

SUMMARY
Obesity with associated metabolic disturbances worsens ischaemic heart disease outcomes, and rodent studies confirm that obesity with insulin-resistance impairs myocardial resistance to ischemia-reperfusion (I-R) injury. However, the effects of obesity per se are unclear, with some evidence for paradoxic cardioprotection (particularly in older subjects). We tested the impact of dietary obesity on I-R tolerance and reperfusion injury salvage kinase (RISK) signalling in hearts from middle-aged (10 months old) insulin-insensitive rats. Hearts from Wistar rats on either a 32-week control (CD) or high carbohydrate obesogenic (OB) diet were assessed for I-R resistance in vivo (45 minutes left anterior descending artery occlusion and 120 minutes reperfusion) and ex vivo (25 minutes ischemia and 60 minutes reperfusion). Expression and δ-opioid receptor (δ-OR) phospho-regulation of pro-survival (Akt/PKB, Erk1/2, eNOS) and pro-injury (GSK3β) enzymes were also examined. OB rats were heavier (764±25 versus 657±22 g for CD; P<0.05), hyperleptinaemic (11.1±0.7 versus 5.0±0.7 for CD; P<0.01) and comparably insulin-insensitive (HOMA-IR of 63.2±3.3 versus 63.2±1.6 for CD). In vivo infarction was more than halved in OB (20±3%) versus CD rats (45±6% P<0.05), as was post-ischaemic lactate dehydrogenase efflux (0.4±0.3 mU/ml versus 5.6±0.5 mU/ml; P<0.02) and ex vivo contractile dysfunction (62±2% versus 44±6% recovery of ventricular force; P<0.05). OB hearts exhibited up to 60% higher Akt expression, with increased phosphorylation of eNOS (+100%), GSK3β (+45%) and Erk1/2 (+15%). Pre-ischaemic δ-OR agonism with BW373U86 improved recoveries in CD hearts in association with phosphorylation of Akt (+40%), eNOS (+75%) and GSK3β (+30%), yet failed to further enhance RISK-NOS activation or I-R outcomes in OB hearts. In summary, dietary obesity in the context of age-related insulin-insensitivity paradoxically improves myocardial I-R tolerance, in association with moderate hyperleptinaemic and enhanced RISK expression and phospho-regulation. However, OB hearts are resistant to further RISK modulation and cardioprotection via acute δ-OR agonism.
INTRODUCTION
Obesity might double the risk of coronary artery disease and heart failure (Kenchaiah et al., 2002) and worsens outcomes from myocardial infarction (Aronson et al., 2010; Clavijo et al., 2006). These negative influences are consistent with experimental evidence of ischaemic intolerance and repressed cardioprotection in hearts from obese, dyslipidaemia and insulin-resistant animals (Bouhidel et al., 2008; Clark et al., 2011; du Toit et al., 2005; du Toit et al., 2008; Essop et al., 2009; Katakam et al., 2007; Maarman et al., 2012; Morel et al., 2003). However, obesity is complex, associated with co-morbidities including dyslipidaemia, inflammation and oxidative stress, insulin-insensitivity or overt diabetes with hyperglycaemia. Contributions of such factors to the overall effects of obesity remain unclear. We have reported that adiposity is not a reliable predictor of ischemia-reperfusion (I-R) tolerance (du Toit et al., 2008), and different potential determinants might be modified independently of obesity (Aubin et al., 2008; Aubin et al., 2010). Pro-inflammatory cytokine signalling could link adiposity to cardio-metabolic complications (Savage et al., 2005) and to I-R intolerance, with increased tumour necrosis factor α (TNF-α), for example, inhibiting insulin receptor substrate 1 (IRS-1) and both insulin (Hirosumi et al., 2002) and protective Akt signalling (Chuang et al., 2011). Dyslipidaemia could be important, though data are mixed: hypercholesterolemia with cholesterol-rich diets can improve I-R tolerance (while impairing post-conditioning) (Girod et al., 1999; Lauzier et al., 2009), exaggerate myocardial infarction (Szucs et al., 2011) or exert no effect (Görbe et al., 2011). Hyperglycaemia, arising from insulin insensitivity, can either promote myocardial cell death (Cai et al., 2002; Fiordaliso et al., 2001; Shizukuda et al., 2002) or inhibit myocardial I-R injury (Champattanachai et al., 2007; Schaffer et al., 2000; Xu et al., 2004). Insulin-sensitivity, on the other hand, consistently impacts I-R tolerance and might well be the primary determinant of negative effects of complex obesity. Resistance to I-R injury is impaired in non-obese models of insulin-resistance (Morel et al., 2003), in dietary obesity with insulin-resistance (du Toit et al., 2005; du Toit et al., 2008; Maarman et al., 2012) and in obese Zucker rats with insulin-resistance (Katakam et al., 2007) or diabetes (Yue et al., 2005). Desrois and colleagues also present evidence linking insulin-resistance to I-R intolerance in diabetic Goto-Kakizaki (GK) rats (Desrois et al., 2010), and we report association between insulin-resistance and cardiac injury in male but not in female rats (Clark et al., 2011). Thus, insulin-resistance consistently impairs cardiac I-R tolerance, which probably underpins the negative cardiac effects of obesity and metabolic syndrome. In contrast, the effects of obesity per se remain unclear.
Though generally considered detrimental (potentially due to associated insulin-resistance and other abnormalities), there is also evidence of cardiovascular benefit with obesity in humans (an ‘obesity paradox’), i.e. reduced morbidity and mortality in overweight or obese patients with ischaemic heart disease or heart failure (Arena and Lavie, 2010; Hall et al., 2005; Lavie et al., 2009; Pingitore et al., 2007). These protective effects appear age-related (as is incidence of ischaemic heart disease), being more evident in older than in younger patients (Dorner and Rieder, 2012). Interestingly, obesity appears to retain this protective efficacy in a setting of insulin-resistant diabetes (Doehner et al., 2012), suggesting a potential balance between negative and positive effects of obesity. Nonetheless, the relevance and indeed existence of obesity-dependent protection remains controversial.
TRANSLATIONAL IMPACT.
Clinical issue
Human obesity is a complex disorder usually associated with many other metabolic abnormalities. Most of these co-morbidities (e.g. dyslipidaemia, insulin resistance, hyperglycaemia, oxidative stress and inflammation) can influence myocardial sensitivity to injury or disease. However, the effects of obesity on the heart have been unclear: for example, some studies reported that obesity confers a cardioprotective effect to ischemia-reperfusion injury. To optimally manage cardiovascular outcomes in obese individuals, it is thus important to identify the effects on the heart of individual obesity-associated factors.
Results
In this study, the authors use an insulin-insensitive rat model of dietary obesity to investigate whether obesity per se is a key factor compromising myocardial tolerance to ischemia. Their findings indicate that obesity with mild hyperleptinaemia does not compromise myocardial tolerance to ischemia; rather, obesity might even be beneficial, at least in this setting of insulin insensitivity. Additional data suggest that cardioprotection conferred by obesity might occur via modulation of reperfusion injury salvage kinase (RISK) signalling. Based on these data and their previous findings, the authors propose that insulin resistance is the primary factor that impairs resistance to myocardial ischemia.
Implications and future directions
These data show that obesity per se does not compromise myocardial tolerance to ischemia in insulin-insensitive rats with dietary obesity. These data provide clues about highly debated influence of human obesity on cardiac health and disease. The delineation of underlying mechanisms in future studies should help to develop therapies that limit the detrimental effects of obesity-associated insulin resistance on the heart.
Having established negative cardiac influences of obesity with insulin-resistance, compared with lean insulin-sensitive rats (du Toit et al., 2005; du Toit et al., 2008; Maarman et al., 2012), we here examine the impact of dietary obesity on myocardial I-R tolerance in older rats exhibiting common age-related insulin-insensitivity. This approach permits analysis of the effects of obesity overlaid upon common insulin-insensitivity in older animals. The majority of ischaemic heart disease patients are elderly (more than 70% of sufferers are >65 years of age), and age also impairs insulin-sensitivity in animals (Gómez-Pérez et al., 2011; Gu et al., 2011) and humans (Pagano et al., 1981). Because myocardial resistance to I-R injury depends in part upon functionality of reperfusion injury salvage kinase (RISK) pathway components (Downey et al., 2007; Halestrap et al., 2007; Hausenloy and Yellon, 2009),, including Akt, which is also key to insulin signalling (Bertrand et al., 2008), and because reversal of the negative effects of insulin-resistance is linked to improved RISK and nitric oxide synthase (NOS) signalling (Bulhak et al., 2009; Huisamen et al., 2011), we tested the hypothesis that obesity modifies myocardial I-R tolerance via shifts in pro-survival RISK-NOS signalling in insulin-insensitive rats. To test the influence of obesity on the functional responsiveness of RISK-NOS signalling, we also contrasted the ability of δ-opioid receptor (δ-OR) agonism, known to engage these signals (Fryer et al., 2001; Gross et al., 2007a), to activate kinases and associated protection in hearts from obese and lean rats.
RESULTS
Baseline characteristics of obese and lean animals
Wistar rats were kept for 32 weeks on control (CD) or high carbohydrate obesogenic (OB) diets. Rats on the OB diet were 16% heavier, with larger visceral fat reserves than CD litter-mates (Table 1). OB rats also exhibited elevated triglycerides, with comparable insulin and glucose compared with lean CD rats. Consistent with prior studies, 10-month-old Wistar rats displayed elevated serum insulin and homeostatic model assessment of insulin resistance (HOMA-IR) levels compared with 3-month-old rats (Gómez-Pérez et al., 2011; Gu et al., 2011). Serum insulin levels in young rats were 50% of those in 10-month-old CD and OB rats (3.04±0.10 versus 6.70±0.18 and 6.86±0.11 ng/ml in young versus old CD and OB rats, respectively). Furthermore, OB rats exhibited normal serum TNF-α and insulin-like growth factor 1 (IGF-1) levels, with significantly elevated leptin levels (Table 1). In vivo arterial pressures and heart rates were comparable between CD and OB groups (Table 2).
Table 1.
Biometric data for rats fed on control (CD) and high carbohydrate (OB) diets
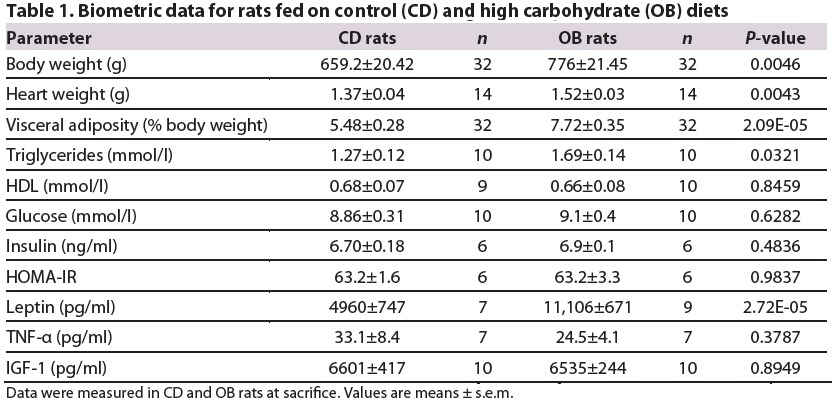
Table 2.
Measures of basal mechanical function in in vivo and x vivo hearts from rats fed on control (CD) and high carbohydrate (OB) diets
Normoxic cardiac performance and coronary perfusion assessed ex vivo did not differ between CD and OB hearts (Fig. 1; Table 2). Left ventricular systolic, diastolic and developed pressures were similar across groups, as was the positive differential of force development (+dP/dt) and coronary flow rate. Absence of changes in baseline function ex vivo was consistent with unaltered arterial diastolic, systolic and mean pressures in vivo (Table 2).
Fig. 1.

Pre- and post-ischaemic contractile function and post-ischaemic LDH efflux in Langendorff perfused hearts from CD, OB, CD+BW373U86 and OB+BW373U86 rats. Hearts were untreated or pre-treated for 10 minutes pre-ischemia with 1 μM BW373U86. (A) Left ventricular developed pressure (LVDevP) (aP<0.05 versus CD); (B) Post-ischaemic recovery of +dP/dt after 25 minutes of ischaemia and 60 minutes reperfusion (**P<0.05 versus CD); and (C) post-ischaemic efflux of myocardial LDH in the first and second minutes of reperfusion (*P<0.05,**P<0.02). Data are means ± s.e.m. (n=4).
Obesity improves cardiac I-R tolerance while eliminating δ-OR cardioprotection
Global I-R significantly depressed contractile function in vitro (Fig. 1), with reduced ventricular pressure development and a sustained increase in diastolic pressure. Post-ischaemic contractile recovery was incomplete in both CD and OB hearts, but significantly better in OB than in CD hearts (Fig. 1A). Recovery of inotropic state, as assessed by +dP/dt, was also significantly improved in hearts from OB rats, whereas post-ischaemic efflux of cardiac lactate dehydrogenase (LDH) was significantly reduced (Fig. 1B,C). Pre-ischaemic δ-OR agonism with BW373U86 had no effect on post-ischaemic mechanical function in either CD or OB hearts (Fig. 1A). However, δ-OR agonism significantly reduced LDH efflux in CD but not OB hearts (Fig. 1C).
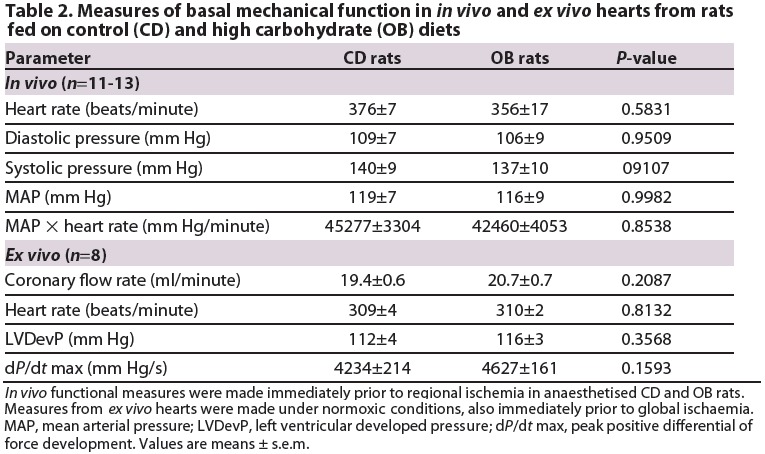
Analysis of myocardial infarction in vivo revealed that ∼45% of the area at risk (AAR) was infarcted in hearts from CD animals, with infarction in OB hearts more than halved to ∼20% (Fig. 2). δ-OR agonism also decreased infarction by ∼40% in CD hearts, yet failed to modify infarct size in OB hearts (Fig. 2). The AAR was similar across all groups (CD, 34.7±5.1%; CD+BW373U86, 31.0±5.3%; OB, 38.1±3.4%; OB+BW373U86, 37.6±3.2%). These data collectively reveal that dietary obesity induces cardioprotection in older insulin-insensitive animals, an effect evident in vivo and ex vivo.
Fig. 2.
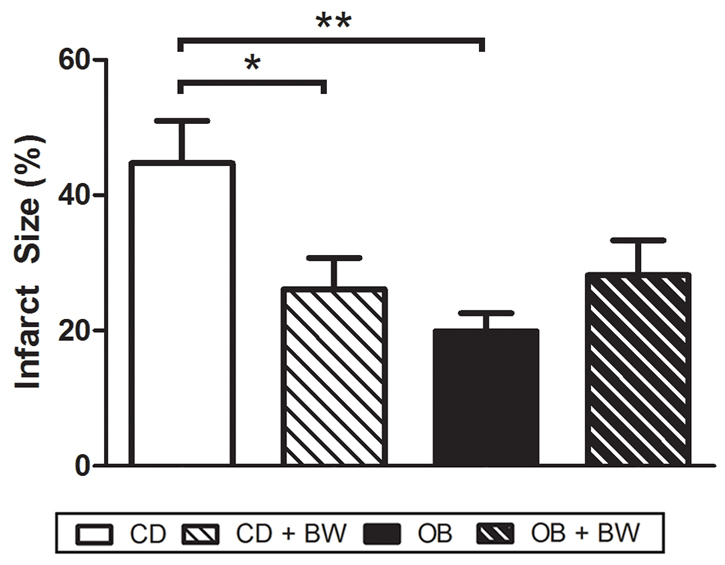
Myocardial infarct size following 45 minutes of LAD coronary artery occlusion and 120 minute reperfusion in situ in hearts of CD and OB, CD+BW373U86 and OB+BW373U86 rats. Animals received either vehicle or 1 mg/kg BW373U86 administered via the right jugular vein for 10 minutes prior to LAD occlusion. Data are means ± s.e.m. (n=6-7); *P<0.05, **P<0.02.
Obesity improves RISK-NOS signalling in hearts from insulin-insensitive rats
Dietary obesity induced significant changes in cardiac signalling proteins. The OB diet increased expression of Akt in both cytosolic and membrane compartments by 40-60%, without modifying total expression levels for other proteins (Fig. 3). Obesity had no effect on the phosphorylation state of Akt (Fig. 4). Furthermore, phosphorylation states of cytosolic glycogen synthase kinase 3β (GSK3β), membrane endothelial nitric oxide synthase (eNOS; only detected in the membrane fraction in all groups), and cytosolic extracellular signal-regulated kinases 1 and 2 (Erk1/2) were increased in OB hearts by 45, 100 and 15%, respectively (Figs 5, 6).
Fig. 3.
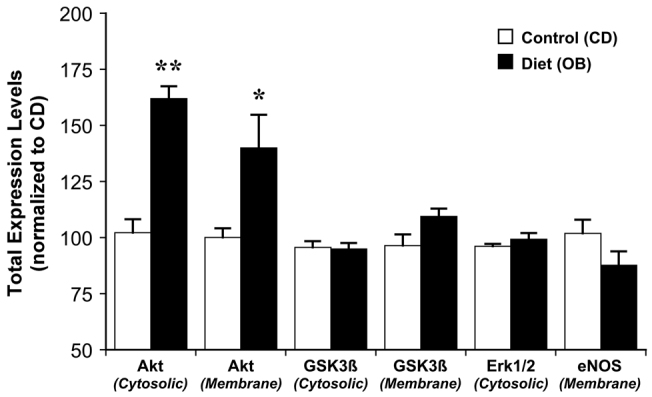
Effects of dietary obesity on total myocardial expression of Akt, GSK3β, Erk1/2 and eNOS. Expression data are normalised to values for control (CD) hearts. Data are means ± s.e.m. (n࿃5); *P<0.05, **P<0.005.
Fig. 4.
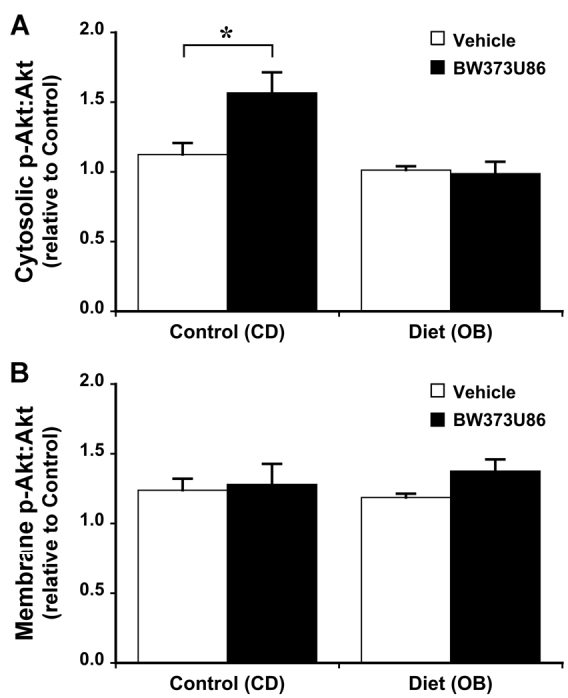
Effects of dietary obesity and BW373U86 pre-treatment on ratio of myocardial phosphorylated Akt to total Akt. Animals were treated with vehicle or 1 mg/kg BW373U86 administered via the right jugular vein for 10 minutes prior removal of the heart. (A) Cytosolic Akt ratios. (B) Membrane Akt ratios. For comparison, phosphorylation ratios are normalised to values from vehicle-treated control (CD) hearts. Data are means ± s.e.m. (n=5); *P<0.05.
Fig. 5.
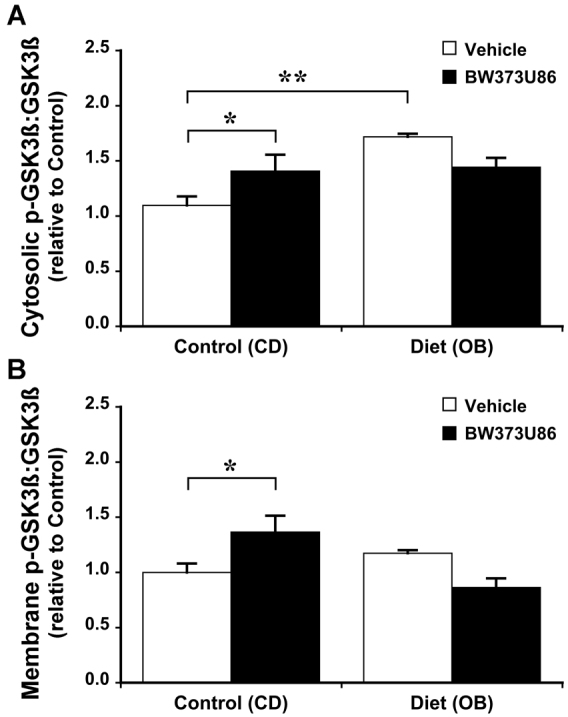
Effects of dietary obesity and BW373U86 pre-treatment on ratio of myocardial phosphorylated GSK3β to total GSK3β. Animals were treated with vehicle or 1 mg/kg BW373U86 administered via the right jugular vein for 10 minutes prior removal of the heart. (A)Cytosolic GSK3β ratios. (B) Membrane GSK3β ratios. For comparison, phosphorylation ratios are normalised to values from vehicle-treated control (CD) hearts. Data are means ± s.e.m. (n=5); *P<0.05, **P<0.005.
Fig. 6.
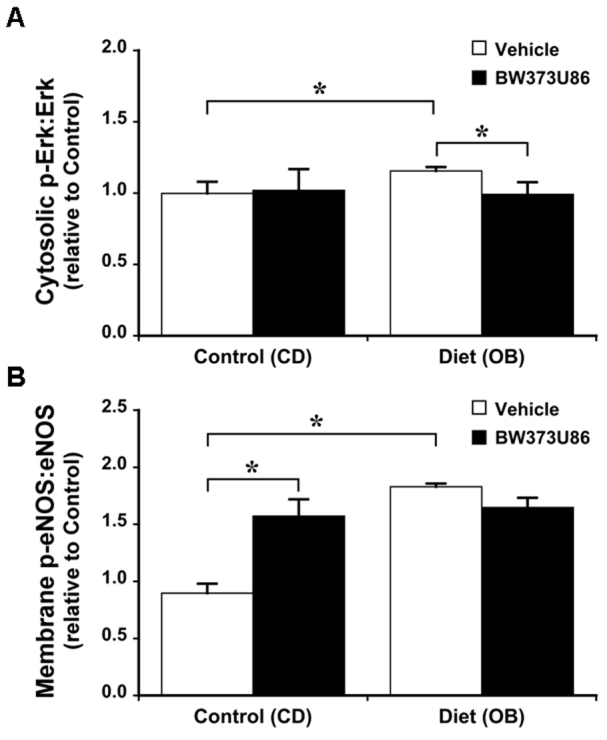
Effects of dietary obesity and BW373U86 pre-treatment on ratio of myocardial phosphorylated Erk1/2 to total Erk1/2. Animals were treated with vehicle or 1 mg/kg BW373U86 administered via the right jugular vein for 10 minutes prior removal of the heart. (A) Cytosolic Erk 1/2 ratios; (B) Membrane eNOS ratios. For comparison, phosphorylation ratios are normalised to values from vehicle-treated control (CD) hearts. Data are means ± s.e.m. (n=5); *P<0.05.
Obesity attenuates δ-OR agonist activation of RISK-NOS signalling
In terms of signalling responsiveness to δ-OR agonism in CD hearts, 1 μM BW373U86 treatment increased phosphorylation of cytosolic (but not membrane) Akt by 40% (Fig. 4); membrane (but not cytosolic) GSK3β by ∼40% (Fig. 5); and membrane eNOS by 75% (Fig. 6). Cytosolic Erk1/2 was insensitive to activation (Fig. 6). Obesity eliminated signalling sensitivity to δ-OR agonism. In contrast to CD hearts, δ-OR agonism failed to increase phosphorylation of Akt, GSK3β or eNOS in OB hearts. Indeed, there was a small 15% decline in apparent phosphorylation of cytosolic Erk1/2 in the OB group (Fig. 6).
DISCUSSION
We have previously reported that obesity with features of metabolic syndrome (obesity, insulin-resistance, dyslipidaemia) decreases myocardial resistance to I-R compared with lean insulin-sensitive animals. Decreased ischaemic tolerance is reflected in worsened infarction (du Toit et al., 2005; du Toit et al., 2008; Essop et al., 2009; Maarman et al., 2012) and mechanical dysfunction (du Toit et al., 2005; du Toit et al., 2008). By contrast, the present study reveals that high-carbohydrate-induced obesity in older insulin-insensitive animals paradoxically improves myocardial I-R tolerance (compared with lean insulin-insensitive animals), an effect apparent vo and ex vivo. Improved I-R tolerance is associated with normal pro-inflammatory cytokine levels and similar glucose levels and/or insulin-insensitivity, moderate hyperleptinaemic, and improved expression and phospho-regulation of myocardial enzymes impacting on cell survival. However, δ-OR-triggered RISK activation and associated cardioprotection are eliminated by obesity in these animals. The data thus collectively indicate that obesity itself improves I-R tolerance in insulin-insensitive older rats, potentially via improved RISK-NOS signalling. However, enhanced basal expression and phospho-activation of RISK-NOS paths might also limit capacity to further protect via stimuli engaging the same signalling (e.g. δ-OR agonism).
Preserved contractile function with dietary obesity
The present data indicate that high-carbohydrate-driven dietary obesity does not negatively influence myocardial contractile function in insulin-sensitive rats (Fig. 1; Table 2). Data regarding contractile effects of obesity are scarce and equivocal, with evidence for and against obesity-dependent dysfunction. Obesity is an established independent risk factor for heart failure (Kenchaiah et al., 2002), though whether obesity directly promotes pump dysfunction is unknown. Some studies support impairment of systolic function (Peterson et al., 2004; Wong et al., 2004), whereas other data support preserved function in obese humans (Stoddard et al., 1992; Zarich et al., 1991), or negative effects due to shifts in cardiac loading (Alpert et al., 1993). Wilson et al. report mechanical dysfunction with an obesogenic Western diet of high fat and reduced carbohydrates (Wilson et al., 2007), whereas our prior work indicates that contractile function is compromised in obese insulin-resistant rats but not in lean insulin-sensitive rats (du Toit et al., 2005; du Toit et al., 2008). Ren et al. recently found that 16 weeks of a high-fat diet reduced isolated myocyte contractility, though presented no data regarding the myocardium (Ren et al., 2010). They also reported impaired contractility in the murine ob/ob model, in association with reduced phospho-activation of Akt and eNOS. Our data for dietary rather than genetically determined obesity, with a high carbohydrate rather than high fat diet, reveals a different outcome, with no shift in contractile function in intact beating hearts (Fig. 1; Table 2). Common arterial pressures in OB and CD rats also support preservation of contractile function in vivo (Table 2). It is possible that negative contractile effects of obesogenic diets arise from repression of Akt and/or NOS signalling (Ren et al., 2010), whereas baseline Akt expression and NOS phospho-activation are enhanced in the current model.
Dietary obesity enhances I-R tolerance in hearts from insulin-insensitive animals
Some clinical studies identify worsened outcomes from myocardial infarction with obesity (Kragelund et al., 2005; Rana et al., 2004; Sharma et al., 2009). Nonetheless, reduced morbidity and mortality and emergence of the ‘obesity paradox’ in overweight and obese patients (Hall et al., 2005; Lavie et al., 2009; Pingitore et al., 2007) indicate that there are situations where obesity can in fact prove protective. This might reflect differing roles of associated metabolic disturbances in determining cardiac outcomes. Myocardial I-R tolerance in insulin-insensitive rats was enhanced by obesity in vivo and ex vivo, supporting conservation of a protected phenotype from in situ to ex vivo blood-free hearts with fixed substrate supply. These data contrast prior studies in hearts from obese insulin-resistant rats exhibiting exaggerated post-ischaemic dysfunction (du Toit et al., 2005; du Toit et al., 2008) and infarction (Clark et al., 2011; du Toit et al., 2005; du Toit et al., 2008; Essop et al., 2009; Maarman et al., 2012). A key difference is that these studies compare obese insulin-resistant with lean insulin-sensitive rats, thus assessing combined effects of obesity and insulin-resistance. Multiple studies confirm that insulin-resistance, isolated (Morel et al., 2003) or obesity-related (Clark et al., 2011; du Toit et al., 2005; du Toit et al., 2008; Essop et al., 2009; Katakam et al., 2007; Maarman et al., 2012), impairs myocardial resistance to I-R injury. The present study, by contrast, compares obese and lean rats with comparable insulin-insensitivity and glucose levels, allowing emergence of an isolated obesity effect. Another difference is the absence of changes in pro-inflammatory TNF-α in the current model (Table 1), whereas TNF-α is typically upregulated by a combination of obesity and insulin-resistance (du Toit et al., 2005; du Toit et al., 2008). Lack of change in TNF-α argues against substantial inflammation in the present model. However, although enhanced inflammation can contribute to the negative effects of obesity, absence of an inflammatory response across both groups cannot explain cardiac protection in the present model.
Although leptin might contribute to post-ischaemic myocardial dysfunction (Moro et al., 2011; Ren et al., 2010), leptin and IGF-1 are also implicated as triggers of cytoprotection (Lisa et al., 2011; Smith and Yellon, 2011). Whereas IGF-1 levels were unaltered, circulating leptin was increased ∼twofold in OB rats (Table 1). Moderate hyperleptinaemic might contribute to cardiac benefit if tissue is not leptin-resistant, because leptin can engage beneficial Erk1/2 and STAT signalling (Smith et al., 2006). Although we detect no differences in Erk1/2 signalling between CD and OB groups (Fig. 6), it remains feasible that elevated leptin contributes to improved I-R tolerance.
Modulation of pro-survival signalling by obesity
Myocardial pro-survival and pro-death kinases play important roles in the effects of obesity on cardiac stress resistance (Bouhidel et al., 2008; Smith and Yellon, 2011; Tsang et al., 2005), and modulation of RISK and NOS signalling reverses the negative cardiac effects of insulin-resistance (Bulhak et al., 2009; Huisamen et al., 2011). Myocardial Akt, Erk1/2 and NOS promote cell growth and development, limit cell death and are implicated in multiple protective responses (Gao et al., 2002; Ji et al., 2010; Yang et al., 2004). Myocardial GSK3β on the other hand plays an injurious role, active (de-phosphorylated) GSK-3β contributing to mitochondrial dysfunction and cell death (Juhaszova et al., 2009), with studies uniformly reporting cardiac protection via GSK3β inhibition.
These proteins are all components of cardioprotective pathways that converge on, and activate, the so-called mitochondrial end effectors that include the mitochondrial KATP channels and the mitochondrial permeability transition pore (mPTP). Opening of the KATP channels and closing of the mPTP protects against I-R injury. Opening of the mitochondrial KATP channel is believed to close the mPTP, which when opened renders the inner mitochondrial membrane non-specifically permeable to water and solutes. Influx of water and solutes through the mPTP causes the collapse of the mitochondrial membrane potential and uncoupling of oxidative phosphorylation, which results in ATP depletion, mitochondrial swelling and necrotic cell death (Downey et al., 2007; Halestrap et al., 2007; Hausenloy and Yellon, 2009).
These signalling proteins and enzymes are sensitive to obesity and diabetes: activation of Akt, Erk1/2 and p70S6K1 is impaired in ob/ob mice (Bouhidel et al., 2008); Erk1/2 and GSK3β dysregulation arise in metabolic syndrome (Wagner et al., 2008); and diabetes inhibits the responsiveness of kinases to conditioning stimuli (Ferdinandy et al., 2007; Gross et al., 2007b). Contrasting detrimental signalling changes in more severe disease states, we show that dietary obesity with mild hyperleptinaemic significantly improves expression and activation of Akt, phospho-activation of eNOS and Erk1/2, and phospho-inhibition of GSK3β (Figs 4-6) in myocardium from insulin-insensitive animals. Observed changes are collectively consistent with involvement in obesity-dependent protection against I-R injury.
Expression of Akt was increased in both cytosol and membrane fractions (Fig. 3). Because Akt activation involves cytosol to membrane translocation, increased expression in both compartments reflects an increase in both intrinsically active Akt and the capacity for further activation of Akt signalling. eNOS expression was largely restricted to the membrane, where phosphorylation was increased in response to obesity and BW373U86 (Fig. 6). This is consistent with known caveolar localisation of cardiac eNOS, essential to its protective functions (caveolae and caveolins regulating protective eNOS activation against injurious uncoupling for example). By contrast, expression of phosphorylated (inactive) GSK3β was increased in the cytosol but not in the membrane fraction (Fig. 5), consistent with concentration of de-phosphorylated (active) GSK3β in nuclear and mitochondrial compartments, where it promotes cell death. Based on their known actions, upregulation of Akt and activation of eNOS and Erk1/2 is predicted to limit myocardial stunning, oxidative stress, mitochondrial dysfunction and apoptotic and oncotic pathways of cell death. Similarly, enhanced inhibition of GSK3β is predicted to limit mitochondrial dysfunction and cell death (Juhaszova et al., 2009). Thus, observed changes in expression and phosphorylation of these molecules are likely to contribute to the obesity-dependent protection against I-R injury reported here.
How might beneficial adaptations in Akt, Erk1/2, NOS and GSK3β signalling be achieved? Improved phospho-regulation could stem from markedly increased expression (and activation) of upstream Akt, which influences multiple pathways (Bertrand et al., 2008; Chang et al., 2010) and is sensitive to and regulates substrate metabolism (Pagano et al., 1981; Smith and Yellon, 2011). Enhanced phosphorylation of Erk1/2, NOS and GSK3β could thus all stem from increased upstream expression of active Akt. As noted above, elevated leptin (Table 1) might also protect via activation of phosphoinositide 3-kinase (PI3K)-Akt and Erk1/2 (Smith and Yellon, 2011; Smith et al., 2006). Although leptin protection might be limited in extreme models of obesity (Smith and Yellon, 2011), benefit could be preserved in obesity with insulin-sensitivity, and absence of elevations in TNF-α could further promote Akt-dependent protection. However, although leptin can contribute to Akt and Erk1/2 phosphorylation, its elevation is an unlikely explanation for increased Akt expression (Ren et al., 2010) or GSK3β phosphorylation in cardiac cells (Palanivel et al., 2006). The basis of these latter effects awaits further investigation.
Dietary obesity renders hearts refractory to δ-OR activation of signalling and protection
In addition to assessing baseline changes in RISK-NOS signalling, we contrasted the responsiveness of RISK-NOS signalling to a conventional protective stimulus engaging these paths, i.e. δ-OR agonism (Fryer et al., 2001; Gross et al., 2007a). Several studies indicate that hearts from obese insulin-resistant (Katakam et al., 2007) or diabetic (Bouhidel et al., 2008) animals are refractory to protective interventions targeting RISK signalling and potentially involving δ-ORs. Wagner and co-workers also report that obese rats with metabolic syndrome are refractory to post-conditioning (also potentially involving ORs) (Wagner et al., 2008). Such studies (Bouhidel et al., 2008; Katakam et al., 2007; Wagner et al., 2008) suggest that altered RISK functionality might contribute to impaired efficacy of conventional protective strategies. We confirm here that hearts from obese insulin-insensitive animals are unresponsive to δ-OR agonism – neither RISK activation nor protection observed in CD hearts was evident in OB tissue. The basis of this insensitivity remains to be more directly assessed, though interestingly we have also shown that OR engagement of myocardial protein kinase signalling is inhibited with aging, in association with impaired cardioprotection (Peart et al., 2007). Age and dietary obesity might thus exert similar inhibitory effects on δ-OR signal transduction. In addition, substantial upregulation and activation of baseline RISK-NOS signalling in OB hearts (e.g. more than a 100% increase in eNOS phospho-activation and 50% increase in GSK3β phospho-inhibition) might render these paths relatively refractory to further activation. The level of protection with dietary activation of RISK-NOS paths could also be near maximal, consistent with the profound 55% reduction in infarct size in OB hearts, comparable to or exceeding protection with δ-OR agonism in CD hearts (Fig. 2), and the protection typically observed with pre- or post-conditioning. Thus, although obesity might induce some benefit, these very effects might also render hearts resistant to further protective intervention.
Limitations
The primary limitation of the current study is that although we assessed cardiac injury and infarction in vivo and in vitro (observing parallel responses), we did not assess longer term outcomes beyond 2 hours of reperfusion. It is thus feasible that differences detected at 2 hours reflect delayed rather than overall reduced cardiac injury. However, this is highly unlikely given that evidence of infarct development in this model is essentially complete within 1-2 hours of reperfusion (Marais et al., 2005; Schwarz et al., 2000), limiting the potential for subsequent emergence of differences in cell death. A second limitation is that we were unable to identify the specific basis of shifts in RISK-NOS signalling in the current work. Although a modest twofold elevation in leptin could contribute to activation of Akt and other signal components, this cannot explain markedly increased Akt expression, nor is unaltered Erk1/2 activation consistent with leptin involvement. Future work should more directly address the molecular mechanisms contributing to improvements in RISK-NOS signalling with obesity.
Conclusions
Dietary obesity (with hyperleptinaemic) protects older insulin-insensitive hearts against I-R injury when compared with hearts from lean insulin-insensitive animals, an effect that could contribute to a so-called ‘obesity paradox’ in humans (Arena and Lavie, 2010; Barbarroja et al., 2010; Dorner and Rieder, 2012; Hall et al., 2005; Lavie et al., 2009; Pingitore et al., 2007). Cardioprotection with obesity in this setting is associated with improved expression and phospho-activation of the PI3K-Akt-eNOS pathway, together with phospho-inhibition of injurious GSK3β. Although these changes might contribute to obesity-dependent protection, they might also render hearts refractory to further protection via receptor stimuli (e.g. δ-OR agonism) targeting the same PI3K-Akt-eNOS signalling. These findings thus support initial beneficial adaptations in I-R tolerance and pro-survival signalling with obesity per se, and implicate transition to insulin-resistance and ultimately diabetes as the key determinant of the negative influences of complex obesity (or metabolic syndrome) on myocardial stress resistance.
MATERIALS AND METHODS
Animals and experimental design
All studies were performed in accordance with the guidelines of the Animal Ethics Committee of Griffith University, which is accredited by the Queensland Government, Department of Primary Industries and Fisheries under the guidelines of The Animal Care and Protection Act 2001, Section 757.
Sixty four 8-week-old male Wistar rats (∼200 g) were placed on either a control (CD) or high carbohydrate obesogenic (OB) diet for 32 weeks. Fresh food was provided daily, with rats having ad libitum access to water and standard rat chow (carbohydrate 73%; protein 18%; fat 9%) for the CD group, or rat chow supplemented with sweetened condensed milk (Nestle, Australia) and raw sugar (carbohydrate 82%; protein 11%; fat 7%) for the OB group. Rats were supplied by the Animal Resources Centre (Canning Vale, Australia), and housed at 23°C with a 12-hour day-night cycle in an on-site animal facility.
Rats were randomly divided into either CD or OB groups (n=32). OB rats become viscerally obese whereas CD rats remained lean (Table 1). Both groups of middle-aged animals were insulin-insensitive (Table 1), consistent with recent observations in the Wistar rat (Gómez-Pérez et al., 2011; Gu et al., 2011). Within each experimental group, 14 rats were randomly selected for assessment of infarct development in vivo and 8 rats for ex vivo assessment of functional responses to I-R. The remaining 10 rats per group were used for biochemical and western blot analysis. Of the 14 rats from each in vivo study group, 7 were pre-treated with vehicle and 7 were pre-treated with a δ-OR agonist (BW373U86). Of the 8 rats from each ex vivo study group, 4 were pre-treated with vehicle and 4 were pre-treated with a δ-OR agonist (BW373U86).
Ex vivo perfused hearts
Rats were anaesthetised (60 mg/kg sodium pentobarbital i.p.) and hearts perfused in a Langendorff mode as described in detail previously (du Toit et al., 2005; du Toit et al., 2008; Essop et al., 2009; Maarman et al., 2012). Briefly, hearts were excised into ice-cold perfusion fluid, the aorta cannulated and coronary vessels perfused at a pressure of 100 cm H2O with modified Krebs-Henseleit buffer containing: 118 mM NaCl, 25 mM NaHCO3, 4.7 mM KCl, 1.75 mM CaCl2, 1.2 mM MgSO4, 11 mM D-glucose and 0.5 mM EDTA. Perfusion fluid was saturated with 95% O2 and 5% CO2 at 37°C, giving a pH of 7.4. A fluid-filled polyvinyl chloride film balloon was inserted into the left ventricle and connected to a P23XL pressure transducer (Viggo-Spectramed, Oxnard, CA) for measurement of ventricular pressure. Balloon volume was adjusted to an initial end-diastolic pressure of 4-6 mm Hg and then left unchanged. Functional data were recorded at 1 KHz on a four-channel MacLab system (ADInstruments, Castle Hill, Australia) connected to an Apple iMac computer. Peak systolic, diastolic and developed pressures, heart rate, and rates of contraction or relaxation over time (dP/dt) were documented. Coronary flow was monitored via volumetric analysis of coronary effluent.
After balloon placement, hearts were immersed in a water-jacketed chamber maintained at 37°C. Temperature of perfusion fluid was monitored by a needle thermometer probe (Model 52II, Fluke Corporation, Everett, Western Australia) in the coronary sinus. Hearts were excluded from study after a 15-minute stabilisation period if they failed to meet any of the following functional criteria: (i) coronary flow >25 ml/minute, (ii) unstable (fluctuating) contractile function, (iii) left ventricular systolic pressure <90 mmHg or (iv) significant cardiac arrhythmias.
After an initial 15-minute period of normoxic stabilisation, hearts were switched to ventricular pacing at 300 beats/minute (Grass S9 stimulator, Quincy, MA), normalising rate to allow comparison of rate-dependent measures of contractile function (systolic and developed ventricular pressures) and inotropic state (dP/dt). Baseline measures were made after an additional 10-minute stabilisation, before subjecting hearts to 25 minutes of normothermic global zero-flow ischemia followed by 60 minutes of aerobic reperfusion. Pacing was terminated on initiation of ischemia and resumed after 5 minutes of reperfusion. As a measure of myocardial injury or oncosis, coronary effluent was collected throughout reperfusion for analysis of total cardiac LDH efflux. Hearts were untreated (0.9% saline vehicle) or pre-treated for 10 minutes of pre-ischemia with 1 μM of δ-OR agonist (BW373U86; Tocris Bioscience, Bristol, UK) in order to activate RISK-NOS signalling and associated protection (Fryer et al., 2001; Gross et al., 2007a; Peart et al., 2007).
In vivo myocardial infarction
Rats were anaesthetised (sodium pentobarbital, 60 mg/kg i.p.), intubated and ventilated (Harvard Instruments, Model 683), and placed on an adjustable heating pad to maintain a core temperature of 36-37°C, as monitored by rectal thermometer (Model 52II, Fluke Corporation, Everett, Western Australia). Maintenance of anaesthesia was monitored by assessing pedal withdrawal reflexes at 10-minute intervals. The carotid artery was cannulated for measurement of heart rate and arterial diastolic, systolic and mean pressures. A thoracotomy and reversible left anterior descending (LAD) artery ligation were performed as outlined previously (Clark et al., 2011). Animals received either 0.9% saline (vehicle) or 1 mg/kg BW373U86 (Tocris Bioscience, Bristol, UK) administered via the right jugular vein for 10 minutes prior to LAD occlusion. The most effective cardioprotective dose was initially determined from preliminary dose-response data (with 0.1, 1.0, 10 and 100 mg/kg infusions), acquired prior to commencement of the study (data not shown). The LAD was occluded for 45 minutes followed by 120 minutes of reperfusion upon ligature release. At the end of reperfusion, hearts were excised, mounted on a Langendorff perfusion system and stained with Evan's Blue dye, frozen overnight before TTC staining, as outlined previously (Clark et al., 2011), to delineate viable and necrotic myocardium. Tissue areas were quantified by volumetric planimetry using a flat-bed scanner for image capture and computer software for image analysis (UTHSCSA Image Tool, V3), with data expressed as percentage of AAR.
Biochemical analyses
Animals set aside for biochemical analyses were fasted for 10 hours before anaesthesia, blood collection and heart excision. Peritoneal and retro-peritoneal fat was removed and weighed. Serum triglycerides, high density lipoprotein (HDL) cholesterol and glucose levels were determined in fresh whole-blood using a Blood-Lipid and Glucose Analyser (L.D.X. Cholestech Analyser, Alere, Australia). For serum analysis, remaining blood samples were placed in serum separation tubes (BD Vacutainer tubes), centrifuged and the serum stored at -80°C for later analysis. Levels of insulin (ALPCO Immunoassays, Salem, NH), leptin (Life Research, Australia), TNF-α (Life Research, Australia) and IGF-1 (Life Research, Australia) were determined in 96-well enzyme-linked immunosorbant assays (ELISAs) according to manufacturer instructions. Coronary effluent LDH activity was determined spectrophotometrically, as described previously (Peart and Headrick, 2003). To assess insulin resistance in lean and obese animals, the HOMA index was measured. Fasting blood glucose and insulin levels were used to determine the HOMA index using the standard formula: [fasting insulin (μIU/ml) × fasting glucose (mmol/l)] / 22.5.
For myocardial tissue sample collection, animals were treated with 0.9% saline (vehicle) or 1 mg/kg BW373U86 administered via the right jugular vein for 10 minutes prior to removal of the heart. Hearts were removed and immediately snap-frozen in liquid nitrogen and stored at −80°C.
Western immunoblot analyses
Left ventricular tissue samples were homogenised in lysis buffer (50 mmol/l HEPES, 150 mmol/l NaCl, 1.5 mmol/l MgCl2, 1 mmol/l EGTA, 10% glycerol plus 8.6 μmol/l leupeptin, 5.8 μmol/l pepstatin A, 4 mmol/l phenylmethylsulfonyl fluoride, 0.6 μmol/l aprotinin, 4 mmol/l sodium fluoride, and 0.8 mmol/l sodium orthovanadate) and centrifuged at 10,000 g for 15 minutes to remove nuclei and debris. Because protein activation processes might involve translocation from soluble to particulate fractions, we investigated expression of total and phosphorylated protein in both membrane and cytosolic fractions. The supernatant was centrifuged at 10,000 g to enrich for the cytosolic fraction, with the pelleted membrane fraction re-suspended by ultrasonication in a lysis buffer containing 1% Triton X-100. Fraction protein concentrations were determined by the Pierce assay. Equal quantities of protein (35 μg) were subsequently loaded onto 10% Tris-HCl gels and after electrophoresis (150 V, 1.5 hours) were transferred to PVDF membranes (50 V, 2 hours). Membranes were blocked with 3% BSA solution followed by probing overnight with antibodies (Cell Signaling Technology, Danvers, MA) for total or phosphorylated Akt (Ser473), Erk1/2 (p42/p44), GSK-3β (Ser9) and eNOS (Ser1177) followed by secondary antibody application (1:2500, Cell Signaling; 1:10,000, Santa Cruz Biotechnology) and enhanced chemiluminescence. Equal sample loading was confirmed by co-analysis of actin or GAPDH. Protein was detected by X-ray film, and densitometry assessed using NIH Image 1.63, with total expression and phosphorylation ratios normalised to values for untreated control (CD) tissue for the purposes of comparison across groups.
Statistical analyses
Unless otherwise stated, all results are expressed as means ± s.e.m. A two-tailed Student's t-test was employed to contrast measures between CD and OB groups. Multi-way ANOVA with Tukey's HSD test was used for comparison of δ-OR responses in CD and OB hearts. Evidence of statistical significance was accepted for P<0.05.
ACKNOWLEDGEMENTS
The authors thank Grant Williams-Pritchard and Nikkie Beckett for excellent technical assistance with western immunoblotting.
FUNDING: This study was supported by funds from the Griffith University Research Grant scheme and the Griffith Health Institute. J.N.P. was supported by a Future Fellowship from the Australian Research Council.
Footnotes
COMPETING INTERESTS: The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS: E.F.D.T., D.D., J.P.H. and J.N.P. conceived and designed the experiments. D.D. and E.F.D.T. performed the experiments. D.D., E.F.D.T. and J.P.H. analysed the data. E.F.D.T., J.P.H., D.D. and J.N.P. interpreted the data. E.F.D.T., D.D., J.P.H. and J.N.P. prepared the figures and manuscript.
References
- Alpert M. A., Terry B. E., Lambert C. R., Kelly D. L., Panayiotou H., Mukerji V., Massey C. V., Cohen M. V. (1993). Factors influencing left ventricular systolic function in nonhypertensive morbidly obese patients, and effect of weight loss induced by gastroplasty. Am. J. Cardiol. 71, 733-737 [DOI] [PubMed] [Google Scholar]
- Arena R., Lavie C. J. (2010). The obesity paradox and outcome in heart failure: is excess bodyweight truly protective? Future Cardiol. 6, 1-6 [DOI] [PubMed] [Google Scholar]
- Aronson D., Nassar M., Goldberg T., Kapeliovich M., Hammerman H., Azzam Z. S. (2010). The impact of body mass index on clinical outcomes after acute myocardial infarction. Int. J. Cardiol. 145, 476-480 [DOI] [PubMed] [Google Scholar]
- Aubin M. C., Lajoie C., Clément R., Gosselin H., Calderone A., Perrault L. P. (2008). Female rats fed a high-fat diet were associated with vascular dysfunction and cardiac fibrosis in the absence of overt obesity and hyperlipidemia: therapeutic potential of resveratrol. J. Pharmacol. Exp. Ther. 325, 961-968 [DOI] [PubMed] [Google Scholar]
- Aubin M. C., Cardin S., Comtois P., Clément R., Gosselin H., Gillis M. A., Le Quang K., Nattel S., Perrault L. P., Calderone A. (2010). A high-fat diet increases risk of ventricular arrhythmia in female rats: enhanced arrhythmic risk in the absence of obesity or hyperlipidemia. J. Appl. Physiol. 108, 933-940 [DOI] [PubMed] [Google Scholar]
- Barbarroja N., López-Pedrera R., Mayas M. D., García-Fuentes E., Garrido-Sánchez L., Macías-González M., El Bekay R., Vidal-Puig A., Tinahones F. J. (2010). The obese healthy paradox: is inflammation the answer? Biochem. J. 430, 141-149 [DOI] [PubMed] [Google Scholar]
- Bertrand L., Horman S., Beauloye C., Vanoverschelde J. L. (2008). Insulin signalling in the heart. Cardiovasc. Res. 79, 238-248 [DOI] [PubMed] [Google Scholar]
- Bouhidel O., Pons S., Souktani R., Zini R., Berdeaux A., Ghaleh B. (2008). Myocardial ischemic postconditioning against ischemia-reperfusion is impaired in ob/ob mice. Am. J. Physiol. Heart Circ. Physiol. 295, H1580-H1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bulhak A. A., Jung C., Ostenson C. G., Lundberg J. O., Sjöquist P. O., Pernow J. (2009). PPAR-alpha activation protects the type 2 diabetic myocardium against ischemia-reperfusion injury: involvement of the PI3-Kinase/Akt and NO pathway. Am. J. Physiol. Heart Circ. Physiol. 296, H719-H727 [DOI] [PubMed] [Google Scholar]
- Cai L., Li W., Wang G., Guo L., Jiang Y., Kang Y. J. (2002). Hyperglycemia-induced apoptosis in mouse myocardium: mitochondrial cytochrome C-mediated caspase-3 activation pathway. Diabetes 51, 1938-1948 [DOI] [PubMed] [Google Scholar]
- Champattanachai V., Marchase R. B., Chatham J. C. (2007). Glucosamine protects neonatal cardiomyocytes from ischemia-reperfusion injury via increased protein-associated O-GlcNAc. Am. J. Physiol. Cell. Physiol. 292, C178-C187 [DOI] [PubMed] [Google Scholar]
- Chang Z., Xiao Q., Feng Q., Yang Z. (2010). PKB/Akt signaling in heart development and disease. Front. Biosci. (Elite Ed.) E2, 1485-1491 [DOI] [PubMed] [Google Scholar]
- Chuang C.C., Bumrungpert A., Kennedy A., Overman A., West T., Dawson B., McIntosh M.K. (2011). Grape powder extract attenuates tumor necrosis factor α-mediated inflammation and insulin resistance in primary cultures of human adipocytes. J. Nutr. Biochem. 22: 89-94 [DOI] [PubMed] [Google Scholar]
- Clark C., Smith W., Lochner A., du Toit E. F. (2011). The effects of gender and obesity on myocardial tolerance to ischemia. Physiol. Res. 60, 291-301 [DOI] [PubMed] [Google Scholar]
- Clavijo L. C., Pinto T. L., Kuchulakanti P. K., Torguson R., Chu W. W., Satler L. F., Kent K. M., Suddath W. O., Pichard A. D., Waksman R. (2006). Metabolic syndrome in patients with acute myocardial infarction is associated with increased infarct size and in-hospital complications. Cardiovasc. Revasc. Med. 7, 7-11 [DOI] [PubMed] [Google Scholar]
- Desrois M., Clarke K., Lan C., Dalmasso C., Cole M., Portha B., Cozzone P. J., Bernard M. (2010). Upregulation of eNOS and unchanged energy metabolism in increased susceptibility of the aging type 2 diabetic GK rat heart to ischemic injury. Am. J. Physiol. Heart Circ. Physiol. 299, H1679-H1686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doehner W., Erdmann E., Cairns R., Clark A. L., Dormandy J. A., Ferrannini E., Anker S. D. (2012). Inverse relation of body weight and weight change with mortality and morbidity in patients with type 2 diabetes and cardiovascular co-morbidity: An analysis of the PROactive study population. Int. J. Cardiol. 162, 20-26 [DOI] [PubMed] [Google Scholar]
- Dorner T. E., Rieder A. (2012). Obesity paradox in elderly patients with cardiovascular diseases. Int. J. Cardiol. 155, 56-65 [DOI] [PubMed] [Google Scholar]
- Downey J. M., Davis A. M., Cohen M. V. (2007). Signaling pathways in ischemic preconditioning. Heart Fail. Rev. 12, 181-188 [DOI] [PubMed] [Google Scholar]
- du Toit E. F., Nabben M., Lochner A. (2005). A potential role for angiotensin II in obesity induced cardiac hypertrophy and ischaemic/reperfusion injury. Basic Res. Cardiol. 100, 346-354 [DOI] [PubMed] [Google Scholar]
- du Toit E. F., Smith W., Muller C., Strijdom H., Stouthammer B., Woodiwiss A. J., Norton G. R., Lochner A. (2008). Myocardial susceptibility to ischemic-reperfusion injury in a prediabetic model of dietary-induced obesity. Am. J. Physiol. Heart Circ. Physiol. 294, H2336-H2343 [DOI] [PubMed] [Google Scholar]
- Essop M. F., Anna Chan W. Y., Valle A., García-Palmer F. J., du Toit E. F. (2009). Impaired contractile function and mitochondrial respiratory capacity in response to oxygen deprivation in a rat model of pre-diabetes. Acta Physiol. (Oxf.) 197, 289-296 [DOI] [PubMed] [Google Scholar]
- Ferdinandy P., Schulz R., Baxter G. F. (2007). Interaction of cardiovascular risk factors with myocardial ischemia/reperfusion injury, preconditioning, and postconditioning. Pharmacol. Rev. 59, 418-458 [DOI] [PubMed] [Google Scholar]
- Fiordaliso F., Leri A., Cesselli D., Limana F., Safai B., Nadal-Ginard B., Anversa P., Kajstura J. (2001). Hyperglycemia activates p53 and p53-regulated genes leading to myocyte cell death. Diabetes 50, 2363-2375 [DOI] [PubMed] [Google Scholar]
- Fryer R. M., Patel H. H., Hsu A. K., Gross G. J. (2001). Stress-activated protein kinase phosphorylation during cardioprotection in the ischaemic myocardium. Am. J.Physiol. Heart Circ. Physiol. 81, H1184-H1192 [DOI] [PubMed] [Google Scholar]
- Gao F., Gao E., Yue T. L., Ohlstein E. H., Lopez B. L., Christopher T. A., Ma X.L. (2002). Nitric oxide mediates the antiapoptotic effect of insulin in myocardial ischemia-reperfusion: the roles of PI3-kinase, Akt, and endothelial nitric oxide synthase phosphorylation. Circulation 105, 1497-1502 [DOI] [PubMed] [Google Scholar]
- Girod W. G., Jones S. P., Sieber N., Aw T. Y., Lefer D. J. (1999). Effects of hypercholesterolemia on myocardial ischemia-reperfusion injury in LDL receptor-deficient mice. Arterioscler. Thromb. Vasc. Biol. 19, 2776-2781 [DOI] [PubMed] [Google Scholar]
- Gómez-Pérez Y., Gianotti M., Proenza A. M., Lladó I. (2011). Age-related decline of skeletal muscle insulin sensitivity in rats: effect of sex and muscle type. Rejuvenation Res. 14, 153-161 [DOI] [PubMed] [Google Scholar]
- Görbe A., Varga Z. V., Kupai K., Bencsik P., Kocsis G. F., Csont T., Boengler K., Schulz R., Ferdinandy P. (2011). Cholesterol diet leads to attenuation of ischemic preconditioning-induced cardiac protection: the role of connexin 43. Am. J. Physiol. Heart Circ. Physiol. 300, H1907-H1913 [DOI] [PubMed] [Google Scholar]
- Gross E. R., Hsu A. K., Gross G. J. (2007a). GSK3β inhibition and K(ATP) channel opening mediate acute opioid-induced cardioprotection at reperfusion. Basic Res. Cardiol. 102, 341-349 [DOI] [PubMed] [Google Scholar]
- Gross E. R., Hsu A. K., Gross G. J. (2007b). Diabetes abolishes morphine-induced cardioprotection via multiple pathways upstream of glycogen synthase kinase-3beta. Diabetes 56, 127-136 [DOI] [PubMed] [Google Scholar]
- Gu Z., Du Y., Liu Y., Ma L., Li L., Gong Y., Tian H., Li C. (2011). Effect of aging on islet beta-cell function and its mechanisms in Wistar rats. AGE 34, 1393-1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halestrap A. P., Clarke S. J., Khaliulin I. (2007). The role of mitochondria in protection of the heart by preconditioning. Biochim. Biophys. Acta 1767, 1007-1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J. A., French T. K., Rasmusson K. D., Vesty J. C., Roberts C. A., Rimmasch H. L., Kfoury A. G., Renlund D. G. (2005). The paradox of obesity in patients with heart failure. J. Am. Acad. Nurse Pract. 17, 542-546 [DOI] [PubMed] [Google Scholar]
- Hausenloy D. J., Yellon D. M. (2009). Preconditioning and postconditioning: underlying mechanisms and clinical application. Atherosclerosis 204, 334-341 [DOI] [PubMed] [Google Scholar]
- Hirosumi J., Tuncman G., Chang L., Görgün C. Z., Uysal K. T., Maeda K., Karin M., Hotamisligil G. S. (2002). A central role for JNK in obesity and insulin resistance. Nature 420, 333-336 [DOI] [PubMed] [Google Scholar]
- Huisamen B., Pêrel S. J., Friedrich S. O., Salie R., Strijdom H., Lochner A. (2011). ANG II type I receptor antagonism improved nitric oxide production and enhanced eNOS and PKB/Akt expression in hearts from a rat model of insulin resistance. Mol. Cell. Biochem. 349, 21-31 [DOI] [PubMed] [Google Scholar]
- Ji L., Fu F., Zhang L., Liu W., Cai X., Zhang L., Zheng Q., Zhang H., Gao F. (2010). Insulin attenuates myocardial ischemia/reperfusion injury via reducing oxidative/nitrative stress. Am. J. Physiol. Endocrinol. Metab. 298, E871-E880 [DOI] [PubMed] [Google Scholar]
- Juhaszova M., Zorov D. B., Yaniv Y., Nuss H. B., Wang S., Sollott S. J. (2009). Role of glycogen synthase kinase-3beta in cardioprotection. Circ. Res. 104, 1240-1252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katakam P. V., Jordan J. E., Snipes J. A., Tulbert C. D., Miller A. W., Busija D. W. (2007). Myocardial preconditioning against ischemia-reperfusion injury is abolished in Zucker obese rats with insulin resistance. Am. J. Physiol. Regul. Integr. Comp. Physiol. 292, R920-R926 [DOI] [PubMed] [Google Scholar]
- Kenchaiah S., Evans J. C., Levy D., Wilson P. W. F., Benjamin E. J., Larson M. G., Kannel W. B., Vasan R. S. (2002). Obesity and the risk of heart failure. N. Engl. J. Med. 347, 305-313 [DOI] [PubMed] [Google Scholar]
- Kragelund C., Hassager C., Hildebrandt P., Torp-Pedersen C., Køber L. (2005). Impact of obesity on long-term prognosis following acute myocardial infarction. Int. J. Cardiol. 98, 123-131 [DOI] [PubMed] [Google Scholar]
- Lauzier B., Delemasure S., Pesant M., Collin B., Duvillard L., Vergely C., Connat J. L., Rochette L. (2009). A cholesterol-rich diet improves resistance to ischemic insult in mouse hearts but suppresses the beneficial effect of post-conditioning. J. Heart Lung Transplant. 28, 821-826 [DOI] [PubMed] [Google Scholar]
- Lavie C. J., Milani R. V., Ventura H. O. (2009). Obesity and cardiovascular disease: risk factor, paradox, and impact of weight loss. J. Am. Coll. Cardiol. 53, 1925-1932 [DOI] [PubMed] [Google Scholar]
- Lisa M., Haleagrahara N., Chakravarthi S. (2011). Insulin-Like Growth Factor-1 (IGF-1) Reduces ischemic changes and increases circulating angiogenic factors in experimentally-induced myocardial infarction in rats. Vasc. Cell 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maarman G., Marais E., Lochner A., du Toit E. F. (2012). Effect of chronic CPT-1 inhibition on myocardial ischemia-reperfusion injury (I/R) in a model of diet-induced obesity. Cardiovasc. Drugs Ther. 26, 205-216 [DOI] [PubMed] [Google Scholar]
- Marais E., Genade S., Salie R., Huisamen B., Maritz S., Moolman J. A., Lochner A. (2005). The temporal relationship between p38 MAPK and HSP27 activation in ischaemic and pharmacological preconditioning. Basic Res. Cardiol. 100, 35-47 [DOI] [PubMed] [Google Scholar]
- Morel S., Berthonneche C., Tanguy S., Toufektsian M. C., Foulon T., de Lorgeril M., de Leiris J., Boucher F. (2003). Insulin resistance modifies plasma fatty acid distribution and decreases cardiac tolerance to in vivo ischaemia/reperfusion in rats. Clin. Exp. Pharmacol. Physiol. 30, 446-451 [DOI] [PubMed] [Google Scholar]
- Moro C., Grauzam S., Ormezzano O., Toufektsian M. C., Tanguy S., Calabrese P., Coll J. L., Bak I., Juhasz B., Tosaki A., et al. (2011). Inhibition of cardiac leptin expression after infarction reduces subsequent dysfunction. J. Cell. Mol. Med. 15, 1688-1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagano G., Cassader M., Diana A., Pisu E., Bozzo C., Ferrero F., Lenti G. (1981). Insulin resistance in the aged: the role of the peripheral insulin receptors. Metabolism 30, 46-49 [DOI] [PubMed] [Google Scholar]
- Palanivel R., Eguchi M., Shuralyova I., Coe I., Sweeney G. (2006). Distinct effects of short- and long-term leptin treatment on glucose and fatty acid uptake and metabolism in HL-1 cardiomyocytes. Metabolism 55, 1067-1075 [DOI] [PubMed] [Google Scholar]
- Peart J. N., Headrick J. P. (2003). Adenosine-mediated early preconditioning in mouse: protective signaling and concentration dependent effects. Cardiovasc. Res. 58, 589-601 [DOI] [PubMed] [Google Scholar]
- Peart J. N., Gross E. R., Headrick J. P., Gross G. J. (2007). Impaired p38 MAPK/HSP27 signaling underlies aging-related failure in opioid-mediated cardioprotection. J. Mol. Cell. Cardiol. 42, 972-980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson L. R., Waggoner A. D., Schechtman K. B., Meyer T., Gropler R. J., Barzilai B., Dávila-Román V. G. (2004). Alterations in left ventricular structure and function in young healthy obese women: assessment by echocardiography and tissue Doppler imaging. J. Am. Coll. Cardiol. 43, 1399-1404 [DOI] [PubMed] [Google Scholar]
- Pingitore A., Di Bella G., Lombardi M., Iervasi G., Strata E., Aquaro G. D., Positano V., De Marchi D., Rossi G., L'Abbate A., et al. (2007). The obesity paradox and myocardial infarct size. J. Cardiovasc. Med. (Hagerstown) 8, 713-717 [DOI] [PubMed] [Google Scholar]
- Rana J. S., Mukamal K. J., Morgan J. P., Muller J. E., Mittleman M. A. (2004). Obesity and the risk of death after acute myocardial infarction. Am. Heart J. 147, 841-846 [DOI] [PubMed] [Google Scholar]
- Ren J., Dong F., Cai G. J., Zhao P., Nunn J. M., Wold L. E., Pei J. (2010). Interaction between age and obesity on cardiomyocyte contractile function: role of leptin and stress signaling. PLoS ONE 5, e10085. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Savage D. B., Petersen K. F., Shulman G. I. (2005). Mechanisms of insulin resistance in humans and possible links with inflammation. Hypertension 45, 828-833 [DOI] [PubMed] [Google Scholar]
- Schaffer S. W., Croft C. B., Solodushko V. (2000). Cardioprotective effect of chronic hyperglycemia: effect on hypoxia-induced apoptosis and necrosis. Am. J. Physiol. Heart Circ. Physiol. 278, H1948-H1954 [DOI] [PubMed] [Google Scholar]
- Schwarz E. R., Somoano Y., Hale S. L., Kloner R. A. (2000). What is the required reperfusion period for assessment of myocardial infarct size using triphenyltetrazolium chloride staining in the rat? J. Thromb. Thrombolysis 10, 181-187 [DOI] [PubMed] [Google Scholar]
- Sharma V., Mustafa S., Patel N., Wambolt R., Allard M. F., McNeill J. H. (2009). Stimulation of cardiac fatty acid oxidation by leptin is mediated by a nitric oxide-p38 MAPK-dependent mechanism. Eur. J. Pharmacol. 617, 113-117 [DOI] [PubMed] [Google Scholar]
- Shizukuda Y., Reyland M. E., Buttrick P. M. (2002). Protein kinase C-delta modulates apoptosis induced by hyperglycemia in adult ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 282, H1625-H1634 [DOI] [PubMed] [Google Scholar]
- Smith C. C., Yellon D. M. (2011). Adipocytokines, cardiovascular pathophysiology and myocardial protection. Pharmacol. Ther. 129, 206-219 [DOI] [PubMed] [Google Scholar]
- Smith C. C., Mocanu M. M., Davidson S. M., Wynne A. M., Simpkin J. C., Yellon D. M. (2006). Leptin, the obesity-associated hormone, exhibits direct cardioprotective effects. Br. J. Pharmacol. 149, 5-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoddard M. F., Tseuda K., Thomas M., Dillon S., Kupersmith J. (1992). The influence of obesity on left ventricular filling and systolic function. Am. Heart J. 124, 694-699 [DOI] [PubMed] [Google Scholar]
- Szucs G., Bester D. J., Kupai K., Csont T., Csonka C., Esterhuyse A. J., Ferdinandy P., Van Rooyen J. (2011). Dietary red palm oil supplementation decreases infarct size in cholesterol fed rats. Lipids Health Dis. 10, 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang A., Hausenloy D. J., Mocanu M. M., Carr R. D., Yellon D. M. (2005). Preconditioning the diabetic heart: the importance of Akt phosphorylation. Diabetes 54, 2360-2364 [DOI] [PubMed] [Google Scholar]
- Wagner C., Kloeting I., Strasser R. H., Weinbrenner C. (2008). Cardioprotection by postconditioning is lost in WOKW rats with metabolic syndrome: role of glycogen synthase kinase 3beta. J. Cardiovasc. Pharmacol. 52, 430-437 [DOI] [PubMed] [Google Scholar]
- Wilson C. R., Tran M. K., Salazar K. L., Young M. E., Taegtmeyer H. (2007). Western diet, but not high fat diet, causes derangements of fatty acid metabolism and contractile dysfunction in the heart of Wistar rats. Biochem. J. 406, 457-467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong C. Y., O'Moore-Sullivan T., Leano R., Byrne N., Beller E., Marwick T. H. (2004). Alterations of left ventricular myocardial characteristics associated with obesity. Circulation 110, 3081-3087 [DOI] [PubMed] [Google Scholar]
- Xu G., Takashi E., Kudo M., Ishiwata T., Naito Z. (2004). Contradictory effects of short- and long-term hyperglycemias on ischaemic injury of myocardium via intracellular signaling pathway. Exp. Mol. Pathol. 76, 57-65 [DOI] [PubMed] [Google Scholar]
- Yang X. M., Krieg T., Cui L., Downey J. M., Cohen M. V. (2004). NECA and bradykinin at reperfusion reduce infarction in rabbit hearts by signaling through PI3K, ERK, and NO. J. Mol. Cell. Cardiol. 36, 411-421 [DOI] [PubMed] [Google Scholar]
- Yue T. L., Bao W., Gu J. L., Cui J., Tao L., Ma X. L., Ohlstein E. H., Jucker B. M. (2005). Rosiglitazone treatment in Zucker diabetic Fatty rats is associated with ameliorated cardiac insulin resistance and protection from ischemia/reperfusion-induced myocardial injury. Diabetes 54, 554-562 [DOI] [PubMed] [Google Scholar]
- Zarich S. W., Kowalchuk G. J., McGuire M. P., Benotti P. N., Mascioli E. A., Nesto R. W. (1991). Left ventricular filling abnormalities in asymptomatic morbid obesity. Am. J. Cardiol. 68, 377-381 [DOI] [PubMed] [Google Scholar]