

SUMMARY
Multiple sclerosis (MS) is the most common CNS-demyelinating disease of humans, showing clinical and pathological heterogeneity and a general resistance to therapy. We first discovered that abnormal myelin hypercitrullination, even in normal-appearing white matter, by peptidylarginine deiminases (PADs) correlates strongly with disease severity and might have an important role in MS progression. Hypercitrullination is known to promote focal demyelination through reduced myelin compaction. Here we report that 2-chloroacetamidine (2CA), a small-molecule, PAD active-site inhibitor, dramatically attenuates disease at any stage in independent neurodegenerative as well as autoimmune MS mouse models. 2CA reduced PAD activity and protein citrullination to pre-disease status. In the autoimmune models, disease induction uniformly induced spontaneous hypercitrullination with citrulline+ epitopes targeted frequently. 2CA rapidly suppressed T cell autoreactivity, clearing brain and spinal cord infiltrates, through selective removal of newly activated T cells. 2CA essentially prevented disease when administered before disease onset or before autoimmune induction, making hypercitrullination, and specifically PAD enzymes, a therapeutic target in MS models and thus possibly in MS.
INTRODUCTION
Multiple sclerosis (MS) is the most common demyelinating disease of human adults. Its therapies have limited effectiveness in decreasing relapse frequencies without affecting disease progression (Steinman and Zamvil, 2006). Treatments are essentially immunosuppressive (Wingerchuk, 2008; Comi and Martino, 2006; Hemmer et al., 2005), reflecting the broad consensus of autoimmune effector mechanisms in MS. However, disease heterogeneity provides evidence for non-autoimmune, biochemical and epigenetic MS abnormalities, whose role in the complex hierarchy of pathogenesis remains unclear. In a study of 286 MS cases (mostly biopsies) (Lucchinetti et al., 2000), MS has been categorized into four different patterns of pathogenesis, which we have recognized in our animal models, which combined show most of the features of MS. In early studies we showed that myelin basic protein (MBP) isolated from normal human brain contained about 20% of the citrullinated MBP (Moscarello et al., 1994). In chronic MS white matter, the citrullinated protein was 45% and in fulminating MS (Marburg's Disease) it accounted for 90% of the MBP (Wood et al., 1996). This less cationic MBP was unable to compact lipid bilayers in model systems, as revealed with several techniques including X-ray diffraction (Brady et al., 1981a; Brady et al., 1981b) electron spin resonance (Boggs et al., 1982) circular dichroism (Epand et al., 1974) and NMR (Deber et al., 1986). We postulated that the decreased protein-lipid interactions resulted in destabilization of the myelin, which could then be more readily degraded.
Citrulline in proteins is generated by a family of enzymes, the peptidylarginine deiminases (PADs) of which five are known to be all localized to a single locus on chromosome 1 (lp36.1). Of the five isozymes, PAD2 and PAD4 are found in brain, localized in myelin and oligodendrocytes. PAD4 is unique because it is the only PAD to carry a nuclear localization signal. We have shown that PAD4 can be translocated to the nucleus in oligodendrocytes in culture in the presence of TNFα where it deiminates histone H3, suggesting a role in apoptosis (Mastronardi et al., 2006). PAD2 was found in myelin, the axons, and the periaxonal space at the contact between myelin and the oligodendrocyte (Wood et al., 2008). Both processes, the decreased myelin compaction and apoptosis of the oligodendrocytes, represent important pathways in the patho-mechanism of demyelination.
Further support for an important role of PAD enzymes and hypercitrullination of proteins in MS was obtained with two transgenic mouse lines that demyelinate spontaneously. In the ND4 line, which contains 70 copies of the cDNA for DM20 (a myelin proteolipid), an increase in PAD enzymes was observed at 2 months of age, 1 month prior to the onset of both clinical signs of demyelination and increased protein citrullination at 3 months of age, suggesting a causative role of PAD enzymes (Moscarello et al., 2002). In the other model, generated by the addition of 30 copies of cDNA for PAD2, signs of demyelination occurred at 6 months of age spontaneously, showing that increased PAD enzymes on their own induced demyelination (Musse et al., 2008). These results, combined with the above mentioned data on citrullinated MBP, provide a compelling case for a prominent role for PAD enzymes and citrullinated proteins in the pathogenesis of MS.
TRANSLATIONAL IMPACT.
Clinical issue
Multiple sclerosis (MS) is the most common demyelinating disease of human adults, affecting ∼2 million people worldwide. It is characterized by fatigue, muscle weakness, and cognitive impairment. These features are the result of degradation of the myelin sheath, which surrounds the nerves and which must be intact for proper nerve conduction. The disease is generally progressive and can develop over many years. Although generally regarded as an autoimmune disease, a neurodegenerative etiology that involves dysregulation of one or more neurobiological processes has also been implicated in MS pathogenesis. The therapeutic needs of MS are currently largely unmet. Most currently available therapies are immune modulatory and, although they can improve relapse rates, they do not affect disease progression.
Results
Previous work by this group showed that myelin basic protein (MBP), a major protein in myelin, is hypercitrullinated in MS, and that the enzymes responsible for converting arginine in proteins to citrulline – the peptidylarginine deiminases (PADs) – are increased in white matter from MS brains. In this study, the authors show that a small molecule PAD inhibitor, 2-chloroacetamidine (2CA), inhibits PADs in vitro and in vivo, and identify the covalent binding site of the drug in the active site of the enzyme. Inhibition of PADs with 2CA also attenuated disease in two neurodegenerative and two autoimmune mouse models of MS. In all four models, 2CA inhibited PAD activity in the brain and spinal cord, and decreased the amount of citrullinated proteins. In addition, remyelination was observed in the treated mice.
Implications and future directions
These findings identify a commonality in four different mouse models of MS. In all four models, protein hypercitrullination resulted from upregulation of PADs, and these enzymes seem to be a therapeutic target that can be regulated by the small-molecule inhibitor 2CA. Small-molecule PAD inhibitors could, therefore, represent a new therapeutic approach for MS. However, because 2CA binds covalently to the active site of PAD, it is not an ideal drug. Future research will aim to develop new, reversible PAD inhibitors that might be suitable for use in patients with MS.
Here we report the use of a small molecule active site PAD inhibitor, 2-chloroacetamidine (2CA) (Stone et al., 2005) in the treatment of four animal models of demyelination, two neurodegenerative and two autoimmune. In all four models, protein citrullination was decreased, PAD activities were decreased, disease was attenuated and remyelination was observed. In addition, 2CA markedly decrease CD3+ T cells in the autoimmune models. 2CA is an irreversible PAD inhibitor and we have exploited this property to localize the adduct (PAD-2CA) in brain. With these studies we have identified a new therapeutic target readily available for intervention by small molecules.
RESULTS
PAD expression and activity in white matter from MS patients
We compared protein citrullination, PAD protein expression and PAD activity in extracts of normal brain and of normal-appearing white matter (NAWM) from MS patient brains using the slot blot method (supplementary material Table S1). PAD protein expression was elevated in NAWM from MS brain (Fig. 1A) with a corresponding increase in PAD activity and protein citrullination (Fig. 1B,C). Following addition of 2CA to NAWM extracts of MS brain, PAD activity declined to normal levels, demonstrating that 2CA was effective in human brain extracts (Fig. 1C). Using recombinant PAD2 and PAD4 enzymes we then measured the residual activity with α-N-benzoyl-L-arginine ethyl ester (BAEE) in the presence of increasing concentrations of 2CA (Fig. 2A). A rapid decrease in activity was observed with both enzymes, more marked with PAD2, yielding an IC50 of 14.4 mM. Human PAD enzymes are well conserved in the C-terminus, which contains the active-site region (Fig. 2A, inset). To identify the 2CA binding site, recombinant human PAD2 was reacted with 2CA. Trypsin digests of 2CA-reacted and unreacted enzyme were compared by LC-ESI-QToF-tandem mass spectrometry analysis (see Materials and Methods; supplementary material Fig. S1). We identified a tryptic peptide in 2CA-reacted PAD2 with a mass of 1386.68 Da, whereas the corresponding unreacted peptide had a mass of only 1330.45 Da. The difference of 56Da corresponds to addition of one molecule of acetamidine (Fig. 2C). As shown in Fig. 2B, we propose that the mechanism of the reaction is acetamidine binding to Cys656 in a covalent bond between the cysteine sulfur and the carbon of the acetamidine in a covalent interaction, as originally described (Stone et al., 2005). Fragmentation analysis (Fig. 2C) mapped the relevant peptide (650FLGEVHCGTNVR) to the C-terminal region containing the active site cysteine (Cys656). A similar analysis identified the same active-site cysteine in recombinant PAD4 (our unpublished results). Fragmentation analysis determined that the y-ions 688-326-17 (NH3) and in PAD2 and PAD4, respectively, contained the cysteine-adduct (Fig. 2C). To determine whether 2CA crosses into the brain, our therapeutic target, we generated monoclonal antibodies to p650FLGEVHC*GTNVR, carrying the *acetamidine adduct on the Cys656 residue. Monoclonal antibody mAb4E12 recognized 2CA-modified, but not unmodified, PAD2 in western blots. We injected 2CA i.p. twice per week into transgenic mice overexpressing PAD2 in white matter (Musse et al., 2008). Brain sections were stained with mAb4E12 and developed with gold-labeled anti-mouse IgG for immuno-electron microscopy (Fig. 3). Drug-PAD2 adducts were scattered in myelin sheaths and nuclei of oligodendrocytes, indicating that the drug reached its target molecules in the brain. Clusters of gold particles were found in the periaxonal region (white arrows) but gold label was absent in similarly stained sections from untreated PAD2 transgenics.
Fig. 1.
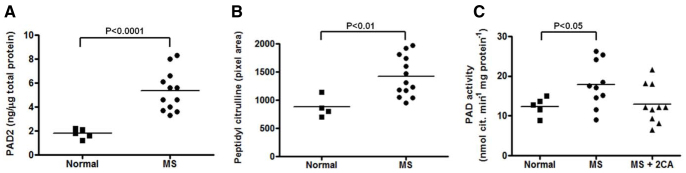
PAD activity in NAWM of MS patients. (A) Quantification of PAD2 protein in white matter from normal and MS brain by immunoslot blot (n=5, P<0.0001). (B) Citrullinated protein in white matter from normal and MS brains by immunoslot blot as pixel density (n=4, P<0.01). (C) PAD enzyme activity in normal and MS tissue, with or without pre-incubation with 2CA (n=5, P<0.05). Each dot represents one patient analysed three times. The means (horizontal bar) for all the MS patients were compared with the normal.
Fig. 2.

Interaction of PAD with 2CA. (A) PAD2 and PAD4 inhibition curves in the presence of increasing 2CA concentrations. Insert: PAD1-4 enzymes contain a common C-terminal active-site cysteine residue (Cys656) bound by 2CA, confirmed by ESI mass spectrometry of tryptic digests of PAD2-acetamidine adducts. (B) Schematic of the nucleophilic reaction between 2CA and the Cys656 residue in the active site of PAD2 (Stone et al., 2005). (C) Tabular summary of peptide fragment atomic masses for 2CA-modified and native PAD2.
Fig. 3.
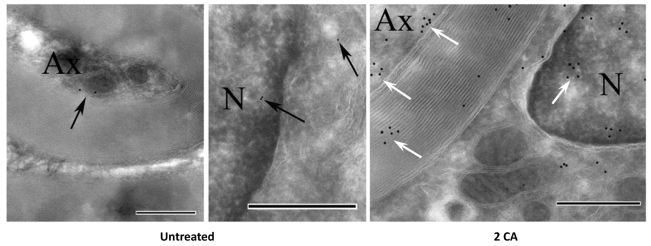
mAb4E12 (anti-2CA adduct) immunogold labeled optic nerve cryosections from control and 2CA-treated PAD2 transgenic mice. Minimal labeling in untreated mice (black arrows). Numerous gold particles (white arrows) in nuclei (N) and cytoplasm of oligodendrocytes, myelin and axonoplasm (Ax) of 2CA-treated mice. Scale bars: 500 nm.
Disease attenuation by 2CA in demyelinating mice
We employed four independent mouse models of demyelination, two non-autoimmune transgenic (Musse et al., 2008; Mastronardi et al., 1993) and two induced autoimmune models of demyelinating disease (Iglesias et al., 2001; Tuohy et al., 1995), thus comparing 2CA responses across different genetic backgrounds, disease causes and progression programs.
The first of the neurodegenerative models was the ND4 transgenic mice (DM20 overexpressors), which are normal from birth until ∼10-12 weeks of age, when they develop a non-autoimmune, primary progressive and ultimately fatal CNS demyelinating condition (Mastronardi et al., 1993; Johnson et al., 1995). Disease progression is associated with increased myelin PAD expression and hypercitrullination of myelin proteins (Moscarello et al., 2002) and histone H3 proteins (Mastronardi et al., 2006). ND4 mice received i.p. phosphate-buffered saline (PBS), 2CA (5 mg/kg) or 2CA plus 10 mg/kg B12 every other day, beginning well before disease onset (2 months) (Fig. 4A) or early after disease onset (3.5 months) (Fig. 4B). In another group with early treatment onset, 2CA injections were stopped after six weeks and B12 injections continued (Fig. 4C). Early and prolonged 2CA injections starting at 2 months of age essentially prevented disease, with or without B12 (Fig. 4A). Most untreated mice were sacrificed with severe disease by 6 months; none died in the treatment groups. Treatment of early disease at 3.5 months prevented progression, but mild disability continued unless animals received 2CA plus B12. However, fully progressive clinical disease re-emerged promptly after therapy cessation at 6 months (Fig. 4B). The temporal link of disease protection by 2CA and relapse after therapy cessation suggests that PAD-mediated citrullination and disease progression are a basic mechanism in the transgene-driven pathogenesis. Abbreviated 2CA treatment (2-3.5 months) was not very effective, but continued B12 injections generated milder disease and delayed progression (Fig. 4C). We previously reported that B12 by itself has little effect in the ND4 mouse (Mastronardi et al., 2007b; Mastronardi et al., 2004), which we confirm here. Overall, 2CA induced dramatic disease attenuation, which did, however, require continued injections due to obvious persistence of pathogenic transgene expression.
Fig. 4.

2CA attenuates demyelinating disease in ND4 mice. (A) ND4 mice treated with PBS, 2CA (5 mg/kg), or 2CA+B12 (5 mg/kg and 10 mg/kg) starting at 2 months before disease onset (n=5, P<0.0001). (B) ND4 mice treated at disease onset (n=4, P<0.0001). (C) Stopping 2CA, but continuing B12 at 3.5 months in ND4 mice (n=5, P<0.0001) demonstrates that B12 alone does not attenuate disease. (D) PAD activity in brains from animals shown in Fig. 3B (n=5, P<0.05). The first four bars represent results from animals at 6 months of age, whereas the post-treatment animals were 8 months of age. (E) PAD2 RT PCR in white matter extracts from normal, PBS-, 2CA- and 2CA+B12-treated ND4 mice (n=9, P<0.05). (F) LFB and hematoxylin stain of cerebella from normal, PBS-, 2CA- and 2CA+B12-treated ND4 mice (40×). WM, white matter; GM, grey matter.
To determine how improved clinical scores were related to PAD activity, 6-month-old mice from the treatment groups in Fig. 4B were examined (Fig. 4D). 2CA normalized the white matter PAD activity in brain homogenates, with considerable overshoot (and rapid disease progression) in such animals after therapy cessation. B12 further reduced PAD hyperactivity, preventing the post-therapy overshoot (Fig. 4D). PAD2 gene expression (Fig. 4E) paralleled PAD activity, suggesting that disease-induced elevations are regulated at the transcriptional level, where PAD promoter hypomethylation could explain PAD overexpression as well as the positive impact of B12 adjunct therapy (Mastronardi et al., 2007b).
Staining of myelin with Luxol Fast Blue (Fig. 4F) demonstrated impressive myelin deficits and pronounced vacuolization in untreated ND4 mice, which were dramatically improved in treated mice. When treatment was removed, myelinolysis re-emerged in both treatment groups, albeit milder in those that received B12. Morphological changes in myelin structure were analyzed by transmission electron microscopy (TEM) of optic nerve cross-sections from 6-month-old mice (described in Fig. 5A-F). In non-transgenic (normal) CD1 littermates (Fig. 5A), axons were well myelinated with myelin of uniform thickness. ND4 mice showed wide areas of myelin loss, degradation, and nude axons were common (Fig. 5B). 2CA treatment (Fig. 5C) clearly improved this morphology, with few axons seriously affected, but many still showed thinning of the myelin thickness. Addition of B12 resulted in a morphological picture that was virtually indistinguishable from normal (Fig. 5D). When treatment stopped at 6 months, analysis at 8 months showed reappearance of disease (Fig. 5E,F) due to the continuing presence of the transgene.
Fig. 5.
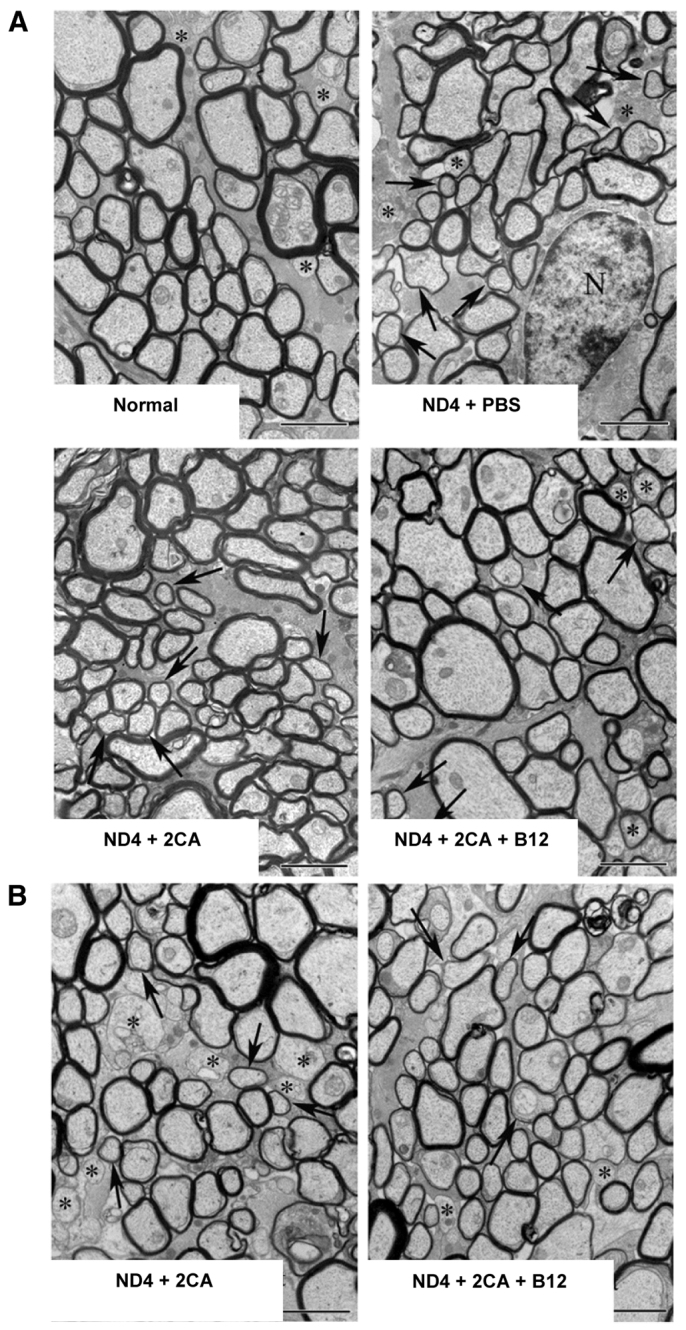
TEM of optic nerves after 2CA treatment. (A) TEM of optic nerve sections from normal, PBS-, 2CA- and 2CA+B12-treated ND4 mice at 6 months. (B) TEM of optic nerve sections from 2CA- and 2CA+B12-treated ND4 mice, 2 months after treatment cessation. Asterisks show nude axons, arrows show thin myelin sheaths. Scale bars: 2 μm.
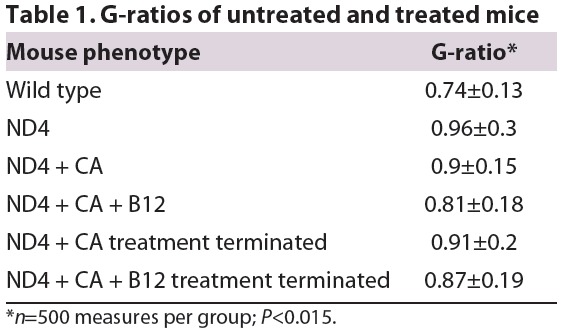
To quantify the above myelin changes, we calculated G-ratios (axon diameter/fiber diameter) from ∼500 non-contiguous semi-thin sections per treatment group (Table 1). Compared with healthy littermates (G-ratio: 0.74±0.13) ND4 mice showed a reduction in optic nerve myelin thickness (G-ratio: 0.96±0.3; P<0.0013). In 2CA-treated ND4, myelin thickness was slightly improved and showed less variation (G ratio: 0.9±0.15; P<0.015). 2CA+B12 treatment nearly normalized myelin thickness (G-ratio: 0.81±0.18; P<0.015). These treatment data are typical for remyelination, where the original myelin thickness is never re-achieved.
Table 1.
G-ratios of untreated and treated mice
Transgenic mice overexpressing PAD2 under control of the MBP promoter (Musse et al., 2008) develop a demyelinating phenotype (Fig. 6). Treatment with 2CA or 2CA+B12 had little effect on early disease, but treatments prevented the second, more aggressive disease phase equally. PAD activity was high in transgenic white matter, and 2CA, with or without B12, normalized tissue enzyme activity levels (Fig. 6). B12 has little effect in this model.
Fig. 6.
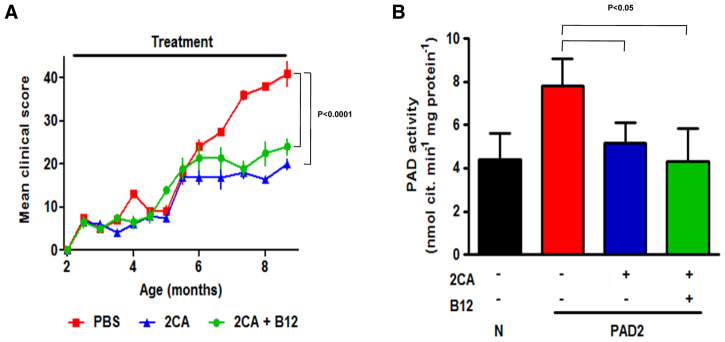
2CA attenuates PAD2 overexpressor. (A) Demyelinating disease in PAD2 transgenic mice treated with PBS, 2CA or 2CA+B12 starting at 6 months of age (n=5, P<0.0001). (B) PAD activity in brain extracts of PAD2 transgenic mice treated with PBS, 2CA or 2CA+B12, and non-transgenic littermates (n=4, P<0.05). PAD activity is reduced to normal levels by treatment.
Survival from acute MOG EAE
Experimental autoimmune encephalomyelitis (EAE) was induced in C57BL/6 mice with 100 μg of myelin oligodendrocyte glycoprotein (MOG)35-55 peptide, emulsified in Freund's complete adjuvant and 300 ng of pertussis toxin. At the earliest sign of disease, usually 9 days post-immunization, groups of mice received carrier (PBS), 2CA or 2CA+B12 as before (Fig. 7A). Untreated mice developed rapidly progressive disease and were sacrificed when moribund (∼day 19). 2CA and 2CA+B12 treatment did not affect the disease course observed in untreated controls until ∼day 14, when progression halted and recovery began, leaving ∼50% survival by day 30 (Fig. 7B). When 2CA treatment was started before immunization, disease lethality was zero (our unpublished observation). Despite the severity of disease, there was relatively little histopathology in the brain. However, vacuolar demyelination and lymphoid infiltration were prominent in the spinal cord (Fig. 7C, arrows). By contrast, surviving 2CA and 2CA+B12 recipients showed much improved, virtually normal spinal histology (Fig. 7C, center and right panels). More sensitive immunofluorescence still detected scattered CD3+ T cells in treated animals, but the heavy T cell clusters seen in PBS-treated controls were absent (Fig. 8A), suggesting that one possible 2CA effect might be suppression of T cell expansion. White matter from PBS-treated mice had elevated PAD activities, which were normalized after 2CA (Fig. 8B). Spinal cord PAD activities of PBS-treated mice reached threefold elevations, but reductions to normal levels were once again observed following treatment (Fig. 8C).
Fig. 7.

2CA attenuates acute EAE. (A) Clinical progression (n=5, P<0.0001) and (B) survival (n=5, P<0.01) during acute MOG EAE in C57BL/6 mice treated with PBS, 2CA or 2CA+B12. (C) H&E stain of spinal cord treated with PBS, 2CA or 2CA+B12 (40×) showing decreased cellular infiltration after treatment (arrows).
Fig. 8.

CD3+ lymphocytes, PAD activity and splenocyte recall. (A) Spinal cord CD3+ lymphocytes in MOG-EAE mice treated with PBS, 2CA or 2CA+B12 (n=12, P<0.05). (B) PAD activity in brain (n=4, P not significant). (C) Spinal cord extracts (n=4, P<0.05) showing threefold increase of PAD in EAE reduced to normal levels by 2CA. (D) Ex vivo recall proliferative splenocytes response of C57BL/6 mice with peak disease stimulated with MOG35-55 and MOG35-55 citrullinated at residue 41 (MOGcit41), residue 46 (MOGcit46), and residues 41 plus 46 (MOGcit41,46) (n=8, P<0.05 and P<0.01).
To seek more mechanistic understanding of the 2CA targets in the disease process, we measured MOG-specific autoreactive T cell pools. All mice developed highly reactive, systemically distributed pools of pMOG35-55-reactive T cells, detected in ex vivo T cell recall assays (Fig. 8D). Drug treatments significantly reduced immunity and recall responses to MOG within the short disease course. There are two arginine residues in the immunodominant MOG peptide and we measured T cell recognition of replacement peptides, carrying citrulline instead of arginine in one or both sites (Fig. 8D, right panels). Single arginine to citrulline replacement peptides were recognized by substantial T cell pools, each about half the size of those recognizing native pMOG35-55. T cell pools recognizing MOG35-55 with both arginine residues replaced by citrulline, were much smaller; testimonial to the stringency of in vivo T cell repertoire selection. Cell responses to citrullinated MOG peptides were reduced in mice receiving 2CA or 2CA+B12, in keeping with the ability of 2CA to block citrullination and the reduced availability of citrullinated peptides to be attacked by the immune system. These data indicated that the disease-related T cell autoreactivity repertoire prominently includes recognition of citrulline-containing epitopes, an observation with precedence in the literature (Carrillo-Vico et al., 2010).
We concluded that the acute 2CA blockade of citrullination in the early phase of this model produced a major reduction in autoreactive T cell pools, providing a mechanistic explanation for the drug-induced failure to generate the massive T cell tissue invasion characteristic of the effector phase of this disease. The remaining infiltrates of scattered CD3+ T cells in treated survivors (Fig. 8A) might be either anergic or non-specific bystanders with little pathogenicity, because there were no relapses after therapy cessation in CD3+ T cell populations. Our conclusion of immune expansion as a major 2CA target could also explain the lack of B12 effects in acute MOG-EAE, where time is short for remyelination.
Attenuation of chronic relapsing EAE by 2CA
Acute MOG-EAE is a rapid and aggressive disease and we decided to include a chronic relapsing EAE (crEAE) model that better mimics the relapsing-remitting course common in MS. Disease was induced in SJL mice with a proteolipid protein peptide (pPLP139-155; 200 μg/mouse) (Tuohy et al., 1995; Mastronardi et al., 2004). Treatments with 2CA and 2CA+B12 were started at disease onset (day 10-11) with different treatment schedules, as indicated in the top bars of Fig. 9. Attenuation of disease was observed in all treated mice (Fig. 9A). In contrast to our previous findings, where B12 showed synergistic effectiveness in interferon-β-treated crEAE (Mastronardi et al., 2004), there was little added benefit when combined with 2CA and it was omitted in some of the subsequent experiments.
Fig. 9.
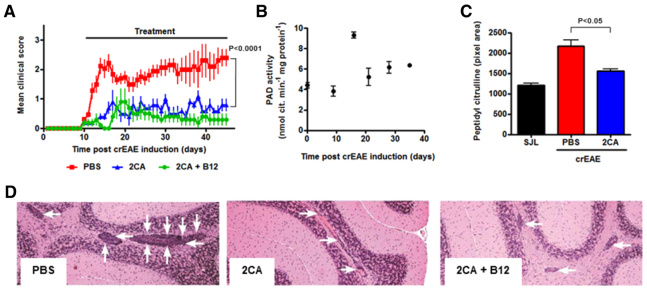
2CA attenuates crEAE. (A) Clinical crEAE progression in SJL mice treated with PBS, 2CA or 2CA+B12 (n=5, P<0.0001). (B) PAD activity in SJL brain extracts during crEAE (n=4). (C) Protein citrullination in brains of SJL animals (n=5, P<0.0001) after termination of the experiment at 42 days. (D) H&E stain of SJL brain (40×) showing infiltration of cells (white arrows). Note the decrease after treatment.
After pilot experiments suggested steep excursions at disease onset, we followed PAD activity over the disease course in 20 mice (Fig. 9B). PAD activities indeed showed a sharp rise at disease onset, peaking at the height of the initial disease phase and declining to still above-background levels thereafter. Protein citrullination in white matter was consequently elevated, a process prevented by 2CA (Fig. 9C). Besides strain differences, the different EAE induction modes crucially reflect the choice of the inducing myelin peptide, thus involving a cognate recognition event in CD4+ and/or CD8+ T cells with potentially encephalitogenic T cell receptors (Zhu et al., 2006). The extent of disease was closely reflected by the extent of cellular infiltration in white matter sections (Fig. 9D), confirmed by measuring CD3+ T cell invasion around blood vessels (Fig. 10A). As with the MOG EAE model, 2CA also prevented and/or reduced (see below) T cell tissue infiltrates in the chronic relapsing model of EAE (Fig. 9D; Fig. 10A).
Fig. 10.

Inhibition of EAE by 2CA. (A) CD3+ lymphocytes in SJL spinal cord (n=12, P<0.01), (B) IFN-γ. (n=3, P<0.05) and (C) IL-17 (n=3, P<0.05) secretion following in vitro stimulation of SJL splenocytes with PLP139-155. (D-F) Progression of crEAE following treatment with PBS or 2CA, beginning on day 20 (D), 25 (E) or 28 (F) (n=5, P<0.0001).
We compared systemically distributed pPLP139-155-specific T cells of treated and untreated mice via their release of ex vivo, peptide-induced effector cytokines IFN-γ and IL-17 (Fig. 10B,C). The dramatic depression of cytokine responses to near background values was interpreted to suggest that 2CA interfered with recruitment and/or expansion of the ‘inciting’ T cell repertoire.
The chronic phase of crEAE allowed us to examine 2CA effects in greater detail (Fig. 10D-F; supplementary material Fig. S2). PAD inhibition was effective in abrogating disease at all stages of disease progression (Fig. 10D,F). The prompt decline of clinical signs following 2CA application in mice with crEAE implies a very rapid local and systemic (Fig. 10B,C) loss of effector cells following inhibition of continued protein citrullination. Tissue half-life times of citrullinated proteins are unknown, as is the persistence of the relevant, processed peptides in local antigen-presenting cells (APCs). The rapidity of 2CA effects therefore makes direct actions of the drug on T cells and/or APCs likely: both cell lineages express PAD (Liu et al., 2006).
DISCUSSION
MS is the most common demyelinating disease of humans and is a heterogeneous disease characterized into four types (Lucchinetti et al., 2000). Pattern 1 shows inflammatory demyelination with macrophage infiltration. Pattern 2 shows demyelination with inflammatory infiltration with T cells. Pattern 3 is characterized by loss of oligodendrocytes by apoptosis. Pattern 4 also shows loss of oligodendrocytes, but is rare.
Understanding the pathogenesis of MS has relied heavily on animal models, of which there are several. Although all models reproduce some of the features of MS, none reproduces all the features. In our studies described here, we have used four animal models to try to bring together as many of the features of MS as possible. Two of the models were inflammatory autoimmune models (acute and chronic relapsing EAE) reflective of pattern 2 MS. The acute EAE was monophasic and rapid, whereas the crEAE was more reflective of the relapsing-remitting course of MS. The ND4 transgenic mouse, which contains 70 copies of the cDNA for the myelin proteolipid protein DM20, is representative of pattern 3 MS. Pathology was found in brain primarily, with none in the spinal cord (Ludwin, 2006). Demyelination was observed in the absence of cellular infiltration (in contrast to EAE where cellular infiltration is a prominent feature). The ND4 was characterized by apoptosis of oligodendrocytes, showing early retraction of the inner tongue process (Ludwin, 2006). In fact, some have suggested that all MS begins in this way by apoptosis of oligodendrocytes (Barnett and Prineas, 2004). The importance of apoptosis was demonstrated in a recent publication, which reported that oligodendrocyte ablation produced extensive myelin loss with myelin fragments in the lymph nodes, but no CNS immunity, supporting a neurodegenerative hypothesis for MS (Locatelli et al., 2012). Patterns 2 and 3 account for 80% of the MS cases. Therefore, our combination of animal models represents a relevant system for the study of MS.
Tissue-selective overexpression of PAD enzymes and consequent local protein hypercitrullination has become hallmark of a growing and diverse group of pro-inflammatory diseases, which share challenging or unmet therapeutic needs (Vossenaar et al., 2003; Ishigami et al., 2005; Ehrlich et al., 2004; Bhattacharya et al., 2006; Jang et al., 2010; Jang et al., 2008). The fact that several animal models of such human disorders show comparable elevations of PAD activity and hypercitrullination (Nicholas et al., 2005; Hill et al., 2008; Kuhn et al., 2008) beg the question of commonalities in the underlying pathomechanisms of hypercitrullination disorders: what causes local PAD overexpression, and is hypercitrullination a marker or mediator of disease progression?
In an effort to understand the importance of PAD enzymes in the EAE model, a PAD2 knockout mouse was generated (Raijmakers et al., 2005). The authors claimed that citrullinated MBP was not present in these knockout mice, as detected with an anti-modified citrulline antibody. Because EAE was still induced in these mice, Raijmakers et al. concluded that citrullination was not required for disease induction in the PAD2 knockout. Using mice from their colony, we isolated MBP from PAD2 knockout mice, trypsin digested the MBP fraction and identified several citrulline-containing MBP peptides by mass spectrometry (Wood et al., 2008). We also detected citrullinated CNPase and MOG. Although PAD2 was absent from the knockout, PAD4 was found at the same levels as found in wild-type mice. Furthermore, we reported that PAD4 deiminated MBP, as well as PAD2 in vitro (Wood et al., 2008). In a recent report, Coudane et al. showed that the presence of citrullinated proteins in the PAD2 knockout mouse was independent of the presence or absence of PAD2 (Coudane et al., 2011). Therefore, citrullination was active in the PAD2 knockout, suggesting that ablation of citrullination requires knockout of all PAD activity.
Over the past few years, we have documented an increased citrullinated MBP in MS tissue and an increase in PAD enzyme activities. In order to place protein hypercitrullination and elevated PAD activities in the important role of the pathogenesis in MS, we sought to establish that increased PAD activity preceded disease, which we demonstrated in the ND4 model of MS (Moscarello et al., 2002).
Hypercitrullination in MS appears to work through several mechanisms involving biochemical change in the CNS and immune system. First, we have shown in our neurodegenerative mouse models that 2CA was able to attenuate clinical signs of demyelination accompanied by improved myelination and normalized PAD activity in CNS. It was also shown in our acute MOG-EAE model that 2CA improves survival rate, possibly by decreasing the amount of citrullinated MOG peptides. Because previous studies have reported that MOG can become citrullinated at Arg41 and Arg46 of peptide 35-55 (both TCR contact residues), which in turn leads to an immune response against the peptides and development of EAE (Carrillo-Vico et al., 2010), we propose that inhibition of PAD prevents citrullination of MOG peptide resulting in less severe autoimmune responses. The molecular mechanism of PAD upregulation in the ND4 mice remains uncertain, although the PAD2 promoter hypomethylation reported in MS (Mastronardi et al., 2007a) might be responsible, as indicated by studies in which decreased methylation was found at several CpG sites in the ND4 mouse (our unpublished data). Because the blood brain barrier is intact in this model, inflammatory infiltrates are absent and cannot contribute to increased PAD enzyme activity.
Our studies with 2CA argue cogently that hypercitrullination is a crucial effector element in the pathogenesis of progressive demyelinating disease affecting both immune and non-immune elements. 2CA has excellent therapeutic efficacy in all phases of these quite diverse model diseases and is able to prevent onset, suppress ongoing and allow considerable reversal of established demyelination, with no adverse toxicities observed at the dose and timeframes employed. Because the active sites of PAD2 and PAD4 are similar, 2CA can bind to both thereby inhibiting citrullination totally (see PAD2 knockout described above). Of the growing number of hypercitrullination syndromes, one targets the eye (Bhattacharya et al., 2006) and four the brain: Alzheimer's disease and MS (Ishigama et al., 2005), sporadic Creutzfeldt-Jakob disease (Jang et al., 2008) and scrapie (Wang et al., 2009). It is tempting to speculate that the clustering of these disorders in neuronal tissue might indicate that PAD overexpression and/or hypercitrullination is a perhaps more predominant natural response factor to tissue stress in these neuronal organs. This idea has been recently supported by spinal cord injury studies whereby inhibition of PAD enzymes by a general PAD inhibitor decreased apoptosis of neural stem cells and improved regeneration of the cord (Lange et al., 2011), suggesting an important role for PAD enzymes in spinal cord regeneration. Improved survival of neural stem cells following PAD inhibition can have a major impact on remyelination in MS.
Concluding remarks
MS, a heterogeneous disease of the CNS, arises from the combination of several pathogenic mechanisms. Two of these have been described earlier, i.e. bilayer instability and apoptosis of oligodendrocytes, to which we now add inhibition of T cell expansion. The first mechanism results in myelin degradation, the second in a failure to repair and the third to the inflammatory response. With the recent demonstration of the importance of PAD4 activity in the production of neutrophil extracellular trap (NET) formation by deiminating histone (Li et al., 2010) and the subsequent demonstration that NETs can directly prime T cells by reducing their activation threshold (Tillak et al., 2012), strong support is provided for an important role of citrullination in T cell activation. In the present manuscript, we demonstrate that the commonality between these diverse processes resides in protein hypercitrullination mediated by PAD enzymes. These processes are attenuated by PAD inhibition using the small molecule PAD active site inhibitor 2CA. PAD inhibition resulted in decreased hypercitrullination, reduced PAD activity, reduced CD3+ T cells, attenuation of disease and remyelination in four relevant animal models of MS, thus validating PAD as a therapeutic target in MS by small molecule intervention. Because the 2CA is a weak general PAD inhibitor, we are now developing a library of highly active small molecule PAD inhibitors (active in the nanomolar range) that are reversible and therefore of therapeutic value.
MATERIALS AND METHODS
Mice
SJL and C57BL/6 mice were purchased from Charles River (Montreal, QUE) and Jackson Laboratories (Bar Harbor, ME) respectively. ND4 (DM20) transgenic mice and homozygous PAD2 transgenic mice both on CD1 background developed in our institution have been described (Musse et al., 2008; Johnson et al., 1995; Mastronardi et al., 1996). All animals were housed in a closed colony in the animal facility at The Hospital for Sick Children (HSC, Toronto, ON) in a controlled environment using a 12-hour light and dark cycle. Experiments were performed using sex- and age-matched mice under approved protocols and in agreement with animal ethics guidelines.
Therapy protocol and clinical scoring
2-Chloroacetamidine hydrochloride, 96% (2CA) (Alfa Aesar, Ward Hill, MA) and Vitamin B12 (Sigma-Aldrich, St Louis, MO) were dissolved in PBS pH 7.4, filter sterilized and administered at 5 mg/kg (2CA) and 10 mg/kg (B12). Injections (i.p.) of 50 μl were performed every other day for the periods indicated. ND4 mice were scored three time a week by two independent observers, with non-transgenic littermates serving as controls (Johnson et al., 1995; Mastronardi et al., 2004). Briefly, animals were observed for clinical signs of demyelinating diseases including general body shaking, hind limb or tail tremor, head tremor, wobbly gait, limp tail, poor balance, weakness, unsteadiness, seizure and activity level. Animals were scored on a scale of 0-4, where 0 represented no signs and 4 represented severe signs. SJL and C57BL/6 mice of 6-8 weeks old were immunized subcutaneously with 200 μg of PLP139-155 (Alpha Diagnostics, San Antonio, TX) or 100 μg of MOG35-55 peptide (Alpha Diagnostics), respectively, emulsified in complete Freunds adjuvant (Sigma-Aldrich). Pertussis toxin (Sigma-Aldrich) 100 or 300 ng (C57BL/6) was administered on the day of immunization and 48 hours later. Mice were scored using a well-established scoring system for EAE signs (Tuohy et al., 1995; Mastronardi et al., 2004), i.e. 0, no signs; 1, limp tail; 2, inability of righting; 3, paralysis of either hind limb or forelimb; 4, full paralysis of both hind and forelimbs; and 5, moribund or death.
In vitro proliferation and cytokine secretion
Splenocytes were isolated 45 days after crEAE or 14 days after acute EAE induction, and restimulated with the appropriate peptide. Cells were cultured for 72 hours, with 1 μCi of [3H]thymidine added for the final 18 hours prior to harvesting and liquid scintillation counting. To quantify cytokine secretion, supernatants were collected after 72 hours of culture and levels of IL-17 (R&D Systems, Minneapolis, MN) and IFN-γ (BD Biosciences, San Jose, CA) were measured by ELISA, according to manufacturer's instructions.
Generation of mAb
A synthetic peptide 650FLGEVHC*GTNVR containing an acetamidine adduct on the active-site cysteine (C*) of the PAD2 enzyme was synthesized in our Biotechnology Center and conjugated to keyhole limpet hemocyanin (KLH). This was then used to generate monoclonal antibodies by standard procedures in our monoclonal antibody service. Two monoclonal antibodies (mAb4B11 and mAb4E12) detected only the adduct-tagged peptide, and the mAb4E12 clone was used for production.
Reverse transcriptase PCR
cDNA from mouse white matter or spinal cord was prepared with a reverse transcriptase kit (Invitrogen, Burlington, ON) and amplicons of primer pairs for GAPDH and PAD2 were separated on agarose gel, stained and analyzed using Image SXM software (http://www.ImageSXM.org.uk).
Immunoslot blotting and PAD enzyme assays
Both PAD2 and citrullinated proteins were quantified from white matter homogenates by immunoslot blots. Membranes were probed with either anti-panPAD antibody (Nishijyo et al., 1997; Rus'd et al., 1999; Takahara et al., 1989), or anti-peptidylcitrulline antibody (F95) (Nicholas et al., 2005) overnight at 4°C and developed with secondary antibodies for 1 hour at 20°C. Images were analyzed as above. PAD activity in aqueous white matter extracts from human and mouse whole brain was determined as previously described (Lamensa and Moscarello, 1993; Watanabe et al., 1988). In summary, 100 μl of each brain homogenate sample was used as enzyme source and incubated in 50 mM HEPES, 5 mM CaCl2, 2 mM dithiothreitol and 12.5 mM α-N-benzoyl-L-arginine ethyl ester (BAEE) at 52°C for 30 minutes. The reaction was stopped by adding 100 μl of 5.0 M HClO4. The reactants were centrifuged at 12,000 g at 4°C for 3 minutes. Then, 450 μl of this supernatant was taken to measure citrulline concentration using 0.5 ml of reagent A (0.5% diacetyl monoxime, 15% NaCl) followed by 1 ml of reagent B (0.1% antipyrine, 0.25% ferric ammonium sulfate, 25% H2SO4, 25% H3PO4). The mixture was boiled for 15 minutes and cooled on ice before the absorbance at 464 nm was measured. The PAD activity level was determined from a standard curve generated with various amounts of L-citrulline (0-20 μg). The PAD activity level was expressed in nanomoles citrulline/minute/ milligram protein.
Electron microscopy
Optic nerves from ND4 mice and non-transgenic littermates were removed at 6 months, immersion-fixed in Karnovsky's solution (Feirabend et al., 1994) as described (Moscarello et al., 2002). For immunogold electron microscopy, optic nerves from untreated and 2CA-treated ND4 mice were fixed in 4% paraformaldehyde and 0.1% glutaraldehyde in 0.1 M phosphate buffer pH 7.4 for 4-6 hours.
Mass spectrometry and determination of peptide sequencing of modified and unmodified PAD
Recombinant PAD2 (5 μg)was reacted with 2CA in the presence of Ca2+ in HEPES buffer pH 7.6 (BioShop Canada, Burlington, ON) at 52°C for one hour, after which it was dialyzed and lyophilized. 2CA-bound and unbound recombinant PAD2 enzyme was digested with 0.05 μg trypsin (Roche Diagnostics, Laval, QUE) in 25 mM ammonium bicarbonate pH 8.6. After overnight incubation at 37°C, 10 μl of sample in 0.1% trifluoroacetic acid was used for liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis on a QSTAR XL electrospray ionization quadrupole time-of-flight (ESI-QToF) mass spectrometer (Applied Biosystems/MDS Sciex, Concord, ON), coupled with an Agilent LC system. The acquired data set was converted into a peak list file using the Mascot script, a component of the Analyst QS 1.1 software (Applied Biosystems). A custom modification on cysteine, called acetamidination, was built into the modification database with composition C(2)H(4)N(2) having a delta mass of 56.0374 (monoisotopic). Protein N-terminal acetylation, methionine oxidation and acetamidination were set as variable modifications in the search. Peptide mass tolerance and MS/MS tolerance were set to 0.2 Da.
Statistics
Statistical significance between means was assessed by unpaired t-tests using Welch's correction where appropriate. Analysis of clinical scoring curves was performed using two-way ANOVA or life tables where appropriate. Statistical significance was two tailed and set at 5%, with error bars showing a single standard deviation.
Supplementary Material
ACKNOWLEDGEMENTS
The immunological input in the EAE studies and the cell proliferation studies by Hans-Michael Dosch and his staff are acknowledged, without whom these studies would not have been possible. In particular we mention Geoffrey Palster and Jason Yantha. We thank Anthony Nicholas for the F-95 antibody. Technical assistance was provided by Teresa Miani (Molecular Structure and Function, HSC), Aina Tilups (Division of Pathology, HSC) and Howard Rosenberg (Division of Pathology, HSC) for preparation of samples for electron microscopy and Sudha Arya (Monoclonal antibody Facility at HSC) for generation and purification of monoclonal antibodies 4E12. We also thank Herman Yeger (HSC) and Harry Schachter (HSC) for valuable suggestions; Rashed Nagra, Wallace Tourtellotte and their team for providing normal and MS samples from the Human Brain and Spinal Fluid Resource Center (VA West Los Angeles Healthcare Center, Los Angeles, CA, jointly sponsored by NINDS/NIMH, National Multiple Sclerosis Society USA and the Department of Veterans Affairs).
FUNDING: This research was funded by grants from the MS Society of Canada (to M.A.M. and F.G.M.).
Footnotes
AUTHOR CONTRIBUTIONS: H.L., an MSc graduate student, performed most of the enzyme assays and peptide citrulline determination. She injected mice with 2CA, followed and scored the mice, both the ND4 transgenics and the acute EAE. F.G.M. was conceptually involved from the beginning of the study and maintained an involvement in injecting and scoring of ND4 and EAE mice. S.W. and H.T., both from the Department of Immunology, provided valuable assistance with the EAE experiments. Z.L. developed the PAD2 transgenic mice. C.A. was responsible for the microscopy, in particular the electron microscopy of optic nerves and the immunogold studies. L.Z. carried out the mass spectrometry (Fig. 2). R.R. provided us with generous amounts of recombinant PAD2 and PAD4 for the in vitro studies. D.D.W. carried out inhibition assays, identified the PAD-2CA adduct and generated the monoclonal antibody.
COMPETING INTERESTS: The authors declare no competing financial interests
SUPPLEMENTARY MATERIAL: Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.010520/-/DC1
References
- Barnett M. H., Prineas J. W. (2004). Relapsing and remitting multiple sclerosis: pathology of the newly forming lesion. Ann. Neurol. 55, 458-468 [DOI] [PubMed] [Google Scholar]
- Bhattacharya S. K., Crabb J. S., Bonilha V. L., Gu X., Takahara H., Crabb J. W. (2006). Proteomics implicates peptidyl arginine deiminase 2 and optic nerve citrullination in glaucoma pathogenesis. Invest. Ophthalmol. Vis. Sci. 47, 2508-2514 [DOI] [PubMed] [Google Scholar]
- Boggs J. M., Stamp D., Moscarello M. A. (1982). Effect of pH and fatty acid chain length on the interaction of myelin basic protein with phosphatidylglycerol. Biochemistry 21, 1208-1214 [DOI] [PubMed] [Google Scholar]
- Brady G. W., Fein D. B., Wood D. D., Moscarello M. A. (1981a). The interaction of basic proteins from normal and multiple sclerosis myelin with phosphatidylglycerol vesicles. FEBS Lett. 125, 159-160 [DOI] [PubMed] [Google Scholar]
- Brady G. W., Murthy N. S., Fein D. B., Wood D. D., Moscarello M. A. (1981b). The effect of basic myelin protein on multilayer membrane formation. Biophys. J. 34,345-350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo-Vico A., Leech M. D., Anderton S. M. (2010). Contribution of myelin autoantigen citrullination to T cell autoaggression in the central nervous system. J.Immunol. 184, 2839-2846 [DOI] [PubMed] [Google Scholar]
- Comi G., Martino G. (2006). MS treatment: new perspectives. Clin. Neurol. Neurosurg. 108, 339-345 [DOI] [PubMed] [Google Scholar]
- Coudane F., Mechin M.C., Hucheng A., Henry J., Nachat R., Ishigami A., Adoue V., Sebbage M., Serre G., Simon M. (2011). Deimination and expression of peptidylarginine deiminases during cutaneous wound healing in mice. Eur. J. Dermatol. 21, 376-384 [DOI] [PubMed] [Google Scholar]
- Deber C. M., Hughes D. W., Fraser P. E., Pawagi A. B., Moscarello M. A. (1986). Binding of human normal and multiple sclerosis-derived myelin basic protein to phospholipid vesicles: effects on membrane head group and bilayer regions. Arch. Biochem. Biophys. 245, 455-463 [DOI] [PubMed] [Google Scholar]
- Ehrlich A., Booher S., Becerra Y., Borris D. L., Figg W. D., Turner M. L., Blauvelt A. (2004). Micellar paclitaxel improves severe psoriasis in a prospective phase II pilot study. J. Am. Acad. Dermatol. 50, 533-540 [DOI] [PubMed] [Google Scholar]
- Epand R. M., Wheeler G. E., Moscarello M. A. (1974). Circular dichroism and proton magnetic resonance studies of random chain poly-L-lysine. Biopolymers 13, 359-369 [DOI] [PubMed] [Google Scholar]
- Feirabend H. K., Kok P., Choufoer H., Ploeger S. (1994). Preservation of myelinated fibers for electron microscopy: a qualitative comparison of aldehyde fixation, microwave stabilisation and other procedures all completed by osmication. J. Neurosci. Methods 55, 137-153 [DOI] [PubMed] [Google Scholar]
- Hemmer B., Stüve O., Kieseier B., Schellekens H., Hartung H. P. (2005). Immune response to immunotherapy: the role of neutralising antibodies to interferon beta in the treatment of multiple sclerosis. Lancet Neurol. 4, 403-412 [DOI] [PubMed] [Google Scholar]
- Hill J. A., Bell D. A., Brintnell W., Yue D., Wehrli B., Jevnikar A. M., Lee D. M., Hueber W., Robinson W. H., Cairns E. (2008). Arthritis induced by posttranslationally modified (citrullinated) fibrinogen in DR4-IE transgenic mice. J. Exp. Med. 205, 967-979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iglesias A., Bauer J., Litzenburger T., Schubart A., Linington C. (2001). T- and B-cell responses to myelin oligodendrocyte glycoprotein in experimental autoimmune encephalomyelitis and multiple sclerosis. Glia 36, 220-234 [DOI] [PubMed] [Google Scholar]
- Ishigami A., Ohsawa T., Hiratsuka M., Taguchi H., Kobayashi S., Saito Y., Murayama S., Asaga H., Toda T., Kimura N., et al. (2005). Abnormal accumulation of citrullinated proteins catalyzed by peptidylarginine deiminase in hippocampal extracts from patients with Alzheimer's disease. J. Neurosci. Res. 80, 120-128 [DOI] [PubMed] [Google Scholar]
- Jang B., Kim E., Choi J. K., Jin J. K., Kim J. I., Ishigami A., Maruyama N., Carp R. I., Kim Y. S., Choi E. K. (2008). Accumulation of citrullinated proteins by up-regulated peptidylarginine deiminase 2 in brains of scrapie-infected mice: a possible role in pathogenesis. Am. J. Pathol. 173, 1129-1142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang B., Jin J. K., Jeon Y. C., Cho H. J., Ishigami A., Choi K. C., Carp R. I., Maruyama N., Kim Y. S., Choi E. K. (2010). Involvement of peptidylarginine deiminase-mediated post-translational citrullination in pathogenesis of sporadic Creutzfeldt-Jakob disease. Acta Neuropathol. 119, 199-210 [DOI] [PubMed] [Google Scholar]
- Johnson R. S., Roder J. C., Riordan J. R. (1995). Over-expression of the DM-20 myelin proteolipid causes central nervous system demyelination in transgenic mice. J. Neurochem. 64, 967-976 [DOI] [PubMed] [Google Scholar]
- Kuhn K. A., Cozine C. L., Tomooka B., Robinson W. H., Holers V. M. (2008). Complement receptor CR2/CR1 deficiency protects mice from collagen-induced arthritis and associates with reduced autoantibodies to type II collagen and citrullinated antigens. Mol. Immunol. 45, 2808-2819 [DOI] [PubMed] [Google Scholar]
- Lamensa J. W., Moscarello M. A. (1993). Deimination of human myelin basic protein by a peptidylarginine deiminase from bovine brain. J. Neurochem. 61, 987-996 [DOI] [PubMed] [Google Scholar]
- Lange S., Gogel S., Leung K. Y., Vernay B., Vocholas A. P., Sausey C. P., Thompson P. R., Greene N. D. D., Feretti P. (2011). Protein deiminases: new players in the developmentally regulated loss of neural regenerative ability. Dev. Biol. 355, 205-214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P., Li M., Lindberg M. R., Kennett M. J., Xiong N., Wang Y. (2010). PAD4 is essential for antibacterial innate immunity mediated by neutrophil extracellular traps. J. Exp. Med. 207, 1853-1862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G. Y., Liao Y. F., Chang W. H., Liu C. C., Hsieh M. C., Hsu P. C., Tsay G. J., Hung H. C. (2006). Overexpression of peptidylarginine deiminase IV features in apoptosis of hematopoietic cells. Apoptosis 11, 183-196 [DOI] [PubMed] [Google Scholar]
- Locatelli G., Wortage S., Buch T., Ingolo B., Frommer F., Sobottka B., Krugger M., Karram K., Buhlmann C., Bechmann I., et al. (2012). Primary oligodendrocyte death does not elicit anti CNS immunity. Nat. Neurosci. 15, 543-550 [DOI] [PubMed] [Google Scholar]
- Lucchinetti C., Brück W., Parisi J., Scheithauer B., Rodriguez M., Lassmann H. (2000). Heterogeneity of multiple sclerosis lesions: implications for the pathogenesis of demyelination. Ann. Neurol. 47, 707-717 [DOI] [PubMed] [Google Scholar]
- Ludwin S. K. (2006). The pathogenesis of multiple sclerosis: relating human pathology to experimental studies. J. Neuropathol. Exp. Neurol. 65, 305-318 [DOI] [PubMed] [Google Scholar]
- Mastronardi F. G., Ackerley C. A., Arsenault L., Roots B. I., Moscarello M. A.(1993). Demyelination in a transgenic mouse: a model for multiple sclerosis. J. Neurosci. Res. 36, 315-324 [DOI] [PubMed] [Google Scholar]
- Mastronardi F. G., Ackerley C. A., Roots B. I., Moscarello M. A. (1996). Loss of myelin basic protein cationicity in DM20 transgenic mice is dosage dependent. J. Neurosci. Res. 44, 301-307 [DOI] [PubMed] [Google Scholar]
- Mastronardi F. G., Min W., Wang H., Winer S., Dosch M., Boggs J. M., Moscarello M. A. (2004). Attenuation of experimental autoimmune encephalomyelitis and nonimmune demyelination by IFN-beta plus vitamin B12: treatment to modify notch-1/sonic hedgehog balance. J. Immunol. 172, 6418-6426 [DOI] [PubMed] [Google Scholar]
- Mastronardi F. G., Wood D. D., Mei J., Raijmakers R., Tseveleki V., Dosch H. M., Probert L., Casaccia-Bonnefil P., Moscarello M. A. (2006). Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: a role for tumor necrosis factor-induced peptidylarginine deiminase 4 translocation. J. Neurosci. 26, 11387-11396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastronardi F. G., Noor A., Wood D. D., Paton T., Moscarello M. A. (2007a). Peptidyl argininedeiminase 2 CpG island in multiple sclerosis white matter is hypomethylated. J. Neurosci. Res. 85, 2006-2016 [DOI] [PubMed] [Google Scholar]
- Mastronardi F. G., Tsui H., Winer S., Wood D. D., Selvanantham T., Galligan C., Fish E. N., Dosch H. M., Moscarello M. A. (2007b). Synergy between paclitaxel plus an exogenous methyl donor in the suppression of murine demyelinating diseases. Mult. Scler. 13, 596-609 [DOI] [PubMed] [Google Scholar]
- Moscarello M. A., Wood D. D., Ackerley C., Boulias C. (1994). Myelin in multiple sclerosis is developmentally immature. J. Clin. Invest. 94, 146-154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moscarello M. A., Pritzker L., Mastronardi F. G., Wood D. D. (2002). Peptidylarginine deiminase: a candidate factor in demyelinating disease. J. Neurochem. 81, 335-343 [DOI] [PubMed] [Google Scholar]
- Musse A. A., Li Z., Ackerley C. A., Bienzle D., Lei H., Poma R., Harauz G., Moscarello M. A., Mastronardi F. G. (2008). Peptidylarginine deiminase 2 (PAD2) overexpression in transgenic mice leads to myelin loss in the central nervous system. Dis. Model. Mech. 1, 229-240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholas A. P., Sambandam T., Echols J. D., Barnum S. R. (2005). Expression of citrullinated proteins in murine experimental autoimmune encephalomyelitis. J. Comp. Neurol. 486, 254-266 [DOI] [PubMed] [Google Scholar]
- Nishijyo T., Kawada A., Kanno T., Shiraiwa M., Takahara H. (1997). Isolation and molecular cloning of epidermal- and hair follicle-specific peptidylarginine deiminase (type III) from rat. J. Biochem. 121, 868-875 [DOI] [PubMed] [Google Scholar]
- Raijmakers R., Vogelzangs J., Croxford J. L., Wesseling P., van Venrooij W. J., Pruijn G. J. (2005). Citrullination of central nervous system proteins during the development of experimental autoimmune encephalomyelitis. J. Comp. Neurol. 486, 243-253 [DOI] [PubMed] [Google Scholar]
- Rus'd A. A., Ikejiri Y., Ono H., Yonekawa T., Shiraiwa M., Kawada A., Takahara H. (1999). Molecular cloning of cDNAs of mouse peptidylarginine deiminase type I, type III and type IV, and the expression pattern of type I in mouse. Eur. J. Biochem. 259, 660-669 [DOI] [PubMed] [Google Scholar]
- Steinman L., Zamvil S. S. (2006). How to successfully apply animal studies in experimental allergic encephalomyelitis to research on multiple sclerosis. Ann. Neurol. 60, 12-21 [DOI] [PubMed] [Google Scholar]
- Stone E. M., Schaller T. H., Bianchi H., Person M. D., Fast W. (2005). Inactivation of two diverse enzymes in the amidinotransferase superfamily by 2-chloroacetamidine: dimethylargininase and peptidylarginine deiminase. Biochemistry 44, 13744-13752 [DOI] [PubMed] [Google Scholar]
- Takahara H., Tsuchida M., Kusubata M., Akutsu K., Tagami S., Sugawara K. (1989). Peptidylarginine deiminase of the mouse. Distribution, properties, and immunocytochemical localization. J. Biol. Chem. 264, 13361-13368 [PubMed] [Google Scholar]
- Terakawa H., Takahara H., Sugawara K. (1991) Three types of mouse peptidylarginine deiminase: characterization and tissue distribution. J. Biochem. 110,661-666 [DOI] [PubMed] [Google Scholar]
- Tillak R., Breden P., Martin R., Sospendra M. (2012). T-Lymphocyte priming of neutrophil extracellular traps links innate and adaptive immune responses. J. Immunol. 188, 3150-3159 [DOI] [PubMed] [Google Scholar]
- Tuohy V. K., Thomas D. M., Haqqi T., Yu M., Johnson J. M. (1995). Determinant-regulated onset of experimental autoimmune encephalomyelitis: distinct epitopes of myelin proteolipid protein mediate either acute or delayed disease in SJL/J mice. Autoimmunity 21, 203-213 [DOI] [PubMed] [Google Scholar]
- Vossenaar E. R., Nijenhuis S., Helsen M. M., van der Heijden A., Senshu T., van den Berg W. B., van Venrooij W. J., Joosten L. A. (2003). Citrullination of synovial proteins in murine models of rheumatoid arthritis. Arthritis Rheum. 48,2489-2500 [DOI] [PubMed] [Google Scholar]
- Wang Y., Li M., Stadler S., Correll S., Li P., Wang D., Hayama R., Leonelli L., Han H., Grigoryev S. A., et al. (2009). Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 184, 205-213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K., Akiyama K., Hikichi K., Ohtsuka R., Okuyama A., Senshu T.(1988). Combined biochemical and immunochemical comparison of peptidylarginine deiminases present in various tissues. Biochim. Biophys. Acta 966, 375-383 [DOI] [PubMed] [Google Scholar]
- Wingerchuk D. M. (2008). Current evidence and therapeutic strategies for multiple sclerosis. Semin. Neurol. 28, 56-68 [DOI] [PubMed] [Google Scholar]
- Wood D. D., Bilbao J. M., O'Connors P., Moscarello M. A. (1996). Acute multiple sclerosis (Marburg type) is associated with developmentally immature myelin basic protein. Ann. Neurol. 40, 18-24 [DOI] [PubMed] [Google Scholar]
- Wood D. D., Ackerley C. A., Brand B., Zhang L., Raijmakers R., Mastronardi F. G., Moscarello M. A. (2008). Myelin localization of peptidylarginine deiminases 2 and 4: comparison of PAD2 and PAD4 activities. Lab. Invest. 88, 354-364 [DOI] [PubMed] [Google Scholar]
- Zhu B., Guleria I., Khosroshahi A., Chitnis T., Imitola J., Azuma M., Yagita H., Sayegh M. H., Khoury S. J. (2006). Differential role of programmed death-ligand 1 [corrected] and programmed death-ligand 2 [corrected] in regulating the susceptibility and chronic progression of experimental autoimmune encephalomyelitis. J. Immunol. 176, 3480-3489 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.