

SUMMARY
Childhood exposure to environmental particulates increases the risk of development of asthma. The underlying mechanisms might include oxidant injury to airway epithelial cells (AEC). We investigated the ability of ambient environmental particulates to contribute to sensitization via the airways, and thus to the pathogenesis of childhood asthma. To do so, we devised a novel model in which weanling BALB/c mice were exposed to both ambient particulate pollutants and ovalbumin for sensitization via the respiratory tract, followed by chronic inhalational challenge with a low mass concentration of the antigen. We also examined whether these particulates caused oxidant injury and activation of AEC in vitro. Furthermore, we assessed the potential benefit of minimizing oxidative stress to AEC through the period of sensitization and challenge by dietary intervention. We found that characteristic features of asthmatic inflammation developed only in animals that received particulates at the same time as respiratory sensitization, and were then chronically challenged with allergen. However, these animals did not develop airway hyper-responsiveness. Ambient particulates induced epithelial injury in vitro, with evidence of oxidative stress and production of both pro-inflammatory cytokines and Th2-promoting cytokines such as IL-33. Treatment of AEC with an antioxidant in vitro inhibited the pro-inflammatory cytokine response to these particulates. Ambient particulates also induced pro-inflammatory cytokine expression following administration to weanling mice. However, early-life dietary supplementation with antioxidants did not prevent the development of an asthmatic inflammatory response in animals that were exposed to particulates, sensitized and challenged. We conclude that injury to airway epithelium by ambient environmental particulates in early life is capable of promoting the development of an asthmatic inflammatory response in sensitized and antigen-challenged mice. These findings are likely to be relevant to the induction of childhood asthma.
INTRODUCTION
Childhood asthma is strongly associated with the development of atopy and T helper type 2 (Th2)-driven allergic airway inflammation (Sly et al., 2008; Holt and Sly, 2012). However, the mechanisms involved in the induction of a Th2-biased immunologic response are poorly understood. Moreover, the cause-and-effect relationship between inflammation, airway remodeling and airway hyper-responsiveness (AHR), as well as the temporal sequence of development of these changes, remain incompletely defined.
What is clear is that both genetic and environmental factors are crucial in the induction of asthma in children. Epidemiological studies have identified that important environmental triggers include early-life lower respiratory viral infection associated with wheezing (Sly et al., 2008; Holt et al., 2009) and exposure to environmental tobacco smoke (Gilmour et al., 2006; Wigle et al.,2008). More recently, a number of large population-based studies, including prospective cohort studies, have demonstrated a significantly increased risk of development of asthma in children exposed to traffic-related particulate pollutants (Gehring et al., 2010; McConnell et al., 2010; Pénard-Morand et al., 2010). Such studies also indicate that, in the past, the burden of exacerbations of asthma among children whose asthma was caused by living near roadways might have been significantly underestimated (Perez et al., 2012). Thus, the adverse respiratory effects of exposure to ambient pollutants, notably diesel exhaust particulates, are increasingly being recognized as a global public health problem of mounting significance (Laumbach and Kipen, 2012). However, the design of appropriate control strategies is hindered by the lack of knowledge about specific mechanisms (Balmes, 2011).
This lack of knowledge is related, at least in part, to the paucity of studies using appropriate experimental models. In particular, there have been few reports involving exposure of neonatal or weanling animals to these environmental triggers in the context of exposure to a sensitizing antigen via the respiratory tract and ongoing antigenic challenge, which is the context in which childhood asthma develops. We recently described a model of the induction phase of childhood asthma following early-life viral infection, involving a self-limiting infection with a murine pneumovirus followed by recurrent inhalational challenge with antigen (Siegle et al., 2010). Using this model, we demonstrated that development of airway inflammation typical of mild asthma, with a Th2-biased immunologic response and recruitment of eosinophils, was dependent on an interaction between early-life infection and exposure to allergen. By contrast, development of AHR was primarily related to the preceding viral infection, which led to persistent changes in the phenotype of epithelial cells in the conducting airways.
TRANSLATIONAL IMPACT.
Clinical issue
Childhood exposure to environmental particulates, particularly traffic-related particulate pollutants, is one of the environmental factors that has been associated with an increased risk of developing asthma. The underlying mechanisms remain poorly understood, in part because of the lack of appropriate animal experimental models, although they might include oxidant injury to airway epithelial cells.
Results
The authors devised a novel animal model to investigate the ability of ambient environmental particulates to contribute to sensitization via the airways, relevant to the pathogenesis of childhood asthma. They found that characteristic features of asthmatic inflammation developed only in weanling mice that received particulates at the same time as respiratory sensitization and were then chronically challenged with allergen. However, these mice did not develop airway hyper-responsiveness. Results show that ambient particulates induce airway epithelial injury in vitro, with evidence of oxidative stress. However, dietary supplementation with antioxidants during sensitization and allergen challenge did not prevent the development of an asthmatic inflammatory response.
Implications and future directions
The finding that injury to airway epithelium by ambient environmental particulates in early life promotes the development of an asthmatic inflammatory response sheds new light on the role of epithelial injury in the induction phase of childhood asthma. This animal model will be useful for further studies of mechanisms of disease, and of potential interventions to prevent the onset of asthma.
As has long been recognized, viral infections have the potential to disrupt the barrier function of the airway epithelium and thus to promote sensitization to inhaled antigens (Holt et al., 1988; Holtzman et al., 2002; Holgate, 2011). The injury to airway epithelial cells (AEC) could in part be a consequence of oxidative stress (Cho et al., 2009; Hosakote et al., 2009). Environmental particulates are also able to cause oxidant injury to AEC (Li et al., 2002; Bleck et al., 2008). Whether this contributes to sensitization via the airways, and thus to the pathogenesis of childhood asthma, is unknown. To address these questions, we devised a novel model of exposure of young mice to ambient particulate matter (PM) and ovalbumin (OVA) as the sensitizing antigen, followed by chronic inhalational challenge with a low mass concentration of antigen. We compared animals that were treated with particulate matter followed by OVA sensitization and challenge (PM/OVA) with mice that were exposed to particulate matter but were not sensitized or challenged (PM/Nil), mice that did not receive particulates but were sensitized and challenged (Sham/OVA), and naïve mice.
This model provides direct experimental evidence of the contribution of ambient particulates to the development of asthmatic inflammation. Together with in vitro studies, we have been able to dissect the potential role of different mechanisms in the induction of asthmatic inflammation and AHR. We have also assessed the potential benefit of minimizing oxidative stress to AEC at the time of exposure to particulates by dietary intervention in early life. Our results indicate that environmental particulates cause oxidative epithelial injury and that such injury concurrent with sensitization to allergen is necessary for development of an asthmatic inflammatory response, but this is not sufficient for development of AHR.
RESULTS
Airway inflammation in sensitized, chronically challenged mice
Only animals that received particulates at the same time as respiratory sensitization and were then chronically challenged, i.e. the PM/OVA group, developed features characteristic of asthmatic inflammation. As shown in Fig. 1A,B, there was significant recruitment of intra-epithelial eosinophils into the conducting airways, and increased numbers of eosinophils in the lungs, as assessed by an eosinophil peroxidase assay, in the PM/OVA group of animals. By contrast, no such increase was observed in PM/Nil animals exposed to particulate matter but not sensitized or challenged, or in Sham/OVA mice that did not receive particulates but were sensitized and challenged.
Fig. 1.

Airway inflammatory response. (A) Number of eosinophils in the airway epithelium, assessed morphologically. (B) Relative accumulation of eosinophils in lung tissue, assessed by peroxidase activity. (C) Percentage of neutrophils in BAL fluid. (D) Number of neutrophils in lung tissue, assessed by immunostaining. Data are mean ± s.e.m. (n=6-8 samples per group). Significant differences are shown as *P<0.05 and **P<0.01 compared with the naïve group and as #P<0.05 and ##P<0.01 compared with the Sham/OVA group.
Significant recruitment of neutrophils as a component of the inflammatory response was also a feature of only the PM/OVA group of animals, both with respect to neutrophils in bronchoalveolar lavage (BAL) fluid and in the lungs (Fig. 1C,D). However, there were no significant increases in the numbers of CD3+ T cells in the lungs in any of the experimental groups (not shown).
Cytokine response
Consistent with the mild inflammatory response, relatively few cytokines exhibited increased concentrations in BAL fluid from sensitized and challenged mice. However, increases were observed for the pro-inflammatory/chemoattractant cytokines CXCL1, CCL3 and granulocyte colony-stimulating factor (G-CSF). Whereas a significant increase in levels of CXCL1 was only observed in samples from the PM/OVA group, levels of CCL3 and G-CSF were elevated in both the Sham/OVA and PM/OVA mice (Fig. 2A-C).
Fig. 2.

Cytokine concentrations in BAL fluid. Concentrations of (A) CXCL1, (B) CCL3 and (C) G-CSF were measured in BAL fluid. Data are mean ± s.e.m. (n=6-8 samples per group). Significant differences are shown as ***P<0.001 compared with the naïve group and as #P<0.05 compared with the Sham/OVA group.
Airway responsiveness
Although sensitized and challenged mice developed minor changes in both transpulmonary resistance (RL) and dynamic compliance (Cdyn), responses were not significantly different from those of control animals. This was true irrespective of whether comparisons were for individual concentrations of inhaled methacholine or for the area under the curve of the dose response, i.e. there was no evidence of AHR in these animals. Representative results for RL and Cdyn at 25 mg/ml of methacholine are shown in Fig. 3A,B.
Fig. 3.

Airway responsiveness to β-methacholine. Airway responsiveness was assessed by measurement of (A) transpulmonary resistance (RL) and (B) dynamic compliance (Cdyn). Data are mean ± s.e.m. (n=6-8 samples per group). Differences are not statistically significant.
Response of airway epithelial cells to particulates
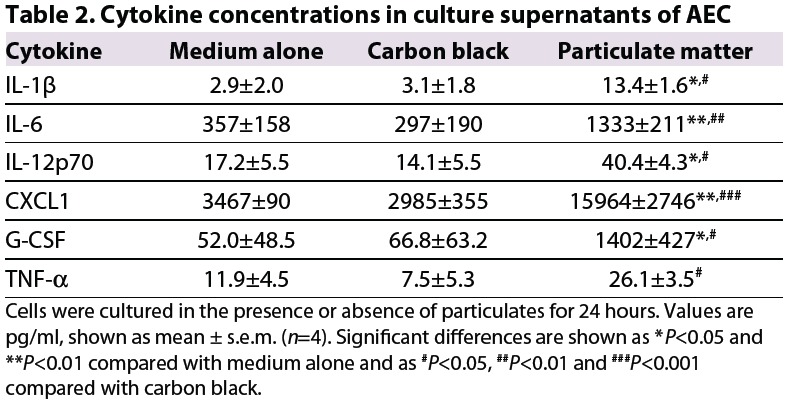
Exposure of AEC to environmental particulates, but not to carbon black, was associated with significant upregulation of expression of mRNA for the pro-inflammatory cytokines interleukin (IL)-6, CXCL1, G-CSF and tumor necrosis factor-α (TNF-α), as well as the Th2-promoting cytokine IL-33 (Table 1). In addition, there was a modest but not statistically significant increase in expression of mRNA for the pro-inflammatory cytokine IL-12p70 and for the Th2-promoting cytokines IL-25 and thymic stromal lymphopoietin, but no increase in expression for IL-1β at the time point assessed (not shown). However, in culture supernatants from cells exposed to environmental particulates, the concentrations of all of these pro-inflammatory cytokines were significantly increased (Table 2).
Table 1.
Expression of mRNA for pro-infammatory cytokines by AEC
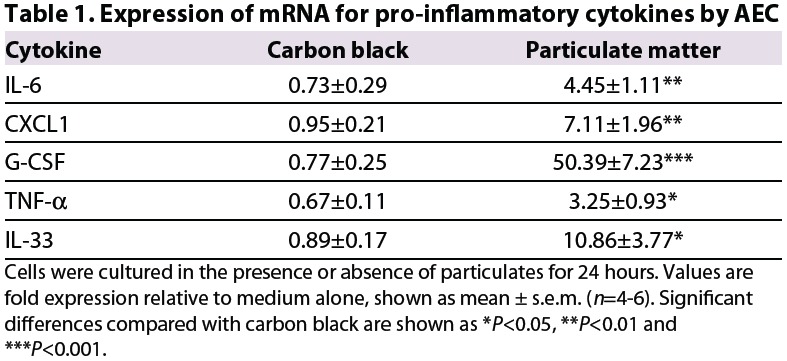
Table 2.
Cytokine concentrations in culture supernatants of AEC
Exposure to environmental particulates significantly enhanced the expression by AEC of mRNA for enzymes involved in the response to the oxidative stress, including heme oxygenase-1 (HO-1), glutathione-S-transferase-P1 (GSTP-1) and glutathione peroxidase 2 (GPx-2) (Fig. 4A-C).
Fig. 4.

Expression of mRNA for enzymes involved in the response to oxidative stress by AEC exposed to medium alone, carbon black or ambient environmental particulates. Expression of (A) HO-1, (B) GSTP-1 and (C) GPx-2. Data are mean ± s.e.m. (n=4 samples per group). Significant differences are shown as *P<0.05 compared with medium alone and as #P<0.05 and ##P<0.01 compared with carbon black.
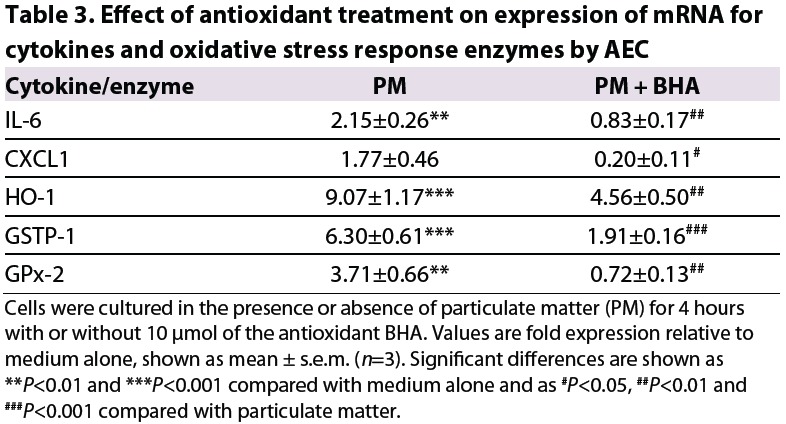
Treatment of AEC with the antioxidant butylated hydroxyanisole (BHA), at a concentration sufficient to prevent induction of the oxidative stress response, inhibited the increase in expression of pro-inflammatory cytokines by the cells in response to environmental particulates (Table 3).
Table 3.
Effect of antioxidant treatment on expression of mRNA for cytokines and oxidative stress response enzymes by AEC
Response of weanling mice to particulates
Administration of either environmental particulates or carbon black elicited a limited inflammatory response at 4 hours. In the lungs of animals that received ambient particulate matter, there was a 1.5-fold increase in the number of neutrophils, as assessed by immunostaining (Fig. 5A), although the magnitude of this change was not statistically significant (0.05<P<0.1). There was also significantly increased expression of mRNA for TNF-α (Fig. 5B). No such increase was observed in animals administered carbon black. The inflammatory response to environmental particulates was accompanied by a 1.7-fold increase in expression of HO-1, which was again not statistically significant when assessed in whole lung tissue (not shown). Levels of expression of other pro-inflammatory cytokines and oxidative stress enzymes were unaltered at 4 hours.
Fig. 5.

Response of 4-week-old mice to intranasal administration of carbon black or ambient particulate matter. (A) Number of neutrophils in lung tissue, assessed by immunostaining. (B) Expression of mRNA for TNF-α. Data are mean ± s.e.m. (n=6-8 samples per group). Significant differences are shown as *P<0.05 compared with the naïve group and as #P<0.05 compared with carbon black.
Effect of early-life treatment of sensitized, chronically challenged mice with antioxidants
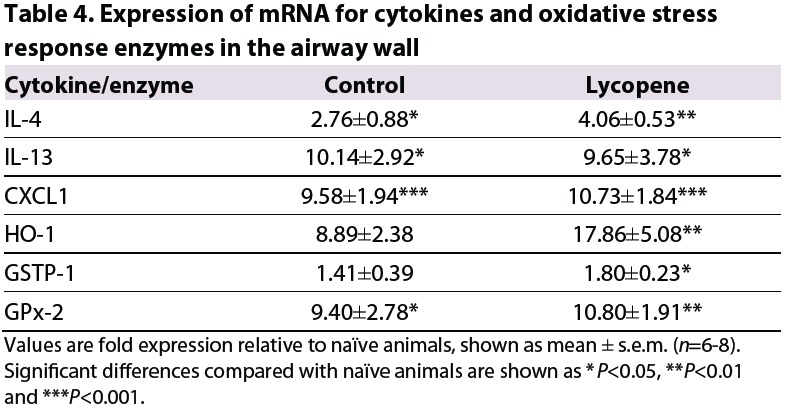
In a subsequent experiment, mice were sensitized with OVA following exposure to environmental particulates, either without or with concurrent treatment with dietary antioxidants. Treatment with antioxidants was continued throughout the period of inhalational challenge with OVA. However, this treatment had no protective effect on the development of airway inflammation, in terms of either neutrophil or eosinophil recruitment (Fig. 6A,B). Antioxidants also had no effect on the upregulated expression of mRNA for pro-inflammatory or Th2 cytokines, as assessed in airway wall tissues (Table 4). This was despite the fact that antioxidant treatment in early life was effective in significantly increasing the expression of mRNA for HO-1, GSTP-1 and GPx-2 compared with naïve animals, whereas such increases were not seen in untreated animals (Table 4).
Fig. 6.

Effect of early-life dietary supplementation with lycopene on inflammatory, cytokine and oxidative stress responses. (A) Percentage of neutrophils in BAL fluid. (B) Relative accumulation of eosinophils in lung tissue, as assessed by peroxidase activity. Significant differences compared with the naïve group are shown as *P<0.05 and **P<0.01.
Table 4.
Expression of mRNA for cytokines and oxidative stress response enzymes in the airway wall
DISCUSSION
In this study, we have demonstrated for the first time that exposure to ambient particulates in early life can contribute to the development of asthmatic inflammation in the setting of repeated low-level inhalational exposure to an allergen. This work builds upon our previous report of a model of the induction phase of childhood asthma, which was based on respiratory sensitization following a self-limiting pneumovirus infection in early life (Siegle et al., 2010). As has been emphasized by others, there is a need for studies of childhood asthma in models that evaluate the effects of concurrent exposure to environmental triggers and allergens, and in which timing and route of exposures are clinically relevant (Kulhankova et al., 2009). We believe that our new model achieves these aims with respect to evaluating the effects of environmental particulates. Furthermore, in the present study we have been able to investigate the potential role of airway epithelial injury in the pathogenesis of asthma associated with exposure to particulate matter at the time of sensitization.
A key goal of this investigation was to simulate the early events in the development of asthma in childhood. Thus, the magnitude of inflammatory changes in our model was modest, similar to that we previously observed in the early-life model of asthma following pneumovirus infection (Siegle et al., 2010). Also analogous to that study, but in contrast to our findings in models involving systemic sensitization, there was limited evidence of increased levels of expression of pro-inflammatory or Th2 cytokines. Nevertheless, we have clearly demonstrated that characteristic features of asthmatic inflammation developed only in animals that received particulates at the same time as respiratory sensitization, and were then chronically challenged with allergen. These features included the accumulation of eosinophils within the airway epithelial layer, a hallmark feature of chronic asthma in humans (Bousquet et al., 1990), and recruitment of eosinophils into the lungs, which as we have previously demonstrated is a component of the inflammation surrounding bronchioles that is triggered by moderate-level allergen challenge in sensitized animals (Siegle et al., 2006). In parallel, we observed a significant increase in the percentage of neutrophils in BAL fluid, which is typical of the response observed at the 4 hour time-point when samples were collected (Siegle et al., 2006; Siegle et al., 2010), as well as increased numbers of neutrophils in lung tissue.
Importantly, respiratory sensitization and allergen challenge alone did not induce these inflammatory changes. Indeed, neonatal exposure to ovalbumin via the respiratory tract is well recognized to induce tolerance to this antigen (Gerhold et al., 2003; Wang and McCusker, 2006). Exposure to environmental particulates was clearly able to break such tolerance. Because concurrent administration of relatively low levels of bacterial endotoxin (100 ng per mouse) has been shown to be able to facilitate sensitization to ovalbumin via the respiratory tract (Eisenbarth et al., 2002) and because airborne particulates might be associated with adsorbed endotoxin (Shoenfelt et al., 2009), we considered the possibility that the effect of environmental particulates was related to the presence of endotoxin. However, based on direct measurement of concentrations in the dispersed suspensions of particulates, only about 0.1 ng of endotoxin would have been delivered in the 40-μl dose that was administered intranasally, which would have been too low to affect sensitization (Eisenbarth et al., 2002).
In preliminary investigations, we had found that exposure of neonatal mice to ambient environmental particulates via the respiratory tract, followed by respiratory sensitization 1-3 weeks later, did not lead to development of an asthmatic inflammatory response (data not shown). Thus, it seemed likely that the acute injurious effect of these particulates at the time of sensitization was important in the subsequent development of an allergic inflammatory response. Therefore, we investigated the effect of ambient particulates on low-passage cultures of primary epithelial cells from the mouse trachea, which most closely resemble human conducting AEC (Harkema et al., 1991). Consistent with previously published investigations of the effects of diesel exhaust particulates on human AEC (Bleck et al., 2008; Bleck et al., 2010), we found that ambient particulates induced epithelial injury in vitro, with evidence of oxidative stress in the form of upregulation of HO-1, GSTP-1 and GPx-2, as well as production of pro-inflammatory cytokines and cytokines such as IL-33, which might be able to induce a Th2-biased host immunologic response. This response could be inhibited by treatment of AEC with an antioxidant in culture.
In further experiments, we found that ambient particulates triggered an inflammatory response and upregulated the expression of at least some relevant pro-inflammatory cytokines and enzymes in weanling mice as early as 4 hours after exposure. Because isolation of airways in these very small animals was difficult, gene expression was assessed in whole lung tissue, which would have diluted any demonstrable changes. Nevertheless, these findings are consistent with a key pathogenetic role for oxidant injury to AEC. It is likely that such injury induced a host response that, when followed by exposure to a sensitizing antigen via the respiratory tract, permitted the development of an allergic inflammatory response. In keeping with this, airway epithelial activation of the NF-κB pathway has recently been shown to break tolerance to inhaled ovalbumin (Ather et al., 2011).
The capacity of adsorbed chemicals associated with particulates to break airway tolerance in young animals has previously been demonstrated in a model in which mice were concurrently exposed to aerosolized ovalbumin and leachate from residual oil fly ash, a surrogate for ambient particulate pollutants (Hamada et al., 2000). In that study, however, the leachate was considered to have an adjuvant effect. Moreover, following the relatively short-term inhalation challenge employed, airway inflammation was dominated by neutrophils and there was little evidence of an allergic asthmatic pattern of response. Our model thus provides novel experimental evidence that, following exposure to ambient particulates to promote respiratory sensitization, chronic allergen challenge then provokes development of asthmatic inflammation.
Other models of neonatal sensitization and challenge, using clinically relevant allergens such as house dust mite or cockroach allergen, have been reported (Kulhankova et al., 2009; Saglani et al., 2009). These models also reproduced various elements of an allergic asthmatic inflammatory response and, in addition, the house dust mite model was associated with AHR (Saglani et al., 2009). However, in that model, the very high dosage of antigen administered intranasally caused severe inflammation around bronchioles and in lung tissue, quite unlike that in human asthma. Moreover, because both models involved sensitization using allergens with intrinsic protease activity, it would not have been possible to separately define the role of epithelial injury in the development of the allergic inflammatory response.
By contrast, the model described in this study uses an innocuous sensitizing protein and, unlike our previously reported early-life asthma model based on pneumovirus infection, is not associated with persistent epithelial injury. Thus, it allows dissection of the relationship between epithelial injury, development of allergic inflammation of the airways and AHR. Significantly, although typical eosinophilic asthmatic inflammation did develop in animals that were exposed to particulates, then sensitized and chronically challenged, AHR was absent. This finding in our model highlights the probable importance of persistent epithelial injury in the development of this characteristic manifestation of asthma (Holtzman et al., 2005).
As noted above, the ambient particulates used in these studies clearly were able to cause oxidative stress. The concept that oxidant injury is a key early event in the pathogenesis of asthma is supported by evidence that functional polymorphisms in oxidant defence genes increase the risk of developing asthma in childhood (Salam et al., 2007; Castro-Giner et al., 2009). Thus, there is some support for the use of dietary interventions with potential antioxidant activity for the primary prevention of asthma, and a recent systematic review and meta-analysis concluded that further experimental studies of this approach were warranted (Nurmatov et al., 2011). Of available dietary antioxidants, lycopene from tomatoes is of particular interest because it can directly inhibit production of inflammatory cytokines by injured AEC (Saedisomeolia et al., 2009) as well as allergic pulmonary inflammation in an animal model (Lee et al., 2008; Hazlewood et al., 2011). Therefore, we attempted to prevent the development of an asthmatic inflammatory response by administration of a dietary supplement rich in lycopene to juvenile mice, prior to and at the time of exposure to environmental particulates. However, this was not successful, even though the host response to oxidant injury appeared to have been enhanced, with clear evidence of an upregulation of antioxidant enzymes that was comparable to that previously demonstrated using this dietary supplement (Sahin et al., 2010).
In conclusion, we have provided direct experimental evidence of the contribution of ambient particulates to the development of asthmatic inflammation. We have shown that these particulates cause oxidative injury to AEC and induce the release of pro-inflammatory and pro-Th2 cytokines by these cells. Such injury, concurrent with allergic sensitization, appears to be essential for development of an asthmatic inflammatory response, but this is not sufficient for development of AHR. These findings shed new light on the role of epithelial injury by environmental particulates in the induction phase of childhood asthma. This novel model will be useful for further studies of mechanisms of disease and of potential interventions to prevent the onset of asthma.
MATERIALS AND METHODS
Animals
Specific pathogen-free female BALB/c mice (either timed pregnant or aged about 8 weeks) were obtained from the Biological Resources Centre, University of New South Wales. Animals were held in individually ventilated cages, exposed to a 12-hour light/dark cycle and provided autoclaved food and water ad libitum. All experimental procedures complied with the requirements of the Animal Care and Ethics Committee of the University of New South Wales (reference number 09/94A).
Particulates
Ambient particulate matter (PM) (median aerodynamic diameter less than 10 μm) was recovered from PM10 sampling filters obtained from the Air Quality Monitoring Unit, Office of Environment and Heritage, New South Wales Government, Sydney, Australia. Dispersed suspensions of particles were prepared by ultrasonication in 0.9% saline for 30 minutes in a water bath. To quantify the mass of released particles, filters were dried for a minimum of 48 hours, weighed on a precision balance before ultrasonication, then dried for a further 48 hours before re-weighing. Particulates were resuspended in saline at 1 mg/ml.
Chemicals associated with particulates were characterized by the Bioanalytical Mass Spectrometry Facility and the Solid State and Elemental Analysis Unit, UNSW. Alkanes (C10-C30, with major peaks in the range C12-C14) and various aromatic hydrocarbons were the dominant classes of organic compounds detected. Among inorganic chemicals, sulfates and nitrates were prominent. The most abundant trace metals associated with the particulates included, in descending order, Si, Mg, Al, Ba, Fe, Zn, B, Ti, Cu, Mn, V and Sr. These findings are similar to published data on the chemical characteristics of ambient particulates (Seagrave et al., 2006). Bacterial endotoxin in the dispersed suspensions was assessed as <3 ng/ml using a Limulus assay kit (ChromoLAL, Associates of Cape Cod, E Falmouth, MA).
Sensitization and challenge of mice
For intranasal sensitization of mice, 100 μg of chicken egg ovalbumin (OVA) (Grade V, ≥98% pure, Sigma Australia) were delivered in 40 μl of saline on days 28 and 29 of life. Animals administered ambient particulates received 20 μg/20 μl intranasally on days 14 and 15 of life, then 40 μg/40 μl on days 28 and 29 of life. To ensure that any observed effects of treatment with particulate matter in OVA-sensitized animals (PM/OVA) were not merely a consequence of adsorption of OVA to the surface of particulates, leading to an adjuvant effect, an interval of 6 hours was maintained between delivery of particulates and OVA on days 28/29. Commencing at day 49 of life, mice were exposed to a controlled low mass concentration of approximately 3 mg/m3 of aerosolized OVA for 30 minutes/day on 3 days/week for 4 weeks, followed by a single moderate challenge of approximately 30 mg/m3 for 30 minutes to induce changes in acute allergic inflammation (Siegle et al., 2006) (Fig. 7).
Fig. 7.
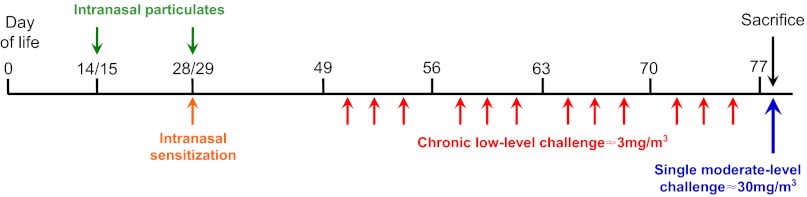
Timeline of exposure to particulates, respiratory sensitization and inhalational challenges in the early-life model. Animals were administered ambient particulates on days 14 and 15 of life, then again on days 28 and 29 of life, and were sensitized with intranasal OVA at the latter time point. Commencing at day 49 of life, mice received repeated low-level challenges with aerosolized OVA for 4 weeks, followed by a single moderate-level challenge.
Exposures were carried out in a whole-body inhalation exposure system (Unifab Corporation, Kalamazoo, MI), in which mice were held in flow-through wire cage racks and filtered air was drawn through the 0.5 m3 chamber at a flow rate of 250 l/minute. A solution of 2.5% OVA in in 0.9% saline was aerosolized by delivery of compressed air to a sidestream jet nebulizer (Niche Medical, Sydney) and injected into the airstream prior to entering the chamber. Particle concentration within the breathing zone of the mice was continuously and cumulatively monitored using a light scatter device (DustTrak 8520; TSI, St Paul, MN) and maintained by controlling the airflow into the nebulizer. Experimental groups typically comprised 8 mice. Control groups included mice that were exposed to particulate matter but were not sensitized or challenged (PM/Nil), mice that did not receive particulates but were sensitized and challenged (Sham/OVA), and naïve mice.
Assessment of inflammatory response
At 4 hours after the final airway challenge, mice were killed by exsanguination following an overdose of sodium pentobarbital. This time point was selected on the basis of our earlier studies using this approach to allergen challenge (Siegle et al., 2006; Herbert et al., 2010). Recruitment of inflammatory cells into the airway lumen was assessed by performing total and differential cell counts following BAL with 3×1 ml of PBS. The inflammatory response in tissues of the conducting airways was quantified in longitudinally oriented 4-μm sections of formalin-fixed, paraffin-embedded tracheas stained with hematoxylin and eosin. Pulmonary tissue accumulation of eosinophils was quantified using a colorimetric assay for eosinophil peroxidase, adapted from previously described methods (Schneider and Issekutz, 1996). Accumulation of neutrophils and of T cells in the lungs was quantified by immunostaining in frozen sections, using rat anti-Gr-1 (RB6-8C5, BD Bioscience, Sydney, Australia) and a rabbit antibody to CD3 (Sigma), as previously described (Ito et al., 2008; Siegle et al., 2010). The validity and reliability of the morphometric techniques we employed have been established in previous reports (Temelkovski et al., 1998; Kumar et al., 2002).
mRNA expression in blunt-dissected airways
Proximal airway tissue was isolated by blunt dissection using two pairs of forceps to separate lung parenchyma from the larger airways and leaving several generations of airway attached to the trachea (Herbert et al., 2008). Tissue was frozen in liquid nitrogen until RNA extraction was performed using TriReagent (Sigma Australia). Extracted RNA samples were treated with DNase (Turbo DNase, Ambion, Scoresby, Australia) and reverse transcribed into cDNA using Superscript III (Invitrogen). Quantitative real time PCR was used to assess expression of cytokines, with detection of amplified products using SYBR Green (BioLine, Tauton, MA). Primers were either custom-designed in house or were obtained from Qiagen Australia. Reactions were performed using an ABI Prism 7700 Sequence Detector (Applied Biosystems, Melbourne, Australia) and expression was normalized to that of the housekeeping gene HPRT.
Cytokine assays
The concentration of various pro-inflammatory cytokines were measured using a bead-based multiplex immunoassay (Bioplex, Mouse 23-Plex panel, Biorad Laboratories, Hercules, CA) according to the manufacturer's instructions.
Airway responsiveness
Airway responsiveness to inhaled β-methacholine was determined in mice 4 hours after the final aerosol challenge. Transpulmonary resistance (RL) and dynamic compliance (Cdyn) were assessed as described (Siegle et al., 2006). Animals were anesthetized with ketamine-xylazine, tracheostomized and mechanically ventilated within a plethysmograph chamber. Volume changes due to thoracic expansion and alterations in tracheal pressure were measured in response to challenge with saline, followed by increasing concentrations of β-methacholine (6.25, 12.5, 25 and 50 mg/ml). Peak values were taken as the maximum response to the concentration of methacholine being tested, and were expressed as the percentage change relative to the saline control.
Effects of particulates in weanling mice
In experiments to confirm the in vivo effects of environmental particulates, mice were administered a single intranasal dose of either particulate matter or carbon black at day 28 of life and sacrificed 4 hours later. Because of the fragility of airway tissues in these weanling mice, RNA extraction was then performed from whole lung tissue.
Antioxidant treatment
The dietary supplement Lyc-O-Mato 6% (200 mg/kg; LycoRed, Beer-Sheva, Israel) (Hazlewood et al,. 2011) was administered by once-weekly gavage commencing on day 18 of life, i.e. two doses before intranasal sensitization and then throughout the period of chronic inhalational challenge. According to the manufacturer's data sheet, the 4 mg dose that was administered contained not less than 0.24 mg of lycopene, 0.008 mg of β-carotene and 0.06 mg of tocopherols. This preparation has previously been demonstrated to exhibit antioxidant activity in mice treated in this manner (Hazlewood et al,. 2011).
Airway epithelial cell culture
Primary cultures of AEC were established under serum-free conditions, as previously described (Kumar et al,. 1997). Tracheas from 8-week-old BALB/c mice were diced into fragments (approximately 0.5×0.5 mm) and dispersed evenly across the base of a 25-cm2 culture flask. Flasks were pre-coated with about 5 mg/cm2 of rat-tail collagen type 1 to promote cell attachment. Complete growth medium consisted of MCDB201 supplemented with BSA, lipoproteins, epidermal growth factor, vitamin E acetate, prostaglandin E1, glutathione, dithiothreitol (all from Sigma Australia), transferrin (Roche Applied Sciences, Sydney, Australia) and antibiotics and antifungals. Medium was routinely changed after 10-12 days. When cells reached 80% confluence, they were detached by trypsinization and reseeded (5×105-7.5×105 cells per 25-cm2 flask). Cells were used at passage 5. AEC were exposed to 2.5 μg/ml of particulate matter, 2.5 μg/ml of carbon black used as a control particulate (Langridge Colours, Yarraville, Victoria, Australia) or media alone. After 24 hours, supernatants were collected and cells were resuspended in 1 ml of TriReagent for subsequent RNA extraction, cDNA synthesis and assessment of gene expression by real-time PCR. In experiments to assess the role of oxidative stress in the response of AEC, cells were exposed to particulate matter in the presence of 10 μmol of the antioxidant BHA for 4 hours (Malhotra et al,. 2008), then collected for assessment of gene expression.
Statistical analysis
Data are presented as arithmetic means ± s.e.m. Results were analyzed by a one-way ANOVA or Kruskal-Wallis test, followed by a Holm-Sidak or Dunn's multiple comparison test as appropriate. The software package GraphPad Prism 6.00 (GraphPad Software, San Diego, CA) was used for data analysis and preparation of graphs.
ACKNOWLEDGEMENTS
We thank John Kirkwood, Barbara Veluppillai and Gunaratnam Gunashanhar of the Office of Environment and Heritage, New South Wales Government, Sydney, for providing the air sampling filters.
FUNDING
Supported by grants from the National Health and Medical Research Council of Australia ( 630501 and 1004267) and the Rebecca Cooper Medical Research Foundation.
Footnotes
COMPETING INTERESTS: The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS: R.K.K., J.S.S., C.H. and P.S.F. conceived and designed the experiments. C.H., J.S.S., A.M.S., S.N., L.G. and N.G.H. performed the experiments and analyzed the data. C.H., J.S.S. and R.K.K. wrote the paper.
References
- Ather J. L., Hodgkins S. R., Janssen-Heininger Y. M., Poynter M. E. (2011). Airway epithelial NF-κB activation promotes allergic sensitization to an innocuous inhaled antigen. Am. J. Respir. Cell Mol. Biol. 44, 631-638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balmes J. R. (2011). How does diesel exhaust impact asthma? Thorax 66, 4-6 [DOI] [PubMed] [Google Scholar]
- Bleck B., Tse D. B., Curotto de Lafaille M. A., Zhang F., Reibman J. (2008). Diesel exhaust particle-exposed human bronchial epithelial cells induce dendritic cell maturation and polarization via thymic stromal lymphopoietin. J. Clin. Immunol. 28, 147-156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleck B., Tse D. B., Gordon T., Ahsan M. R., Reibman J. (2010). Diesel exhaust particle-treated human bronchial epithelial cells upregulate Jagged-1 and OX40 ligand in myeloid dendritic cells via thymic stromal lymphopoietin. J. Immunol. 185, 6636-6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bousquet J., Chanez P., Lacoste J. Y., Barnéon G., Ghavanian N., Enander I., Venge P., Ahlstedt S., Simony-Lafontaine J., Godard P., et al. (1990). Eosinophilic inflammation in asthma. N. Engl. J. Med. 323, 1033-1039 [DOI] [PubMed] [Google Scholar]
- Castro-Giner F., Künzli N., Jacquemin B., Forsberg B., de Cid R., Sunyer J., Jarvis D., Briggs D., Vienneau D., Norback D., et al. (2009). Traffic-related air pollution, oxidative stress genes, and asthma (ECHRS). Environ. Health Perspect. 117, 1919-1924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H. Y., Imani F., Miller-DeGraff L., Walters D., Melendi G. A., Yamamoto M., Polack F. P., Kleeberger S. R. (2009). Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 179, 138-150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenbarth S. C., Piggott D. A., Huleatt J. W., Visintin I., Herrick C. A., Bottomly K. (2002). Lipopolysaccharide-enhanced, toll-like receptor 4-dependent T helper cell type 2 responses to inhaled antigen. J. Exp. Med. 196, 1645-1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring U., Wijga A. H., Brauer M., Fischer P., de Jongste J. C., Kerkhof M., Oldenwening M., Smit H. A., Brunekreef B. (2010). Traffic-related air pollution and the development of asthma and allergies during the first 8 years of life. Am. J. Respir. Crit. Care Med. 181, 596-603 [DOI] [PubMed] [Google Scholar]
- Gerhold K., Bluemchen K., Franke A., Stock P., Hamelmann E. (2003). Exposure to endotoxin and allergen in early life and its effect on allergen sensitization in mice. J. Allergy Clin. Immunol. 112, 389-396 [DOI] [PubMed] [Google Scholar]
- Gilmour M. I., Jaakkola M. S., London S. J., Nel A. E., Rogers C. A. (2006). How exposure to environmental tobacco smoke, outdoor air pollutants, and increased pollen burdens influences the incidence of asthma. Environ. Health Perspect. 114, 627-633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada K., Goldsmith C. A., Goldman A., Kobzik L. (2000). Resistance of very young mice to inhaled allergen sensitization is overcome by coexposure to an air-pollutant aerosol. Am. J. Respir. Crit. Care Med. 161, 1285-1293 [DOI] [PubMed] [Google Scholar]
- Harkema J. R., Mariassay A., St George J., Hyde D. M., Plopper C. G. (1991). Epithelial cells of the conducting airways: a species comparison. In The Airway Epithelium: Physiology, Pathophysiology and Pharmacology (ed. Farmer S. G., Hay D. W. P.), pp. 3-39 New York, NY: Marcel Dekker [Google Scholar]
- Hazlewood L. C., Wood L. G., Hansbro P. M., Foster P. S. (2011). Dietary lycopene supplementation suppresses Th2 responses and lung eosinophilia in a mouse model of allergic asthma. J. Nutr. Biochem. 22, 95-100 [DOI] [PubMed] [Google Scholar]
- Herbert C., Hettiaratchi A., Webb D. C., Thomas P. S., Foster P. S., Kumar R. K. (2008). Suppression of cytokine expression by roflumilast and dexamethasone in a model of chronic asthma. Clin. Exp. Allergy 38, 847-856 [DOI] [PubMed] [Google Scholar]
- Herbert C., Scott M. M., Scruton K. H., Keogh R. P., Yuan K. C., Hsu K., Siegle J. S., Tedla N., Foster P. S., Kumar R. K. (2010). Alveolar macrophages stimulate enhanced cytokine production by pulmonary CD4+ T-lymphocytes in an exacerbation of murine chronic asthma. Am. J. Pathol. 177, 1657-1664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holgate S. T. (2011). The sentinel role of the airway epithelium in asthma pathogenesis. Immunol. Rev. 242, 205-219 [DOI] [PubMed] [Google Scholar]
- Holt P. G., Sly P. D. (2012). Viral infections and atopy in asthma pathogenesis: new rationales for asthma prevention and treatment. Nat. Med. 18, 726-735 [DOI] [PubMed] [Google Scholar]
- Holt P. G., Vines J., Bilyk N. (1988). Effect of influenza virus infection on allergic sensitization to inhaled antigen in mice. Int. Arch. Allergy Appl. Immunol. 86, 121-123 [DOI] [PubMed] [Google Scholar]
- Holt P. G., Strickland D. H., Bosco A., Jahnsen F. L. (2009). Pathogenic mechanisms of allergic inflammation: atopic asthma as a paradigm. Adv. Immunol. 104, 51-113 [DOI] [PubMed] [Google Scholar]
- Holtzman M. J., Morton J. D., Shornick L. P., Tyner J. W., O'Sullivan M. P., Antao A., Lo M., Castro M., Walter M. J. (2002). Immunity, inflammation, and remodeling in the airway epithelial barrier: epithelial-viral-allergic paradigm. Physiol. Rev. 82, 19-46 [DOI] [PubMed] [Google Scholar]
- Holtzman M. J., Tyner J. W., Kim E. Y., Lo M. S., Patel A. C., Shornick L. P., Agapov E., Zhang Y. (2005). Acute and chronic airway responses to viral infection: implications for asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 2, 132-140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosakote Y. M., Liu T., Castro S. M., Garofalo R. P., Casola A. (2009). Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir. Cell Mol. Biol. 41, 348-357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K., Herbert C., Siegle J. S., Vuppusetty C., Hansbro N., Thomas P. S., Foster P. S., Barnes P. J., Kumar R. K. (2008). Steroid-resistant neutrophilic inflammation in a mouse model of an acute exacerbation of asthma. Am. J. Respir. Cell Mol. Biol. 39, 543-550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulhankova K., George C. L., Kline J. N., Snyder J. M., Darling M., Field E. H., Thorne P. S. (2009). Early-life co-administration of cockroach allergen and endotoxin augments pulmonary and systemic responses. Clin. Exp. Allergy 39, 1069-1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R. K., Maronese S. E., O'Grady R. (1997). Serum-free culture of mouse tracheal epithelial cells. Exp. Lung Res. 23, 427-440 [DOI] [PubMed] [Google Scholar]
- Kumar R. K., Herbert C., Yang M., Koskinen A. M. L., McKenzie A. N. J., Foster P. S. (2002). Role of interleukin-13 in eosinophil accumulation and airway remodelling in a mouse model of chronic asthma. Clin. Exp. Allergy 32, 1104-1111 [DOI] [PubMed] [Google Scholar]
- Laumbach R. J., Kipen H. M. (2012). Respiratory health effects of air pollution: update on biomass smoke and traffic pollution. J. Allergy Clin. Immunol. 129, 3-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. M., Chang J. H., Moon D. O., Choi Y. H., Choi I. W., Park Y. M., Kim G. Y. (2008). Lycopene suppresses ovalbumin-induced airway inflammation in a murine model of asthma. Biochem. Biophys. Res. Commun. 374, 248-252 [DOI] [PubMed] [Google Scholar]
- Li N., Wang M., Oberley T. D., Sempf J. M., Nel A. E. (2002). Comparison of the pro-oxidative and proinflammatory effects of organic diesel exhaust particle chemicals in bronchial epithelial cells and macrophages. J. Immunol. 169, 4531-4541 [DOI] [PubMed] [Google Scholar]
- Malhotra J. D., Miao H., Zhang K., Wolfson A., Pennathur S., Pipe S. W., Kaufman R. J. (2008). Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc. Natl. Acad. Sci. USA 105, 18525-18530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell R., Islam T., Shankardass K., Jerrett M., Lurmann F., Gilliland F., Gauderman J., Avol E., Künzli N., Yao L., et al. (2010). Childhood incident asthma and traffic-related air pollution at home and school. Environ. Health Perspect. 118, 1021-1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurmatov U., Devereux G., Sheikh A. (2011). Nutrients and foods for the primary prevention of asthma and allergy: systematic review and meta-analysis. J. Allergy Clin. Immunol. 127, 724-733, e1-e30 [DOI] [PubMed] [Google Scholar]
- Pénard-Morand C., Raherison C., Charpin D., Kopferschmitt C., Lavaud F., Caillaud D., Annesi-Maesano I. (2010). Long-term exposure to close-proximity air pollution and asthma and allergies in urban children. Eur. Respir. J. 36, 33-40 [DOI] [PubMed] [Google Scholar]
- Perez L., Lurmann F., Wilson J., Pastor M., Brandt S. J., Künzli N., McConnell R. (2012). Near-roadway pollution and childhood asthma: implications for developing “win-win” compact urban development and clean vehicle strategies. Environ. Health Perspect. 120, 1619-1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saedisomeolia A., Wood L. G., Garg M. L., Gibson P. G., Wark P. A. (2009). Lycopene enrichment of cultured airway epithelial cells decreases the inflammation induced by rhinovirus infection and lipopolysaccharide. J. Nutr. Biochem. 20, 577-585 [DOI] [PubMed] [Google Scholar]
- Saglani S., Mathie S. A., Gregory L. G., Bell M. J., Bush A., Lloyd C. M. (2009). Pathophysiological features of asthma develop in parallel in house dust mite-exposed neonatal mice. Am. J. Respir. Cell Mol. Biol. 41, 281-289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahin K., Tuzcu M., Sahin N., Ali S., Kucuk O. (2010). Nrf2/HO-1 signaling pathway may be the prime target for chemoprevention of cisplatin-induced nephrotoxicity by lycopene. Food Chem. Toxicol. 48, 2670-2674 [DOI] [PubMed] [Google Scholar]
- Salam M. T., Lin P. C., Avol E. L., Gauderman W. J., Gilliland F. D. (2007). Microsomal epoxide hydrolase, glutathione S-transferase P1, traffic and childhood asthma. Thorax 62, 1050-1057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T., Issekutz A. C. (1996). Quantitation of eosinophil and neutrophil infiltration into rat lung by specific assays for eosinophil peroxidase and myeloperoxidase. Application in a brown Norway rat model of allergic pulmonary inflammation. J. Immunol. Methods 198, 1-14 [DOI] [PubMed] [Google Scholar]
- Seagrave J., McDonald J. D., Bedrick E., Edgerton E. S., Gigliotti A. P., Jansen J. J., Ke L., Naeher L. P., Seilkop S. K., Zheng M., et al. (2006). Lung toxicity of ambient particulate matter from southeastern U.S. sites with different contributing sources: relationships between composition and effects. Environ. Health Perspect. 114, 1387-1393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoenfelt J., Mitkus R. J., Zeisler R., Spatz R. O., Powell J., Fenton M. J., Squibb K. A., Medvedev A. E. (2009). Involvement of TLR2 and TLR4 in inflammatory immune responses induced by fine and coarse ambient air particulate matter. J. Leukoc. Biol. 86, 303-312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegle J. S., Hansbro N., Herbert C., Yang M., Foster P. S., Kumar R. K. (2006). Airway hyperreactivity in exacerbation of chronic asthma is independent of eosinophilic inflammation. Am. J. Respir. Cell Mol. Biol. 35, 565-570 [DOI] [PubMed] [Google Scholar]
- Siegle J. S., Hansbro N., Herbert C., Rosenberg H. F., Domachowske J. B., Asquith K. L., Foster P. S., Kumar R. K. (2010). Early-life viral infection and allergen exposure interact to induce an asthmatic phenotype in mice. Respir. Res. 11, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sly P. D., Boner A. L., Björksten B., Bush A., Custovic A., Eigenmann P. A., Gern J. E., Gerritsen J., Hamelmann E., Helms P. J., et al. (2008). Early identification of atopy in the prediction of persistent asthma in children. Lancet 372, 1100-1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temelkovski J., Hogan S. P., Shepherd D. P., Foster P. S., Kumar R. K. (1998). An improved murine model of asthma: selective airway inflammation, epithelial lesions and increased methacholine responsiveness following chronic exposure to aerosolised allergen. Thorax 53, 849-856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y., McCusker C. (2006). Neonatal exposure with LPS and/or allergen prevents experimental allergic airways disease: development of tolerance using environmental antigens. J. Allergy Clin. Immunol. 118, 143-151 [DOI] [PubMed] [Google Scholar]
- Wigle D. T., Arbuckle T. E., Turner M. C., Bérubé A., Yang Q., Liu S., Krewski D. (2008). Epidemiologic evidence of relationships between reproductive and child health outcomes and environmental chemical contaminants. J. Toxicol. Environ. Health B Crit. Rev. 11, 373-517 [DOI] [PubMed] [Google Scholar]