

SUMMARY
Mutations in the ATP6V0A4 gene lead to autosomal recessive distal renal tubular acidosis in patients, who often show sensorineural hearing impairment. A first Atp6v0a4 knockout mouse model that recapitulates the loss of H+-ATPase function seen in humans has been generated and recently reported (Norgett et al., 2012). Here, we present the first detailed analysis of the structure and function of the auditory system in Atp6v0a4−/− knockout mice. Measurements of the auditory brainstem response (ABR) showed significantly elevated thresholds in homozygous mutant mice, which indicate severe hearing impairment. Heterozygote thresholds were normal. Analysis of paint-filled inner ears and sections from E16.5 embryos revealed a marked expansion of cochlear and endolymphatic ducts in Atp6v0a4−/− mice. A regulatory link between Atp6v0a4, Foxi1 and Pds has been reported and we found that the endolymphatic sac of Atp6v0a4−/− mice expresses both Foxi1 and Pds, which suggests a downstream position of Atp6v0a4. These mutants also showed a lack of endocochlear potential, suggesting a functional defect of the stria vascularis on the lateral wall of the cochlear duct. However, the main K+ channels involved in the generation of endocochlear potential, Kcnj10 and Kcnq1, are strongly expressed in Atp6v0a4−/− mice. Our results lead to a better understanding of the role of this proton pump in hearing function.
INTRODUCTION
The a4 subunit is a component of the multi-subunit proton pump (H+-ATPase), which is known to be widely expressed, including in all vertebrate cells. Among other functions, proton pumps have a crucial role in acid-base homeostasis. These pumps consist of two main domains, V0 (membrane-bound) and V1 (catalytic). Each of these domains is composed of several subunits encoded by separate genes located in different chromosomes (Miranda et al., 2010). Some H+-ATPase subunits are ubiquitously expressed whereas others, like ATP6V1B1 and ATP6V0A4, encoding the B1 and a4 subunits, are tissue-specific and when mutated can cause human disease (Karet et al., 1999; Stover et al., 2002).
Mutations in ATP6V0A4 cause distal renal tubular acidosis (dRTA) in humans, often accompanied by sensorineural hearing loss. Although it was previously reported that mutations in the human ATP6V0A4 gene lead to late-onset hearing loss or normal hearing (Stover et al., 2002), some patients develop severe early sensorineural hearing loss (Vargas-Poussou et al., 2006).
The first Atp6v0a4 knockout mouse has been recently reported, showing a severe renal phenotype accompanied by deafness (Norgett et al., 2012) A previous study showed that murine Atp6v0a4 is specifically expressed in kidney, epididymis and inner ear (Vidarsson et al., 2009), although we have recently reported that Atp6v0a4 is transiently expressed during development in other tissues such as bone and skin (Norgett et al., 2012). The a4-containing proton pumps in kidney are essential for the acidification of urine and pH homeostasis, and this might be important for normal hearing as well.
The mammalian inner ear consists of three different fluid-filled compartments: cochlea, vestibular labyrinth and endolymphatic sac (ES). The cochlea contains the hair cells of the organ of Corti, separating two different chambers containing the endolymph and perilymph. Endolymph bathes the apical surface of the hair cells and is high in K+ and low in Na+, whereas perilymph, underneath the basilar membrane, is high in Na+ and low in K+. The endolymph in the cochlea is maintained at a high positive resting potential, the endocochlear potential (EP), of around +100 mV in mice. This high voltage contributes to a strong electrochemical gradient between the positive EP and the negative potential within the hair cell, enhancing the flow of cations into the cells when the transduction channels are opened in response to sound. This depolarises the hair cells and triggers synaptic activity. The stria vascularis in the lateral wall of the cochlea is a highly vascularised tissue consisting of three main types of cells: marginal cells (facing the cochlear duct), intermediate cells (melanocytes-like cells) and basal cells (adjacent to the spiral ligament). The stria vascularis pumps K+ into the endolymph and generates the EP. Consequently, proper ionic composition of endolymph is vital for normal hearing.
The vestibular system contains the utricle, saccule and semicircular canals, which are essential for detection of acceleration and balance. The ES is connected to the vestibular labyrinth and the cochlear duct, and its epithelium has a crucial role in endolymph homeostasis (Salt, 2001). The H+-ATPase a4 subunit is expressed in the ES epithelium (Dou et al., 2004), suggesting a role in endolymph homeostasis.
Here, we report the severe inner ear phenotype of Atp6v0a4 knockout mice and present the first description of the inner ear pathophysiology that recapitulates the ATP6V0A4 loss of function seen in the human disease.
TRANSLATIONAL IMPACT.
Clinical issue
The kidney and the inner ear are often both affected in syndromic disorders. In the kidney, defects can affect renal anatomy, or glomerular or tubular function. In the ear, the defect is usually hearing impairment, which can result from structural or physiological abnormalities. Distal renal tubular acidosis (dRTA) is an example of a severe human disorder in which acidosis, renal tract calcification and metabolic bone disease are often, but not universally, accompanied by deafness. Mutations in ATP6V1B1 or ATP6V0A4 (which encode the B1 and a4 subunits, respectively, of the H+-ATPase that is essential for acid base homeostasis) underlie recessive dRTA. A mouse model that recapitulates the loss of function of this proton pump can contribute to understanding its role in normal cochlear physiology.
Results
Here, the authors present the first detailed analysis of the structure and function of the auditory system in Atp6v0a4 knockout mice, which faithfully recapitulates the human renal dRTA phenotype. These mutants display a headtilt and slow circling behaviour, suggesting an inner ear defect. Measurements of auditory brainstem responses showed significantly elevated thresholds in Atp6v0a4-null animals, indicating severe hearing impairment. Analyses of the morphology of the inner ears of embryos and newborn mice showed marked expansion of endolymphatic compartments in mutants. This expansion did not affect the development of hair cells of the organ of Corti, or their hair bundles, which are crucial for normal hearing. The authors also examined links between the a4 subunit encoded by Atp6v0a4, Foxi1 and pendrin, because of similarities in the respective mutant mouse phenotypes. They show that Atp6v0a4 mutants express both Foxi1 and pendrin, suggesting that a deficiency in these proteins does not contribute to the hearing defect in Atp6v0a4 mutants. Importantly, Atp6v0a4 mutants lack an endocochlear potential (EP), the voltage generated by the stria vascularis in the lateral wall of the cochlea. However, Kcnj10 and Kcnq1, the main K+ channels involved in the generation of EP, are strongly expressed in Atp6v0a4 mutants.
Implications and future directions
This first accurate model of human dRTA with associated hearing loss is an important tool for dissecting the underlying mechanisms that cannot be addressed directly in humans. Understanding the role of Atp6v0a4 expression in the ear will contribute to the development of effective treatments for dRTAassociated deafness, which is currently progressive and irreversible in affected individuals.
RESULTS
Generation of the Atp6v0a4 knockout mice and phenotype
The generation of Atp6v0a4−/− mice and breeding strategy have been recently published (Norgett et al., 2012). These mutants are small and have lower body weight than their control littermates. In addition, some mutants show a head-tilt and slow circling behaviour, suggesting an inner ear defect. Atp6v0a4−/− mice also lack a Preyer reflex (visible ear flick response to noise), an indication of hearing impairment. Homozygotes rarely survive beyond 21 days, unless they are treated with oral alkali (Norgett et al., 2012). These Atp6v0a4−/− mice are real knockouts without any leaky or residual expression of a4 subunit (see below).
Hearing sensitivity and endocochlear potential in Atp6v0a4 mutants
Auditory brainstem responses (ABR) were recorded in mice at postnatal day 14 (P14). Mean ABR thresholds are shown in Fig. 1A for wild-type (n=5, green), heterozygous (n=20, blue) and homozygous mutant (n=9, red) mice. All mutant mice were severely hearing-impaired and although ABRs were not completely abolished, they were often only apparent in the 12-24 kHz frequency range. Results at P20 were similar (Norgett et al., 2012).
Fig. 1.
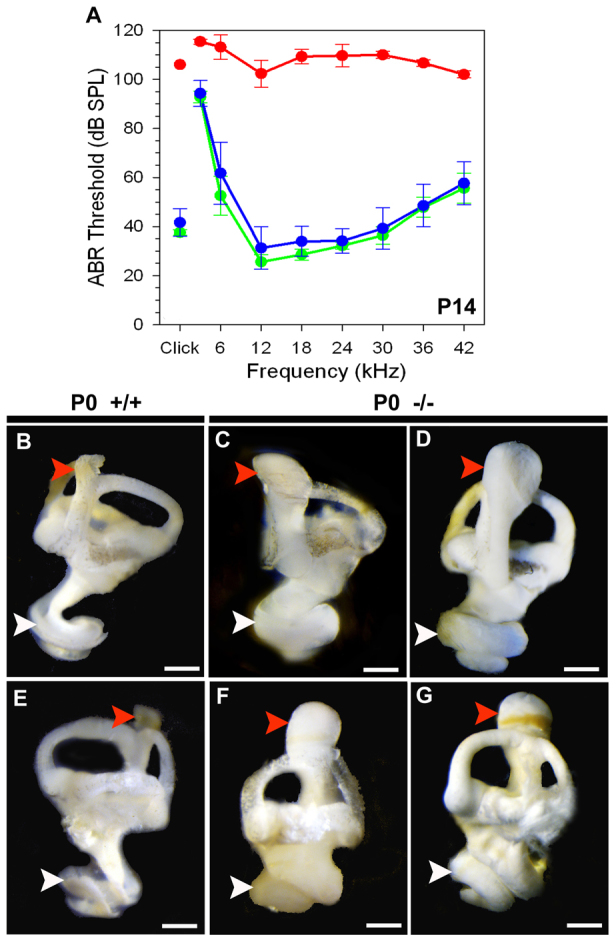
Hearing sensitivity and inner ear morphology in Atp6v0a4 mice. (A)=Mean ABR thresholds (± s.d.) for wild-type (green), heterozygous (blue) and homozygous mutant (red) mice aged P14. Atp6v0a4-/- mice show elevated thresholds compared with control littermates. (B-G) Paint-filled inner ears at P0. Homozygous mutants (C,D,F,G) show an expansion of ES (red arrowheads) and cochlear ducts (white arrowheads) compared with wild types (B,E). Scale bars: 100 μm.
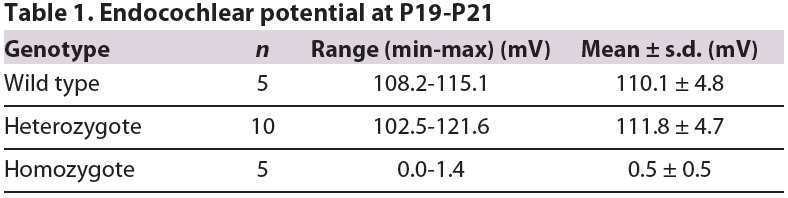
EP was also measured in Atp6v0a4−/− mice and, as we reported previously (Norgett et al., 2012), EP is reduced to near zero in homozygous mutants, whereas it has a normal value of around +110 mV in wild-type and heterozygous mice (Table 1).
Table 1.
Endocochlear potential at P19-P21
Expansion of cochlear and endolymphatic ducts in Atp6v0a4−/− mice
Inner ear clearing showed an abnormal dilation of endolymphatic compartments and total absence of otoconial crystals in Atp6v0a4−/− mice (Norgett et al., 2012). To assess this further, we used the paint-filling method (Kiernan, 2006) at P0 (Fig. 1B-G). This technique revealed a morphological defect in the inner ear of mutants, with severely enlarged ES and abnormally expanded cochleae (Fig. 1C,D,F,G), compared with control mice (Fig. 1B,E).
In addition, we carried out histological analysis of serial sections stained with haematoxylin and eosin (H&E) at embryonic day 16.5 (E16.5; Fig. 2A,D) and P0 (Fig. 2B,C,E,F). In Atp6v0a4−/− mice there is a clear expansion of the scala media throughout the cochlear duct, from base to apex, at both timepoints (Fig. 2D,E); the stria vascularis looks thinner and the Reissner's membrane is distended towards the scala vestibuli (Fig. 2F). The tectorial membrane is uncovered by any cellular layer (Fig. 2F), indicating that Reissner's membrane is not collapsed or fused to the organ of Corti. We also noted that in null animals, the spiral ligament appeared less populated by fibrocytes compared with controls (Fig. 2C,F). In the mutants the modiolus (central bony portion of the cochlea) looked slightly malformed and the spiral ganglion appeared distorted (Fig. 2, compare D,E with A,B).
Fig. 2.

Cochlear duct of Atp6v0a4 mutants is severely enlarged. (A-F) H&E staining of mid-modiolar Atp6v0a4 inner ear at E16.5 (A,D) and P0 (B,C,E,F). At E16.5 and P0 homozygous mutants (D-F) show an enlargement of the entire cochlear duct compared with controls (A-C). A higher magnification of the organ of Corti (C,F) shows the abnormal expansion of the scala media. Reissner's membrane is distended towards the scala vestibuli and the tectorial membrane is observed uncovered by any cellular layer (F), indicating that the membrane is not collapsed or fused to the organ of Corti. CO, cochlea; SG, spiral ganglion; SL, spiral ligament; SV, stria vascularis; OC, organ of Corti; RM, Reissner's membrane; TM, tectorial membrane. Scale bars: 200 μm (A,B,D,E); 100 μm (C,F).
To check whether the hair cells within the organ of Corti were affected by this expansion, we examined the expression of myosin7a (Myo7a) in inner ear sections of Atp6v0a4 mutant mice at E16.5 and P0 (see below) and at P20 by scanning electron microscopy (SEM) (Fig. 3A-F). We found that all three genotypes had the typical arrangement of three rows of outer hair cells (OHCs) and one row of inner hair cells (IHCs). Homozygous mutants showed a normal arrangement of stereocilia at the top of the OHCs and IHCs, although a few OHCs were missing from the most lateral row (Fig. 3B). Stereocilia of OHCs in mutants looked slightly disorganised. We also examined the stereocilia of Atp6v0a4−/− mice at P7, when the abnormal expansion of the scala media is already detectable, but we found no significant variations in the arrangement of hair cells or in the organisation of stereocilia, compared with controls (data not shown). For this reason, we consider these mild defects in mutants at P20 to be secondary to the endolymph homeostasis defect, which is observed much earlier.
Fig. 3.
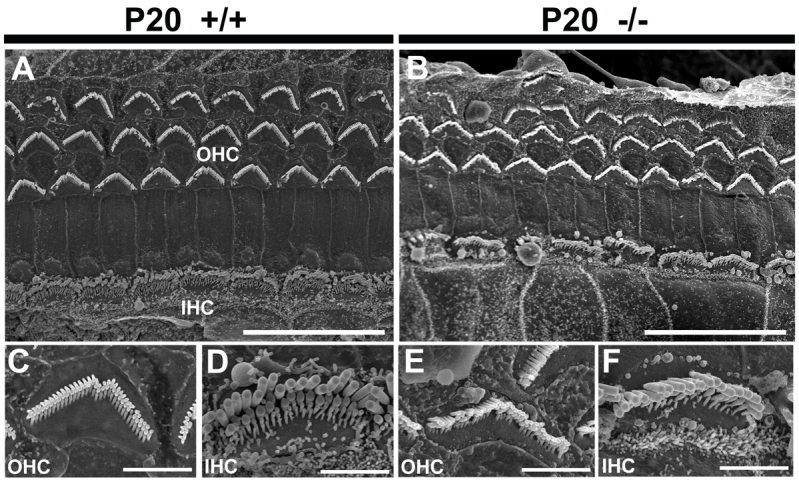
Analysis of hair cells and stereocilia of the organ of Corti. (A-F) SEM images of the organ of Corti at 30% distance from the base of the cochlea at P20. The typical arrangement of three rows of outer hair cells (OHC) and one row of inner hair cells (IHC) is observed in the Atp6v0a4 wild type (A,C,D) and mutants (B,E,F). The stereocilia are mildly disorganised and some OHCs are absent in mutants. The higher magnifications showed only mild disorganisation of stereocilia in mutants (E,F) compared with controls (C,D). Scale bars: 20 μm (A,B); 3 μm (C-F).
Analysis of histological sections also revealed prominent expansion of the endolymphatic duct and sac epithelium in Atp6v0a4−/− mice at E16.5 (Fig. 4B) and P0 (Fig. 4D,F) compared with controls (Fig. 4A,C,E). Sections at P0 showed that the typical infolded ES epithelium, seen in controls, becomes flatter and thinner in Atp6v0a4−/− mice (Fig. 4, compare E with F).
Fig. 4.

Endolymphatic duct and sac of Atp6v0a4 mutants is abnormally expanded. (A-F) H&E staining of sections at E16.5 (A,B) and P0 (C-F). Mutant mice show a clear expansion of the endolymphatic duct and sac at E16.5 (A,B) and in newborn mice (C-F). Higher magnification at P0 (E,F) shows an abnormal enlargement of the endolymphatic duct and sac in mutants (F) without the typical infolded epithelium seen in the wild types (E). ED, endolymphatic duct; CO, cochlea. Scale bars: (A-D) 300 μm; (E,F) 200 μm.
Analysis of the stria vascularis
The severe expansion of the scala media, a flattened and thinner stria vascularis, and the lack of endocochlear potential shown by Atp6v0a4−/− mice, all point to defects in stria vascularis function.
To further investigate the presence and integrity of the hair cells in the organ of Corti and the structure of the stria vascularis in Atp6v0a4 mutants at E16.5 and P0, we analysed the expression of the unconventional myosin, Myo7a, in inner ear sections using immunohistochemistry. In wild-type mice at P0 we found strong labelling of Myo7a in hair cells (HCs) of the organ of Corti (Fig. 5A,B) and also in the stria vascularis, in melanocyte-like cells (Fig. 5C, arrowheads) that are considered precursors of strial intermediate cells (Cable and Steel, 1991). Similar expression of Myo7a was found in Atp6v0a4−/− mice, despite the severe expansion of the scala media (Fig. 5D-F). Myo7a expression was also detected in the hair cells and melanocyte-like cells of the immature stria vascularis of Atp6v0a4−/− at E16.5 (data not shown). These results suggest that the lack of EP in mutant mice is not due to a lack of intermediate cells within the stria vascularis (Fig. 5F, arrowheads).
Fig. 5.

Myo7a is expressed in intermediate cells of stria vascularis in Atp6v0a4 mutant mice. (A-F) Expression of Myo7a in sections of Atp6v0a4 wild-type and homozygous mutant mice at P0, as shown by immunohistochemistry. (A,D) Mid-modiolar sections of the cochlea. (B,E) Higher magnification of the boxed regions in A and D, respectively. (C,F) Higher magnifications of the stria vascularis from B and E, respectively. In both wild types and mutants, Myo7a is strongly expressed in the hair cells of the organ of Corti (B,E) and in the melanocyte-like cells within the stria vascularis (B,E; arrowheads in C,F). HC, hair cells; SV, stria vascularis. Scale bars: 200 μm (A,D); 400 μm (B,E); 20 μm (C,F).
The EP is mainly generated by the K+ channel, Kcnj10, expressed by intermediate cells of the stria vascularis (Hibino et al., 1997; Marcus et al., 2002; Takeuchi and Ando, 1999; Wangemann et al., 2004). We therefore analysed the expression of Kcnj10 in Atp6v0a4−/− mice at different time points (E16.5, P0, P5 and P20). At P20, Kcnj10 is expressed in intermediate cells of the mature stria vascularis, with a similar pattern in Atp6v0a4 mutants and controls (Fig. 6B,D, black arrowheads). The expression of Kcnj10 is restricted to intermediate cells, whereas strial marginal and basal cells are devoid of expression (Fig. 6B,D, red arrowheads). Similar results were obtained at P5 (data not shown), when the stria vascularis is still in an immature state and no EP can be recorded (Steel and Barkway, 1989). However, Kcnj10 expression was not detected at E16.5 or P0 (data not shown), when the stria vascularis is still in an immature state, and Myo7a was used instead as a marker of the presence of melanocyte-like cells in the stria (see above). Our results suggest that the absence of normal EP in Atp6v0a4mutants is not caused either by absence of intermediate cells or by lack of Kcnj10 expression in the stria vascularis.
Fig. 6.

Kcnj10 and Kcnq1 are expressed in the stria vascularis of Atp6v0a4 mutant mice. (A,C) Mid-modiolar sections of the inner ear of control (A) and mutant (C) mice at P20. Regions in boxes in A and C are magnified in B and D, respectively. In wild types and mutants, a similar Kcnj10 expression pattern is observed. Especially strong expression is found in the intermediate cells of the stria vascularis (B,D, black arrowheads), whereas the marginal cells are negative (B,D, red arrowheads). It is also noticeable that mutants examined (n=5) did not seem to show Reissner's membrane so much distended towards the scala vestibuli as at P0 and P5. We think this might be due to the decalcification process used to facilitate the sectioning of these samples, or to the resolution of any pressure difference across Reissner's membrane by rupture. (E,F) Expression of Kcnq1 is specifically found in the marginal cells of the stria vascularis at P20 in both wild types and mutants (E,F, red arrowheads). RM, Reissner's membrane; SV, stria vascularis. Scale bars: 100 μm (A,C); 20 μm (B,D-F).
Kcnq1 is another potassium channel that contributes to the generation of EP and is expressed in the apical membranes of marginal cells of the stria vascularis (Wangemann, 2002). To assess whether the expansion of the scala media in Atp6v0a4 mutants affects the integrity and function of marginal cells of the stria vascularis, we examined the expression of Kcnq1 in Atp6v0a4−/− and controls at different time points. At P20, Kcnq1 was similarly expressed in marginal cells of stria vascularis of Atp6v0a4 mutants and controls (Fig. 6E,F), although the surface of the marginal cells looked slightly irregular (Fig. 6F, arrowheads). Expression of Kcnq1 was also detected earlier, at E16.5 and P0, when the stria is still immature (data not shown). Therefore, the absence of EP in Atp6v0a4−/− mice is not caused by absence of marginal cells or by lack of Kcnq1 expression in the stria vascularis.
Taken together, our results suggest that the lack of EP in Atp6v0a4−/− mice is not caused by the lack of expression of the two main K+ channels that contribute to the generation of EP in the stria vascularis (Kcnj10 and Kcnq1).
Atp6v0a4 is expressed specifically in ES epithelium at P0 and P20
We examined the expression pattern of a4 subunit protein in wild types and Atp6v0a4−/− at P0 and P20. In newborn wild-type mice we found expression specifically in a subpopulation of cells within the typically infolded ES epithelium and no stain was observed within the cochlea (Fig. 7A,B). A similar pattern was found at P20 (Fig. 7C). In Atp6v0a4−/− mice there was an abnormal expansion of the ES epithelium and no expression of the protein was detected (Fig. 7D-F), indicating that these mice are complete knockouts without any leaky or residual expression of the a4 subunit.
Fig. 7.
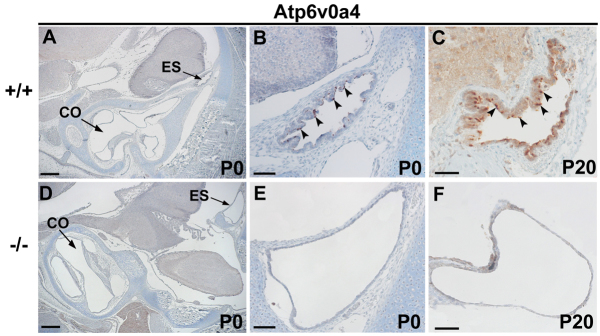
Expression of Atp6v0a4 in inner ear. (A-F) Sections of controls (A-C) and mutants (D-F) at P0 (A,B,D,E) and P20 (C,F) stained by immunohistochemistry. In wild-type mice at both ages, the expression of a4 subunit was specifically found in a subpopulation of cells within the ES epithelium (B,C, arrowheads). No expression was observed anywhere else within the inner ear (A). In Atp6v0a4 knockouts, no expression of this protein was detected (E,F). CO, cochlea. Scale bars: 300 μm (A,D); (B,E) 200 μm; 50 μm (C,F).
Foxi1 and pendrin are expressed in the ES of Atp6v0a4 mutant mice at P0
Atp6v0a4−/− mice show a phenotype similar to that of Foxi1−/− mice (Hulander et al., 2003). We checked the expression of transcription factor Foxi1 in wild-type sections at P0 using immunohistochemistry. We found that Foxi1 is expressed in a subpopulation of cells within the ES epithelium (Fig. 8A,B, arrowheads). In Atp6v0a4−/− mice, Foxi1 is still strongly expressed in the same pattern as in wild types, in a subset of the epithelial ES cells (Fig. 8C,D, arrowheads).
Fig. 8.

Foxi1 and Pds are expressed in the ES of Atp6v0a4 mutants. (A-D) Expression of Foxi1 in the endolymphatic duct and sac of wild-type and mutant mice at P0. (B,D) Higher magnification of the boxed regions in A and C, respectively. In control mice, Foxi1 is strongly expressed in a subpopulation of cells within the infolded epithelium of the ES (B, arrowheads). A similar pattern is observed in the flattened epithelium of the ES in mutants (D, arrowheads). (E-H) Expression of Pds in the ES of wild-type and mutant mice at P0. (F,H) Higher magnification of the boxed regions in E and G, respectively. In wild types, Pds expression is found in a few scattered cells within the ES (F, arrowheads). A similar pattern is found in the flattened ES seen in mutants (H, arrowheads). ED, endolymphatic duct. Scale bars: 100 μm (A,C,E,G); 20 μm (B,D,F,H).
The phenotype of Atp6v0a4 mutant mice is also similar to that described for Slc26a4 (Pds) knockout mice (Everett et al., 2001), a mouse model of human Pendred syndrome. To assess whether the expression of pendrin (Pds) was affected in Atp6v0a4−/− mice we performed immunohistochemistry on inner ear sections at P0. Pds is expressed in a few scattered cells within the epithelium of the ES and endolymphatic duct in mutants (Fig. 8G,H, arrowheads) as well as in controls (Fig. 8E,F, arrowheads).
DISCUSSION
Atp6v0a4 mutant mice are deaf, with an expansion of endolymphatic compartments
In this study we analysed the ear structure and hearing function of the Atp6v0a4 knockout mice. These mutants show a renal metabolic phenotype accompanied by sensorineural hearing loss (Norgett et al., 2012).
We have shown severe expansion of the cochlear and endolymphatic ducts from embryonic stages. In the cochlea, the abnormal expansion of the scala media leads to distension of Reissner's membrane towards the scala vestibuli, and the stria vascularis in the lateral wall looks thinner and flatter compared with wild types. However, this distension did not seem to affect the hair cells of the organ of Corti or their stereocilia bundles, which are essential for the mechanotransduction process underlying normal hearing function. Therefore, the deafness we found in Atp6v0a4 mutants is unlikely to be caused by defects in the hair cells or abnormal structure of stereocilia.
Atp6v0a4−/− mice are not only deaf but also lacked a normal highly positive EP. The EP is essential for normal auditory function and it is generated by active pumping of K+ into the scala media by the stria vascularis (Tasaki and Spyropoulos, 1959). The stria vascularis is a multi-layered epithelium adjacent to the spiral ligament in the lateral wall of the cochlea which contains three main cell types: marginal cells (facing the cochlear duct); intermediate cells (melanocyte-like cells), and basal cells (adjacent to the spiral ligament). The EP is mainly generated by Kcnj10, a K+ channel produced by the intermediate cells (Hibino et al., 1997; Takeuchi and Ando, 1999), and it has been shown that Kcnj10 knockout mice lack an EP (Marcus et al., 2002). The expression pattern of Kcnj10 in Atp6v0a4−/− mice was similar to wild types at P20 (and even in 5-day-old mice before any EP can be measured). However, Kcnj10 expression was not detected at E16.5 or P0, when the expansion of the scala media is already evident in Atp6v0a4−/− mice. For these early ages Myo7a is a good marker of melanocyte-like cells, which are precursors of strial intermediate cells. These results indicate that the lack of EP seen later in Atp6v0a4 mutants is not due to structural defects in the stria vascularis because intermediate cells were present in the stria indicated by Myo7a labelling and Kcnj10 was highly expressed as well.
Another K+ channel that contributes to the generation of the EP is Kcnq1, which is expressed in marginal cells of the stria vascularis. It has been reported that Kcnq1 knockout mice are deaf due to defects in the production and homeostasis of endolymph, associated with atrophy of the stria vascularis (Casimiro et al., 2001; Neyroud et al., 1997; Rivas and Francis, 2005). We also checked the expression of Kcnq1 in Atp6v0a4 mice and found strong expression of this K+ channel in the strial marginal cells of both homozygotes and controls, although in the mutants the marginal cells had an irregular surface and looked stretched, possibly due to high pressure in the scala media. The expression of this K+ channel was also observed at E16.5 and P0 (data not shown), when the stria vascularis is still in immature state, thus Kcnq1 expression can be followed from early stages. In summary, the lack of EP in Atp6v0a4 mutants is not caused by structural defects of the stria vascularis or by lack of expression of the K+ channels Kcnj10 and Kcnq1. Other ion channels such as Na+,K+, 2Cl– co-transporter and Na+,K+-ATPase, which are also involved in the generation and/or maintenance of the EP (Hibino et al., 2010) might underlie the lack of EP in Atp6v0a4 mutant mice.
Foxi1 and pendrin are expressed in the ES of Atp6v0a4 mutants
The phenotype of Atp6v0a4 knockout mice shows similarities to that of the Foxi1 null mutant mice (Hulander et al., 2003). Foxi1 is a transcription factor that interacts with the Atp6v0a4 promoter sequence and regulates its expression in the inner ear (Vidarsson et al., 2009). Furthermore, the expression of Atp6v0a4 in the ES epithelium co-localises with Foxi1 expression (Vidarsson et al., 2009). Our results indicate that the lack of Atp6v0a4 expression in mutants does not affect Foxi1 expression, supporting the suggestion that Atp6v0a4 is a downstream target of Foxi1.
The phenotype shown by Atp6v0a4 homozygotes is also similar to that described for Slc26a4/Pds knockout mice (Everett et al., 2001), a model for human Pendred syndrome, which is caused by mutations in the PDS gene (Everett et al., 1997). The lack of Pds expression in mice leads to abnormal expansion of the ES with hearing loss (Everett et al., 1999; Hulander et al., 2003). It has been previously reported that Pds and Atp6v0a4 are coexpressed in the same subpopulation of cells within the ES epithelium (Dou et al., 2004), which suggests that both proteins have a crucial role in the acid-base balance in the ES. However, the lack of Atp6v0a4 expression in the current mutants does not affect the expression of Pds in the ES epithelium.
Interestingly, Foxi1 is a transcription factor that not only regulates expression of Atp6v0a4 in the inner ear (Vidarsson et al., 2009) but also regulates expression of Pds, as indicated by the lack of Pds expression in the ES epithelium of Foxi1 null mice (Hulander et al., 2003). Consequently, there is coexpression of Foxi1, Pds and Atp6v0a4 in the same subpopulation of cells of the ES epithelium, indicating that they are important for acid-base balance of the ES. Our results clearly demonstrated that both Foxi1 and Pds are present in the ES epithelium, suggesting a downstream position of Atp6v0a4 in relation to Foxi1 and Pds. Therefore, they are unlikely to be contributing to the Atp6v0a4 knockout phenotype. Our data also suggest that the phenotype of Foxi1mutants might be mediated by Atp6v0a4 as well as by Pds.
Expression of Atp6v0a4 proton pump in the ES suggests a role in pH homeostasis
Atp6v0a4 is expressed in different tissues, including the inner ear (Dou et al., 2003; Stover et al., 2002), where its expression is restricted to a subpopulation of cells within the ES epithelium but no staining was found in the cochlea or vestibular system. These H+-ATPases secrete H+ into the endolymph, which is probably crucial for proper ionic composition of the endolymph and pH homeostasis. The coexpression of Foxi1, Pds and Atp6v0a4 in the same subtype of cells within the ES epithelium supports their involvement in acid-base homeostasis (Dou et al., 2004; Salt, 2001; Vidarsson et al., 2009). For instance, in mice lacking Pds there are changes in endolymphatic pH (Kim and Wangemann, 2011; Wangemann et al., 2007) but the abnormal enlargement of the endolymphatic compartments precedes the luminal acidification in the cochlea and the ES (Kim and Wangemann, 2011). Furthermore, in Pds mutants there is also epithelial cell stretching that might alter cell-to-cell communication, leading to retarded development of the stria vascularis and absence of EP (Kim and Wangemann, 2011). In Atp6v0a4 mutants, no abnormalities in the stria vascularis were detected but these mutants lack an EP. Therefore, there must be other mechanism(s) underlying the hearing impairment.
For instance, abnormal expansion of the ES epithelium might lead to expansion in the connected compartments (cochlear duct and vestibular labyrinth). The enlargement of the scala media possibly causes stretching of the epithelial cells lining the compartment, which in turn might disrupt tight junctions throughout the scala media (and stria vascularis), leading to a ‘leaky’ epithelium that is unable to maintain a high potential difference (EP) with external tissues. Alternatively, any changes to endolymphatic pH caused by dysfunctional H+-ATPases (e.g. Atp6v0a4 knockout) might affect various electrochemical gradients around the cochlear duct. It is likely that such changes would have widespread consequences for ionic homeostasis in the inner ear and that altered osmolalities between compartments might lead to expansion of the endolymphatic space seen in the Atp6v0a4 mutants at an early developmental stage.
The extreme reduction of EP to zero (∼100 mV change) in Atp6v0a4 mutants should result in a reduced driving potential across the top of the hair cells to about 40% of the potential difference in the normal cochlea, assuming that the intracellular potential of hair cells remains at its usual negative level. This would be predicted to lead to a threshold increase of ∼40 dB, mostly mediated by effects on outer hair cell amplifier activity (Gow et al., 2004; Patuzzi, 2011; Patuzzi et al., 1989). However, we found larger threshold shifts than this, ranging from 60 to 80 dB depending on frequency (where ABRs could be recorded; see Fig. 1A). Similar results have been reported for other mutants with severely reduced endocochlear potentials, such as E-cadherin conditional mutants (Trowe et al., 2011) and viable dominant spotting mutants (KitW-v) (Cable et al., 1994). It is likely that other effects associated with the reduced EP, such as ionic imbalance in endolymph, might account for thresholds that are raised beyond those predicted if there were only a lack of a high resting potential in scala media in the Atp6v0a4 mutants.
The Atp6v0a4 null mouse is a unique model that resembles human recessive dRTA
The Atp6v0a4 knockout mouse is the first good model of human dRTA. The auditory phenotype in Atp6v0a4 mutant mice is more severe than that described in humans carrying mutations in the same gene, where not all patients suffer severe sensorineural hearing loss (Stover et al., 2002; Vargas-Poussou et al., 2006). Furthermore, in humans, the hearing loss often only becomes apparent in adults, whereas mice are severely impaired from a young age (P14), when the auditory system is still undergoing maturation. It has been difficult to establish a clear correlation between the hearing thresholds and the presence of enlarged vestibular aqueduct in humans with mutations in ATP6V0A4 or ATP6V1B1, due to clinical and genetic heterogeneity (Andreucci et al., 2009). However, the lack of EP can account for the severe hearing impairment in the mouse mutants, and a similar mechanism probably explains the deafness observed in affected patients.
MATERIALS AND METHODS
All animal experiments were carried out in full compliance with UK Home Office regulations.
Auditory brainstem response recordings
ABRs were recorded (Ingham et al., 2011) in mice at P14 (+/+, n=5; −/+, n=20; −/−, n=9). Mice were anaesthetised using Ketamine (100mg/kg, Ketaset, Fort Dodge Animal Health) and Xylazine (10mg/kg, Rompun, Bayer Animal Health). Subcutaneous needle electrodes were inserted in the skin on the vertex (active) and overlying the ventral region of the left (reference) and right (ground) bullae to record responses of the left ear. Calibrated stimuli were presented as free-field clicks (0.01 milliseconds duration) and tone pips (3, 6, 18, 24, 30, 36 and 42 kHz of 5 milliseconds duration, 1 milliseconds rise/fall time) over a range of intensity levels of 10- 100 dB sound pressure level in 3 dB steps. Averaged responses to 256 stimuli, presented at 42.2/second, were analysed and thresholds established as the lowest sound intensity giving a visually detectable ABR response.
Measurement of EP
Mice aged P19-P21 (+/+, n=5; −/+, n=11; −/−, n=5) were anaesthetised with 0.01 ml/g body weight of 20% urethane. A tracheal cannula was inserted and the bulla opened to reveal the cochlea. Body temperature was maintained at 37°C using a homeothermic blanket. A small hole was made in the bony wall of the cochlea over the basal turn of the scala media. A micropipette electrode filled with 150 mM KCl was advanced through the hole and through the spiral ligament of the lateral wall into the scala media. The EP was recorded as the potential difference between the scala media and a reference silver/silver chloride pellet under the skin on the animal's neck (Steel and Barkway, 1989).
Paint-filling
Newborn mice (+/+, n=5; −/−, n=7) and E16.5 (+/+, n=3; −/−, n=4) embryos were used for paint-filling of the inner ear. Animals were decapitated and half-heads were fixed in Bodian's fixative overnight followed by washes (minimum of 2 hours each) twice in 75% ethanol, twice in 95% ethanol, twice in 100% ethanol, and cleared overnight by placing specimens in methyl salicylate. Inner ears were filled with 1% white gloss paint in methyl salicylate using a Hamilton syringe, with a pulled glass capillary needle broken to a tip diameter of 20-40 μm, which was then inserted into the endolymphatic compartment through the common crus and the saccule under a dissecting microscope (Kiernan, 2006).
Histology and immunohistochemistry
Specimens at P0 (+/+, n=5; −/−, n=5) and E16.5 embryos (+/+, n=5; −/−, n=5) were collected and fixed overnight in 4% paraformaldehyde in PBS at 4°C. For histological examination, samples were dehydrated and embedded in paraffin wax and 8-μm sections were cut using a microtome. Tissue sections were stained with H&E. Sections selected for immunohistochemistry (+/+, n=5; −/−, n=5 for each age examined) were processed using the Ventana Discovery System (Ventana Medical Systems, Illkirch, France) according to manufacturer's instructions. Antibodies used were: rabbit anti-H+-ATPase subunit a4 (RA2922, 1:400) (Smith et al., 2000), and subunit B1 (1:400, a gift from Ge-Hong Sun Wada, Doshisha Women's College, Kyotanabe, Japan), goat anti-Foxi1 (1:50; Abcam), goat anti-Pds (1:50; Santa Cruz Biotechnology), rabbit anti-Kcnj10 (1:300; Alomone) and rabbit anti-Kcnq1 (1:200; Santa Cruz Biotechnology). Secondary antibodies used were donkey anti-goat and donkey anti-rabbit (both at 1:100; Jackson ImmunoResearch Laboratories).
Scanning electron microscopy
Mice at P20 (+/+, n=4; -/+, n=7; −/−, n=4) were selected for SEM. After cervical dislocation, inner ears were dissected and fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer for 3 hours at room temperature, followed by washes in 0.1 M phosphate buffer. Samples were then prepared using the osmium tetroxidethiocarbohydrazide method (OTOTO). Processed cochleae were dehydrated followed by critical point drying using pressurised carbon dioxide. Samples were mounted and examined under a scanning electron microscope (Hitachi 4800 FE) at 5 kV.
Acknowledgments
FUNDING: This work was supported by the Wellcome Trust [grant numbers 098051 (to K.P.S.) and 088489 (to F.E.K.F)] and the Medical Research Council (K.P.S.).
Footnotes
AUTHOR CONTRIBUTIONS: B.L.-C. performed histological analyses. N.I. performed physiological measurements. E.E.N. and Z.J.G. genotyped the mice and contributed experimental reagents. B.L.-C., N.I., F.E.K.F. and K.P.S. conceived the experiments. B.L.-C., N.I. and K.P.S. wrote the paper. All authors edited the manuscript prior to submission.
Present address: Wolfson Centre for Age-Related Diseases, King's College London, Guy's Campus, London, SE1 1UL, UK
(to F.E.K.F)] and the Medical Research Council (K.P.S.).
COMPETING INTERESTS: The authors declare that they do not have any competing or financial interests.
References
- Andreucci E., Bianchi B., Carboni I., Lavoratti G., Mortilla M., Fonda C., Bigozzi M., Genuardi M., Giglio S., Pela I. (2009). Inner ear abnormalities in four patients with dRTA and SNHL: clinical and genetic heterogeneity. Pediatr. Nephrol. 24, 2147-2153 [DOI] [PubMed] [Google Scholar]
- Cable J., Steel K. P. (1991). Identification of two types of melanocyte within the stria vascularis of the mouse inner ear. Pigment Cell Res. 4, 87-101 [DOI] [PubMed] [Google Scholar]
- Cable J., Huszar D., Jaenisch R., Steel K. P. (1994). Effects of mutations at the W locus (c-kit) on inner ear pigmentation and function in the mouse. Pigment Cell Res. 7, 17-32 [DOI] [PubMed] [Google Scholar]
- Casimiro M. C., Knollmann B. C., Ebert S. N., Vary J. C., Jr, Greene A. E., Franz M. R., Grinberg A., Huang S. P., Pfeifer K. (2001). Targeted disruption of the Kcnq1 gene produces a mouse model of Jervell and Lange-Nielsen Syndrome. Proc. Natl. Acad. Sci. USA 98, 2526-2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H., Finberg K., Cardell E. L., Lifton R., Choo D. (2003). Mice lacking the B1 subunit of H+-ATPase have normal hearing. Hear. Res. 180, 76-84 [DOI] [PubMed] [Google Scholar]
- Dou H., Xu J., Wang Z., Smith A. N., Soleimani M., Karet F. E., Greinwald J. H., Jr, Choo D. (2004). Co-expression of pendrin, vacuolar H+-ATPase alpha4- subunit and carbonic anhydrase II in epithelial cells of the murine endolymphatic sac. J. Histochem. Cytochem. 52, 1377-1384 [DOI] [PubMed] [Google Scholar]
- Everett L. A., Glaser B., Beck J. C., Idol J. R., Buchs A., Heyman M., Adawi F., Hazani E., Nassir E., Baxevanis A. D., et al. (1997). Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nat. Genet. 17, 411-422 [DOI] [PubMed] [Google Scholar]
- Everett L. A., Morsli H., Wu D. K., Green E. D. (1999). Expression pattern of the mouse ortholog of the Pendred's syndrome gene (Pds) suggests a key role for pendrin in the inner ear. Proc. Natl. Acad. Sci. USA 96, 9727-9732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everett L. A., Belyantseva I. A., Noben-Trauth K., Cantos R., Chen A., Thakkar S. I., Hoogstraten-Miller S. L., Kachar B., Wu D. K., Green E. D. (2001). Targeted disruption of mouse Pds provides insight about the inner-ear defects encountered in Pendred syndrome. Hum. Mol. Genet. 10, 153-161 [DOI] [PubMed] [Google Scholar]
- Gow A., Davies C., Southwood C. M., Frolenkov G., Chrustowski M., Ng L., Yamauchi D., Marcus D. C., Kachar B. (2004). Deafness in Claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J. Neurosci. 24, 7051-7062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H., Horio Y., Inanobe A., Doi K., Ito M., Yamada M., Gotow T., Uchiyama Y., Kawamura M., Kubo T., et al. (1997). An ATP-dependent inwardly rectifying potassium channel, KAB-2 (Kir4. 1), in cochlear stria vascularis of inner ear: its specific subcellular localization and correlation with the formation of endocochlear potential. J. Neurosci. 17, 4711-4721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibino H., Nin F., Tsuzuki C., Kurachi Y. (2010). How is the highly positive endocochlear potential formed? The specific architecture of the stria vascularis and the roles of the ion-transport apparatus. Pflugers Arch. 459, 521-533 [DOI] [PubMed] [Google Scholar]
- Hulander M., Kiernan A. E., Blomqvist S. R., Carlsson P., Samuelsson E. J., Johansson B. R., Steel K. P., Enerbäck S. (2003). Lack of pendrin expression leads to deafness and expansion of the endolymphatic compartment in inner ears of Foxi1 null mutant mice. Development 130, 2013-2025 [DOI] [PubMed] [Google Scholar]
- Ingham N. J., Pearson S., Steel K. P. (2011). Using the auditory brainstem response (abr) to determine sensitivity of hearing in mutant mice. Curr. Protoc. Mouse Biol., 9, 279–287 [DOI] [PubMed] [Google Scholar]
- Karet F. E., Finberg K. E., Nelson R. D., Nayir A., Mocan H., Sanjad S. A., Rodriguez-Soriano J., Santos F., Cremers C. W., Di Pietro A., et al. (1999). Mutations in the gene encoding B1 subunit of H+-ATPase cause renal tubular acidosis with sensorineural deafness. Nat. Genet. 21, 84-90 [DOI] [PubMed] [Google Scholar]
- Kiernan A. E. (2006). The paintfill method as a tool for analyzing the threedimensional structure of the inner ear. Brain Res. 1091, 270-276 [DOI] [PubMed] [Google Scholar]
- Kim H. M., Wangemann P. (2011). Epithelial cell stretching and luminal acidification lead to a retarded development of stria vascularis and deafness in mice lacking pendrin. PLoS ONE 6, e17949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus D. C., Wu T., Wangemann P., Kofuji P. (2002). KCNJ10 (Kir4.1) potassium channel knockout abolishes endocochlear potential. Am. J. Physiol. Cell. Physiol. 282, C403-C407 [DOI] [PubMed] [Google Scholar]
- Miranda K. C., Karet F. E., Brown D. (2010). An extended nomenclature for mammalian V-ATPase subunit genes and splice variants. PLoS ONE 5, e9531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyroud N., Tesson F., Denjoy I., Leibovici M., Donger C., Barhanin J., Fauré S., Gary F., Coumel P., Petit C., et al. (1997). A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nat. Genet. 15, 186-189 [DOI] [PubMed] [Google Scholar]
- Norgett E. E., Golder Z. J., Lorente-Cánovas B., Ingham N., Steel K. P., Karet Frankl F. E.(2012). Atp6v0a4 knockout mouse is a model of distal renal tubular acidosis with hearing loss, with additional extrarenal phenotype. Proc. Natl. Acad. Sci. USA 109, 13775-13780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patuzzi R. (2011). Ion flow in cochlear hair cells and the regulation of hearing sensitivity. Hear. Res. 280, 3-20 [DOI] [PubMed] [Google Scholar]
- Patuzzi R. B., Yates G. K., Johnstone B. M. (1989). Changes in cochlear microphonic and neural sensitivity produced by acoustic trauma. Hear. Res. 39, 189-202 [DOI] [PubMed] [Google Scholar]
- Rivas A., Francis H. W. (2005). Inner ear abnormalities in a Kcnq1 (Kvlqt1) knockout mouse: a model of Jervell and Lange-Nielsen syndrome. Otol. Neurotol. 26, 415-424 [DOI] [PubMed] [Google Scholar]
- Salt A. N. (2001). Regulation of endolymphatic fluid volume. Ann. N. Y. Acad. Sci. 942, 306-312 [DOI] [PubMed] [Google Scholar]
- Smith A. N., Skaug J., Choate K. A., Nayir A., Bakkaloglu A., Ozen S., Hulton S. A., Sanjad S. A., Al-Sabban E. A., Lifton R. P., et al. (2000). Mutations in ATP6N1B, encoding a new kidney vacuolar proton pump 116-kD subunit, cause recessive distal renal tubular acidosis with preserved hearing. Nat. Genet. 26, 388-389 [DOI] [PubMed] [Google Scholar]
- Steel K. P., Barkway C. (1989). Another role for melanocytes: their importance for normal stria vascularis development in the mammalian inner ear. Development 107, 453-463 [DOI] [PubMed] [Google Scholar]
- Stover E. H., Borthwick K. J., Bavalia C., Eady N., Fritz D. M., Rungroj N., Giersch A. B., Morton C. C., Axon P. R., Akil I., et al. (2002). Novel ATP6V1B1 and ATP6V0A4 mutations in autosomal recessive distal renal tubular acidosis with new evidence for hearing loss. J. Med. Genet. 39, 796-803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi S., Ando M. (1999). Voltage-dependent outward K(+) current in intermediate cell of stria vascularis of gerbil cochlea. Am. J. Physiol. 277, C91-C99 [DOI] [PubMed] [Google Scholar]
- Tasaki I., Spyropoulos C. S. (1959). Stria vascularis as source of endocochlear potential. J. Neurophysiol. 22, 149-155 [DOI] [PubMed] [Google Scholar]
- Trowe M. O., Maier H., Petry M., Schweizer M., Schuster-Gossler K., Kispert A. (2011). Impaired stria vascularis integrity upon loss of E-cadherin in basal cells. Dev. Biol. 359, 95-107 [DOI] [PubMed] [Google Scholar]
- Vargas-Poussou R., Houillier P., Le Pottier N., Strompf L., Loirat C., Baudouin V., Macher M. A., Déchaux M., Ulinski T., Nobili F., et al. (2006). Genetic investigation of autosomal recessive distal renal tubular acidosis: evidence for early sensorineural hearing loss associated with mutations in the ATP6V0A4 gene. J. Am. Soc. Nephrol. 17, 1437-1443 [DOI] [PubMed] [Google Scholar]
- Vidarsson H., Westergren R., Heglind M., Blomqvist S. R., Breton S., Enerbäck S. (2009). The forkhead transcription factor Foxi1 is a master regulator of vacuolar H-ATPase proton pump subunits in the inner ear, kidney and epididymis. PLoS ONE 4, e4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P. (2002). K+ cycling and the endocochlear potential. Hear. Res. 165, 1-9 [DOI] [PubMed] [Google Scholar]
- Wangemann P., Itza E. M., Albrecht B., Wu T., Jabba S. V., Maganti R. J., Lee J. H., Everett L. A., Wall S. M., Royaux I. E., et al. (2004). Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2, 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P., Nakaya K., Wu T., Maganti R. J., Itza E. M., Sanneman J. D., Harbidge D. G., Billings S., Marcus D. C. (2007). Loss of cochlear HCO3-secretion causes deafness via endolymphatic acidification and inhibition of Ca2+ reabsorption in a Pendred syndrome mouse model. Am. J. Physiol. Renal Physiol. 292, F1345-F1353 [DOI] [PMC free article] [PubMed] [Google Scholar]