGACGGAGCCGCTGCGGCAGACGG-3' and 5'-CCGTCTGCCGCAGCGGCTCCGTCTTGCACCAGTCACT-3'. Successful cloning of wild-type GREM2 and GREM2-Q76E was confirmed by restriction enzyme digests and direct sequencing.
Cell culture
Human embryonic kidney 293 (HEK293) cells were transiently transfected with 1, 25, 50 and 100 ng of wild-type GREM2 or GREM2-Q76E plasmids using Lipofectamine 2000 reagent (Invitrogen) and treated with 5 ng/ml BMP4 for 6 hours. RNA was then extracted and analyzed for ID2 expression using quantitative RT-PCR. Lipofectamine 2000 (Invitrogen) was used to stably transfect undifferentiated CGR8 mouse embryonic stem cells with 8 μg of linearized wild-type GREM2 and GREM2-Q76E plasmids. Positive clones were selected with 200 μg/ml G418 for 10 days and single colonies were picked, expanded, and genotyped. CGR8 cells were cultured and differentiated using the hanging-drop method (Rai et al, 2012).
RNA preparation and qRT-PCR analysis
Total RNA was extracted from mouse embryonic stem cells at day 8 of differentiation using the RNeasy kit (Qiagen) and reverse-transcribed into cDNA (Beck et al, 2008). For quantitative PCR, 20 ng of cDNA was amplified with the iQ SYBR Green Supermix kit on an iCycler (BioRad) using β-actin or GAPDH as controls. In addition, we normalized qPCR values in Fig. 6 to the relative expression levels of variant and wild-type GREM2. Relative gene expression levels were quantified using the formula 2(-ΔΔCt) as described (Beck et al., 2008). Primer sequences are included in supplementary material Table S2.
Protein modeling
GREM2 structure was previously modeled on human sclerostin (Rider and Mulloy, 2010). We used the UCSF Chimera system for visualization of the GREM2 structure (Pettersen et al., 2004).
Statistical analysis
Statistical analyses on continuous data were performed using unpaired two-tailed Student's t-test; analysis on categorical data were performed using two-tailed Fisher's exact test. qPCR results are shown as mean ± s.e.m. of data from at least three separate experiments, each performed with triplicate samples. Differences between groups were analyzed for statistical significance using one-way ANOVA with Bonferroni post-hoc test (GraphPad Prism software). P-values <0.05 were accepted as statistically significant.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Cory Guthrie and Kirill Zavalin for excellent zebrafish care and Marcia Blair for excellent technical assistance. We are indebted to H. Scott Baldwin and Daniel Levic for critically reading and editing the manuscript.
FUNDING: This work has been supported in part by the zebrafish initiative of the Vanderbilt University Academic Venture Capital Fund; by National Institutes of Health (NIH) grants from the National Institute of Dental and Craniofacial Research (NIDCR) [grant number DE018477 to E.W.K.], the National Heart, Lung, and Blood Institute (NHLBI) [grant numbers HL083958 and HL100398 to A.K.H., HL65962 to D.M.R., and HL09221 to D.D.], an American Heart Association Established Investigator Award [grant number 0940116N to D.D.], and the National Institute of General Medical Sciences (NIGMS) [grant number T32 GM008554]; and by the Cellular, Biochemical and Molecular Sciences Training Program at Vanderbilt (to D.B.M.).
Footnotes
AUTHOR CONTRIBUTIONS: E.W.K. and A.K.H. designed and directed the research. I.I.M., D.B.M., V.T., W.M.R., A.M., W.-D.W. and J.A.S. performed experiments in zebrafish and mouse embryonic stem cells. D.D., D.M.R. and M.B.S. provided and analyzed clinical data. E.W.K., A.K.H., I.I.M., D.B.M., V.T. and W.-D.W. analyzed data. E.W.K. and A.K.H. wrote the manuscript, with contributions from D.D., D.B.M, M.B.S., J.A.S. and V.T.
Present address: Eberhard-Karls-Universität Tübingen, Klinik für Herzkreislauf-erkrankungen, Tübingen, Germany
These authors contributed equally to this work
Present address: Cincinnati Children's Hospital Medical Center, Cincinnati, OH 45229, USA
Present address: Department of Bioagricultural Science, National Chiayi University, Chiayi 60004, Taiwan
Present address: Stanford University Medical Center, Stanford, CA 94305, USA
COMPETING INTERESTS: The authors declare that they do not have any competing or financial interests.
SUPPLEMENTARY MATERIAL: Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.010488/-/DC1
References
- Abraham R. L., Yang T., Blair M., Roden D. M., Darbar D. (2010). Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J. Mol. Cell. Cardiol. 48, 181-190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antzelevitch C., Pollevick G. D., Cordeiro J. M., Casis O., Sanguinetti M. C., Aizawa Y., Guerchicoff A., Pfeiffer R., Oliva A., Wollnik B., et al. (2007). Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442-449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avsian-Kretchmer O., Hsueh A. J. (2004). Comparative genomic analysis of the eight-membered ring cystine knot-containing bone morphogenetic protein antagonists. Mol. Endocrinol. 18, 1-12 [DOI] [PubMed] [Google Scholar]
- Beck H., Semisch M., Culmsee C., Plesnila N., Hatzopoulos A. K. (2008). Egr-1 regulates expression of the glial scar component phosphacan in astrocytes after experimental stroke. Am. J. Pathol. 173, 77-92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgrove B. W., Essner J. J., Yost H. J. (1999). Regulation of midline development by antagonism of lefty and nodal signaling. Development 126, 3253-3262 [DOI] [PubMed] [Google Scholar]
- Chocron S., Verhoeven M. C., Rentzsch F., Hammerschmidt M., Bakkers J. (2007). Zebrafish Bmp4 regulates left-right asymmetry at two distinct developmental time points. Dev. Biol. 305, 577-588 [DOI] [PubMed] [Google Scholar]
- Clarke L., Zheng-Bradley X., Smith R., Kulesha E., Xiao C., Toneva I., Vaughan B., Preuss D., Leinonen R., Shumway M., et al. (2012). The 1000 Genomes Project: data management and community access. Nat. Methods 9, 459-462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbar D. (2008). Genetics of atrial fibrillation: rare mutations, common polymorphisms, and clinical relevance. Heart Rhythm 5, 483-486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbar D., Motsinger A. A., Ritchie M. D., Gainer J. V., Roden D. M. (2007). Polymorphism modulates symptomatic response to antiarrhythmic drug therapy in patients with lone atrial fibrillation. Heart Rhythm 4, 743-749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbar D., Parvez B., Abraham R. (2012). Repolarization recipes for atrial fibrillation: beyond single channel variants. J. Am. Coll. Cardiol. 59, 1026-1028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinor P. T., Lunetta K. L., Glazer N. L., Pfeufer A., Alonso A., Chung M. K., Sinner M. F., de Bakker P. I., Mueller M., Lubitz S. A., et al. (2010). Common variants in KCNN3 are associated with lone atrial fibrillation. Nat. Genet. 42, 240-244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinor P. T., Lunetta K. L., Albert C. M., Glazer N. L., Ritchie M. D., Smith A. V., Arking D. E., Müller-Nurasyid M., Krijthe B. P., Lubitz S. A., et al. (2012). Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat. Genet. 44, 670-675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essner J. J., Branford W. W., Zhang J., Yost H. J. (2000). Mesendoderm and left-right brain, heart and gut development are differentially regulated by pitx2 isoforms. Development 127, 1081-1093 [DOI] [PubMed] [Google Scholar]
- Feinberg W. M., Blackshear J. L., Laupacis A., Kronmal R., Hart R. G. (1995). Prevalence, age distribution, and gender of patients with atrial fibrillation. Analysis and implications. Arch. Intern. Med. 155, 469-473 [PubMed] [Google Scholar]
- Firouzi M., Ramanna H., Kok B., Jongsma H. J., Koeleman B. P., Doevendans P. A., Groenewegen W. A., Hauer R. N. (2004). Association of human connexin40 gene polymorphisms with atrial vulnerability as a risk factor for idiopathic atrial fibrillation. Circ. Res. 95, e29-e33 [DOI] [PubMed] [Google Scholar]
- Furtado M. B., Solloway M. J., Jones V. J., Costa M. W., Biben C., Wolstein O., Preis J. I., Sparrow D. B., Saga Y., Dunwoodie S. L., et al. (2008). BMP/SMAD1 signaling sets a threshold for the left/right pathway in lateral plate mesoderm and limits availability of SMAD4. Genes Dev. 22, 3037-3049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollob M. H., Jones D. L., Krahn A. D., Danis L., Gong X. Q., Shao Q., Liu X., Veinot J. P., Tang A. S., Stewart A. F., et al. (2006). Somatic mutations in the connexin 40 gene (GJA5) in atrial fibrillation. N. Engl. J. Med. 354, 2677-2688 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson D. F., Arnar D. O., Helgadottir A., Gretarsdottir S., Holm H., Sigurdsson A., Jonasdottir A., Baker A., Thorleifsson G., Kristjansson K., et al. (2007). Variants conferring risk of atrial fibrillation on chromosome 4q25. Nature 448, 353-357 [DOI] [PubMed] [Google Scholar]
- Gudbjartsson D. F., Holm H., Gretarsdottir S., Thorleifsson G., Walters G. B., Thorgeirsson G., Gulcher J., Mathiesen E. B., Njølstad I., Nyrnes A., et al. (2009). A sequence variant in ZFHX3 on 16q22 associates with atrial fibrillation and ischemic stroke. Nat. Genet. 41, 876-878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haïssaguerre M., Jaïs P., Shah D. C., Takahashi A., Hocini M., Quiniou G., Garrigue S., Le Mouroux A., Le Métayer P., Clémenty J. (1998). Spontaneous initiation of atrial fibrillation by ectopic beats originating in the pulmonary veins. N. Engl. J. Med. 339, 659-666 [DOI] [PubMed] [Google Scholar]
- Hao J., Daleo M. A., Murphy C. K., Yu P. B., Ho J. N., Hu J., Peterson R. T., Hatzopoulos A. K., Hong C. C. (2008). Dorsomorphin, a selective small molecule inhibitor of BMP signaling, promotes cardiomyogenesis in embryonic stem cells. PLoS ONE 3, e2904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson-Zingman D. M., Karst M. L., Zingman L. V., Heublein D. M., Darbar D., Herron K. J., Ballew J. D., de Andrade M., Burnett J. C., Jr, Olson T. M. (2008). Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. N. Engl. J. Med. 359, 158-165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirchhof P., Kahr P. C., Kaese S., Piccini I., Vokshi I., Scheld H. H., Rotering H., Fortmueller L., Laakmann S., Verheule S., et al. (2011). PITX2c is expressed in the adult left atrium, and reducing Pitx2c expression promotes atrial fibrillation inducibility and complex changes in gene expression. Circ. Cardiovasc. Genet. 4, 123-133 [DOI] [PubMed] [Google Scholar]
- Kriebitz N. N., Kiecker C., McCormick L., Lumsden A., Graham A., Bell E. (2009). PRDC regulates placode neurogenesis in chick by modulating BMP signalling. Dev. Biol. 336, 280-292 [DOI] [PubMed] [Google Scholar]
- Levin M. D., Lu M. M., Petrenko N. B., Hawkins B. J., Gupta T. H., Lang D., Buckley P. T., Jochems J., Liu F., Spurney C. F., et al. (2009). Melanocyte-like cells in the heart and pulmonary veins contribute to atrial arrhythmia triggers. J. Clin. Invest. 119, 3420-3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078-2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C., Liu W., Palie J., Lu M. F., Brown N. A., Martin J. F. (2002). Pitx2c patterns anterior myocardium and aortic arch vessels and is required for local cell movement into atrioventricular cushions. Development 129, 5081-5091 [DOI] [PubMed] [Google Scholar]
- Lubitz S. A., Yi B. A., Ellinor P. T. (2010). Genetics of atrial fibrillation. Heart Fail. Clin. 6, 239-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville D. B., Montero-Balaguer M., Levic D. S., Bradley K., Smith J. R., Hatzopoulos A. K., Knapik E. W. (2011). The feelgood mutation in zebrafish dysregulates COPII-dependent secretion of select extracellular matrix proteins in skeletal morphogenesis. Dis. Model. Mech. 4, 763-776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills R. E., Walter K., Stewart C., Handsaker R. E., Chen K., Alkan C., Abyzov A., Yoon S. C., Ye K., Cheetham R. K., et al. (2011). Mapping copy number variation by population-scale genome sequencing. Nature 470, 59-65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka Y., Barnes M. E., Gersh B. J., Cha S. S., Bailey K. R., Abhayaratna W. P., Seward J. B., Tsang T. S. (2006). Secular trends in incidence of atrial fibrillation in Olmsted County, Minnesota, 1980 to 2000, and implications on the projections for future prevalence. Circulation 114, 119-125 [DOI] [PubMed] [Google Scholar]
- Mommersteeg M. T., Brown N. A., Prall O. W., de Gier-de Vries C., Harvey R. P., Moorman A. F., Christoffels V. M. (2007). Pitx2c and Nkx2-5 are required for the formation and identity of the pulmonary myocardium. Circ. Res. 101, 902-909 [DOI] [PubMed] [Google Scholar]
- Mommersteeg M. T., Christoffels V. M., Anderson R. H., Moorman A. F. (2009). Atrial fibrillation: a developmental point of view. Heart Rhythm 6, 1818-1824 [DOI] [PubMed] [Google Scholar]
- Monteiro R., van Dinther M., Bakkers J., Wilkinson R., Patient R., ten Dijke P., Mummery C. (2008). Two novel type II receptors mediate BMP signalling and are required to establish left-right asymmetry in zebrafish. Dev. Biol. 315, 55-71 [DOI] [PubMed] [Google Scholar]
- Müller I. I., Knapik E. W., Hatzopoulos A. K. (2006). Expression of the protein related to Dan and Cerberus gene – prdc – during eye, pharyngeal arch, somite, and swim bladder development in zebrafish. Dev. Dyn. 235, 2881-2888 [DOI] [PubMed] [Google Scholar]
- Parvez B., Darbar D. (2010). Lone AF - etiologic factors and genetic insights into pathophysiolgy. J. Atr. Fibrillation 1, 675-684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearce J. J., Penny G., Rossant J. (1999). A mouse cerberus/Dan-related gene family. Dev. Biol. 209, 98-110 [DOI] [PubMed] [Google Scholar]
- Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E. (2004). UCSF Chimera – a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605-1612 [DOI] [PubMed] [Google Scholar]
- Rai M., Walthall J. M., Hu J., Hatzopoulos A. K. (2012). Continuous antagonism by Dkk1 counter activates canonical Wnt signaling and promotes cardiomyocyte differentiation of embryonic stem cells. Stem Cells Dev. 21, 54-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiter J. F., Alexander J., Rodaway A., Yelon D., Patient R., Holder N., Stainier D. Y. (1999). Gata5 is required for the development of the heart and endoderm in zebrafish. Genes Dev. 13, 2983-2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rider C. C., Mulloy B. (2010). Bone morphogenetic protein and growth differentiation factor cytokine families and their protein antagonists. Biochem. J. 429, 1-12 [DOI] [PubMed] [Google Scholar]
- Ritchie M. D., Rowan S., Kucera G., Stubblefield T., Blair M., Carter S., Roden D. M., Darbar D. (2012). Chromosome 4q25 variants are genetic modifiers of latent familial atrial fibrillation. J. Am. Coll. Cardiol. 60, 1173-1181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan A. K., Blumberg B., Rodriguez-Esteban C., Yonei-Tamura S., Tamura K., Tsukui T., de la Pena J., Sabbagh W., Greenwald J., Choe S., et al. (1998). Pitx2 determines left-right asymmetry of internal organs in vertebrates. Nature 394, 545-551 [DOI] [PubMed] [Google Scholar]
- Sarmah S., Barrallo-Gimeno A., Melville D. B., Topczewski J., Solnica-Krezel L., Knapik E. W. (2010). Sec24D-dependent transport of extracellular matrix proteins is required for zebrafish skeletal morphogenesis. PLoS ONE 5, e10367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlange T., Arnold H. H., Brand T. (2002). BMP2 is a positive regulator of Nodal signaling during left-right axis formation in the chicken embryo. Development 129, 3421-3429 [DOI] [PubMed] [Google Scholar]
- Schultheiss T. M., Burch J. B., Lassar A. B. (1997). A role for bone morphogenetic proteins in the induction of cardiac myogenesis. Genes Dev. 11, 451-462 [DOI] [PubMed] [Google Scholar]
- Shanmugam M., Molina C. E., Gao S., Severac-Bastide R., Fischmeister R., Babu G. J. (2011). Decreased sarcolipin protein expression and enhanced sarco(endo)plasmic reticulum Ca2+ uptake in human atrial fibrillation. Biochem. Biophys. Res. Commun. 410, 97-101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart S., Hart C. L., Hole D. J., McMurray J. J. (2002). A population-based study of the long-term risks associated with atrial fibrillation: 20-year follow-up of the Renfrew/Paisley study. Am. J. Med. 113, 359-364 [DOI] [PubMed] [Google Scholar]
- Sudo S., Avsian-Kretchmer O., Wang L. S., Hsueh A. J. (2004). Protein related to DAN and cerberus is a bone morphogenetic protein antagonist that participates in ovarian paracrine regulation. J. Biol. Chem. 279, 23134-23141 [DOI] [PubMed] [Google Scholar]
- Wang W. D., Melville D. B., Montero-Balaguer M., Hatzopoulos A. K., Knapik E. W. (2011). Tfap2a and Foxd3 regulate early steps in the development of the neural crest progenitor population. Dev. Biol. 360, 173-185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellcome Trust Case Control Consortium (2007). Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661-678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuniga E., Rippen M., Alexander C., Schilling T. F., Crump J. G. (2011). Gremlin 2 regulates distinct roles of BMP and Endothelin 1 signaling in dorsoventral patterning of the facial skeleton. Development 138, 5147-5156 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.