

SUMMARY
Mutations in the WAS gene cause Wiskott-Aldrich syndrome (WAS), which is characterized by eczema, immunodeficiency and microthrombocytopenia. Although the role of WASP in lymphocytes and myeloid cells is well characterized, its role on megakaryocyte (MK) development is poorly understood. In order to develop a human cellular model that mimics the megakaryocytic-derived defects observed in WAS patients we used K562 cells, a well-known model for study of megakaryocytic development. We knocked out the WAS gene in K562 cells using a zinc-finger nuclease (ZFN) pair targeting the WAS intron 1 and a homologous donor DNA that disrupted WASP expression. Knockout of WASP on K562 cells (K562WASKO cells) resulted in several megakaryocytic-related defects such as morphological alterations, lower expression of CD41ɑ, lower increments in F-actin polymerization upon stimulation, reduced CD43 expression and increased phosphatidylserine exposure. All these defects have been previously described either in WAS-knockout mice or in WAS patients, validating K562WASKO as a cell model for WAS. However, K562WASPKO cells showed also increased basal F-actin and adhesion, increased expression of CD61 and reduced expression of TGFβ and Factor VIII, defects that have never been described before for WAS-deficient cells. Interestingly, these phenotypic alterations correlate with different roles for WASP in megakaryocytic differentiation. All phenotypic alterations observed in K562WASKO cells were alleviated upon expression of WAS following lentiviral transduction, confirming the role of WASP in these phenotypes. In summary, in this work we have validated a human cellular model, K562WASPKO, that mimics the megakaryocytic-related defects found in WAS-knockout mice and have found evidences for a role of WASP as regulator of megakaryocytic differentiation. We propose the use of K562WASPKO cells as a tool to study the molecular mechanisms involved in the megakaryocytic-related defects observed in WAS patients and as a cellular model to study new therapeutic strategies.
INTRODUCTION
Wiskott-Aldrich syndrome (WAS) is an X-linked inherited disease that combines immunodeficiency and platelet dysfunction. The immune defects are of varying clinical severity, depending on the mutation in the WAS gene. However, the platelet defects (profound thrombocytopenia with small platelets) are a consistent feature of this monogenic disease (Sullivan et al., 1994). The role of WASP in immune-related defects is well characterized. In leucocytes, WASP binds the actin-related protein complex 2/3 (Arp2/3), allowing actin nucleation and the generation of new actin filaments (Gallego et al., 1997). Therefore, WASP-deficient leucocytes are impaired in responses requiring actin filament remodeling such as directed migration, immune synapse formation and proliferative responses (Ochs, 1998).
In contrast to leucocytes, the function of WASP in megakaryocytes (MKs) and platelets is controversial. The absence of a mouse model that mimics the MKs and platelet defects found in WAS patients, combined with the limited availability of patients' bone marrow and their small number of platelets (Gröttum et al., 1969) have precluded further advances in the understanding of WASP function in MK physiology (Strom, 2009). Different groups have found different results in almost all aspects of MK physiology. Several studies describe normal MK development in WAS patients, arguing platelet clearance through recognition of phosphatidylserine (PS) in WASP-deficient platelets as the main mechanism explaining thrombocytopenia (Gröttum et al., 1969; Haddad et al., 1999; Rengan et al., 2000; Shcherbina et al., 1999). However, platelet clearance cannot explain all platelet-derived defects observed in WAS patients. Indeed, splenectomy-treated patients can partially restored platelet defects but bleeding still remains and it does not restore completely the numbers, size or function of platelets (Litzman et al., 1996). Sabri et al. found increased numbers of MKs in the bone marrow that produced irregular proplatelets, indicating a premature differentiation of MK precursors (Sabri et al., 2006). Other groups have also reported abnormal proplatelet formation (Luthi et al., 2003; Schulze et al., 2006) that generates smaller platelets with lower number of granules and mitochondria. The role of WASP in platelet activation and function is also controversial. Several groups have reported that WASP-deficient platelets have normal agonist-induced responses (shape change and actin polymerization) as well as normal elaboration of filopodia and lamellipodia (Gross et al., 1999; Rengan et al., 2000). Other groups have reported low adhesion and aggregation (Gröttum et al., 1969; Tsuboi et al., 2006) whereas yet others have found normal shape changes but increased aggregation and increased microparticle release (Gross et al., 1999; Shcherbina et al., 2001; Shcherbina et al., 1999). Understanding the role of WASP in MK physiology has been made more difficult by these seemingly contradictory results.
RESOURCE IMPACT.
Background
Microthrombocytopenia (a decrease in the number and size of platelets) is an invariable characteristic of Wiskott-Aldrich Syndrome (WAS), a primary immunodeficiency caused by mutations in the WAS gene that cause the absence or inactivity of the WASP protein. WASP is a hematopoietic-specific signaling molecule that integrates extracellular signals with actin cytoskeleton rearrangements. Although the role of WASP in lymphocytes and myeloid cells is well characterized, its role in the development of megakaryocytes (the bone marrow cells that gives rise to platelets) is poorly understood, in part because WAS-knockout mice models exhibit only mild thrombocytopenia and have platelets of a normal size.
Results
To obtain a human cellular model that mimics the megakaryocytic-derived defects observed in patients with WAS, the authors knocked out the WAS gene in K562 cells, a human leukemia cell line that produces megakaryocytes on activation with PMA. Specifically, the authors used zinc finger nucleases (ZFNs) to introduce several modifications into the WAS gene that block WASP expression. WASP-knockout K562 cells show several megakaryocytic-related defects previously described in mice models and in patients with WAS, including morphological alterations and altered F-actin re-organization on activation with PMA. WASP-knockout K562 cells also show some additional phenotypes not previously associated with WASP deficiency, such as increased basal levels of polymerized F-actin and enhanced adhesion in the absence of PMA activation. Finally, the authors show that expression of WASP in WASP-knockout K562 cells using lentiviral vectors alleviates all of these phenotypic alterations.
Implications and future directions
These findings introduce WASP-knockout K562 cells as a new model for studying the role of WASP in megakaryocytic development. This cellular model mimics the defects previously found in megakaryocytes isolated from patients with WAS and provides data suggesting that WASP has two separate roles in megakaryocytic differentiation – as a protein required to complete normal terminal megakaryocytic differentiation, and as a negative regulator of megakaryocytic activation and differentiation in the absence of the appropriate signals. Further investigations using WASP-knockout K562 cells will contribute to the understanding of the molecular mechanism involved in these processes. In addition, these findings also introduce the first ZFNs to target the WAS gene. These ZFNs provide a new tool to model WAS in other human cell lines, including embryonic stem cells, and should allow the development of human cellular models of any WAS mutation described previously in patients.
The human leukemia cell line K562, derived from a chronic myelogenous leukemia patient, resembles a bipotent megakaryocytic-erythroid progenitor (MEP) (Alitalo, 1990). Activation of K562 cells with phorbol esters such as phorbol 12-myristate 13-acetate (PMA) induces MK differentiation, whereas hemin or sodium butyrate induces differentiation to erythrocytes. Upon activation with PMA, K562 cells undergo biochemical and cytological changes similar to those observed during MK differentiation in the bone marrow. These changes include increased cell size, cell growth inhibition, cytoplasmic vacuolation, endomitosis, expression of MK surface markers such as integrin αIIbβ3 (CD41α and CD61) and integrin 2α, increased adhesive capacity and secretion of factors such as platelet-derived growth factor (PDGF) and Factor VIII (Herrera et al., 1998; Whalen et al., 1997). The accessibility of K562 cells to the scientific community and its easy culture have made PMA-treated K562 cells an important model for studying MK development. K562 cells have been fundamental in uncovering molecular mechanisms involved in megakaryocytic differentiation (Chang et al., 2010; Navarro et al., 2009; Randrianarison-Huetz et al., 2010; Sardina et al., 2010). For instance, K562 cells have been instrumental in uncovering the role of ERK1/2, p38 and PI3K signaling pathways in the development of mature MKs (Conde et al., 2010; Chang et al., 2010).
Modeling human genetic disease by inactivating specific genes or introducing specific mutations has been very limited due to the low rates of homologous recombination (10−6) in human cells (Vasquez et al., 2001). The introduction of a targeted double-strand DNA break by zinc-finger nucleases (ZFNs) to increase the rate of homologous recombination has boosted the gene targeting field (Urnov et al., 2005). The specific targeting provided by the ZFN allows gene disruption, gene correction or gene addition in cell lines and primary cells (Hockemeyer et al., 2009; Holt et al., 2010; Moehle et al., 2007), providing an excellent tool for modeling human disease. Here, we have used ZFNs specifically targeting the WAS locus to develop WASP-knockout K562 (K562WASPKO) cells. K562WASPKO cells mimic several of the defects found in MKs from WAS patients. Indeed, PMA-driven induction of megakaryocytic differentiation proceeded abnormally in K562WASPKO cells, as evidenced by alterations in cell morphology and cytoskeleton reorganization. We also observed lower expression of CD41, PDGF and Factor VIII. Interestingly, we have also found upregulation of CD61, increased adhesion, downregulation of CD43 and enhanced PS exposure on the surface of PMA-treated K562WASPKO cells compared with PMA-treated parental K562 cells. All these phenotypic alterations were restored upon expression of WASP by lentiviral transduction, demonstrating that WASP plays a direct role in the alterations observed in K562WASPKO cells.
RESULTS
Generation of K562WASPKO cells by homologous directed repair using ZFNs
In order to generate a human cell-based model in which to study the role of WASP in megakaryocytic development we edited the WAS locus of K562 cells using a pair of ZFNs targeting WAS intron 1 (Fig. 1A, top) and (supplementary material Fig. S1) and a homologous donor DNA (Fig. 1A, middle; supplementary material Fig. S2) that, when used to repair the locus by HDR, disrupts WASP expression by introducing mutations in exons 1 and 2 (Fig. 1A, bottom) (see Materials and Methods for details). The donor DNA also includes a neo expression cassette to allow selection of successfully targeted cells. K562 cells were nucleofected with both WAS-specific ZFNs, each expressed from a separate plasmid, and a third plasmid encoding the donor template DNA (Fig. 1A). Homology-directed repair (HDR)-driven modification of WAS was evaluated in the bulk population and in nine clones resistant to the antibiotic G418 (Fig. 1B). PCR analysis of both the bulk population and G418-resistant clones showed that the neo cassette had inserted in to the WAS locus in the correct orientation, as evidenced by the amplification of a 1.7-kb band using the WASF1/NeoR1 primer pair (Fig. 1B, top panel). Because K562 cells are derived from a female donor and contain two X-chromosomes, we analyzed whether modification occurred on one (heterozygous) or both (homozygous) copies of the WAS locus on the X-chromosome. As shown in Fig. 1B (bottom panel), two out of nine clones (clones 3 and 8) were homozygous, as demonstrated by the appearance of the 3.5-kb band (indicative of insertion of the donor DNA in the WAS locus) using WASF1/WASR1 primers pair (see Fig. 1A, bottom). In the PCR conditions that were used to evaluate whether cells were mono- or biallelically modified, smaller PCR fragments are favored for amplification, therefore, only the smaller 2-kb band is detectable in the heterozygous clones, corresponding to the wild-type locus. Analysis of other clones (30 in total) demonstrated that 20% of targeted K562 clones were homozygous, containing the desired modifications (supplementary material Figs S3, S4). The selection with G418 could in theory allow the selection of cells that have the donor DNA inserted in loci other than the WAS locus due to random integration. However, we did not detect any G418-positive clones other than those that have proper integration at least in one X chromosome. In one clone (clone 5), we observed a band that was one third of the expected size when using the WASF1/WASR1 pair for amplification (Fig. 1B, bottom panel), although the pair WASF1/NeoR1 rendered the expected size (Fig. 1B, top panel). This data indicates that clone 5 is heterozygous, with a neo cassette integrated in the WAS locus of one X chromosome via homologous recombination (HR) with the donor DNA, and a large deletion (>1 kb) in the other X chromosome (probably due to non-homologous end joining, NHEJ).
Fig. 1.

Efficient targeting of the WAS locus by ZFNs. (A) Strategy to disrupt WAS expression. Upper diagram illustrates the first 6 exons of the WAS locus (E1-E6), the sequences recognized by the ZFN pair (ZFN1 and ZFN2, in gray), the homologous sequences used to promote homologous recombination (H.arm5′ and Harm3′) and the primer pairs used to identify the wild-type locus (rendering a 2-kb fragment). The middle diagram illustrates the donor DNA used to disrupt WAS expression. Exon 1 has a deletion in the ATG codon used to initiate translation (E1*). A neo expression cassette (SV40-neo-pA) has been introduced to allow antibiotic selection. Lower diagram shows the ‘edited’ WAS locus after homologous recombination with the donor DNA. Arrows indicate the primers used to identify clones modified by HDR. (B) Successful gene editing of K562 cells. The upper agarose gel shows the analysis of nine neomycin-resistant K562 clones (out of 30 analyzed), wild-type K562 cells (C-) and bulk population (Bulk) using the WASF/NeoR primers. A band of 1.7 kb appears in all clones, indicating that homologous recombination has occurred in at least one X chromosome. In the lower gel, the same samples were analyzed using the WASF/WASR primer pair to identify whether the clones where homozygous (HR has occurred in both X chromosomes) or heterozygous (HR has occurred only in one X chromosome). Clones 3 and 8 were homozygous, all other clones were heterozygous as shown by the appearance of a 2-kb band. (C) Homozygous WAS-targeted clones do not express WAS mRNA. Graph shows WAS mRNA levels in selected homozygous clones. Bulk populations, clones 3 and 8 from B and three other homozygous clones (12, 22 and 27; see supplementary material Fig. S3) were analyzed by RT-qPCR. Results were normalized with GAPDH and compared with WAS expression levels in wild-type K562 cells (WT). (D) Homozygous WAS-targeted clones do not express WASP protein. Western blot showing complete knockout of WASP protein in the five homozygous clones analyzed. Wild-type K562 cells were used as positive control (C+). The membranes were incubated with mouse anti-human monoclonal antibody anti-WASP (D1 clone) and rabbit anti-human anti-ERK-1 monoclonal antibody.
The modifications encoded by the WAS donor template should cause premature termination of transcription due to the insertion of a polyadenylation site downstream of the neomycin-resistance gene (supplementary material Fig. S2). Therefore, WASP expression in homozygous clones should be completely abrogated. We verified the absence of WASP expression using real-time quantitative polymerase chain reaction (RT-qPCR) and western blot analysis (Fig. 1C,D) in all clones homozygous for HDR-driven modification. WASP expression in the bulk population was reduced by ∼55% (as determined by qPCR; Fig. 1C), presumably due to the combined effects of a reduction or elimination of WASP expression in heterozygous and unmodified cell populations, respectively.
WASP-deficient K562 cells exhibit impaired actin reorganization, enhanced adhesion and downregulation of CD43
Although the role of WASP in cytoskeleton reorganization in lymphocytes and myeloid cells is well established, its role in megakaryocytic physiology is controversial. We therefore analyzed changes in polymerized actin in K562 and K562WASPKO cells activated with PMA (Fig. 2A,B). Fluorescence microphotographs of PMA-treated cells demonstrated clear differences in F-actin distribution in WASP-deficient cells compared with wild-type K562 cells (Fig. 2A, compare top panels with bottom panels). K562WASPKO cells have a more irregular shape and a higher number of F-actin protrusions (hairy cell surface) than wild-type K562 cells (Fig. 2A, bottom panels). Interestingly, FACS analysis of F-actin content showed that K562WASPKO cells also have increased basal levels of polymerized actin compared with K562 cells (Fig. 2B, left graph, No PMA). However, the increase in polymerized actin upon PMA treatment was lower in K562WASPKO cells (Fig. 2B, right graph) than in the parental K562 cell line. The data indicate that K562WASPKO cells could have weaker responses to those stimuli that require an increment in polymerized actin.
Fig. 2.

WASP deficiency produces aberrant F-actin distribution, increased adhesion and downregulation of CD43 during megakaryocytic differentiation. (A) Aberrant F-actin localization in WASP-deficient K562 cells. Wild-type (WT) and K562WASPKO clones (Hm1, Hm2) were cultured at 2×105 cells/ml into chamber slides and stimulated with 30 nM of PMA for 96 hours. The cells were stained with rhodamine-phalloidin to visualize F-actin filaments. Arrows indicate F-actin protrusions. Pictures were acquired using the EVOS fluorescence microscope at 40×. (B) Altered F-actin polymerization in WASP-deficient K562 cells. Wild-type K562 (WT) and K562WASPKO (Hm1) cells were stained with rhodamine-phalloidin and analyzed by flow cytometry before and 96 hours after PMA stimulation. Left graph shows mean fluorescence intensity of wild-type K562 and K562WASPKO cells before and after PMA treatment as indicated. Right graph shows the increase in polymerized actin in wild-type K562 and K562WASPKO cells after PMA treatment. (C) K562WASPKO cells (Hm1 and Hm2) have increased unspecific adhesion. Adhesion of PMA-stimulated cells was evaluated by the RT-CES system. WT, Hm1 and Hm2 clones (5×104 cells) were plated into untreated 16-well plates, stimulated with PMA (30 nM) and cell adhesion was measured over 96 hours. The graph shows the cell index over time as a measure of the adhesiveness of the cells. (D) PMA-stimulated K562WASPKO cells have increased adhesion to fibrinogen-coated plates. WT, Hm1 and Hm2 cells (5×104) were stimulated for 96 hours with PMA and then plated onto fibrinogen-coated RT-CES plates. Cell index was monitored over 17 hours as for C. (E) PMA-stimulated and ADP-activated K562WASPKO cells have increased adhesion to fibrinogen-coated plates. WT, Hm1 and Hm2 cells (5×104) were stimulated for 96 hours with PMA, activated with ADP (50 μM, added at the time of plating), plated onto fibrinogen-coated RT-CES plates and the cell index monitored for 17 hours. (F) Minimal response of K562 (WT) and K562WASKO (Hm1 and Hm2) cells to ADP. The graph shows the increment in fibrinogen binding after addition of ADP. (G) K562WASPKO cells have decreased levels of surface CD43. Histograms show mean fluorescence intensity (MFI) of CD43 staining in PMA-stimulated cells (96 hours) (top). The graph shows the ratio between the MFI of CD43 in Hm1 and Hm2 clones related to wild-type cells. Data represent mean ± s.d. for three independent experiments; *P<0.05.
Altered F-actin reorganization could lead to defective adhesion to plastic or fibrinogen, one of the physiological changes occurring during megakaryocytic differentiation (Huang et al., 2004). We therefore studied the potential effect of knocking out WASP function on K562 adhesion upon megakaryocytic differentiation via PMA treatment. We used the Real-Time Cell Electronic Sensing system (RT-CES) that allows in vivo quantification of cell adhesion (Fig. 2C-F). To analyze unspecific adhesion, K562 cells and K562WASPKO clones Hm1 and Hm2 were treated with PMA and incubated for 96 hours in untreated RT-CES plates (Fig. 2C). K562WASPKO cells had higher cell index (indicative of higher adhesion) from the earliest times points, indicating that the enhanced adhesion did not require megakaryocytic differentiation. We next studied fibrinogen binding activity of the PMA-stimulated K562WASPKO clones in the absence or presence of ADP (an activator of αIIbβ3 integrin through inside-out signaling). The data showed that K562WASPKO cells bind surface-bound fibrinogen more efficiently than K562 cells in the absence (Fig. 2D) or presence (Fig. 2E) of activator. We could not detect a significant increase in binding to fibrinogen upon addition of ADP in either K562WASKO or K562 cells (Fig. 2F), probably indicating a drawback of this model in that it cannot mimic the inside-out activation of the αIIbβ3 integrin occurring in MK and platelets. However, we observed a four- to fivefold increment in cell adhesion in the presence of fibrinogen versus no coating in K562WASPKO and K562 cells (Fig. 2C-E; supplementary material Fig. S5). Together, these results suggest an inhibitory role for WASP on cell adhesion, affecting both plastic and fibrinogen-mediated adhesion.
Because it has been previously described that WAS patients had altered CD43 expression (Rocca et al., 1996; Shelley et al., 1989), a mucin involved in blocking cellular adhesion (Park et al., 2012; Remold-O'Donnell and Rosen, 1990), we studied whether the K562WASPKO cells also had altered CD43 expression. Interestingly, we observed a decrease in the expression levels of CD43 in K562WASPKO cells compared with K562 cells (Fig. 2G). This downregulation of CD43 expression coincides with the phenotype found in lymphocytes from WAS patients and could explain the increased adhesion observed in K562WASPKO cells.
K562WASPKO cells have impaired megakaryocytic differentiation and increased sensitivity to apoptosis
The megakaryocytic differentiation of K562 cells upon PMA stimulation is a well-characterized process in which the cells stop dividing, change morphology and start to express MK-specific genes. To study the potential role of WASP in megakaryocytic differentiation we treated K562WASPKO with PMA and analyzed the presence of differentiated cells (Fig. 3A; supplementary material Fig. S6). Differentiated K562 cells showed a marked increase in cell size and a nucleus located in the periphery (Fig. 3A, left panel). We observed a reduction in the number of MK-like cells in K562WASPKO cells (Fig. 3A, middle and right panels). Quantification of MK-like cells in wild-type cells and K562WASPKO clones confirmed that the differentiation potential of the WASP-deficient K562 cells was compromised (Fig. 3B). Cell cycle analysis of PMA-treated and untreated cells showed no significant differences, indicating that the morphological defects observed were not due to increased cell death under this conditions (supplementary material Fig. S7). In order to corroborate the impaired megakaryocytic differentiation of the WASP-deficient K562WASPKO cells, we analyzed other characteristic MK markers that are upregulated upon PMA treatment in wild-type K562 cells such as PDGF, Factor VIII, CD41α and CD61 (Fig. 3). All MK markers except CD61 were downregulated in K562WASPKO clones compared with wild-type K562 cells, supporting the morphological data and confirming the involvement of WASP in K562-MK differentiation. In contrast to CD41, PDGF and Factor VIII, we observed an increase in CD61 expression levels (Fig. 3, top-right graph) as well as in the percentage of cells expressing CD61 (Fig. 3E; supplementary material Fig. S8).
Fig. 3.
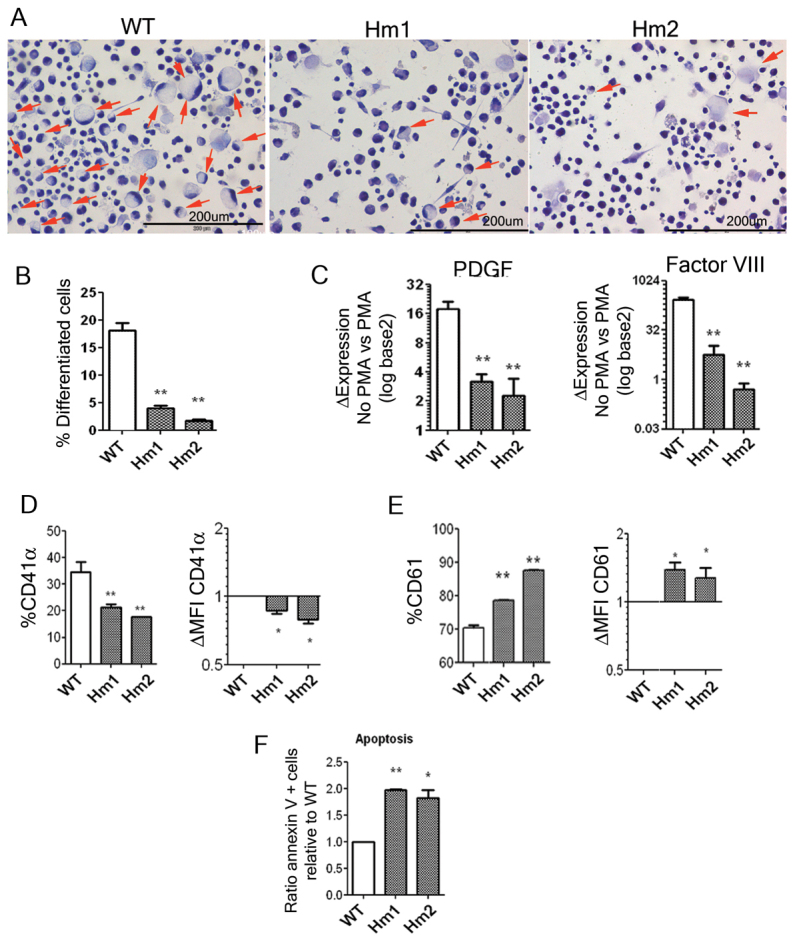
WASP-deficient K562 have impaired megakaryocytic differentiation. (A) Morphological analysis of PMA-treated K562WASPKO shows defective MK differentiation. Wild-type K562 cells (left panel) and K562WASPKO clones (middle and right panels) were treated with PMA and stained with Papanicolaou to evaluate cell morphology. Differentiated MK-like cells (arrows) were identified as bigger, round cells with a clear cytoplasm and edge-displaced nuclei. Pictures were acquired at 20× magnification. (B) Morphological quantification of megakaryocytic differentiation. (C) Defective expression of PDGF and Factor VIII in PMA-treated K562WASPKO cells. The expression of PDGF and Factor VIII upon PMA stimulation was analyzed by RT-qPCR in wild-type K562 cells (WT) and in K562WASPKO clones (Hm1 and Hm2). The graph shows increments in PDGF (left graph) and Factor VIII (right graph) in each cell line upon PMA treatment relative to basal conditions (without PMA). (D) Defective CD41 induction in K562WASPKO cells. K562 wild-type and K562WASPKO cells (homozygous clones, Hm1 and Hm2) were analyzed for CD41α expression by flow cytometry at 96 hours after PMA stimulation (30 nM). Left graph represents the percentage of CD41α-positive cells and right graph represent the increment in CD41 expression in K562WASPKO cells relative to wild-type K562 cells. (E) Enhanced CD61 induction in K562WASPKO cells. K562 (WT) and K562WASPKO cells (clones Hm1 and Hm2) were stimulated for 24 hours with 0.5 nM PMA and analyzed for CD61 expression by flow cytometry as in D. (F) Enhanced apoptosis in serum-starved K562WASPKO cells. Cells were grown in 1% FCS media for 7 days. PS exposure (apoptosis) was measured by flow cytometry after staining with annexin-V PE and 7AAD. The graph shows the increment in apoptosis of K562WASPKO cells (Hm1 and Hm2) relative to wild type (ratio of annexin-V-positive cells relative to wild type). Data represent mean ± s.d. for three independent experiments; *P<0.05, **P<0.005.
PS exposure is a marker for apoptosis and a signal for macrophage clearance of cells and cell debris. However, in normal platelets PS is a procoagulant signal in the late phase of plug formation. Platelets and MK from WAS patients have increased levels of PS that has been implicated in the high turnover of platelets from the bloodstream. We therefore analyzed PS exposure in K562 cells and K562WASPKO clones under serum deprivation. Interestingly, K562WASPKO cells were more sensitive and showed enhanced PS exposure compared with wild-type K562 cells, indicating an increased susceptibility to apoptosis (Fig. 3F). These data are in agreement with previous studies linking the loss of WASP to increased apoptosis and the premature cell death of MKs, leading to PS exposure in platelets.
Morphological and functional alterations of K562WASPKO cells are corrected by de novo expression of WASP
Differences in cellular behavior between wild-type K562 cells and the isolated WASP-knockout clonal lines could be due to clonal variability and/or potential side effects of drug selection. In order to confirm that the observed defects are due to the absence of WASP protein we restored its expression in a K562WASPKO line and analyzed the phenotype. The WASP-deficient cells were transduced with a lentiviral vector expressing WAS (WW) (Frecha et al., 2008) at a multiplicity of infection (MOI) of 1. All transduced cell lines were grown for 2 weeks before performing any further cell studies to avoid potential issues due to vector toxicity that could interfere with data collection. We evaluated morphology, adhesion, expression of PDGF, Factor VIII, CD41α and CD43 and apoptosis (Fig. 4). WW lentiviral transduction of K562WASPKO Hm1 cells (Hm1-WW) restored all the defects observed due to the disruption of WASP expression. The percentage of Hm1-WW cells that acquired MK-like morphology upon PMA treatment went to normal levels (Fig. 4A). Following transduction with the WW lentiviral vector, the adhesiveness of K562WASPKO cells returned to within the normal range, as can be observed by the drop in cell index to near wild-type levels (Fig. 4B). Interestingly, CD43 expression levels also increased in Hm1-WW cells to levels similar to those in wild-type cells (Fig. 4D). The expression levels of CD41α, PDGF and Factor VIII upon PMA stimulation were also restored in WW-transduced K562WASPKO cells (Fig. 4C). Finally, we analyzed PS exposure because it provides a measure of the rate of apoptosis and platelet clearance, which are two important aspects of MK physiology. WW-transduced K562WASPKO cells show an increased resistance to apoptosis, similar to that seen in parental K562 cells under conditions of serum starvation (Fig. 4E), corroborating the role of WASP as a negative regulator of apoptosis and/or PS exposure.
Fig. 4.

De novo WASP expression restores the defective MK differentiation of WASP-KO cells. Hm1 K562WASPKO cells were transduced at MOI 1 with lentiviral vectors expressing WAS(WW) and analyzed for the restoration of the alterations observed. (A) WW transduction restored the ability of K562WASPKO cells to form MK-like cells. Wild-type K562 cells (WT), K562WASPKO cells (Hm1) and WW-transduced K562WASPKO cells (Hm1-WW) were treated with PMA and stained with Papanicolaou to evaluate cell morphology. Arrows indicate nuclei located in the periphery. Pictures were acquired at 20× magnification. The graph shows the quantification of MK-like cells. (B) The expression of WASP reduces adhesion of K562WASPKO cells to normal levels. Adhesion of PMA-stimulated cells was evaluated by the RT-CES system. Wild-type K562 cells (WT), K562WASPKO cells (Hm1) and WW-transduced K562WASPKO cells (Hm1-WW) were plated into untreated 16-well RT-CES plates, stimulated with PMA and cell adhesion monitored in vivo over 96 hours (top graph). The graph on the right shows the increment in cell adhesion of untransduced (−) and WW-transduced (+) K562WASKO Hm1 cells related to K562 (WT). In a parallel experiment, the same cells were previously stimulated with PMA for 96 hours, activated with ADP (50 μM, added at the time of plating) and plated onto fibrinogen-coated RT-CES plates to monitor adhesion for 17 hours (bottom graphs). (C) WW transduction restored normal MK differentiation of K562WASPKO cells. Wild-type K562 (WT), untransduced (−) and WW-transduced (+) K562WASPKO cells were analyzed for the expression of PDGF and Factor VIII by RT-qPCR (top and middle graph, respectively) as well as for CD41 expression by flow cytometry (bottom graph). The top and bottom graphs show increments in PDGF and Factor VIII in each cell line upon PMA treatment relative to basal conditions (without PMA). The bottom graph shows the ratio between the percentage of CD41+ cells in Hm1 (−) and HM1-WW (+) related to wild-type cells. (D) WW transduction restored normal expression levels of CD43. The graph shows the ratio between the MFI of CD43 in Hm1 (−) and Hm1-WW (+) related to wild-type cells. (E) WW transduction restored the resistance of K562WASPKO cells to apoptosis. Wild-type K562 (WT), untransduced (−) and WW transduced (+) K562WASPKO cells were grown in 1% FCS media for 7 days. PS exposure (apoptosis) was measured by flow cytometry after staining with annexin-V PE and 7AAD. The graph shows the levels of annexin-V-positive cells of WW-transduced and untransduced K562WASPKO cells relative to wild type. Data represent mean ± s.d. for three independent experiments; *P<0.05.
DISCUSSION
Although the role of WASP in leukocytes has been very well characterized (Blundell et al., 2010), its role in megakaryocytic development and platelet physiology has remained controversial. The development of a suitable human cellular model capable of recapitulating the MK differentiation defects found in WAS patients will certainly help in elucidating the functional role that WASP plays in this process. To address this issue, we aimed to knock out the WAS gene in K562 cells, a well-established cell model that has been instrumental in uncovering the molecular mechanisms involved in MK differentiation (Alitalo, 1990; Conde et al., 2010; Navarro et al., 2009).
Modeling human genetic diseases by inactivating specific genes or introducing particular mutations has been limited due to the low efficiency of homologous recombination in human cells (Vasquez et al., 2001). ZFNs have been shown to stimulate the rates of HDR by inducing the creation of a double-strand break (DSB) at a specified location in the genome (Urnov et al., 2010). ZFN-driven genome editing has allowed the rapid development of cell lines and animal models harboring almost any desired alteration to the genome (Hockemeyer et al., 2009; Holt et al., 2010; Moehle et al., 2007; Urnov et al., 2005). A DSB originated by ZFNs can be repaired by non-homologous end-joining (NHEJ) or by homologous recombination (HR) depending on the absence or presence of homologous DNA sequences. Both systems can be used for gene disruption; however, error-prone NHEJ introduces a range of insertion or deletion mutations whereas HDR can be harnessed to make specific nucleotide changes encoded by the donor DNA template (Urnov et al., 2010).
We have designed a donor DNA that, when used by the HDR machinery as the template for repairing the endogenous WAS locus, introduces mutations that abrogate WAS transcription and translation. In addition, the donor template also encoded for the insertion of a neomycin-resistance gene at the WAS locus that allowed for the selection of K562 cells modified by HDR. Using this methodology, we have generated a WASP-deficient bulk population of K562 cells, 26 heterozygous clones (with only one X-chromosome targeted) and six homozygous clones with both copies of the WAS gene mutated in cells. All K562WASPKO homozygous and some of the heterozygous lines (data not shown) have complete abrogation of WAS expression at both the mRNA and protein levels. We generated cell lines from the homozygous WASP-knockout clones in order to study the role of WASP in MK physiology.
We first studied alterations in F-actin reorganization and adhesion properties upon PMA stimulation, a treatment that induces MK differentiation in K562 cells. The results confirmed a direct role of WASP protein in F-actin reorganization during K562 MK differentiation, because the PMA-induced increase in polymerized F-actin was reduced in WASP-deficient K562 cells as compared with wild-type K562 cells. However, the exact function of WASP in this process remains unknown because K562WASPKO cells are still able to respond to PMA treatment, leading to an increase in actin polymerization, presumably through one of the WASP-homologous proteins such as N-WASP or WAVE. Interestingly, our results also showed that untreated K562WASPKO cells showed an increase in the basal level of polymerized F-actin. This could indicate a possible role of WASP in K562 MK differentiation, blocking actin polymerization in the absence of proper stimuli. Our hypothesis is that, in basal conditions, inactive WASP sequesters different components of the actin-polymerization machinery. Therefore, the absence of WASP protein in basal conditions will free these factors, increasing actin polymerization. However, the absence of WASP will affect the way that the cell polymerizes actin. In line with this hypothesis, fluorescence images from PMA-stimulated K562WASPKO cells show that F-actin is distributed differently in these cells and that they possess a higher number of protrusions compared with wild-type K562 cells. Interestingly, similar differences in F-actin distribution have been reported in MK from WAS patients (Haddad et al., 1999).
Altered F-actin reorganization could lead to defective control of adhesion and aggregation. Indeed, as expected by the observation of increased F-actin content, PMA-stimulated K562WASPKO cells showed enhanced adhesiveness from very early stages after PMA stimulation. In order to study the mechanisms involved in the increased adhesion, we studied the expression levels of CD43, a mucin involved in the pathophysiology of WAS (Remold-O'Donnell and Rosen, 1990) that can confer anti-adherent properties to different cell types (Manjunath et al., 1993; Park et al., 2012; Sánchez-Mateos et al., 1995). We showed reduced surface expression of CD43 in K562WASPKO cells, indicating that this could be a factor contributing to the increased adhesion in these cells. The restoration of normal levels of CD43 and normal adhesiveness of K562WASPKO cells upon transduction with WW lentiviral vectors further point to a role of CD43 in maintaining low adhesiveness of K562 cells. All these data indicate a role for WASP in preventing actin polymerization and unspecific adhesion of K562 cells in the absence of the appropriated signals. These results open the possibility that WASP could play a similar role in MKs and platelets. However, this hypothesis should be confirmed with WAS cells and/or with other WASKO models.
The function of WASP in megakaryocytic differentiation is unknown and controversial. Some groups have reported normal megakaryocytic differentiation in WAS patients (Gröttum et al., 1969; Haddad et al., 1999) whereas others have found several alterations including abnormal proplatelet formation (Luthi et al., 2003; Schulze et al., 2006) and premature MK differentiation (Sabri et al., 2006). Kajiwara et al. showed that CD34+ cells from WAS patients generate lower numbers of MK colonies compared with CD34+ cells from healthy donors (Kajiwara et al., 1999). The same authors also showed a significant reduction in proplatelets formation from MK generated from these cells. All these data favor the hypothesis of a relevant role for WASP in normal MK development. In line with this theory, we observed several signs of impaired MK differentiation in WASP-deficient K562 cells upon PMA treatment. PMA-treated K562WASPKO cells had a reduced number of MK-like cells (as determined by their morphology) and decreased levels of MK markers such as CD41α (αIIb), PDGF and Factor VIII. Interestingly, CD41α downregulation has been observed in some WAS patients (Semple et al., 1997) although others have reported normal expression (Shcherbina et al., 2010; Tsuboi et al., 2006). Our results with the K562WASPKO model are in agreement with a role for WASP in megakaryocytic differentiation. This conclusion is further supported by the restoration of normal CD41α, PDGF and Factor VIII expression in K562WASPKO cells transduced with WW lentiviral vectors expressing WASP.
In contrast to the downregulation of CD41α, expression of CD61 (β3) was upregulated in K562WASPKO cells. This could be in line with the theory proposed by Prislovsky et al., where the CD61-mediated clearance of platelets could be an additional mechanism responsible for the thrombocytopenia phenotype (Prislovsky et al., 2008). Importantly, we also found increased PS exposure (apoptosis) after serum starvation and enhanced adhesion. These alterations could be interpreted as premature megakaryocytic differentiation and/or activation of K562WASPKO cells. Several groups have observed increased numbers of premature MKs in the bone marrow of WAS patients (Baldini, 1972; Sabri et al., 2006), hypothesizing that the absence of WASP could be required to negatively regulate megakaryocytic differentiation and/or activation. Sabri et al. showed that binding to fibrillar collagen type I (an important component of the bone marrow microenvironment) of wild-type MKs blocked proplatelet formation via a WASP-dependent mechanism (Sabri et al., 2006). Other publications favoring the hypothesis of a role for WASP in negatively regulating megakaryocytic differentiation are those describing the hyper-susceptibility of platelets to late phase activation events such as microparticle release and PS exposure (Gross et al., 1999; Rengan et al., 2000; Shcherbina et al., 2001). Interestingly, work from several groups has revealed that WASP is linked to the membrane skeleton in resting platelets and that, upon activation with pro-coagulating factors (thrombin and stirring), WASP is phosphorylated on Tyr291 and degraded through a calpain-dependent mechanism (Lutskiy et al., 2007; Shcherbina et al., 2001). This activation and subsequent degradation of WASP leads to a pro-coagulant phenotype (increased PS exposure, microparticle release and aggregation) that resembles the hyper-susceptibility phenotype seen in WAS-patient derived platelets and that observed in K562WASPKO cells. Therefore, we favor the hypothesis that the absence of WASP in K562 cells makes them hyper-susceptible to activation and/or differentiation. The increased apoptosis rate observed in K562WASPKO cells compared with wild-type cells is also consistent with the notion that WASP-deficiency can cause the premature death of MKs and with the exposure of PS in platelets, an important factors involved in thrombocytopenia.
MATERIALS AND METHODS
K562WASPKO cells
K562WASPKO cell lines Hm1 (K562WASPKO-1) and Hm2 (K562WASPKO-2) can be obtained from the SSPA Biobank (Biobanco del Sistema Sanitario Público de Andalucía, Armilla, Granada, Spain).
Plasmids encoding ZFN and donor plasmids
ZFNs targeting the WAS locus (pVax N2A-3FN-15724-FokEL-S4 WAS ZFN-L; pVax N2A-3FN-15755-FokKK-S4 WAS ZFN-R) were designed using an archive of prevalidated zinc finger modules and validated for genome editing activity by transient transfection into K562 cells and measurement via the Surveyor Nuclease Assay for endogenous activity as previously described (Moehle et al, 2007) (supplementary material Fig. S1). Donor plasmids were designed by M.G.T. and F.M. and synthesized by GeneArt (Life Technologies, Carlsbad, CA). All plasmids were produced using the Qiagen Endotoxin-Free Maxi Kit (Qiagen, Valencia, CA).
Cell lines and culture media and transfection
The K562 cell (chronic myelogenous leukemia) line was purchased from the ATCC (American Type Culture Collection, Manassas, VA; CCL-243), cultured in RPMI 1640 (Gibco-BRL, Middlesex, UK) supplemented with 10% fetal bovine serum (PAA, Pasching, Austria). ZFN (6 μg per ZFN) and donor plasmids (12-18 μg) were nucleo-transferred into 1×106 K562 cellsf using the Amaxa nucleofector II and solution V (Lonza, Basel, Switzerland) applying the program T16. The bulk population was first placed under antibiotic selection for 2 weeks (G418 at 50 μg/ml) followed by limited dilution to isolate clones. Identification of homozygous and heterozygous clones is shown in Fig. 1B and in supplementary material Figs S3 and S4.
Vector production and transduction of K562WASPKO cells
The human immunodeficiency virus (HIV) packaging (pCMVΔR8.91) and VSV-G (pMD2.G) plasmids (http://www.addgene.org/Didier_Trono) are described elsewhere (Zufferey et al, 1997). The lentiviral vector expressing WASP (WW) has been described previously (Martín et al., 2005). Vector production and concentration was performed as previously described (Toscano et al, 2008; Toscano et al, 2004). For transduction, K562WASPKO cells were incubated with WW lentiviral particles at MOI=1. The vector genome per cell of transduced human embryonic stem cells was calculated from genomic DNA of 100,000 transduced K562WASPKO cells and tenfold increasing amounts of plasmid DNA (from 102 to 107 copies) to determine the standard curve. The qPCR (Mx3005P, Stratagene, La Jolla, CA) reaction consisted of 40 cycles at 94°C (15 seconds), followed by 60°C (30 seconds) and 72°C (30 seconds). Primers used are depicted in supplementary material Table S1.
RNA preparation and quantitative real-time PCR
Genomic DNA was isolated as previously described. Total RNA was isolated using TRIzol (Invitrogen, Carlsbad, CA) following the manufacturer's instructions. Then, 2 μg of total RNA was reverse transcribed into cDNA using the MultiScribe Reverse Transcriptase (Applied Biosystems, Foster City, CA). Quantitative real-time PCR was performed in the Mx3005P Stratagene thermal cycler (Agilent Technologies, Santa Clara, CA). The real time PCR reactions were performed using the QuantiTect SYBR Green PCR Kit (Qiagen). Primers used are annotated in supplementary material Table S1.
Flow cytometry
For surface staining, 4×105 cells were collected and washed with cold PBS containing 0.1% sodium azide, stained with antibodies for CD41α (eBiosciences, San Diego, CA), CD61 (Dako, Glostrup, Denmark) and CD43 (Biolegend, San Diego, CA) and analyzed using a FACSCanto II flow cytometer (Becton Dickinson, Franklin Lakes, NJ). To measure PS exposure in K562 cells, we used the annexin-V PE apoptosis detection kit from Becton Dickinson (Becton Dickinson), following the manufacturer's instructions.
Apoptosis
In order to determine sensitivity to apoptosis, K562WASKO and K562 cells were grown in starving conditions (1% FCS in normal medium) for 7 days. The different cells were stained with annexin-V PE (PS-positive cells) and 7AAD (necrotic cells). We took as apoptotic those cells that were positive for annexin-V and negative for 7AAD.
Western blot
Cells were lysed with 0.2% NP-40 lysis buffer containing protease inhibitor cocktail (Sigma, St Louis, MO), resolved by SDS-PAGE (10% polyacrylamide gels, reducing conditions) and electrotransferred to nitrocellulose membranes (Amersham, UK). Membranes were blocked with 5% nonfat milk and probed for 1 hour at room temperature with 1 μg/ml of the mouse anti-human WASP monoclonal antibody D1 (Santa Cruz Biotechnology, Santa Cruz, CA) followed by incubation with goat anti-mouse antibody conjugated with the infrared dye 800CW (1:10,000 dilution) (Li-Cor Biotechnologies, Lincoln, NE). The blot was developed by the infrared detection system Odyssey (Li-Cor Biotechnologies). Loading controls were carried out by rehybridization of stripped membranes with a rabbit anti-human Erk polyclonal antibody (anti-MAP kinase 1/2; Upstate Biotechnology, Dundee, UK), followed by incubation with goat anti-rabbit antibody conjugated with the infrared dye 680LT at (1:20,000) (Li-Cor Biotechnologies).
Cytochemistry and immunostaining
To perform the cytological evaluation, the cells were grown in chamber slides, desiccated, fixed with 2% paraformaldehyde and stained using the Papanicolaou staining. The slides were analyzed under optical microscopy and the images acquired with an Olympus digital camera (Olympus, Japan).
For F-actin staining, K562 were desiccated and fixed in PBS containing 2% paraformaldehyde, permeabilized with 0.1% Triton X-100 and blocked in PBS containing 1% BSA for 30 minutes. Cells were incubated with rhodamine-labeled phalloidin (1:50 dilution) (Invitrogen) for 20 minutes to visualize F-actin. Preparations were mounted using the fluorescence protector Vectashield plus DAPI solution (Vector Laboratories, Burlingame, CA). Analysis and image acquisition were performed using the EVOS fl (AMG, Bothell, WA).
Real-time monitoring of K562 cell adhesion
To perform adhesion experiments, we used the Roche Real-Time Cell Electronic Sensing (RT-CES) system (Roche Applied Science, Penzberg, Germany). Cells (5×104) were plated into untreated or fibrinogen-coated (100 μg/ml) 16-well E-plates. For untreated plates, cells were stimulated with 30 nM of PMA and the experiment run for 96 hours in an incubator at 37°C, without change of media. For fibrinogen-coated plates, cells were pretreated with PMA for 96 hours to allow differentiation, plated in the absence or presence of ADP (50 μM) and monitored for 17 hours. Changes in cell adhesion were monitored and quantified by detecting the electrical impedance. Prior to adding the cells, 100 μl of complete media was added to obtain background readings, followed by the addition of 100 μl of cell suspension containing the indicated number of cells. The E-plates containing the cells were incubated at room temperature for 40 minutes before being placed on the device station in the incubator for continuous recording of impedance, as reflected by cell index.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Enrique Rubio (Roche Diagnostics SL., Sevilla, Spain) for his help with the Real-Time Cell Electronic Sensing (RT-CES) system.
FUNDING: This work has been financed by Fondo de Investigaciones Sanitarias ISCIII (Spain) and Fondo Europeo de Desarrollo Regional (FEDER) from the European Union [grant number PS09/00340], by the Consejería de Innovación Ciencia y Empresa [grant numbers P09-CTS-04532 and PAIDI-Bio-326] and by the Consejería de Salud [grant number PI0001/2009] from the Junta de Andalucía and FEDER/Fondo de Cohesion Europeo (FSE) de Andalucía 2007-2013 to F.M. M.C. and F.M. are funded by Fundación Progreso y Salud (Consejería de Salud, Junta de Andalucía). K.B. and M.G.T. are financed by the Consejería de Innovación Ciencia y Empresa [grant number P09-CTS-04532] and the Consejería de Salud [grant number PI0001/2009], respectively. P.A. and P.M. have a Miguel Servet [CP09/00228] and Sara Borrell contracts, respectively, from the Fondo de Investigaciones Sanitarias (FIS) – ISCIII.
Footnotes
Statistics: All the statistical analysis was performed with the GraphPad Prism 5 software. We applied the two-tail unpaired Student t-test.
COMPETING INTERESTS: The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS: F.M. conceived and coordinated the study and wrote the manuscript. M.G.T. performed most of the experiments and wrote the manuscript. P.A., P.M., G.L., M.C. and K.B. performed experiments under F.M. and M.G.T.'s supervision. P.D.G. and M.C.H. developed the ZFNs and helped with the gene targeting experiments. All authors read and approved the final manuscript.
SUPPLEMENTARY MATERIAL: Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.010652/-/DC1
References
- Alitalo R. (1990). Induced differentiation of K562 leukemia cells: a model for studies of gene expression in early megakaryoblasts. Leuk. Res. 14, 501–514 [DOI] [PubMed] [Google Scholar]
- Baldini M. G. (1972). Nature of the platelet defect in the Wiskott-Aldrich syndrome. Ann. N. Y. Acad. Sci. 201, 437–444 [DOI] [PubMed] [Google Scholar]
- Blundell M. P., Worth A., Bouma G., Thrasher A. J. (2010). The Wiskott-Aldrich syndrome: The actin cytoskeleton and immune cell function. Dis. Markers 29, 157– 175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y. I., Hua W. K., Yao C. L., Hwang S. M., Hung Y. C., Kuan C. J., Leou J. S., Lin W. J. (2010). Protein-arginine methyltransferase 1 suppresses megakaryocytic differentiation via modulation of the p38 MAPK pathway in K562 cells. J. Biol. Chem. 285, 20595–20606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conde I., Pabón D., Jayo A., Lastres P., González-Manchón C. (2010). Involvement of ERK1/2, p38 and PI3K in megakaryocytic differentiation of K562 cells. Eur. J. Haematol. 84, 430–440 [DOI] [PubMed] [Google Scholar]
- Frecha C., Toscano M. G., Costa C., Saez-Lara M. J., Cosset F. L., Verhoeyen E., Martin F. (2008). Improved lentiviral vectors for Wiskott-Aldrich syndrome gene therapy mimic endogenous expression profiles throughout haematopoiesis. Gene Ther. 15, 930–941 [DOI] [PubMed] [Google Scholar]
- Gallego M. D., Santamaría M., Peña J., Molina I. J. (1997). Defective actin reorganization and polymerization of Wiskott-Aldrich T cells in response to CD3-mediated stimulation. Blood 90, 3089–3097 [PubMed] [Google Scholar]
- Gross B. S., Wilde J. I., Quek L., Chapel H., Nelson D. L., Watson S. P. (1999). Regulation and function of WASp in platelets by the collagen receptor, glycoprotein VI. Blood 94, 4166–4176 [PubMed] [Google Scholar]
- Gröttum K. A., Hovig T., Holmsen H., Abrahamsen A. F., Jeremic M., Seip M. (1969). Wiskott-Aldrich syndrome: qualitative platelet defects and short platelet survival. Br. J. Haematol. 17, 373–388 [DOI] [PubMed] [Google Scholar]
- Haddad E., Cramer E., Rivière C., Rameau P., Louache F., Guichard J., Nelson D. L., Fischer A., Vainchenker W., Debili N. (1999). The thrombocytopenia of Wiskott Aldrich syndrome is not related to a defect in proplatelet formation. Blood 94, 509–518 [PubMed] [Google Scholar]
- Herrera R., Hubbell S., Decker S., Petruzzelli L. (1998). A role for the MEK/MAPK pathway in PMA-induced cell cycle arrest: modulation of megakaryocytic differentiation of K562 cells. Exp. Cell Res. 238, 407–414 [DOI] [PubMed] [Google Scholar]
- Hockemeyer D., Soldner F., Beard C., Gao Q., Mitalipova M., DeKelver R. C., Katibah G. E., Amora R., Boydston E. A., Zeitler B., et al. (2009). Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat. Biotechnol. 27, 851–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N., Wang J., Kim K., Friedman G., Wang X., Taupin V., Crooks G. M., Kohn D. B., Gregory P. D., Holmes M. C., et al. (2010). Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 28, 839–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C. L., Cheng J. C., Liao C. H., Stern A., Hsieh J. T., Wang C. H., Hsu H. L., Tseng C. P. (2004). Disabled-2 is a negative regulator of integrin alpha(IIb)beta(3)-mediated fibrinogen adhesion and cell signaling. J. Biol. Chem. 279, 42279–42289 [DOI] [PubMed] [Google Scholar]
- Kajiwara M., Nonoyama S., Eguchi M., Morio T., Imai K., Okawa H., Kaneko M., Sako M., Ohga S., Maeda M., et al. (1999). WASP is involved in proliferation and differentiation of human haemopoietic progenitors in vitro. Br. J. Haematol. 107, 254–262 [DOI] [PubMed] [Google Scholar]
- Litzman J., Jones A., Hann I., Chapel H., Strobel S., Morgan G. (1996). Intravenous immunoglobulin, splenectomy, and antibiotic prophylaxis in Wiskott-Aldrich syndrome. Arch. Dis. Child. 75, 436–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luthi J. N., Gandhi M. J., Drachman J. G. (2003). X-linked thrombocytopenia caused by a mutation in the Wiskott-Aldrich syndrome (WAS) gene that disrupts interaction with the WAS protein (WASP)-interacting protein (WIP). Exp. Hematol. 31, 150–158 [DOI] [PubMed] [Google Scholar]
- Lutskiy M. I., Shcherbina A., Bachli E. T., Cooley J., Remold-O'Donnell E. (2007). WASP localizes to the membrane skeleton of platelets. Br. J. Haematol. 139, 98–105 [DOI] [PubMed] [Google Scholar]
- Manjunath N., Johnson R. S., Staunton D. E., Pasqualini R., Ardman B. (1993). Targeted disruption of CD43 gene enhances T lymphocyte adhesion. J. Immunol. 151, 1528–1534 [PubMed] [Google Scholar]
- Martín F., Toscano M. G., Blundell M., Frecha C., Srivastava G. K., Santamaría M., Thrasher A. J., Molina I. J. (2005). Lentiviral vectors transcriptionally targeted to hematopoietic cells by WASP gene proximal promoter sequences. Gene Ther. 12, 715–723 [DOI] [PubMed] [Google Scholar]
- Moehle E. A., Rock J. M., Lee Y. L., Jouvenot Y., DeKelver R. C., Gregory P. D., Urnov F. D., Holmes M. C. (2007). Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc. Natl. Acad. Sci. USA 104, 3055–3060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro F., Gutman D., Meire E., Cáceres M., Rigoutsos I., Bentwich Z., Lieberman J. (2009). miR-34a contributes to megakaryocytic differentiation of K562 cells independently of p53. Blood 114, 2181–2192 [DOI] [PubMed] [Google Scholar]
- Ochs H. D. (1998). The Wiskott-Aldrich syndrome. Springer Semin. Immunopathol. 19, 435–458 [DOI] [PubMed] [Google Scholar]
- Park W. S., Kim H. J., Lee G. K., Son H. S., Bae Y. (2012). Anti-adhesive functions of CD43 expressed on colon carcinoma cells through the modulation of integrins. Exp. Mol. Pathol. 92, 82–89 [DOI] [PubMed] [Google Scholar]
- Prislovsky A., Marathe B., Hosni A., Bolen A. L., Nimmerjahn F., Jackson C. W., Weiman D., Strom T. S. (2008). Rapid platelet turnover in WASP(-) mice correlates with increased ex vivo phagocytosis of opsonized WASP(-) platelets. Exp. Hematol. 36, 609-623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randrianarison-Huetz V., Laurent B., Bardet V., Blobe G. C., Huetz F., Duménil D. (2010). Gfi-1B controls human erythroid and megakaryocytic differentiation by regulating TGF-beta signaling at the bipotent erythro-megakaryocytic progenitor stage. Blood 115, 2784–2795 [DOI] [PubMed] [Google Scholar]
- Remold-O'Donnell E., Rosen F. S. (1990). Sialophorin (CD43) and the Wiskott-Aldrich syndrome. Immunodefic. Rev. 2, 151–174 [PubMed] [Google Scholar]
- Rengan R., Ochs H. D., Sweet L. I., Keil M. L., Gunning W. T., Lachant N. A., Boxer L. A., Omann G. M. (2000). Actin cytoskeletal function is spared, but apoptosis is increased, in WAS patient hematopoietic cells. Blood 95, 1283–1292 [PubMed] [Google Scholar]
- Rocca B., Bellacosa A., De Cristofaro R., Neri G., Della Ventura M., Maggiano N., Rumi C., Landolfi R. (1996). Wiskott-Aldrich syndrome: report of an autosomal dominant variant. Blood 87, 4538–4543 [PubMed] [Google Scholar]
- Sabri S., Foudi A., Boukour S., Franc B., Charrier S., Jandrot-Perrus M., Farndale R. W., Jalil A., Blundell M. P., Cramer E. M., et al. (2006). Deficiency in the Wiskott-Aldrich protein induces premature proplatelet formation and platelet production in the bone marrow compartment. Blood 108, 134–140 [DOI] [PubMed] [Google Scholar]
- Sánchez-Mateos P., Campanero M. R., del Pozo M. A., Sánchez-Madrid F. (1995). Regulatory role of CD43 leukosialin on integrin-mediated T-cell adhesion to endothelial and extracellular matrix ligands and its polar redistribution to a cellular uropod. Blood 86, 2228–2239 [PubMed] [Google Scholar]
- Sardina J. L., López-Ruano G., Sánchez-Abarca L. I., Pérez-Simón J. A., Gaztelumendi A., Trigueros C., Llanillo M., Sánchez-Yagüe J., Hernández-Hernández A. (2010). p22phox-dependent NADPH oxidase activity is required for megakaryocytic differentiation. Cell Death Differ. 17, 1842–1854 [DOI] [PubMed] [Google Scholar]
- Schulze H., Korpal M., Hurov J., Kim S. W., Zhang J., Cantley L. C., Graf T., Shivdasani R. A. (2006). Characterization of the megakaryocyte demarcation membrane system and its role in thrombopoiesis. Blood 107, 3868–3875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semple J. W., Siminovitch K. A., Mody M., Milev Y., Lazarus A. H., Wright J. F., Freedman J. (1997). Flow cytometric analysis of platelets from children with the Wiskott-Aldrich syndrome reveals defects in platelet development, activation and structure. Br. J. Haematol. 97, 747–754 [DOI] [PubMed] [Google Scholar]
- Shcherbina A., Rosen F. S., Remold-O'Donnell E. (1999). Pathological events in platelets of Wiskott-Aldrich syndrome patients. Br. J. Haematol. 106, 875–883 [DOI] [PubMed] [Google Scholar]
- Shcherbina A., Miki H., Kenney D. M., Rosen F. S., Takenawa T., Remold-O'Donnell E. (2001). WASP and N-WASP in human platelets differ in sensitivity to protease calpain. Blood 98, 2988–2991 [DOI] [PubMed] [Google Scholar]
- Shcherbina A., Cooley J., Lutskiy M. I., Benarafa C., Gilbert G. E., Remold-O'Donnell E. (2010). WASP plays a novel role in regulating platelet responses dependent on alphaIIbbeta3 integrin outside-in signalling. Br. J. Haematol. 148, 416– 427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelley C. S., Remold-O'Donnell E., Davis A. E., 3rd, Bruns G. A., Rosen F. S., Carroll M. C., Whitehead A. S. (1989). Molecular characterization of sialophorin (CD43), the lymphocyte surface sialoglycoprotein defective in Wiskott-Aldrich syndrome. Proc. Natl. Acad. Sci. USA 86, 2819–2823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom T. S. (2009). The thrombocytopenia of WAS: a familial form of ITP? Immunol. Res. 44, 42–53 [DOI] [PubMed] [Google Scholar]
- Sullivan K. E., Mullen C. A., Blaese R. M., Winkelstein J. A. (1994). A multiinstitutional survey of the Wiskott-Aldrich syndrome. J. Pediatr. 125, 876–885 [DOI] [PubMed] [Google Scholar]
- Toscano M. G., Frecha C., Ortega C., Santamaría M., Martín F., Molina I. J. (2004). Efficient lentiviral transduction of Herpesvirus saimiri immortalized T cells as a model for gene therapy in primary immunodeficiencies. Gene Ther. 11, 956– 961 [DOI] [PubMed] [Google Scholar]
- Toscano M. G., Frecha C., Benabdellah K., Cobo M., Blundell M., Thrasher A. J., García-Olivares E., Molina I. J., Martin F. (2008). Hematopoietic-specific lentiviral vectors circumvent cellular toxicity due to ectopic expression of Wiskott-Aldrich syndrome protein. Hum. Gene Ther. 19, 179–198 [DOI] [PubMed] [Google Scholar]
- Tsuboi S., Nonoyama S., Ochs H. D. (2006). Wiskott-Aldrich syndrome protein is involved in alphaIIb beta3-mediated cell adhesion. EMBO Rep. 7, 506-511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urnov F. D., Miller J. C., Lee Y. L., Beausejour C. M., Rock J. M., Augustus S., Jamieson A. C., Porteus M. H., Gregory P. D., Holmes M. C. (2005). Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature 435, 646–651 [DOI] [PubMed] [Google Scholar]
- Urnov F. D., Rebar E. J., Holmes M. C., Zhang H. S., Gregory P. D. (2010). Genome editing with engineered zinc finger nucleases. Nat. Rev. Genet. 11, 636–646 [DOI] [PubMed] [Google Scholar]
- Vasquez K. M., Marburger K., Intody Z., Wilson J. H. (2001). Manipulating the mammalian genome by homologous recombination. Proc. Natl. Acad. Sci. USA 98, 8403–8410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen A. M., Galasinski S. C., Shapiro P. S., Nahreini T. S., Ahn N. G. (1997). Megakaryocytic differentiation induced by constitutive activation of mitogen-activated protein kinase kinase. Mol. Cell. Biol. 17, 1947–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R., Nagy D., Mandel R. J., Naldini L., Trono D. (1997). Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat. Biotechnol. 15, 871–875 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.