

SUMMARY
Holoprosencephaly (HPE) is a common congenital defect that results from failed or incomplete forebrain cleavage. HPE is characterized by a wide clinical spectrum, with inter- and intrafamilial variability. This heterogeneity is not well understood and it has been suggested that HPE involves a combination of multiple gene mutations. In this model, several mutated alleles or modifying factors are presumed to act in synergy to cause and determine the severity of HPE. This could explain the various clinical phenotypes. Screening for HPE-associated genes in humans suggests the involvement of NODAL or SHH signaling, or both. To test this multigenic hypothesis, we investigated the effects of chemical inhibition of these two main HPE signaling pathways in a chick embryo model. SB-505124, a selective inhibitor of transforming growth factor-B type I receptors was used to inhibit the NODAL pathway. Cyclopamine was used to inhibit the SHH pathway. We report that both inhibitors caused HPE-like defects that were dependent on the drug concentration and on the developmental stage at the time of treatment. We also investigated double inhibition of NODAL and SHH pathways from the onset of gastrulation by using subthreshold inhibitor concentrations. The inhibitors of the NODAL and SHH pathways, even at low concentration, acted synergistically to promote an HPE-like phenotype. These findings support the view that genetic heterogeneity is important in the etiology of HPE and may contribute to the phenotypic variability.
INTRODUCTION
Holoprosencephaly (HPE) is the most common congenital forebrain defect in humans. It results from failed or incomplete forebrain cleavage between days 18 and 28 of gestation (Dubourg et al., 2007; Marcorelles and Laquerriere, 2010).
The clinical presentation of HPE is remarkably variable, and the severity of the defects observed is evenly distributed along the HPE spectrum. The etiology is very complex and heterogeneous, involving chromosomal anomalies, multiple malformation syndromes and environmental factors. Fourteen genes are known to be involved in non-syndromic human HPE (SHH, ZIC2, SIX3, TGIF, DISP1, NODAL, GLI2, PTCH1, TDGF1, FOXH1, FGF8, GAS1, DLL1 and CDON), but they explain only 30% of HPE cases (Mercier et al., 2011). All mutations are found in the heterozygous state in HPE patients, and most are loss-of-function mutations (Dubourg et al., 2004; Arauz et al., 2010; Ribeiro et al., 2010; Roessler and Muenke, 2010; Bae et al., 2011; Dupé et al., 2011). The correlation between the HPE phenotype and genotype is poor, and both sporadic cases and pedigrees display the extensive HPE phenotype variability. This suggests that heterozygous mutations of HPE genes might be insufficient to produce severe anomalies, and that HPE is the consequence of a complex interplay of developmental, genetic and environmental factors. To date, given the relatively low percentage of cases for which causative mutations have been identified, additional studies are undoubtedly warranted.
From animal model studies, multifactorial inheritance disorder has been proposed to contribute to the phenotype heterogeneity of the disorder. For example, heterozygous mutation of several genes belonging to the same or different pathways could lead to phenotypic heterogeneity. Mouse models have provided evidence, by implicating either one or two different signaling pathways [Nodal+/−;Smad2+/− (Nomura and Li, 1998); Cdo−/−;Boc−/− (Zhang et al., 2011); and Chrd−/−;Nodal+/− (Yang et al., 2010)]. The genetic background also plays an important role in determining the severity of the phenotype, suggesting the existence of modifier genes (Cole and Krauss, 2003; Krauss, 2007; Geng et al., 2008; Pei and Feldman, 2009). Furthermore, observation of some HPE patients with two different genetic alterations are consistent with a multigenic process (Mercier et al., 2011; Ming and Muenke, 2002). Thus, the diversity of combinations of mutations in HPE genes and/or modifier genes belonging to different pathways could contribute to the wide spectrum of HPE phenotypes.
HPE genes belong to various different signaling pathways regulating forebrain development, including the Hedgehog (HH), NODAL, BMP and FGF pathways. The role of these pathways during forebrain development has been extensively studied, especially the SHH and NODAL pathways, the two major pathways implicated in the pathogenesis of HPE (Roessler and Muenke, 2010). However, the interactions between these pathways, in terms of spatial and temporal regulation during the development of HPE, need to be clarified.
The NODAL signaling pathway has been implicated in cases of human HPE with or without congenital heart defects and/or left-right disturbances (Roessler et al., 2008; Roessler et al., 2009). NODAL is a transforming growth factor β (TGF-β)-related signaling molecule that is essential during the initiation of gastrulation. NODAL signals act in the formation of mesoderm and are required later for correct positioning of the anteroposterior axis, midline patterning and left-right specification (Iannaccone et al., 1992; Zhou et al., 1993; Conlon et al., 1994; Lowe et al., 2001; Roessler et al., 2008; Roessler et al., 2009). The NODAL signaling pathway has been extensively studied during cleavage and blastula stages in zebrafish; NODAL acts in a dose-dependent and time-dependent manner during the mid-to-late blastula stage to specify most mesodermal and endodermal cell types (Hagos and Dougan, 2007; Gritsman et al., 2000). The total absence of NODAL is embryonically lethal, whereas a hypomorphic allele of Nodal or chemical inactivation results in anterior patterning defects including cyclopia in mice, zebrafish and Xenopus (Lowe et al., 2001; Vincent et al., 2003; Hagos and Dougan, 2007; Luxardi et al., 2010). These defects are strictly dependent on the degree of NODAL inactivation and on the embryonic stage. Consistent with the importance of this pathway during development, inactivation at early stages leads to very severe phenotypes, and the critical time-window of requirement for NODAL activity during forebrain formation is difficult to define (Shen, 2007; Luxardi et al., 2010).
TRANSLATIONAL IMPACT.
Clinical issue
Holoprosencephaly (HPE), a remarkably common human birth defect, is caused by a failure to form the midline of the forebrain and midface. Its clinical presentation is extremely variable, ranging from alobar HPE (where there is a complete failure to divide the forebrain) to microform (where there are mild craniofacial features but no forebrain defects). Various craniofacial defects (from none to cyclopia) and other extra-craniofacial defects are observed. HPE is probably caused by both environmental and genetic factors; with respect to the latter, heterozygous mutations in components or regulators of the Sonic Hedgehog (SHH) signaling pathway are often associated. The phenotypic heterorogeneity seen in carriers of SHH pathway mutations, ranging from no clinical manifestation to alobar HPE, implicates other modifier genes in forebrain development.
Results
In addition to SHH signaling, several other pathways regulate forebrain development, such as the NODAL pathway. Here, the authors established a chick embryo culture model to investigate the effects of chemical inhibition of SHH and NODAL pathways during forebrain development. They report that inhibition of either pathway caused HPE-like defects with phenotypic variability. Inhibition of both pathways using subthreshold inhibitor concentrations acted synergistically to promote severe HPE-like phenotypes. These findings support the view that genetic heterogeneity plays a key role in HPE etiology and contributes to the phenotypic variability.
Implications and future directions
Because HPE arises from a complex interplay of developmental, genetic and environmental factors, it is difficult to study the condition using genetic approaches in a mouse model. By comparison, it is simpler to use cultured chick embryos for testing the multi-genetic hypothesis of human HPE and for validating new candidate genes. The identification of new modifier genes by whole exome sequencing will aid in the understanding HPE etiology, and provide new direction to mechanistic studies in the chick and in other model organisms.
Disruption of SHH signaling is a common major event in the development of HPE. Mice lacking SHH function have a severe HPE-like phenotype involving the absence of ventral forebrain structures and cyclopia (Chiang et al., 1996). Studies with model organisms demonstrate that SHH produced by the prechordal mesendoderm is required for initiating the development of the midline of the forebrain and the midface (Rubenstein and Beachy, 1998; Muenke and Beachy, 2000; Kiecker and Niehrs, 2001). Abnormal activity of the SHH signaling pathway at particular periods of embryonic development might account for the phenotypical spectrum of HPE (Cordero et al., 2004). There is a progressive mechanism whereby SHH produced by one midline structure induces Shh expression in successive midline structures. Most human HPE genes either encode components of the SHH pathway, or directly or indirectly affect Shh expression in either the prechordal mesendoderm or the developing ventral forebrain (Hu and Helms, 1999; Marcucio et al., 2005; Dubourg et al., 2007; Geng and Oliver, 2009).
SHH and NODAL signals interact and Hedgehog signaling acts downstream from NODAL activity to pattern the ventral telencephalon (Müller et al., 2000; Rohr et al., 2001). Indeed, expression of Shh is severely reduced in mutants for the NODAL pathway, particularly in anterior regions (Strähle et al., 1997; Hagos and Dougan, 2007). The mechanisms by which NODAL signaling regulates SHH activity in axial tissues have yet to be determined, but NODAL signals can cell-autonomously promote floorplate-specific expression of Shh (Müller et al., 2000). The interaction between SHH and NODAL pathways takes place during gastrulation as the floorplate is established. The description of heterozygous mutations of SHH and TGIF (which interact with TGF-β-activated Smad proteins) in the same HPE patient is further evidence for there being interaction between SHH and NODAL (Nanni et al., 1999). However, it is not known whether the association of effects in each of these two pathways can cause HPE.
Here, we studied partial chemical inhibition of these two pathways during chick forebrain development. Our aim was to evaluate the interaction between NODAL and SHH pathways and to investigate the HPE multiple-genetic process in an animal model using a dose- and time-dependent approach. We demonstrate that phenotypic severity was dependent on the stage of treatment and on the concentration of the chemical inhibitors of the NODAL and SHH signaling pathways. Subthreshold concentrations of the two inhibitors in combination caused severe effects. These investigations show that the partial inhibition of two pathways can have a cumulative effect in determining an HPE-like phenotype and support a genetic heterogeneity hypothesis for HPE.
RESULTS
Inactivation of the NODAL signaling pathway from the stage of gastrulation onset leads to severe forebrain defects
The chemical inhibitor SB-505124 was used to inactivate NODAL signaling (Hagos and Dougan, 2007). This compound has been used previously to dissect the complex pathway of the TGF-β superfamily and has been validated for inhibition of the activin receptor-like kinase (ALK)4/5/7-dependent activation (DaCosta Byfield et al., 2004).
To analyze the effect of SB-505124 on chick forebrain during its formation, we studied three molecular markers: Pax6, Shh and Nkx2.1. The Pax6 homeobox gene is specifically expressed in developing optic vesicles, making it a good indicator of eye defects, from hypotelorism to cyclopia, one of the main features of the HPE spectrum (Incardona et al., 1998). Shh and Nkx2.1 are ventral markers of the developing forebrain (Crossley et al., 2001): they are targets of NODAL signaling during forebrain development and are specifically downregulated in animal models of HPE (Müller et al., 2000; Rohr et al., 2001).
We used ex ovo chick embryo cultures (so-called roller cultures) to test the effects of NODAL signaling inhibition during early brain formation (Dupé and Lumsden, 2001). The NODAL signaling pathway was inactivated in whole embryos from the onset of gastrulation (between stage 4 and stage 6 in the chick) until the primary regionalization of the forebrain (stage 12). The NODAL pathway is required during early gastrulation (Shen, 2007), so a concentration and a stage of treatment allowing gastrulation to occur but causing a forebrain phenotype had to be determined.
Whole chick embryos (n=317) from stage 4 to stage 6 were exposed to one of a series of SB-505124 concentrations: 10, 25, 50 or 75 μM. At least five different experiments were run for each condition. Because we clearly observed a phenotypic heterogeneity, we individualized three distinct phenotypes on the basis of the morphology and gene expression (arbitrarily classified as categories 1, 2 and 3) (Fig. 1A).
Fig. 1.
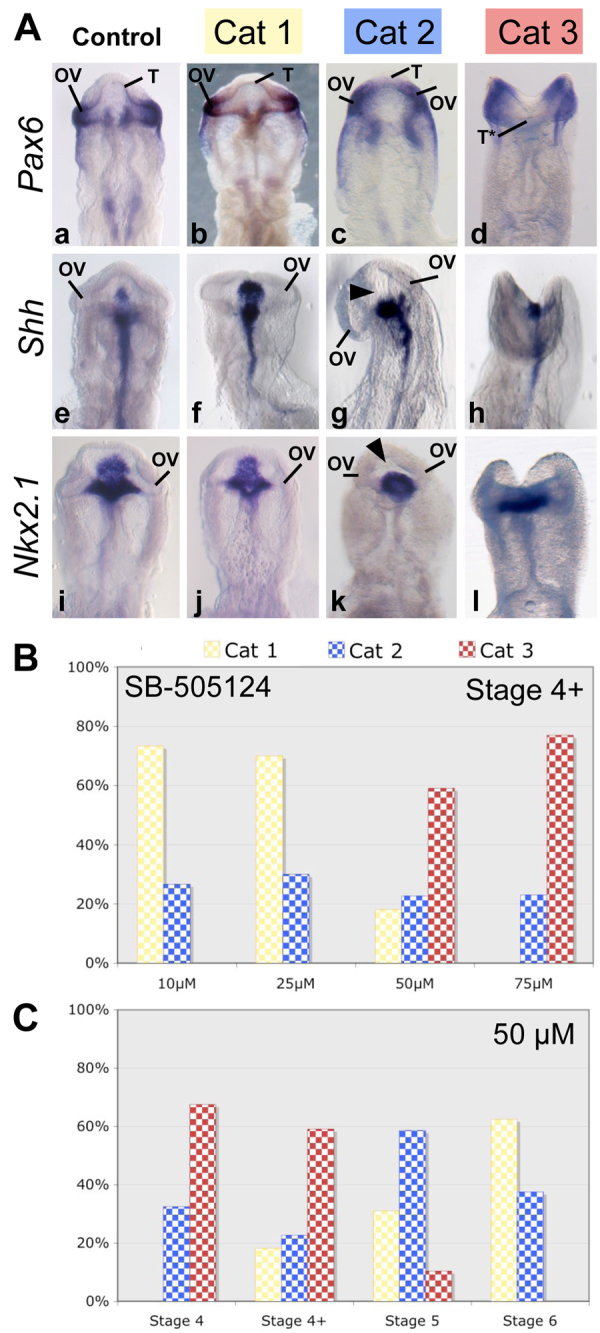
NODAL signal inhibition by SB-505124 treatment leads to severe forebrain defects; the dose-response relationship is nonlinear. (A) Ventral views of chick embryo heads at stage 12. Control embryos (n=150) were treated with DMSO (Aa, Ae, Ai). Other embryos were treated with one of a series of SB-505124 concentrations (10-75 μM) between stage 4 and stage 6 (n=317). Resulting phenotypes were classified into three categories according to morphology and in situ hybridization for Pax6 (n=111) (Aa-Ad), Shh (n=107) (Ae-Ah) and Nkx2.1 (n=99) (Ai-Al). The order of increasing severity was: category 1, category 2 and category 3. Arrowheads show the downregulation of Shh (Ag) and Nkx2-1 (Ak) expression in the ventral forebrain. (B) Distribution of the phenotypes of stage 4+ embryos treated with one of a series of SB-505124 concentrations: 10 μM (n=15), 25 μM (n=20), 50 μM (n=22) or 75 μM (n=26). (C) Distribution of the phenotypes of embryos treated with 50 μM SB-505124 starting at various stages: from stage 4 (n=40), stage 4+ (n=22), stage 5 (n=29) and stage 6 (n=16). OV, optic vesicles; T, telencephalon; T*, non-fused telencephalon; Cat, category.
At the onset of the gastrulation (Stage 4+), most embryos (70%) treated with 25 μM SB-505124 were similar to controls (DMSO-treated embryos) in terms of morphology and expression of Pax6, Shh and Nkx2.1 (Fig. 1A, category 1). However, 30% of the embryos displayed a mild phenotype, involving forebrain hypoplasia (Fig. 1A, category 2). In these category 2 embryos, optic vesicles were formed, as shown by in situ hybridization for Pax6, but they were in a more ventral position than in controls. They were fused only in rare cases. Importantly, expression of both Nkx2.1 and Shh was specifically downregulated in the rostral diencephalic ventral midline (Fig. 1A, arrowheads) similar to that described in several HPE models (Allen et al., 2007; Higashiyama et al., 2007), including hypomorph mutants for NODAL signaling (Lowe et al., 2001; Vincent et al., 2003). At SB-505124 concentrations of 50 μM or 75 μM administered from stage 4+ (Fig 1B), forebrain defects were severe (category 3) and characterized by anterior truncation with non-fused neural folds (Fig. 1Ad, T*), no formation of optic vesicles and downregulation of the expression of Shh and Nkx2.1 in the rostral midline (Fig. 1Ah,Al). This category 3 phenotype was observed in 59% and 77% of the embryos treated with 50 μM and 75 μM, respectively, from stage 4+.
We tested the response of chick embryos to SB-505124 according to the stage of treatment. The category 3 phenotype was frequent among embryos treated with 50 μM from stage 4 (68%) or 4+ (59%) (Fig 1C), but this severe phenotype was significantly less frequent when the treatment was started at stage 5 (10%). After stage 5, severe phenotypes were not observed: category 2 was more frequent for stage 5 (59%) and category 1 for stage 6 (63%).
In conclusion, category 3 (severe) phenotypes were mainly observed at a drug concentration over 25 μM initiated at stages 4 or 4+, just at the onset of gastrulation.
Cyclopamine causes progressive cyclopia in a dose-dependent manner
Cyclopamine is a well-characterized chemical inhibitor of the SHH signaling pathway (Cooper et al., 1998; Incardona et al., 1998): it inhibits the morphogenetic activity of the SHH pathway by binding to and preventing activation of the transmembrane protein, Smoothened (Smo) (Chen et al., 2002; Lipinski et al., 2008). First, as for SB-505124, we identified the stages and concentrations at which cyclopamine caused HPE-like defects.
Chick embryos were exposed to cyclopamine concentrations between 0.006 μM and 2.5 μM at stage 4+ (Fig. 2). After 24 hours of culture, the embryos were individually scored for morphological abnormalities and Pax6 expression. A phenotypic index was calculated and the phenotypes were classified into three categories, from normal embryo, category I to embryos with fused optic vesicles, category III.
Fig. 2.
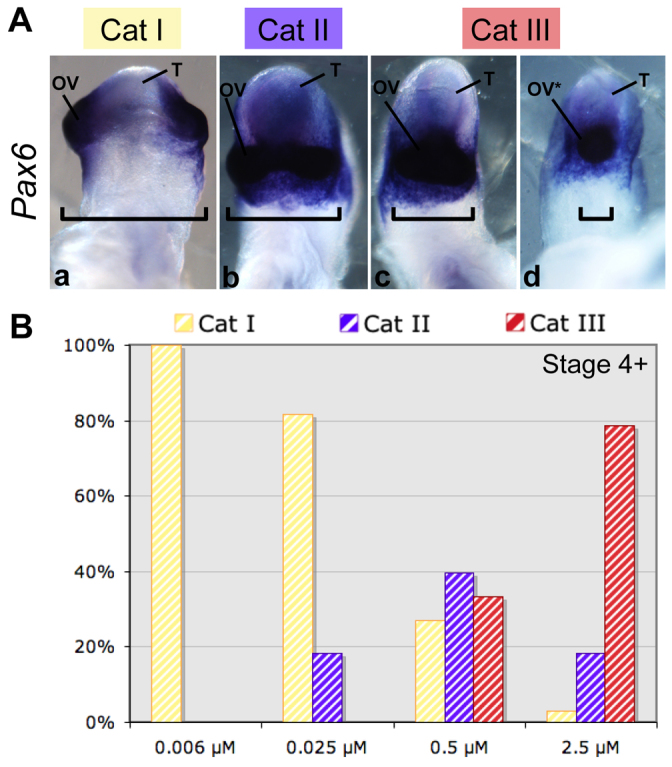
Cyclopamine causes cyclopia in a dose-dependent manner. (A) Ventral views of chick embryo heads at stage 12. Three phenotypes were distinguished morphologically. Pax6 mRNA was used as a marker for optic vesicles (n=70). The order of increasing severity was: category I, category II and category III. (B) Distribution of the phenotypes observed for embryos treated at stage 4+ by one of a series of concentrations of cyclopamine: 0.006 μM (n=30), 0.025 μM (n=38), 0.5 μM (n=48) or 2.5 μM (n=33). OV, optic vesicles; OV*, single optic vesicle; T, telencephalon; Cat, category. Brackets highlight the distance between optic vesicles.
All embryos treated with 0.006 μM of cyclopamine at stage 4+, showed normal head development (category I). Normal head development was similarly observed in 85% of the embryos treated with 0.025 μM cyclopamine. The other 15% showed partial fusion of the optic evaginations ventrally, as confirmed by analysis of Pax6 expression. This was defined as the category II phenotype (Fig. 2A). At a higher cyclopamine concentration (0.5 μM), we observed a new phenotype (category III) characterized by optic vesicle fusion, from partial fusion to cyclopia, associated with an elongated forebrain (35%) (Fig. 2A). At the highest concentration of cyclopamine tested (2.5 μM), 75% of the embryos displayed a category III phenotype (Fig. 2B).
To determine when Shh-dependent forebrain patterning occurs, embryos were treated with 2.5 μM cyclopamine at various stages. Exposure at stage 5 or 6 resulted in a high proportion of cyclopic embryos (data not shown). However, following treatment with 2.5 μM cyclopamine starting at stage 7, none of the embryos developed cyclopia.
These experiments describe the relationship between cyclopia and both the stage of treatment and cyclopamine concentration. They confirms the involvement of SHH in the ventralization of the forebrain during early gastrulation (stages 5 and 6) (Pera and Kessel, 1997). These experiments also identified a subthreshold concentration (0.006 μM) of cyclopamine that has no apparent morphological effect on either head development or fusion of the optic vesicles.
Combined partial inhibition of SHH and NODAL signals leads to cyclopic embryos
We studied the effects of combined treatment with the inhibitors of the NODAL and SHH signaling pathways, at subthreshold concentrations. SHH signal inactivation between stages 4 and 6 was required to obtain cyclops (see above). NODAL inactivation before stage 5 was required to cause a forebrain phenotype. Therefore, we used stage 4+ embryos for these experiments.
Stage 4+ chick embryos (total n=187) in roller cultures were exposed to 0.006 μM cyclopamine or 25 μM SB-505124 or both. Pax6 expression was studied in these embryos.
We defined three new phenotypic categories (I* to III*) to score the degree of fusion of optic vesicles. These categories are similar to those defined for cyclopamine treatment, but include progressive forebrain hypoplasia (Fig. 3A). Thus, category I* was defined as embryos with non-fused expression of Pax6 in the optic vesicles (Fig. 3Aa), and included morphologically normal embryos and embryos with slightly ectopic optic vesicles. Category II* (Fig. 3Ab) embryos displayed a phenotype with ectopic optic vesicles (ventral position) with continuous expression of Pax6. Category III* was a severely affected phenotype with partial or complete fusion of the optic vesicle (cyclopia) associated with marked forebrain hypoplasia (Fig. 3Ac,Ad). We scored the severity of the phenotypes after the three treatments (Fig. 3B).
Fig. 3.
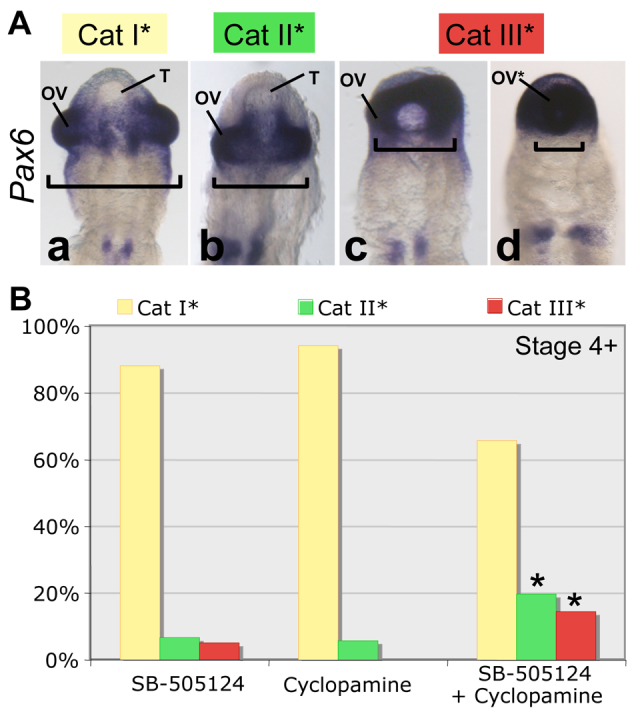
Simultaneous partial inhibition of NODAL and SHH causes HPE-like anterior neural defects in chick embryo. (A) Ventral views of chick embryo heads at stage 12. Embryos were treated with SB-505124 (25 μM) and cyclopamine (0.006 μM) at stage 4+ (n=187). Three phenotypes were distinguished using Pax6 in situ hybridization. The order of increasing severity was: category I*, category II* and category III*. (B) Phenotype distributions among embryos treated with cyclopamine (n=52), SB-505124 (n=59) or both (n=76) at stage 4+. The asterisks show the significant cumulative effect of the double treatment leading to the HPE-like phenotype (*P=4×10−4). OV, optic vesicles; OV*, single optic vesicle; T, telencephalon; Cat, category.
The most severe phenotype, category III*, was never observed following treatment with 0.006 μM cyclopamine alone, and only 6% of these embryos displayed a category II* phenotype (Fig. 3B). Following treatment with 25 μM SB-505124 alone, 7% of the embryos were category II* and 5% category III*. About 20% of the embryos subjected to the double treatment were category II* and 14% category III*. The proportions of categories II* and III* differed significantly between combined and single inactivation groups (P=4×10−4).
Therefore, combination of the two inhibitors at subthreshold concentrations caused more severe midline abnormalities than either inhibitor alone. These findings suggest that abnormal NODAL and SHH signals interact to produce various HPE-spectrum phenotypes. Similar double treatment started at stage 5 (n=49) or stage 6 (n=20) did not have a greater effect than single treatments (data not shown).
DISCUSSION
The molecular basis for the variable expressivity and reduced penetrance of HPE has not yet been identified. However, the digenism described in animal models for HPE supports the existence of a heterogeneous and multigenic mechanism in some human cases, in which the penetrance and expressivity of heterozygous loss-of-function mutations can be modulated by one or more other mutations (Mercier et al., 2011; Roessler et al., 2012). Here, we report the development of an animal model in which various signaling pathways can be simultaneously and partially inhibited. We used this model to inactivate the NODAL and SHH pathways, both major pathways implicated in human HPE, during forebrain development.
In knockout models, the loss of NODAL signaling blocks gastrulation and is embryonically lethal (Lowe et al., 2001). However, analysis of Nodal-insufficient embryos revealed that although the gastrulation takes place, eye field and forebrain fail to develop properly and a HPE-like phenotype develops (Lowe et al., 2001; Vincent et al., 2003; Andersson et al., 2006; Yang et al., 2010). Here, we used the ALK4/5/7 receptor inhibitor, SB-505124, for partial inhibition of the NODAL pathway during chick development. It was previously shown that this drug added to developing zebrafish at various stages of development causes a spectrum of phenotypes, including cyclopia, that are phenocopies of NODAL component mutants (Hagos and Dougan, 2007). Even though multiple ligands can activate the ALK4/5/7 receptors, our phenotypes resemble those resulting from reductions in nodal-related gene function (Vincent et al., 2003; Andersson et al., 2006; Yang et al., 2010). This suggests that the other activin-like ligands are either not implicated during the stages we examined or act downstream of Nodal signals.
It has not yet been established when NODAL signals contribute to the anterior midline specification in chick embryos. In our experiments, embryos were treated in roller culture with various concentrations of SB-505124 at the onset of gastrulation. The severity of the resulting defects in forebrain development depended both on the concentration of the inhibitor and the stage at which treatment started. NODAL is essential for gastrulation through the regulation of several major genes; however, the difficulty was to find a window and concentration at which the inhibition was strong enough to induce an HPE-like phenotype (category 2) without causing a phenotype that was too severe (category 3). During our SB-505124 treatment, analysis of Pax6 expression did not reveal any typical cyclopic phenotype, whereas in situ hybridization evidenced reduced, or loss of, Shh and Nkx2.1 expression in the rostral midline region. Despite the lack of the typical HPE phenotype, this absence of expression of Shh and Nkx2.1 at the anterior boundary of expression support the idea that these category 2 embryos display a HPE-like phenotype (Lowe et al., 2001; Anderson et al., 2002; Vincent et al., 2003; Allen et al., 2007; Higashiyama et al., 2007). The crucial stage for NODAL signaling in our model is before stage 5, which is consistent with the description of NODAL expression: Nodal is expressed in the middle three-quarters of the primitive streak, excluding Hensen's node, up to stage 5; Nodal expression then ceases in the midline (Lawson et al., 2001).
Cyclopamine treatment caused, in a dose-dependent manner, fusion of the optic vesicles up to cyclopia, the most severe typical craniofacial defect in HPE. Interestingly, in HPE patients with altered SHH signaling, midline malformation ranges from hypotelorism to complete cyclopia (Muenke and Beachy, 2000). The features of our model are consistent with the demonstration by Cordero et al. of a relationship between the timing of Hedgehog disruption and the severity of the facial phenotype (Cordero et al., 2004). Interestingly, embryos subjected to cyclopamine treatment starting as late as stage 7 developed a cyclopic phenotype. This is in agreement with the continuous expression of Shh throughout the node at stage 4 and subsequently in the prechordal mesendoderm at stage 7, the crucial structure for ventral forebrain patterning (Crossley et al., 2001).
We investigated whether minor defects in NODAL and SHH pathways can synergize to produce HPE disorders. Our preliminary experiments indicated that stage 4+ embryos were suitable for these experiments for two reasons. First, SB-505124 had the largest effect on head development when administrated at stage 4 and 4+. Second, the stage 4+ is easy to distinguish, but also very transient, leading to less variability in the results. We used concentrations of inhibitors that in isolation did not cause any severe forebrain defect or severe optic vesicle fusion (25 μM SB-505124 and 0.006 μM cyclopamine). The proportion of embryos with ventralized optic vesicles (category II*) and fusion (category III*) was substantially higher following double treatment than single treatment. An optic vesicle phenotype was observed in 34% of double-treated embryos, and optic vesicle fusion up to cyclopia in 15%. This effect was not reproduced when embryos were treated slightly later, at stage 5. These experiments show that minor inhibition of both SHH and NODAL pathways can lead to abnormal development of the forebrain midline, whereas similar inhibition of one or other in isolation does not. The penetrance of the phenotypes obtained was incomplete and variable, which is a common feature of most HPE models described (Lowe et al., 2001; Allen et al., 2007; Zhang et al., 2011; Cordero et al., 2004).
Cyclopamine and SB-505124 single treatments as a combination of both inhibitors generated phenotypic heterogeneity. This variability, from no morphological anterior deficit to severe anterior truncation, is greater with intermediate concentration of inhibitors (50 μM for SB-505124 and 0.5 μM for cyclopamine at stage 4+); it reflects the importance of the quantitative requirement of the level of NODAL and SHH signaling during forebrain development.
NODAL and SHH signaling are decisive in forebrain fate determination and the corresponding genes are expressed simultaneously in the primitive streak and the prechordal plate. The mechanism by which NODAL signaling regulates SHH activity during gastrulation has yet to be determined. However, several studies have indicated that the NODAL pathway regulates ventral forebrain patterning through SHH-independent mechanisms (Rohr et al., 2001; Monuki, 2007). SHH might have to reach a threshold concentration to ensure proper forebrain development (Young et al., 2010). The dose- and stage-dependent relationship we observe here is consistent with a threshold effect: slightly reduced NODAL signaling might cause a small downregulation of SHH, which produces a HPE phenotype only when associated with another small decrease in SHH signaling.
Overall, we demonstrate that weak inhibition of two signaling pathways, one downstream of the other, can lead to an HPE-like phenotype. A similar interaction between NODAL and BMP has also been demonstrated in forebrain development (Yang et al., 2010). Therefore, several independent genetic events influencing different signaling pathways involved in forebrain development, acting at the same or different embryonic stages, can cause the HPE phenotype, most probably through regulation of the main HPE signaling pathway, SHH. This could be the basis for the phenotypic heterogeneity observed in families segregating mutations in SHH. This is coherent with the systematic decrease of Shh in animal models with partial inhibitions of various signaling pathways (Krauss, 2007; Fernandes and Hébert, 2008; Geng and Oliver, 2009; Yang et al., 2010). These observations are consistent with a ‘multisignaling pathways-hit’ model. It is likely that as-yet unidentified polymorphisms contribute to the hypomorphic effects involved in HPE. Further work with whole exome sequencing would help confirm this theory.
MATERIALS AND METHODS
Roller-tube culture and chemical inhibition in chick
Fertile hens' eggs were incubated in a humidified room at 38°C. The embryos were staged according to Hamburger and Hamilton (Hamburger and Hamilton, 1992). They were collected at stages 4, 4+, 5 and 6 and cultured as described elsewhere (Dupé and Lumsden, 2001). Five embryos, folded and sealed along the longitudinal axis, were transferred to a 5-ml plastic bottle containing 500 μl of Liebovitz medium. The bottles were placed on a roller apparatus rotating at 30 revolutions/minute, inclined at an angle of about 10° in a 38°C incubator. Development was then allowed to proceed for 24 hours. The embryos were treated with the following compounds: cyclopamine (Sigma) from a stock solution of 1 mg/ml in 2-hydroxypropyl-β-cyclodextrin (HBC, Sigma); and SB-505124 (Sigma-Aldrich), from a stock solution of 24 mg/ml in DMSO. No toxicity was observed up to 100 μM for SB-505124 (DaCosta Byfield et al., 2004). Embryos were treated with cyclopamine, SB-505124 or both included in the culture medium continuously, starting from stage 4 to stage 6. Control embryos were treated with DMSO (n=352) or HBC (n=115). In our culture conditions, about 90% of the embryos were morphologically normal; the only visible abnormalities in the other 10% were localized failures of neural tube closure. However, the anatomical regionalization and gene expression of Pax6, Nkx2.1 and Shh were in all cases apparently normal.
Whole-mount in situ hybridization
Antisense digoxigenin-labeled riboprobes were used for whole-mount in situ hybridization on chicken embryos as previously described (Chapman et al., 2002). Plasmids carrying chick Shh, Nkx2.1 and Pax6 were used as templates to generate the antisense riboprobes.
Statistical analysis
Variables are expressed as numbers and percentages. Fisher's exact tests were used for comparisons. We used a type I error of 0.05, by convention.
Acknowledgments
FUNDING: This work was supported by the Centre National de la Recherche Scientifique (CNRS), the Institut National de la Santé et de la Recherche Médicale (INSERM) and the University of Rennes.
Footnotes
COMPETING INTERESTS: The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS: L.R., I.G. and V. Dupé set up and designed the experiment. S.M. and I.G. performed the experiments. S.M., V. Dupé and V. David wrote the manuscript, and all authors read, discussed and edited the manuscript.
References
- Allen B. L., Tenzen T., McMahon A. P. (2007). The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 21, 1244-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson R. M., Lawrence A. R., Stottmann R. W., Bachiller D., Klingensmith J. (2002). Chordin and noggin promote organizing centers of forebrain development in the mouse. Development 129, 4975-4987 [DOI] [PubMed] [Google Scholar]
- Andersson O., Reissmann E., Jörnvall H., Ibánez C. F. (2006). Synergistic interaction between Gdf1 and Nodal during anterior axis development. Dev. Biol. 293, 370-381 [DOI] [PubMed] [Google Scholar]
- Arauz R. F., Solomon B. D., Pineda-Alvarez D. E., Gropman A. L., Parsons J. A., Roessler E., Muenke M. (2010). A hypomorphic allele in the FGF8 gene contributes to holoprosencephaly and is allelic to gonadotropin-releasing hormone deficiency in humans. Mol. Syndromol. 1, 59-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae G. U., Domené S., Roessler E., Schachter K., Kang J. S., Muenke M., Krauss R. S. (2011). Mutations in CDON, encoding a hedgehog receptor, result in holoprosencephaly and defective interactions with other hedgehog receptors. Am. J. Hum. Genet. 89, 231-240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman S. C., Schubert F. R., Schoenwolf G. C., Lumsden A. (2002). Analysis of spatial and temporal gene expression patterns in blastula and gastrula stage chick embryos. Dev. Biol. 245, 187-199 [DOI] [PubMed] [Google Scholar]
- Chen J. K., Taipale J., Cooper M. K., Beachy P. A. (2002). Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 16, 2743-2748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang C., Litingtung Y., Lee E., Young K. E., Corden J. L., Westphal H., Beachy P. A. (1996). Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature 383, 407-413 [DOI] [PubMed] [Google Scholar]
- Cole F., Krauss R. S. (2003). Microform holoprosencephaly in mice that lack the Ig superfamily member Cdon. Curr. Biol. 13, 411-415 [DOI] [PubMed] [Google Scholar]
- Conlon F. L., Lyons K. M., Takaesu N., Barth K. S., Kispert A., Herrmann B., Robertson E. J. (1994). A primary requirement for nodal in the formation and maintenance of the primitive streak in the mouse. Development 120, 1919-1928 [DOI] [PubMed] [Google Scholar]
- Cooper M. K., Porter J. A., Young K. E., Beachy P. A. (1998). Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 280, 1603-1607 [DOI] [PubMed] [Google Scholar]
- Cordero D., Marcucio R., Hu D., Gaffield W., Tapadia M., Helms J. A. (2004). Temporal perturbations in sonic hedgehog signaling elicit the spectrum of holoprosencephaly phenotypes. J. Clin. Invest. 114, 485-494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossley P. H., Martinez S., Ohkubo Y., Rubenstein J. L. (2001). Coordinate expression of Fgf8, Otx2, Bmp4, and Shh in the rostral prosencephalon during development of the telencephalic and optic vesicles. Neuroscience 108, 183-206 [DOI] [PubMed] [Google Scholar]
- DaCosta Byfield S., Major C., Laping N. J., Roberts A. B. (2004). SB-505124 is a selective inhibitor of transforming growth factor-beta type I receptors ALK4, ALK5, and ALK7. Mol. Pharmacol. 65, 744-752 [DOI] [PubMed] [Google Scholar]
- Dubourg C., Lazaro L., Pasquier L., Bendavid C., Blayau M., Le Duff F., Durou M. R., Odent S., David V. (2004). Molecular screening of SHH, ZIC2, SIX3, and TGIF genes in patients with features of holoprosencephaly spectrum: Mutation review and genotype-phenotype correlations. Hum. Mutat. 24, 43-51 [DOI] [PubMed] [Google Scholar]
- Dubourg C., Bendavid C., Pasquier L., Henry C., Odent S., David V. (2007). Holoprosencephaly. Orphanet J. Rare Dis. 2, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupé V., Lumsden A. (2001). Hindbrain patterning involves graded responses to retinoic acid signalling. Development 128, 2199-2208 [DOI] [PubMed] [Google Scholar]
- Dupé V., Rochard L., Mercier S., Le Pétillon Y., Gicquel I., Bendavid C., Bourrouillou G., Kini U., Thauvin-Robinet C., Bohan T. P., et al. (2011). NOTCH, a new signaling pathway implicated in holoprosencephaly. Hum. Mol. Genet. 20, 1122-1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes M., Hébert J. M. (2008). The ups and downs of holoprosencephaly: dorsal versus ventral patterning forces. Clin. Genet. 73, 413-423 [DOI] [PubMed] [Google Scholar]
- Geng X., Oliver G. (2009). Pathogenesis of holoprosencephaly. J. Clin. Invest. 119, 1403-1413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng X., Speirs C., Lagutin O., Inbal A., Liu W., Solnica-Krezel L., Jeong Y., Epstein D. J., Oliver G. (2008). Haploinsufficiency of Six3 fails to activate Sonic hedgehog expression in the ventral forebrain and causes holoprosencephaly. Dev. Cell 15, 236-247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gritsman K., Talbot W. S., Schier A. F. (2000). Nodal signaling patterns the organizer. Development 127, 921-932 [DOI] [PubMed] [Google Scholar]
- Hagos E. G., Dougan S. T. (2007). Time-dependent patterning of the mesoderm and endoderm by Nodal signals in zebrafish. BMC Dev. Biol. 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamburger V., Hamilton H. L. (1992). A series of normal stages in the development of the chick embryo. 1951. Dev. Dyn. 195, 231-272 [DOI] [PubMed] [Google Scholar]
- Higashiyama D., Saitsu H., Komada M., Takigawa T., Ishibashi M., Shiota K. (2007). Sequential developmental changes in holoprosencephalic mouse embryos exposed to ethanol during the gastrulation period. Birth Defects Res. A Clin. Mol. Teratol. 79, 513-523 [DOI] [PubMed] [Google Scholar]
- Hu D., Helms J. A. (1999). The role of sonic hedgehog in normal and abnormal craniofacial morphogenesis. Development 126, 4873-4884 [DOI] [PubMed] [Google Scholar]
- Iannaccone P. M., Zhou X., Khokha M., Boucher D., Kuehn M. R. (1992). Insertional mutation of a gene involved in growth regulation of the early mouse embryo. Dev. Dyn. 194, 198-208 [DOI] [PubMed] [Google Scholar]
- Incardona J. P., Gaffield W., Kapur R. P., Roelink H. (1998). The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 125, 3553-3562 [DOI] [PubMed] [Google Scholar]
- Kiecker C., Niehrs C. (2001). The role of prechordal mesendoderm in neural patterning. Curr. Opin. Neurobiol. 11, 27-33 [DOI] [PubMed] [Google Scholar]
- Krauss R. S. (2007). Holoprosencephaly: new models, new insights. Expert Rev. Mol. Med. 9, 1-17 [DOI] [PubMed] [Google Scholar]
- Lawson A., Colas J. F., Schoenwolf G. C. (2001). Classification scheme for genes expressed during formation and progression of the avian primitive streak. Anat. Rec. 262, 221-226 [DOI] [PubMed] [Google Scholar]
- Lipinski R. J., Hutson P. R., Hannam P. W., Nydza R. J., Washington I. M., Moore R. W., Girdaukas G. G., Peterson R. E., Bushman W. (2008). Dose- and route-dependent teratogenicity, toxicity, and pharmacokinetic profiles of the hedgehog signaling antagonist cyclopamine in the mouse. Toxicol. Sci. 104, 189-197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe L. A., Yamada S., Kuehn M. R. (2001). Genetic dissection of nodal function in patterning the mouse embryo. Development 128, 1831-1843 [DOI] [PubMed] [Google Scholar]
- Luxardi G., Marchal L., Thomé V., Kodjabachian L. (2010). Distinct Xenopus Nodal ligands sequentially induce mesendoderm and control gastrulation movements in parallel to the Wnt/PCP pathway. Development 137, 417-426 [DOI] [PubMed] [Google Scholar]
- Marcorelles P., Laquerriere A. (2010). Neuropathology of holoprosencephaly. Am. J. Med. Genet. 154C, 109-119 [DOI] [PubMed] [Google Scholar]
- Marcucio R. S., Cordero D. R., Hu D., Helms J. A. (2005). Molecular interactions coordinating the development of the forebrain and face. Dev. Biol. 284, 48-61 [DOI] [PubMed] [Google Scholar]
- Mercier S., Dubourg C., Garcelon N., Campillo-Gimenez B., Gicquel I., Belleguic M., Ratié L., Pasquier L., Loget P., Bendavid C., et al. (2011). New findings for phenotype-genotype correlations in a large European series of holoprosencephaly cases. J. Med. Genet. 48, 752-760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monuki E. S. (2007). The morphogen signaling network in forebrain development and holoprosencephaly. J. Neuropathol. Exp. Neurol. 66, 566-575 [DOI] [PubMed] [Google Scholar]
- Muenke M., Beachy P. A. (2000). Genetics of ventral forebrain development and holoprosencephaly. Curr. Opin. Genet. Dev. 10, 262-269 [DOI] [PubMed] [Google Scholar]
- Müller F., Albert S., Blader P., Fischer N., Hallonet M., Strähle U. (2000). Direct action of the nodal-related signal cyclops in induction of sonic hedgehog in the ventral midline of the CNS. Development 127, 3889-3897 [DOI] [PubMed] [Google Scholar]
- Nanni L., Ming J. E., Bocian M., Steinhaus K., Bianchi D. W., Die-Smulders C., Giannotti A., Imaizumi K., Jones K. L., Campo M. D., et al. (1999). The mutational spectrum of the sonic hedgehog gene in holoprosencephaly: SHH mutations cause a significant proportion of autosomal dominant holoprosencephaly. Hum. Mol. Genet. 8, 2479-2488 [DOI] [PubMed] [Google Scholar]
- Nomura M., Li E. (1998). Smad2 role in mesoderm formation, left-right patterning and craniofacial development. Nature 393, 786-790 [DOI] [PubMed] [Google Scholar]
- Pei W., Feldman B. (2009). Identification of common and unique modifiers of zebrafish midline bifurcation and cyclopia. Dev. Biol. 326, 201-211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pera E. M., Kessel M. (1997). Patterning of the chick forebrain anlage by the prechordal plate. Development 124, 4153-4162 [DOI] [PubMed] [Google Scholar]
- Ribeiro L. A., Quiezi R. G., Nascimento A., Bertolacini C. P., Richieri-Costa A. (2010). Holoprosencephaly and holoprosencephaly-like phenotype and GAS1 DNA sequence changes: Report of four Brazilian patients. Am. J. Med. Genet. 152A, 1688-1694 [DOI] [PubMed] [Google Scholar]
- Roessler E., Muenke M. (2010). The molecular genetics of holoprosencephaly. Am. J. Med. Genet. 154C, 52-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E., Ouspenskaia M. V., Karkera J. D., Vélez J. I., Kantipong A., Lacbawan F., Bowers P., Belmont J. W., Towbin J. A., Goldmuntz E., et al. (2008). Reduced NODAL signaling strength via mutation of several pathway members including FOXH1 is linked to human heart defects and holoprosencephaly. Am. J. Hum. Genet. 83, 18-29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E., Pei W., Ouspenskaia M. V., Karkera J. D., Veléz J. I., Banerjee-Basu S., Gibney G., Lupo P. J., Mitchell L. E., Towbin J. A., et al. (2009). Cumulative ligand activity of NODAL mutations and modifiers are linked to human heart defects and holoprosencephaly. Mol. Genet. Metab. 98, 225-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E., Vélez J. I., Zhou N., Muenke M. (2012). Utilizing prospective sequence analysis of SHH, ZIC2, SIX3 and TGIF in holoprosencephaly probands to describe the parameters limiting the observed frequency of mutant gene×gene interactions. Mol. Genet. Metab. 105, 658-664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohr K. B., Barth K. A., Varga Z. M., Wilson S. W. (2001). The nodal pathway acts upstream of hedgehog signaling to specify ventral telencephalic identity. Neuron 29, 341-351 [DOI] [PubMed] [Google Scholar]
- Rubenstein J. L., Beachy P. A. (1998). Patterning of the embryonic forebrain. Curr. Opin. Neurobiol. 8, 18-26 [DOI] [PubMed] [Google Scholar]
- Shen M. M. (2007). Nodal signaling: developmental roles and regulation. Development 134, 1023-1034 [DOI] [PubMed] [Google Scholar]
- Strähle U., Jesuthasan S., Blader P., Garcia-Villalba P., Hatta K., Ingham P. W. (1997). one-eyed pinhead is required for development of the ventral midline of the zebrafish (Danio rerio) neural tube. Genes Funct. 1, 131-148 [DOI] [PubMed] [Google Scholar]
- Vincent S. D., Dunn N. R., Hayashi S., Norris D. P., Robertson E. J. (2003). Cell fate decisions within the mouse organizer are governed by graded Nodal signals. Genes Dev. 17, 1646-1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y. P., Anderson R. M., Klingensmith J. (2010). BMP antagonism protects Nodal signaling in the gastrula to promote the tissue interactions underlying mammalian forebrain and craniofacial patterning. Hum. Mol. Genet. 19, 3030-3042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. M., Chong H. J., Hu D., Hallgrímsson B., Marcucio R. S. (2010). Quantitative analyses link modulation of sonic hedgehog signaling to continuous variation in facial growth and shape. Development 137, 3405-3409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W., Hong M., Bae G. U., Kang J. S., Krauss R. S. (2011). Boc modifies the holoprosencephaly spectrum of Cdo mutant mice. Dis. Model. Mech. 4, 368-380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X., Sasaki H., Lowe L., Hogan B. L., Kuehn M. R. (1993). Nodal is a novel TGF-beta-like gene expressed in the mouse node during gastrulation. Nature 361, 543-547 [DOI] [PubMed] [Google Scholar]