

Abstract
Rac-dependent NADPH oxidases generate reactive oxygen species used in cell signaling and microbial killing or both. Whereas the mechanisms leading to NADPH oxidase activation are fairly well studied, the mechanisms that control downregulation of this enzyme complex remain unclear. We hypothesized that reactive oxygen species produced by NADPH oxidase may autoregulate the complex by inhibiting Rac activity. To this end, we searched for binding partners of Rac1 and identified a tyrosine-phosphorylated fragment of MKK6 that bound to Rac1 under redox-stress conditions. Constitutively active MKK6 interacted directly with Rac1 in vitro, and this interaction was enhanced when MKK6 was phosphorylated on tyrosine 219. Both Rac1 and Rac2 immunoprecipitated an MKK6 fragment under conditions that elevate cellular peroxide levels in 293 and RAW cells, respectively. Constitutively active and wild-type MKK6 enhanced Rac-GTPase activity in vitro, and their overexpression inhibited PMA-induced NADPH oxidase activation in RAW cells. In contrast, a Y219F mutant of MKK6 only partially enhanced Rac1 GTPase activity, and its overexpression did not alter PMA-induced NADPH oxidase activation in RAW cells. Last, MKK6 deficiency led to an increase in Rac1-GTP levels in brain tissue. Our findings suggest that MKK6 downregulates NADPH oxidase activity by enhancing Rac-GTPase activity.
INTRODUCTION
Invading pathogens are quickly cleared from bodily fluids by the innate immune system. The first cells to respond to invading pathogens include phagocytes such as neutrophils and monocytes in the blood and macrophages in tissues (4). These cells are equipped with the phagocytic NADPH oxidase (Nox2gp91phox) that produces reactive oxygen species (ROS) to help kill pathogens. The phagocytic NADPH oxidase is a multi-subunit enzyme complex with membrane and cytosolic components (25). The cytochrome b558 resides in the membrane and is composed of two subunits, gp91phox and p22phox. The cytosolic subunits include p47phox, p67phox, p40phox, and the small GTPase Rac1/2. In the resting state, the cytosolic components remain quiescent in the cytoplasm, and the membrane-bound cytochrome b558 complex is inactive. On stimulation, the cytosolic subunits are translocated to the membrane to bind the cytochrome b558 components, leading to activation of the NADPH oxidase complex. Included in this activation process is the phosphorylation of p47phox and p67phox, and the conversion of GDP-bound Rac1/2 into GTP-bound forms through the activation of a Rac guanine nucleotide exchange factor (RacGEF) (32, 41).
The activated NADPH oxidase complex reduces molecular oxygen by using an electron derived from NADPH to form superoxide (O2•−) and NADP+ (35). O2•− in turn is converted to hydrogen peroxide (H2O2), either by spontaneous or facilitated dismutation via the enzymatic activity of superoxide dismutase (SOD). H2O2 is believed to act as a second messenger to facilitate signal transduction of certain pathways through the inactivation of tyrosine phosphatases. H2O2 oxidizes cysteines in the catalytic sites of tyrosine phosphatases, leading to their inactivation and a net increase in tyrosine phosphorylation of their target proteins (24, 42).
NADPH oxidase expression was believed for a long time to be confined to phagocytic cells. However, several studies demonstrated expression of the phagocytic NADPH oxidase in non-phagocytic cell types (1, 2, 10, 11, 27, 34, 37). Expression of the phagocytic NADPH oxidase in nonphagocytic cells appears to be important for redox-mediated signal transduction. For example, NADPH oxidase regulates elements of T cell–receptor signaling (23). Clearance of NADPH oxidase–derived H2O2 by peroxiredoxin II has also been shown to act as a negative regulator of PDGF signaling in vascular smooth muscle cells by controlling the activity of protein tyrosine phosphatases important in PDGF-receptor inactivation (13, 38). Recently, it was demonstrated that Nox2 is activated in endosomes after IL-1β stimulation. In this context, H2O2 derived from superoxide generated inside the endosomal vesicles regulates the recruitment of redox-dependent subunits to the IL-1β–receptor complex on the cytosolic face of the endosome (27).
The increasing identification of functional roles for NADPH oxidases in cell signaling suggests that this complex is likely under tight regulatory control. Although the mechanisms that lead to the activation of NADPH oxidases are fairly well studied (25), the mechanisms that control their inactivation remain poorly understood (17). The p190 and p50 RhoGAPs are thus far the only GTPase-activating proteins identified capable of inactivating Rac and inhibiting NADPH oxidase activity (22, 33, 44, 46). However, these RhoGAPs lack specificity for Rac and also have the ability to associate with and inhibit other Rho GTPases (3).
In the current study, we sought to investigate whether negative regulators of Rac exist that influence NADPH oxidase activity in response to H2O2. To this end, we searched for proteins that associate with Rac in response to cellular treatment with H2O2. We identified a fragment of MKK6 that is tyrosine phosphorylated after H2O2 treatment and directly associates with Rac1/2 when phosphorylated on Y219. Interestingly, a constitutively active mutant of MKK6 had the ability to autophosphorylate Y219 as a bacterial purified fusion protein and also significantly enhanced Rac-GTP hydrolysis. Wild-type MKK6 had the same effect on Rac1 GTPase activity in vitro. In vivo expression of either constitutively active or wild-type MKK6 led to a significant inhibition of PMA-induced O2•− production in RAW 264.7 cells, whereas the Y219F-CA-MKK6 mutant failed to downregulate NADPH oxidase activation in vivo. In addition, brain tissues from MKK6-deficient mice had elevated levels of activated Rac1 compared with those from MKK6 heterozygous littermates. These findings strongly suggest that MKK6 is a novel redox-regulated Rac-binding protein that inhibits NADPH oxidase activation after phosphorylation on Y219 by enhancing Rac-GTPase activity.
MATERIALS AND METHODS
Materials
GTP, GDP, Rac1, imidazole, phorbol myristate acetate (PMA), and cellulose PEI matrix TLC plates were purchased from Sigma-Aldrich Corporation (St. Louis, MO). Dulbecco’s modified Eagle’s medium (DMEM), penicillin/streptomycin (P/S), 0.25% trypsin-EDTA, L-glutamine, and fetal bovine serum (FBS) were purchased from Invitrogen Corporation (Carlsbad, CA). Radioactive nucleotides, glutathione-sepharose beads, liquid scintillation fluid, and nitrocellulose protein transfer membrane were purchased from Amersham Biosciences (Piscataway, NJ). Protease-inhibitor cocktail (PIC), EDTA-free PIC, GTPγS, and GDPβS were purchased from Roche Applied Science (Indianapolis, IN). Histidine-tagged Rac1 (His-Rac1) and glutathione transferase–tagged (GST) p50-Rho-GAP catalytic domain (p29GAP) were purchased from Cytoskeleton, Inc. (Denver, CO). Dynabeads Talon and Dynabeads Protein-G were purchased from Dynal Biotech (Lake Success, NY). Bovine copper/zinc superoxide dismutase (SOD1) was purchased from Oxis Research (Portland, OR). The Rac1 activation assay kit was purchased from Upstate Cell Signaling Solutions (Lake Placid, NY).
Cell culture and treatment
Cells were grown in DMEM with 200 mM L-glutamine, 10% FBS, and 1% penicillin/streptomycin in 150-mm tissue-culture plates. Treatment of RAW cells with H2O2 was conducted by adding H2O2 to 1 mM in the media, and the cells were incubated overnight before harvest.
Recombinant adenoviral vector infection and plasmid transfections
Adenoviral infections were performed in serum-free medium for 2 h at a multiplicity of infection (MOI) of 0.5–1 × 104 particles/cell, as indicated, followed by the addition of an equal volume of fresh media containing 20% FBS. Cells were fed with fresh media at 24 h and then analyzed at the indicated times after infection. Five different previously reported recombinant adenoviruses were used, including Ad.LacZ (control vector that expresses LacZ transgene) (18), Ad.SOD1 (copper/zinc superoxide dismutase wild-type enzyme) (51), Ad.GPx-1 (GPx-1 tagged with a c-Myc epitope at the N-terminus) (28), Ad.catalase (wild-type catalase enzyme) (9), and Ad.wt-Rac1 (wild-type Rac1 GTPase HA-tagged) (54). Three new adenoviral vectors were generated, including Ad.CA-MKK6 (constitutively active form of MKK6 HA-tagged), Ad.Y219F-CA-MKK6, and Ad.WT-MKK6 (wild-type MKK6). The details of construction of these viruses are described later. All plasmid transfections were carried out by using Lipofectamin reagent according to the instructions of the manufacturer, Invitrogen.
Adenoviral vector construction
Adenoviral vectors encoding HA-tagged WT-MKK6, constitutively active MKK6 (CA-MKK6, incorporating two mutations in the kinase active site S207E and T211E), or Y219F-CA-MKK6 (incorporating three mutations, S207E, T211E, and Y219F) were constructed by PCR amplification of cDNA sequences by using primers that were designed to contain an N-terminal HA-tag for all constructs. The HA-tagged mutant cDNA fragments were inserted into the pAd.CMVLink containing the CMV promoter and an SV40 polyadenylation sequence for constitutive transgene expression. Recombinant adenoviruses were generated by co-transfection of the NheI-cut pAd plasmid, containing CA-MKK6, Y219F-CA-MKK6, or WT-MKK6, with Pac1-digested Ad5.sub360 (E3-deleted) viral DNA.
Assay for O2•− production by RAW 264.7 cells
The rate of O2•− production by RAW 264.7 cells was measured by using a cytochrome c reduction assay, as previously described (52). RAW 264.7 cells were scraped off tissue-culture plates in Hank’s balanced salt solution (HBSS), and cell densities were normalized for all samples in a given data set (range for all experiments was from 1 to 4 × 106 cells/ml). Then 50 μl of each cell suspension was used per well of a 96-well plate and stimulated with 1 μM phorbol myristate acetate (PMA) or with vehicle (0.005% DMSO final concentration in HBSS) for 5 min at 37°C in the presence of 125 μM ferricytochrome c (100 μl total reaction volume). O2•− generation was measured in real time as the SOD-inhibitable reduction of ferricytochrome c. The assays were conducted in 96-well plates with two wells for each experimental sample (one well with 30 μg SOD1 and one well without SOD). Reduction of ferricytochrome c was detected by an absorbance change at 550 nm. The linear portion of the curve was used to calculate the reaction rate by linear regression analysis. Superoxide concentrations were calculated by using an extinction coefficient for reduced cytochrome c at 550 nm of 21 mM/cm (14).
SDS-PAGE and Western blotting
Cells were washed with ice-cold PBS and lysed in RIPA buffer containing 150 mM NaCl, 50 mM Tris pH 7.2, 1% DOC, 1% Triton-X100, 0.1% SDS, and protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Protein concentrations were measured by using a BioRad Kit (BioRad, Philadelphia, PA). Equal microgram quantities of protein extract were fractionated by 10% sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred to nitrocellulose membrane and then blocked overnight at 4°C in blocking buffer containing 4% wt/vol non-fat dried milk and 0.3% Tween 20 in PBS. Membranes were then incubated with primary antibodies and then with a peroxidase-conjugated or infrared dye–conjugated secondary antibodies. Proteins were detected with either an enhanced chemiluminescence (ECL) system (Amersham Pharmacia Biotech, Little Chalfont, Buckinghamshire, England) or the Odyssey Infrared Imaging System (LI-COR Biotech, Lincoln, NE). For antiphosphotyrosine blots, Odyssey blocking buffer was used in place of nonfat milk (LI-COR Biotech). Antibodies to phosphotyrosine (PY20) were purchased from Transduction Labs (Lexington, KY), anti-Rac1 antibody from Upstate Cell Signaling Solutions (Lake Placid, NY), anti-MKK6 antibody from Invitrogen, anti-HA antibody from Roche Applied Science (Indianapolis, IN), anti-GST and anti-Rac2 from Santa Cruz Biotech (Santa Cruz, CA).
Immunoprecipitation
Cell samples were washed with ice-cold PBS twice followed by lysis with RIPA buffer at 4°C for 15 min. Cell lysates were cleared by centrifugation at 10,000 g for 10 min at 4°C. Protein concentrations were determined by using a BioRad Kit, and 600 μg cellular protein and 4 μg primary antibody were mixed with 0.5 ml RIPA buffer at 4°C for 1 h. Then 50 μl Protein-A Agarose Beads (Santa Cruz Biotech, Santa Cruz, CA) was then added to the mixture and rotated for 2 h. The beads were spun down at 5,000 rpm for 5 min at 4°C and washed with ice-cold PBS 3 times before SDS-PAGE analysis of immunoprecipitates.
Pull-down assays with His-tagged proteins
His-Rac1 (25 picomoles) was incubated with Dynabeads Talon in PBS at room temperature for 30 min with intermittent gentle agitation. The proteins were either used directly or preloaded with 1 mM GTPγS or GDPβS in GTP-binding buffer containing 50 mM HEPES, pH 7.6, 150 mM NaCl, and 0.1 mM EDTA for 10 min at room temperature followed by the addition of MgCl2 to 10 mM final concentration. Immobilized proteins were then washed in 10 mM MgCl2/PBS, and 25 pmoles of the various GST-MKK6 fusions was then added to each tube in 10 mM MgCl2/PBS. Samples were incubated at RT for 30 min, with intermittent gentle agitation. Beads were then washed 3 times with 10 mM MgCl2/PBS to remove unbound protein. The fourth wash was carried out in 50 mM Tris, pH 6.8. Proteins were eluted by boiling in SDS-PAGE reducing loading buffer and separated by SDS-PAGE for Western blotting.
Rac1 GTPase assay
Rac1 GTPase assays were performed as previously described, with modifications (26). Twenty-five pmoles of Rac1 (Sigma) was incubated with 25 pmoles 32P-labeled γ-GTP in GTPase buffer containing 50 mM HEPES, pH 7.6, 150 mM NaCl, and 0.1 mM EDTA for 10 min at room temperature in a total volume of 192 μl and then placed in ice water. A 1-μl aliquot was then taken for TLC analysis as time zero, before initiating the GTPase reaction. GST-tagged MKK6 proteins or p29GAP was diluted in GTPase buffer with supplemented MgCl2. To initiate the GTPase reaction, 10 μl GTP-loaded Rac1 was mixed with 30 μl of the MKK6 fusion proteins or p29GAP to give a 40-μl reaction volume containing a final concentration of 5 nM of each protein and 10 mM MgCl2 at 15°C. One microliter aliquot from each sample were spotted on the TLC plate at different time points. The TLC was run for 90 min at room temperature in 1 M acetic acid with 0.8 M LiCl running buffer. To quantify GTP hydrolysis, the free phosphate (Pi) bands were cut out along with the corresponding GTP bands. Each was put in liquid scintillation fluid and counted by liquid scintillation spectrometry. Percentage of GTP hydrolyzed was calculated by the equation, Pi/(Pi + GTP) × 100.
Generation of GST-MKK6 mutants and HA-tagged MKK6 and Rac1 constructs
Bacterial expression construct for GST-tagged CA-MKK6 (constitutively active MKK6b mutant, 334 amino acids) was a generous gift from Dr. Jiahuai Han (Professor of Immunology, the Scripps Research Institute, La Jolla, CA). Wild-type and Y219F-CA-MKK6 GST-fusion constructs were generated by using the Gene Editor in vitro site-directed mutagenesis system (Promega, Madison, WI). All bacterial fusion construct mutations were confirmed by complete DNA sequencing. DNA sequence encoding an HA-tag (YPYDVPDYA) was inserted at the N-terminus of MKK6 mutants, wild-type MKK6, or L61Rac1 by PCR. The constructs were then inserted into a CMV-driven mammalian expression plasmid (pAdCMVLink).
Expression and purification of bacterial GST-tagged proteins
The GST-tagged expression constructs were transformed into Escherichia coli by using ampicillin selection. Bacterial colonies harboring the wild-type and mutant constructs were grown in LB medium containing 100 μg/ml ampicillin in 1-L flasks at 37°C until the cell density reached A600 = 0.6. Isopropyl-D-thiogalactopyranoside (IPTG) was then added to 1 mM to induce the expression of GST-tagged proteins and cultures were grown for 6 h at 37°C. The bacteria were collected by a 4,000 g spin for 15 min at 4°C and resuspended in PBS on ice. The bacteria were then lysed on ice by five 30-sec sonicator pulses by using a virsonic cell disruptor (VirTis, Gardiner, NY). The bacterial lysate was then centrifuged at 30,000 g for 30 min to pellet debris. The fusion proteins were purified from cellular extracts with an ÄKTA FPLC high-performance liquid chromatography system with a GSTPrep FF 16/10 Column (Amersham Biosciences), according to the manufacturer’s instructions. GST-fusion proteins were eluted with 15 mM glutathione, 50 mM Tris-HCl, pH 7.5, and 120 mM NaCl. The purity of fusion proteins was assessed by Coomassie stained SDS-PAGE and protein concentrations were normalized by using the Bradford method. Finally, all purified proteins were dialyzed against PBS.
In vitro phosphatase and kinase assays
Twenty-five pmoles of purified GST-CA-MKK6 was incubated with 0.1 unit alkaline phosphatase in phosphatase buffer containing 50 mM Tris-HCl, pH 7.9, 100 mM NaCl, 10 mM MgCl2, and 1 mM dithiothreitol for 15 min at 37°C. The phosphatase reaction was stopped by incubating samples on ice. GST-CA-MKK6 was then captured on glutathione-sepharose beads (Amersham Biosciences) for 1 h at 4°C. The beads bearing the GST-tagged protein were pelleted and washed 3 times in PBS by centrifugation. Finally, samples were then resuspended in SDS-PAGE loading buffer or kinase buffer containing 30 mM HEPES pH 7.4, 20 mM β-glycerol phosphate, 10 mM magnesium sulfate, 1 mM DTT, 100 μM sodium orthovanadate, and 400 μM ATP. The kinase reactions were incubated at 37°C for 30 min, and the reaction was terminated on ice followed by re-suspension of the beads in SDS-PAGE loading buffer.
Detection of cellular ROS production by using H2DCFDA
Samples were infected with adenoviral vectors 48 h before treatment. Stock solutions of H2DCFDA (Molecular Probes) were generated in DMSO at a concentration of 50 μg/ml immediately before use. Cells were washed twice with PBS before treatment with H2DCFDA (10 μM) for 10 min in PBS at 37°C in the dark. Cells were then washed in PBS then fixed for 5 min in 4% paraformaldehyde in PBS. Cells were subsequently mounted in antifadent and were examined by fluorescent microscopy for DCF signal. Exposure settings were constant for all experimental samples.
MKK6 knockout mouse model
Transgenic MKK6 knockout mice were a generous gift from Dr. Roger Davis (Professor of Biochemistry and Molecular Pharmacology, University of Massachusetts Medical School, Worcester, MA) (45). The strain was maintained on a C57BL/6 background, and offspring were derived from breeding heterozygous carrier females to homozygous MKK6 knockout males. Genotypes were confirmed by PCR with the following primers: (a) forward primer for the wild-type MKK6 allele 5′-GAAAACCCAAATAGATGTGTGGTC-3′, (b) forward primer for the knockout MKK6 allele 5′-GTGGGGTGGGATTAGATAAATGC-3′, and (c) reverse primer for both wild-type and knockout alleles 5′-GAAAAGAGGATGTTCGGCACTG-3′.
Rac1 activation assays
Rac1 activation assays were performed by using a kit purchased from Upstate Cell Signaling Solutions (Lake Placid, NY) following the manufacturer’s instructions. In brief, this assay uses a GST-PBD–binding domain of PAK specifically to bind GTP-Rac1 (PBD encodes the p21 binding domain of Pak1). Tissue lysates were normalized for protein concentration by using the Bradford method. GTP-bound Rac1 was precipitated from 3 mg of lysate with GST-PBD immobilized on agarose beads. The precipitated pellet was evaluated with Western blotting for Rac1 and compared with the level of Rac1 in the starting sample crude lysate. The intensity of Rac1 immunoreactivity correlates with the level of GTP-bound Rac1 in the sample.
RESULTS
MKK6 associates with Rac1 after redox stress
We hypothesized that enhanced production of intracellular H2O2 may signal negative regulators of Rac1 to downregulate the activity of NADPH oxidase and hence intracellular ROS production through this complex. To identify such factors, we searched for proteins that associated with Rac1 in response to cellular treatment with H2O2. We used the murine monocyte/macrophage cell line RAW 264.7 to address this hypothesis. HA-tagged wild-type Rac1 expressed from a replication-deficient recombinant adenovirus was used as bait for redox-induced Rac regulators. Immunoprecipitated (IP) HA-Rac1 from H2O2-treated RAW cells led to the identification of a unique protein band in Coomassie-stained SDS-PAGE. With matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF) of the trypsin-digested protein band, we identified an apparently cleaved fragment of MKK6 as a binding partner for Rac1 after H2O2 treatment (data not shown). Interestingly, this MKK6 fragment matched only when a potential phosphorylated amino acid was present on MKK6.
Given this interesting but preliminary finding, we sought to confirm that Rac indeed binds to MKK6 in cells after redox stress. Additionally, we sought to provide a more physiologic means of cellular H2O2 stress by overexpressing copper/zinc superoxide dismutase (SOD1) from a recombinant adenoviral vector. Overexpression of SOD1 was shown previously to up-regulate cellular ROS levels by enhancing dismutation of cellular superoxide to H2O2 (40). Indeed, SOD1 overexpression upregulated cellular ROS, as detected by DCFH-DA fluorescent staining, which was inhibited by co-expression of glutathione peroxidase 1 (GPx-1) or catalase (both enzymes scavenge H2O2) (Fig. 1). These findings confirmed that SOD1 overexpression elevates cellular H2O2. Overexpression of SOD1 in 293 cells significantly promoted MKK6 association with ectopically expressed HA-tagged wild-type Rac1 (Fig. 2A). Similarly, overexpression of SOD1 in RAW cells also promoted MKK6 association with endogenous Rac2 (see Fig. 2A). These findings confirmed that an increase in cellular hydrogen peroxide results in enhanced MKK6 binding to Rac1/2, as suggested by MALDI-TOF findings. To determine whether MKK6 preferentially bound to active (GTP bound) or inactive (GDP bound) forms of Rac1 or both, we performed pull-down experiments in the presence of ectopically expressed HA-tagged wild-type, L61-Rac1 (constitutively activated locked in the GTP-bound state), or N17-Rac1 (dominant-negative locked in the GDP-bound state). Results from these experiments in 293 cells demonstrated that immunoprecipitated L61-Rac1 most avidly associated with MKK6, whereas N17 Rac1 failed to bind MKK6 (see Fig. 2B). Hence, MKK6 binds to wild-type Rac1 only under redox stress and preferentially binds to the activated GTP-bound form of Rac1.
FIG. 1. Overexpression of SOD1 elevates cellular hydrogen peroxide levels.
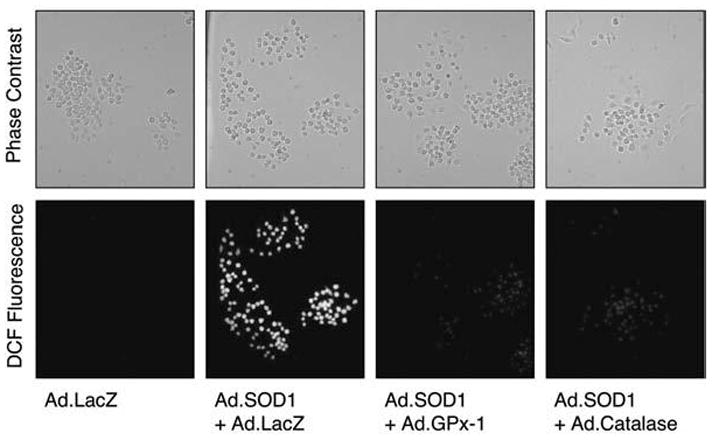
RAW 264.7 cells were infected with the indicated recombinant adenoviral vectors at a multiplicity of infection (MOI) equal to 5,000 particles/cell for each vector. In the condition infected with Ad.LacZ alone, a 10,000 particles/cell MOI was used to normalize vector dose to an equivalent level in all conditions. At 48 h after infection, cells were stained for reactive oxygen species (ROS) by using DCFH-DA fluorescent staining. Phase-contrast (top panel) and fluorescent (bottom panel) photomicrographs are shown. Exposure settings were the same for all conditions, and photomicrographs are representative of three independent experiments.
FIG. 2. The 18-kDa MKK6 fragment binds Rac in 293 cells and RAW cells under conditions that elevate cellular hydrogen peroxide.
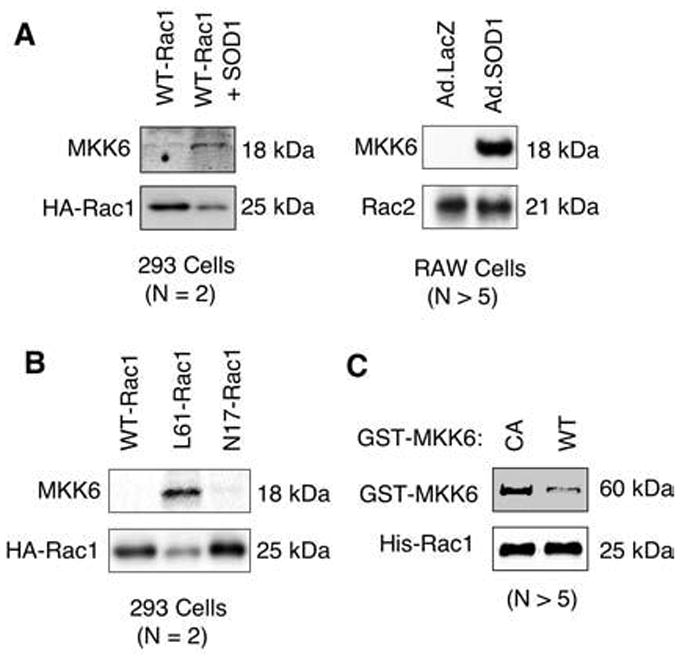
(A, B) The 293 cells were transfected with the following plasmids expressing HA-tagged WT-Rac1, L61-Rac1, N17-Rac1, or WT-Rac1+SOD1. HA-Rac1 was then immunoprecipitated from the 293 cell lysates by using an anti-HA antibody, and the proteins that associated with Rac1 were separated by SDS-PAGE and characterized by Western blotting with the indicated antibodies. In a similar set of experiments, association of endogenous Rac2 with MKK6 was assessed in RAW cells infected with Ad.LacZ or Ad.SOD1. Cell lysate from each condition was immunoprecipitated with anti-Rac2 antibody and evaluated with Western blot. (C) In vitro association of purified GST-CA-MKK6 and GST-WT-MKK6 with His-tagged Rac1. His-tagged Rac1 was immobilized on Dynabeads, and pull-down assays were performed with an equal amount of purified CA or WT GST-MKK6. Proteins bound to Rac1 were evaluated with Western blot by using anti-GST and anti-Rac1 antibodies. All panels are representative for n = 2–5 independent experiments as indicated.
To test whether Rac1 and MKK6 proteins can interact directly, we performed in vitro association assays by using bacterial purified His-tagged Rac1 and GST-tagged forms of the constitutively active (CA-MKK6) or wild-type (WT-MKK6) MKK6 protein. CA-MKK6 has two mutations in the active site, S207E and T211E. These studies demonstrated that Rac1 bound CA-MKK6 to a significantly greater extent than WT-MKK6 (see Fig. 2C). These experiments suggested that the full-length constitutively active form of MKK6 (CA-MKK6) directly binds to Rac1 most efficiently.
Tyrosine 219 is autophosphorylated on constitutively active CA-MKK6
Given that our MALDI-TOF findings suggested that tyrosine-phosphorylated MKK6 appeared to associate with Rac1 in vivo, we sought to determine whether bacterial purified fusions of MKK6 had the capacity to be tyrosine phosphorylated in vitro. Surprisingly, GST-tagged CA-MKK6 purified from bacteria was constitutively phosphorylated on one or more tyrosines (Fig. 3A, lane 1). Because E. coli lack the machinery for posttranslational modification of proteins, we hypothesized that CA-MKK6 is autophosphorylated on Y219. We conducted an in vitro phosphatase reaction by using alkaline phosphatase (AP) to dephosphorylate the tyrosine-phosphorylated GST-CA-MKK6. As shown in Fig. 3A, AP treatment of the isolated CA-MKK6 indeed removed all tyrosine phosphorylation from the purified GST fusion (lane 3). In an attempt to determine whether CA-MKK6 indeed has the ability to be autophosphorylated on one or more tyrosines, we immobilized AP-dephosphorylated GST-CA-MKK6 on glutathione sepharose beads and conducted in vitro kinase assays. Results from these experiments demonstrated that dephosphorylated GST-CA-MKK6 undergoes tyrosine autophosphorylation in the presence of ATP (Fig. 3A, lane 4).
FIG. 3. Tyrosine 219 phosphorylation on MKK6 is important for binding to Rac1.
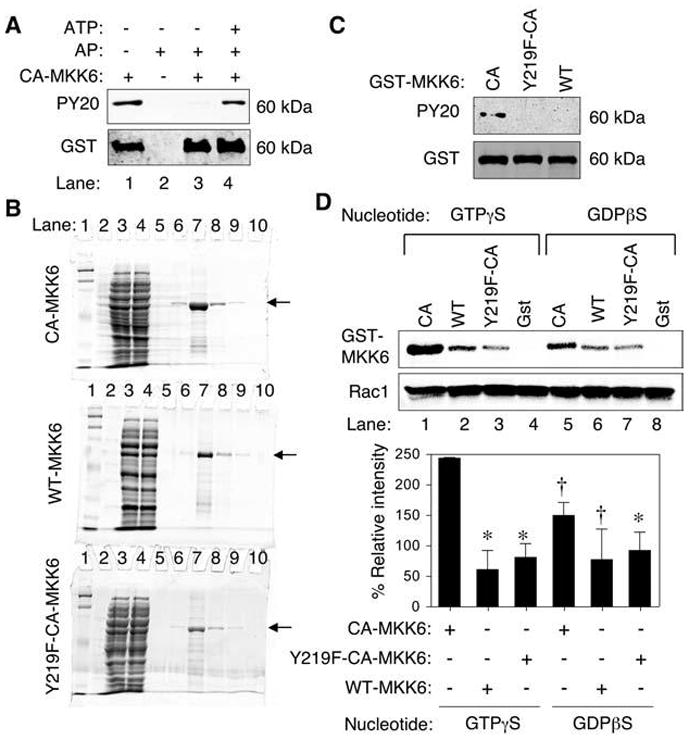
(A) Phosphotyrosine (PY20) Western blot analysis of purified GST-CA-MKK6 demonstrating that bacteria expressed CA-MKK6 is tyrosine phosphorylated (lane 1). Western blots were probed with anti-phosphotyrosine antibody (PY20) or anti-GST as a loading control. Purified GST-CA-MKK6 was treated with alkaline phosphatase (AP) before Western blotting (lane 3) and then reincubated with ATP after removal of alkaline phosphatase (lane 4). (B) Purity of GST-tagged MKK6 proteins. Coomassie-stained SDS-PAGE for GST-MKK6–purified proteins (arrow). Lanes are as follows: 1 and 2 (MW markers), 3 (crude bacterial lysate), 4 (GST-column flow through), 5–10 (purification fractions with lane 7 containing the peak protein fraction). (C) Western blot of MKK6 fusion proteins probed with PY20 and anti-GST antibodies. Both the GST-Y219F-CA-MKK6 mutant and GST-WT-MKK6 failed to demonstrate tyrosine phosphorylation in comparison to GST-CA-MKK6. (D) In vitro pull-down of purified His-tagged Rac1 in the presence of purified constitutively active (CA), wild-type (WT), tyrosine mutant (Y219F-CA) GST-MKK6 proteins followed by Western blotting for GST and Rac1 (upper panel). Free GST protein was also used as a negative control in these studies. The His-tagged Rac1 was preloaded with the indicated nucleotide analogues before incubation with MKK6 or GST proteins. Lower panel, quantification of GST-MKK6 bands relative to Rac1 bands from three independent experiments. The relative intensity was normalized for each experiment to values obtained from CA-MKK6 (GTP) before calculating the mean ± SEM for the three experiments. Marked values are significantly different from the CA-MKK6 + GTPγS-Rac1 group; *p < 0.01; †p < 0.05; Student’s t test.
Based on the MALDI-TOF MKK6 tryptic fragment identified from the 18-kDa phosphotyrosine band that associated with Rac1 after H2O2 treatment (amino acids 195–224 peptide of the full-length MKK6 protein; peak mass 3349.259; sequence MD-CGFISFYLVDSVAKTIDAGCKPYMAPER), two candidate tyrosine residues (Y203 and Y219) for phosphorylation were present. By using a phosphorylation-site prediction tool (Net-Phos server 2.0) (5), we found that tyrosine 219 (Y219) on MKK6 was more likely to be phosphorylated. To test whether Y219 was indeed one of the sites of tyrosine phosphorylation on MKK6, we generated a Y219F mutant on the CA-MKK6 (GST-Y219F-CA-MKK6) backbone and purified the fusion protein from bacteria (Fig. 3B). Interestingly, neither Y219F-CA-MKK6 nor WT-MKK6 was tyrosine phosphorylated in bacteria as compared with CA-MKK6 (Fig. 3C). This result confirmed that tyrosine 219 on MKK6 was indeed the site of tyrosine phosphorylation, as predicted. Furthermore, it suggested that only the activated form of MKK6 undergoes autophosphorylation on Y219, because the wild-type MKK6 protein failed to acquire tyrosine phosphorylation.
Y219 phosphorylation of CA-MKK6 enhances its association with Rac1
Next we sought to evaluate whether tyrosine phosphorylation of MKK6 might control binding to Rac1. To this end, we performed in vitro association studies by using purified Rac1 and the various MKK6 mutants (CA, WT, or Y219F-CA). Furthermore, we sought to determine whether GTP- or GDP-bound Rac1 had a preference for MKK6 binding in vitro. Results from these experiments demonstrated that phosphorylated CA-MKK6 bound to Rac1 GTPγS more avidly than Rac1-GDPβS (Fig. 3D, compare lanes 1 and 5). These findings supported earlier observations that constitutively active L61-Rac1 most effectively bound endogenous MKK6 (see Fig. 2B). Furthermore, unphosphorylated WT-MKK6 and Y219F-CA-MKK6 binding to Rac1 was significantly reduced compared with phosphorylated CA-MKK6, regardless of the nucleotide loaded on Rac1. These results suggested that MKK6 binding to Rac1 is higher when Rac1 is bound to GTP and MKK6 is phosphorylated on Y219. Similar nucleotide preferences for binding of GTPase-activating proteins (GAPs) to GTPases have been previously observed. For example, p120Ras-GAP and neurofibromin each has a 100-fold preference for binding the GTP form of Ras over the GDP-bound form (6). In contrast, Rho-GAP has only a slightly higher affinity for the GTP-bound form relative to the GDP-bound form of Rac1, RhoA, and G25K GTPases (43).
Tyrosine-phosphorylated CA-MKK6 enhances Rac-GTPase activity in vitro
Given that the active form of MKK6 associated more avidly with GTP-Rac1, we hypothesized that MKK6 might regulate Rac1 activation by enhancing Rac1 GTPase activity. To test this hypothesis, we conducted a Rac1 GTPase assay in vitro using purified proteins. In these experiments, Rac1 was loaded with [32P]γ-GTP, followed by a timed assessment of GTP hydrolysis at 15°C in the presence or absence of phosphorylated GST-CA-MKK6. As a positive control for Rac-GTPase–enhancing activity, the p50 RhoGAP catalytic domain (P29GAP) was also evaluated. At different time points, aliquots were taken from the GTPase reaction, and the free phosphate (Pi) was separated from GTP by using thin-layer chromatography (TLC) (Fig. 4A). Results from these studies demonstrate that both GST-CA-MKK6 and p29GAP effectively activated Rac1 GTP hydrolysis (see Fig. 4B). However, denaturing GST-CA-MKK6 by boiling before the GTPase assay prevented the observed enhancement of Rac-GTPase activity. We next evaluated whether WT-MKK6 and Y219F-CA-MKK6 might also influence Rac1 GTPase activity in vitro (see Fig. 4C). Findings from these experiments demonstrated that both CA-MKK6 and WT-MKK6 equally enhanced Rac1-GTPase activity. In contrast, Y219F-CA-MKK6 had a significantly reduced ability to enhance Rac1-GTPase activity as compared with CA-MKK6 and WT-MKK6, confirming the importance of tyrosine phosphorylation for this function. Given that Y219F-CA-MKK6 did retain some ability to enhance Rac1-GTPase activity (albeit at lower levels than CA-MKK6 or WT-MKK6), these data suggest that the regulatory effect of MKK6 on Rac1 is most likely mediated by differential binding affinities of the phosphorylated and unphosphorylated forms of MKK6 for Rac1, as opposed to inherent differences in catalytic activity.
FIG. 4. CA-MKK6 and WT-MKK6 enhance Rac-GTPase activity in vitro.
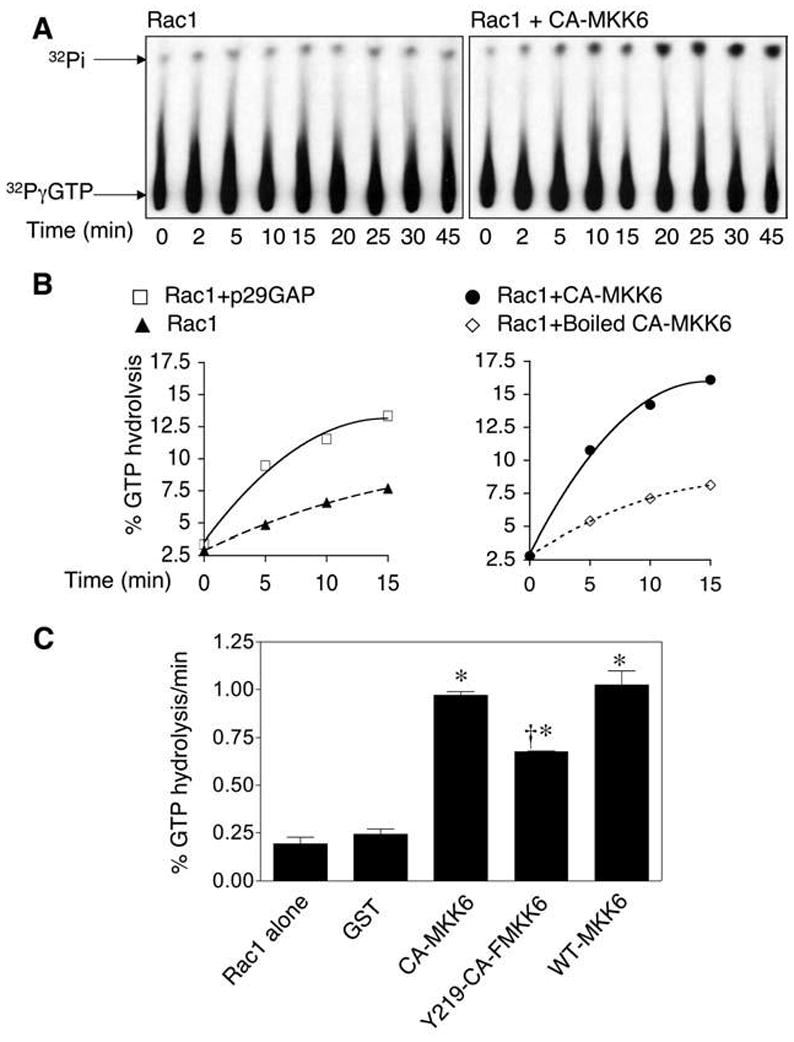
(A) Autoradiography of a representative thin-layer chromatography plate (TLC) separating the free 32phosphate [32Pi] from the [32P]γ-labeled GTP after hydrolysis by Rac1. Rac1 GTPase assays were performed in the presence or absence of GST-tagged CA-MKK6. Rac1 was preloaded with [32P]γ-GTP, and aliquots of the reaction were analyzed at various time points by thin-layer chromatography for GTP hydrolysis by assessing the percentage [32Pi] liberated from [32P]γ-GTP by Rac1. (B) Quantification of the percentage of GTP hydrolysis by Rac1 over time in the presence or absence of GST-tagged p50Rho-GAP (p29GAP), CA-MKK6, or boiled CA-MKK6. The graphs shown are representative of three independent experiments. (C) Quantification of the rate of GTP hydrolysis by Rac1 in the presence or absence of GST or the indicated GST-tagged MKK6 mutants. Values depict the mean ± SEM (n = 3). *Significantly different from Rac1 alone group; †significantly different from CA-MKK6 and WT-MKK6 groups (for all comparisons, p < 0.01; Student’s t test).
CA-MKK6 and WT-MMK6 downregulate NADPH-dependent superoxide production, whereas Y219F-CA-MKK6 does not
Rac1 plays a major role in the regulation of cellular reactive oxygen species (ROS) through the activation of several NADPH oxidases (12, 25). We hypothesized that MKK6 might inhibit NADPH oxidase activity via its ability to enhance Rac-GTPase activity. To test this hypothesis, we infected RAW 264.7 cells with replication-defective adenoviral vectors that expressed LacZ (as a negative control), CA-MKK6, WT-MKK6, or Y219F-CA-MKK6 (Fig. 5A). The cells were then evaluated for the level of PMA-induced O2•− generation by using a cytochrome c reduction assay at 48 h after infection. Results from these NADPH oxidase assays demonstrated that overexpression of CA-MKK6 and WT-MKK6, but not Y219F-CA-MKK6, significantly reduced the rate of PMA-induced O2•− generation by RAW 264.7 cells (Fig. 5B). These results substantiate in vitro Rac1 GTPase assays demonstrating that WT-MKK6 and CA-MKK6 have a greater ability to enhance GTP hydrolysis by Rac1 and support a mechanism whereby MKK6 can inhibit NADPH oxidase activity through Rac1. They also provide in vivo evidence that Y219 on MKK6 is important for this inhibitory process.
FIG. 5. CA-MKK6 and WT-MKK6, but not Y219F-CA-MKK6, inhibit NADPH oxidase activity.
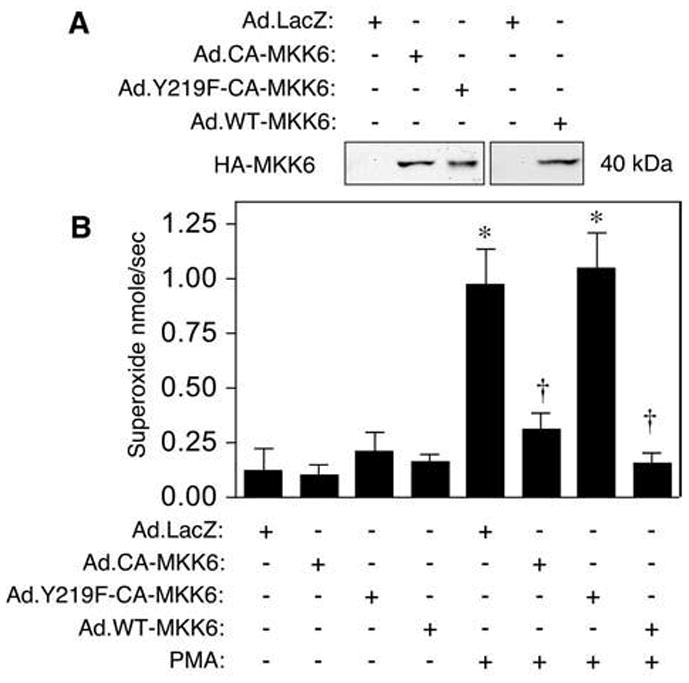
(A) Western blot confirmation for the overexpression of the indicated proteins in RAW 264.7 cells after infection with the indicated recombinant adenoviral vectors. (B) Assay for superoxide production with the cytochrome c reduction method. Adenoviruses expressing CA-MKK6, WT-MKK6, Y219F-CA-MKK6, or LacZ were used to infect RAW 264.7 cells. At 48 h after infection, the cells were challenged with PMA, and superoxide generation was measured in real time by using a cytochrome c reduction assay. The rate of superoxide production in nmole/sec is plotted and was calculated from the SOD inhibitable rate of cytochrome c reduction by using linear regression analysis (slope of the line fitting the data points). Results depict the mean ± SEM rate of superoxide production for n = 5. *Significantly different from LacZ group. †Significantly different from LacZ+PMA group. *, †, marked comparisons are significantly different by using the Student’s t test (p < 0.001).
MKK6 gene deletion enhances Rac1 activity in vivo
To demonstrate a physiologic role for MKK6 in the regulation of Rac1, we used an MKK6 knockout mouse model (45). MKK6 is expressed in brain, heart, liver, muscle, and pancreas, and several alternatively spliced mRNA species are differentially expressed in these tissues (20). We chose to focus our analysis on muscle and brain because these two tissues together express all the known isoforms of MKK6, including the MKK6b isoform we have used for our functional studies (20). These two tissues also express divergent levels of MKK6 (muscle being much higher than brain) (20) that might reflect tissue-specific functions of MKK6. Additionally, muscle and brain express very high levels of Rac1 (31). To this end, we evaluated the effect of MKK6 gene deletion on Rac1 activity in brain and skeletal muscle tissue lysates by using a PAK-binding domain (PBD) pull-down assay. In this assay, GST-PBD binds only to Rac1-GTP and can be used in a pull-down assay to evaluate the ratio of Rac1-GTP to total Rac1 levels in tissue lysates. Results from these experiments (Fig. 6) demonstrated that Rac1-GTP levels were significantly higher in mouse brain tissue of MKK6-deficient mice as compared with heterozygous littermates. In contrast, no differences in Rac1-GTP levels were seen in skeletal muscle (data not shown). These findings provide further support that MKK6 inhibits Rac1 activity in vivo.
FIG. 6. MKK6 deletion enhances Rac1-GTP levels in the brain.
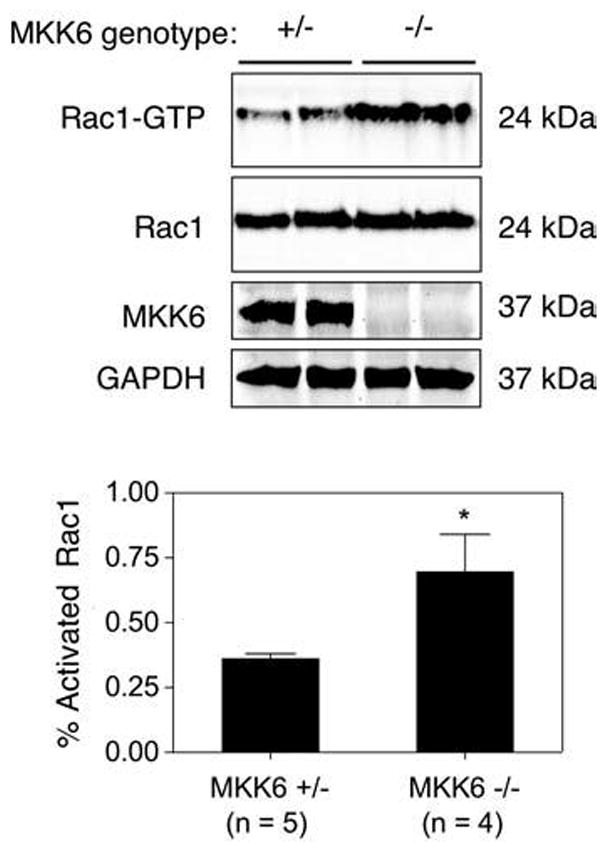
Rac1-GTP levels in brain lysates from MKK6-heterozygous (+/−) vs. MKK6-deficient (−/−) mice were determined by using a PAK-binding domain pull-down assay. (Top panel) A Western blot of representative samples demonstrating GTP-Rac1 levels after the PAK pull-down assay and total Rac1, MKK6, and GAPDH in crude lysates representing 4% of the starting material used in the GTP-Rac1 PAK pull-down assay. (Lower panel) Quantification of the band intensity ratios for Rac1-GTP relative to total Rac1 for each sample. These values were used to calculate the percentage of activated Rac1-GTP levels (mean ± SEM for n independent animals). Rac1-GTP levels were significantly elevated in brain lysates from MKK6−/− mice compared with MKK6+/− mice (p < 0.05; Student’s t test).
DISCUSSION
Although the mechanisms of NADPH oxidase activation have been fairly well elucidated (25), the mechanism(s) that regulate the inactivation of this complex remain poorly understood. Our data suggest that redox-dependent tyrosine phosphorylation of MKK6 plays an active role in downregulating the NADPH oxidase complex by promoting Rac-GTPase activity. Based on these findings, we hypothesize that Rac-GTP–mediated activation of the NADPH oxidase complex leads to production of O2•− and H2O2 that can then autoregulate the Nox complex by reducing Rac-GTP levels. In this context, the accumulation of H2O2, by either spontaneous or SOD-mediated dismutation of O2•−, may act to facilitate association of MKK6 with Rac1/2. Tyrosine phosphorylation of MKK6 on Y219 enhanced its binding to Rac in vitro and was also required for the ability of MKK6 to inhibit NADPH oxidase activation in vivo, suggesting that this phosphorylation event may be a key step in the redox regulation of MKK6. Increasing evidence points to the importance of spatially controlled Nox complex activation in cell signaling at the plasma membrane and within intracellular vesicles (47). Although a physiologic stimulus for regulation of MKK6 Y219-phosphorylation has yet to be identified, we anticipate that this pathway may be most relevant to regulation of Rac-dependent Nox complexes within intracellular membranes, because local concentration changes of H2O2 would occur at sites where Rac/Nox complexes are most active.
Based on our proposed model, one might hypothesize that the loss of MKK6 would lead to higher levels of NADPH oxidase activity. Although elevated Rac1-GTP levels in brain lysates from MKK6-deficient mice were consistent with MKK6 being a Rac1-GAP, we did not observe enhanced NADPH-dependent O2•− production in fractionated endosomal membranes prepared from brain homogenates of MKK6 knockout as compared with heterozygous littermates (data not shown). This may be due to compensatory regulation of Nox gene expression or other regulator pathways that control Nox activators other than Rac1 (25). Additionally, MKK6 may predominantly play a regulatory role in Nox activation after certain cell stimuli that are yet to be determined. The finding that Rac1-GTP levels were altered in brain, but not muscle, also supports cell specificity to the function of MKK6 as a potential Rac1-GAP. In this context, it is interesting to note that MKK6 activation has been linked to a number of neurodegenerative diseases with known components of oxidative stress (21, 39, 53).
H2O2 is known to act as a second messenger of tyrosine phosphorylation signaling pathways by reversibly inactivating tyrosine phosphatases (42). Given that MKK6 can autophosphorylate Y219, it is plausible that H2O2-mediated inactivation of a yet-to-be-identified tyrosine phosphatase may mediate the redox activation of MKK6 into a competent Rac-GAP. This change in MKK6 structure appears to facilitate its association with Rac-GTP, leading to GTP hydrolysis by Rac1 and subsequent inactivation of the NADPH oxidase complex. The importance of tyrosine phosphorylation of MKK6 in mediating GAP activation is supported by related previous findings in the literature. For example, p190Rho-GAP is a predominant tyrosine-phosphorylated protein in the brain (8), and tyrosine phosphorylation of p190Rho-GAP increases GAP activity of this protein (50). In contrast, tyrosine phosphorylation of ASAP1 (an Arf-1-GAP) inhibits GTPase-activating function of this protein. It is worth noting that although tyrosine phosphorylation of MKK6 regulated Rac1 binding to MKK6, it did not destroy intrinsic MKK6 Rac1-GAP activity in vitro. This finding suggests that the regulatory effect of MKK6 on Rac1 is most likely mediated by differential binding affinities of the phosphorylated and unphosphorylated forms of MKK6 for Rac1, as opposed to inherent differences in GAP catalytic activity. Although our studies suggest that MKK6 acts as an Rac1-GAP, we did not find homology between MKK6 amino acid sequence and GAP domains. However, GAP functions have previously been observed for other proteins that lacked an obvious homology to GAP domains (29).
Our studies demonstrate that activated full-length CA-MKK6 can associate with Rac1 in vitro when phosphorylated on Y219. However, in vivo, a non–full-length form of MKK6 (~18 kDa) appears to be the selective target of Y219 phosphorylation. The mechanism by which this shorter form of MKK6 is generated remains unclear. MEKK1, a related mitogen-activated protein kinase kinase kinase family member, has been shown to have caspase-3 cleavage sites important in Fas-mediated apoptosis (16). Given that MKK6 is a downstream target of MEKK1, we searched for known caspase 1 through 10 cleavage sites in MKK6 by using the ExPASy proteomics server PeptideCutter; however, none was found. Other studies have demonstrated specific proteolysis of mitogen-activated protein kinase kinase family members (including MKK6) with anthrax lethal factor peptidase at a conserved site (48). However, whether endogenous factors can process MKK6 in a similar fashion remain to be determined. Because many of the mitogen-activated protein kinase kinase genes are alternatively spliced (49), this is also one potential mechanism to generate a shorter form of MKK6. It is worth noting that when HA-tagged MKK6 was overexpressed in 293 cells, we observed expression of both the full-length HA-tagged MKK6 protein (40 kDa) and the cleaved HA-tagged MKK6 fragment (20 kDa) (data not shown). This suggests that the likely mechanism for generating the MKK6 fragment is an intracellular protein-cleavage event liberating the c-terminal end of the MKK6 protein. However, the protease involved in this cleavage remains to be determined.
MKK6 is an upstream activator of the p38 mitogen-activated protein kinase, a redox-regulated signaling factor activated by many environmental stimuli and proinflammatory cytokines (15, 36). For example, oxidative stress plays an important role in pathogenesis of cardiac ischemia/reperfusion and hypertrophy; both processes involve the MKK6 and p38MAPK pathways (7, 19, 30). Despite suggestive evidence in the literature, the links between the MKK6/p38MAPK pathway and ROS have remained indirect. Here we show that MKK6 directly binds to Rac1/2 and regulates NADPH oxidase ROS production. Moreover, we show for the first time that MKK6 is tyrosine phosphorylated, an event that is intrinsically regulated by the autokinase activity of MKK6 and important for regulating ROS generation through NADPH oxidase. Hence, our findings demonstrate a mechanism of direct crosstalk between the MKK6 pathway and Rac/NADPH oxidase–mediated ROS production.
Acknowledgments
This work was supported by NIDDK (DK51315, DK067928), the vector core funded through the Center for Gene Therapy (P30 DK54759), and the Roy J. Carver Chair in Molecular Medicine. We also gratefully thank Dr. Roger Davis for providing us with the MKK6 knockout mice, Dr. Jiahuai Han for providing us with the GST-MKK6E construct, Jennifer Marden for proofreading this manuscript, and Dr. William Nauseef for his helpful comments during the course of these studies.
ABBREVIATIONS
- Ad.CA-MKK6
adenoviral vector that expresses CA MKK6 HA-tagged on the N-terminus incorporating two mutations in the kinase active site S207E and T211E
- Ad.Y219F-CA-MKK6
adenoviral vector that expresses Y219F CA MKK6 HA-tagged on the N-terminus incorporating three mutations S207E, T211E, and Y219F
- ASAP-1
ARF-GAP containing SH3, ANK repeats, and PH domain
- CA-MKK6
constitutively active mitogen activated protein kinase kinase 6
- DCFH-DA
dichlorofluorescin diacetate
- Expasy
expert protein analysis system
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- Gpx-1
glutathione peroxidase 1
- GST
glutathione-S-transferase
- GST-CA-MKK6
(glutathione-S-transferase)–tagged constitutively active (S207E and T211E) MKK6-purified protein
- GST-Y219F-CA-MKK6
(glutathione-S-transferase)–tagged Y219F CA MKK6 (incorporating three mutations, S207E, T211E, and Y219F)
- HA
influenza A virus hemagglutinin (HA) tag
- H2O2
hydrogen peroxide
- IL-1β
interleukin-1beta
- LF
lethal factor
- MALDI-TOF
matrix-assisted laser desorption/ionization-time of flight
- MEKK1
mitogen-activated protein kinase kinase kinase 1
- MKK6
mitogen-activated protein kinase kinase 6
- NADPH
reduced nicotin-amide–adenine dinucleotide phosphate
- Nox2
phagocytic NADPH oxidase
- O2•−
superoxide
- PBD
PAK binding domain
- PDGF
platelet-derived growth factor
- PMA
phorbol myristate acetate
- ROS
reactive oxygen species
- SOD
super-oxide dismutase
- TLC
thin-layer chromatography
References
- 1.Arbault S, Pantano P, Sojic N, Amatore C, Best-Belpomme M, Sarasin A, Vuillaume M. Activation of the NADPH oxidase in human fibroblasts by mechanical intrusion of a single cell with an ultramicroelectrode. Carcinogenesis. 1997;18:569–574. doi: 10.1093/carcin/18.3.569. [DOI] [PubMed] [Google Scholar]
- 2.Bayraktutan U, Draper N, Lang D, Shah AM. Expression of functional neutrophil-type NADPH oxidase in cultured rat coronary microvascular endothelial cells. Cardiovasc Res. 1998;38:256–262. doi: 10.1016/s0008-6363(98)00003-0. [DOI] [PubMed] [Google Scholar]
- 3.Bernards A. GAPs galore! A survey of putative Ras superfamily GTPase activating proteins in man and Drosophila. Biochim Biophys Acta. 2003;1603:47–82. doi: 10.1016/s0304-419x(02)00082-3. [DOI] [PubMed] [Google Scholar]
- 4.Blasi F, Tarsia P, Aliberti S. Strategic targets of essential host-pathogen interactions. Respiration. 2005;72:9–25. doi: 10.1159/000083394. [DOI] [PubMed] [Google Scholar]
- 5.Blom N, Gammeltoft S, Brunak S. Sequence and structure-based prediction of eukaryotic protein phosphorylation sites. J Mol Biol. 1999;294:1351–1362. doi: 10.1006/jmbi.1999.3310. [DOI] [PubMed] [Google Scholar]
- 6.Bollag G, McCormick F. Differential regulation of rasGAP and neurofibromatosis gene product activities. Nature. 1991;351:576–579. doi: 10.1038/351576a0. [DOI] [PubMed] [Google Scholar]
- 7.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin-NFAT signaling. J Clin Invest. 2003;111:1475–1486. doi: 10.1172/JCI17295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brouns MR, Matheson SF, Settleman J. p190 RhoGAP is the principal Src substrate in brain and regulates axon outgrowth, guidance and fasciculation. Nat Cell Biol. 2001;3:361–367. doi: 10.1038/35070042. [DOI] [PubMed] [Google Scholar]
- 9.Brown MR, Miller FJ, Jr, Li WG, Ellingson AN, Mozena JD, Chatterjee P, Engelhardt JF, Zwacka RM, Oberley LW, Fang X, Spector AA, Weintraub NL. Overexpression of human catalase inhibits proliferation and promotes apoptosis in vascular smooth muscle cells. Circ Res. 1999;85:524–533. doi: 10.1161/01.res.85.6.524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chamseddine AH, Miller FJ., Jr Gp91phox contributes to NADPH oxidase activity in aortic fibroblasts but not smooth muscle cells. Am J Physiol Heart Circ Physiol. 2003;285:H2284–H2289. doi: 10.1152/ajpheart.00459.2003. [DOI] [PubMed] [Google Scholar]
- 11.Chamulitrat W, Stremmel W, Kawahara T, Rokutan K, Fujii H, Wingler K, Schmidt HH, Schmidt R. A constitutive NADPH oxidase-like system containing gp91phox homologs in human keratinocytes. J Invest Dermatol. 2004;122:1000–1009. doi: 10.1111/j.0022-202X.2004.22410.x. [DOI] [PubMed] [Google Scholar]
- 12.Cheng G, Diebold BA, Hughes Y, Lambeth JD. Nox1-dependent reactive oxygen generation is regulated by Rac1. J Biol Chem. 2006;281:17718–17726. doi: 10.1074/jbc.M512751200. [DOI] [PubMed] [Google Scholar]
- 13.Choi MH, Lee IK, Kim GW, Kim BU, Han YH, Yu DY, Park HS, Kim KY, Lee JS, Choi C, Bae YS, Lee BI, Rhee SG, Kang SW. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005;435:347–353. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]
- 14.Clifford DP, Repine JE. Measurement of oxidizing radicals by polymorphonuclear leukocytes. Methods Enzymol. 1984;105:393–398. doi: 10.1016/s0076-6879(84)05054-0. [DOI] [PubMed] [Google Scholar]
- 15.Das DK, Maulik N, Engelman RM. Redox regulation of angiotensin II signaling in the heart. J Cell Mol Med. 2004;8:144–152. doi: 10.1111/j.1582-4934.2004.tb00270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Deak JC, Cross JV, Lewis M, Qian Y, Parrott LA, Distelhorst CW, Templeton DJ. Fas-induced proteolytic activation and intracellular redistribution of the stress-signaling kinase MEKK1. Proc Natl Acad Sci U S A. 1998;95:5595–5600. doi: 10.1073/pnas.95.10.5595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Decoursey TE, Ligeti E. Regulation and termination of NADPH oxidase activity. Cell Mol Life Sci. 2005;62:2173–2193. doi: 10.1007/s00018-005-5177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engelhardt JF, Yang Y, Stratford-Perricaudet LD, Allen ED, Kozarsky K, Perricaudet M, Yankaskas JR, Wilson JM. Direct gene transfer of human CFTR into human bronchial epithelia of xenografts with E1-deleted adenoviruses. Nat Genet. 1993;4:27–34. doi: 10.1038/ng0593-27. [DOI] [PubMed] [Google Scholar]
- 19.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Invest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han J, Lee JD, Jiang Y, Li Z, Feng L, Ulevitch RJ. Characterization of the structure and function of a novel MAP kinase kinase (MKK6) J Biol Chem. 1996;271:2886–2891. doi: 10.1074/jbc.271.6.2886. [DOI] [PubMed] [Google Scholar]
- 21.Hartzler AW, Zhu X, Siedlak SL, Castellani RJ, Avila J, Perry G, Smith MA. The p38 pathway is activated in Pick disease and progressive supranuclear palsy: a mechanistic link between mitogenic pathways, oxidative stress, and tau. Neurobiol Aging. 2002;23:855–859. doi: 10.1016/s0197-4580(02)00029-5. [DOI] [PubMed] [Google Scholar]
- 22.Heyworth PG, Knaus UG, Settleman J, Curnutte JT, Bokoch GM. Regulation of NADPH oxidase activity by Rac GTPase activating protein(s) Mol Biol Cell. 1993;4:1217–1223. doi: 10.1091/mbc.4.11.1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jackson SH, Devadas S, Kwon J, Pinto LA, Williams MS. T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol. 2004;5:818–827. doi: 10.1038/ni1096. [DOI] [PubMed] [Google Scholar]
- 24.Kamata H, Honda SI, Maeda S, Chang L, Hirata H, Karin M. Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell. 2005;120:649–661. doi: 10.1016/j.cell.2004.12.041. [DOI] [PubMed] [Google Scholar]
- 25.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 26.Lancaster CA, Taylor-Harris PM, Self AJ, Brill S, van Erp HE, Hall A. Characterization of rhoGAP, a GTPase-activating protein for rho-related small GTPases. J Biol Chem. 1994;269:1137–1142. [PubMed] [Google Scholar]
- 27.Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi B, Engelhardt JF. Nox2 and Rac1 Regulate H2O2-dependent recruitment of TRAF6 to endosomal interleukin-1 receptor complexes. Mol Cell Biol. 2006;26:140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Q, Sanlioglu S, Li S, Ritchie T, Oberley L, Engelhardt JF. GPx-1 gene delivery modulates NFkappaB activation following diverse environmental injuries through a specific subunit of the IKK complex. Antioxid Redox Signal. 2001;3:415–432. doi: 10.1089/15230860152409068. [DOI] [PubMed] [Google Scholar]
- 29.Low BC, Seow KT, Guy GR. The BNIP-2 and Cdc42GAP homology domain of BNIP-2 mediates its homophilic association and heterophilic interaction with Cdc42GAP. J Biol Chem. 2000;275:37742–37751. doi: 10.1074/jbc.M004897200. [DOI] [PubMed] [Google Scholar]
- 30.Martindale JJ, Wall JA, Martinez-Longoria DM, Aryal P, Rockman HA, Guo Y, Bolli R, Glembotski CC. Overexpression of mitogen-activated protein kinase kinase 6 in the heart improves functional recovery from ischemia in vitro and protects against myocardial infarction in vivo. J Biol Chem. 2005;280:669–676. doi: 10.1074/jbc.M406690200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Matos P, Skaug J, Marques B, Beck S, Verissimo F, Gespach C, Boavida MG, Scherer SW, Jordan P. Small GTPase Rac1: structure, localization, and expression of the human gene. Biochem Biophys Res Commun. 2000;277:741–751. doi: 10.1006/bbrc.2000.3743. [DOI] [PubMed] [Google Scholar]
- 32.Mizrahi A, Molshanski-Mor S, Weinbaum C, Zheng Y, Hirshberg M, Pick E. Activation of the phagocyte NADPH oxidase by Rac guanine nucleotide exchange factors in conjunction with ATP and nucleoside diphosphate kinase. J Biol Chem. 2005;280:3802–3811. doi: 10.1074/jbc.M410257200. [DOI] [PubMed] [Google Scholar]
- 33.Moskwa P, Paclet MH, Dagher MC, Ligeti E. Autoinhibition of p50 Rho GTPase-activating protein (GAP) is released by prenylated small GTPases. J Biol Chem. 2005;280:6716–6720. doi: 10.1074/jbc.M412563200. [DOI] [PubMed] [Google Scholar]
- 34.Moulton PJ, Goldring MB, Hancock JT. NADPH oxidase of chondrocytes contains an isoform of the gp91phox subunit. Biochem J. 1998;329:449–451. doi: 10.1042/bj3290449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nauseef WM. Assembly of the phagocyte NADPH oxidase. Histochem Cell Biol. 2004;122:277–291. doi: 10.1007/s00418-004-0679-8. [DOI] [PubMed] [Google Scholar]
- 36.Nebreda AR, Porras A. p38 MAP kinases: beyond the stress response. Trends Biochem Sci. 2000;25:257–260. doi: 10.1016/s0968-0004(00)01595-4. [DOI] [PubMed] [Google Scholar]
- 37.Pagano PJ, Clark JK, Cifuentes-Pagano ME, Clark SM, Callis GM, Quinn MT. Localization of a constitutively active, phagocyte-like NADPH oxidase in rabbit aortic adventitia: enhancement by angiotensin II. Proc Natl Acad Sci U S A. 1997;94:14483–14488. doi: 10.1073/pnas.94.26.14483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park HS, Lee SH, Park D, Lee JS, Ryu SH, Lee WJ, Rhee SG, Bae YS. Sequential activation of phosphatidylinositol 3-kinase, beta Pix, Rac1, and Nox1 in growth factor-induced production of H2O2. Mol Cell Biol. 2004;24:4384–4394. doi: 10.1128/MCB.24.10.4384-4394.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Peel AL, Sorscher N, Kim JY, Galvan V, Chen S, Bredesen DE. Tau phosphorylation in Alzheimer’s disease: potential involvement of an APP-MAP kinase complex. Neuromol Med. 2004;5:205–218. doi: 10.1385/NMM:5:3:205. [DOI] [PubMed] [Google Scholar]
- 40.Peled-Kamar M, Lotem J, Wirguin I, Weiner L, Hermalin A, Groner Y. Oxidative stress mediates impairment of muscle function in transgenic mice with elevated level of wild-type Cu/Zn superoxide dismutase. Proc Natl Acad Sci U S A. 1997;94:3883–3887. doi: 10.1073/pnas.94.8.3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Price MO, Atkinson SJ, Knaus UG, Dinauer MC. Rac activation induces NADPH oxidase activity in transgenic COSphox cells, and the level of superoxide production is exchange factor-dependent. J Biol Chem. 2002;277:19220–19228. doi: 10.1074/jbc.M200061200. [DOI] [PubMed] [Google Scholar]
- 42.Rhee SG, Kang SW, Jeong W, Chang TS, Yang KS, Woo HA. Intracellular messenger function of hydrogen peroxide and its regulation by peroxiredoxins. Curr Opin Cell Biol. 2005;17:183–189. doi: 10.1016/j.ceb.2005.02.004. [DOI] [PubMed] [Google Scholar]
- 43.Self AJ, Hall A. Measurement of intrinsic nucleotide exchange and GTP hydrolysis rates. Methods Enzymol. 1995;256:67–76. doi: 10.1016/0076-6879(95)56010-6. [DOI] [PubMed] [Google Scholar]
- 44.Szaszi K, Korda A, Wolfl J, Paclet MH, Morel F, Ligeti E. Possible role of RAC-GTPase-activating protein in the termination of superoxide production in phagocytic cells. Free Radic Biol Med. 1999;27:764–772. doi: 10.1016/s0891-5849(99)00126-4. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka N, Kamanaka M, Enslen H, Dong C, Wysk M, Davis RJ, Flavell RA. Differential involvement of p38 mitogen-activated protein kinase kinases MKK3 and MKK6 in T-cell apoptosis. EMBO Rep. 2002;3:785–791. doi: 10.1093/embo-reports/kvf153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tatsis N, Lannigan DA, Macara IG. The function of the p190 Rho GTPase-activating protein is controlled by its N-terminal GTP binding domain. J Biol Chem. 1998;273:34631–34638. doi: 10.1074/jbc.273.51.34631. [DOI] [PubMed] [Google Scholar]
- 47.Ushio-Fukai M. Localizing NADPH oxidase-derived ROS. Sci STKE. 2006;2006:re8. doi: 10.1126/stke.3492006re8. [DOI] [PubMed] [Google Scholar]
- 48.Vitale G, Bernardi L, Napolitani G, Mock M, Montecucco C. Susceptibility of mitogen-activated protein kinase kinase family members to proteolysis by anthrax lethal factor. Biochem J. 2000;352:739–745. [PMC free article] [PubMed] [Google Scholar]
- 49.Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- 50.Wolf RM, Wilkes JJ, Chao MV, Resh MD. Tyrosine phosphorylation of p190 RhoGAP by Fyn regulates oligodendrocyte differentiation. J Neurobiol. 2001;49:62–78. doi: 10.1002/neu.1066. [DOI] [PubMed] [Google Scholar]
- 51.Zhang Y, Zhao W, Zhang HJ, Domann FE, Oberley LW. Overexpression of copper zinc superoxide dismutase suppresses human glioma cell growth. Cancer Res. 2002;62:1205–1212. [PubMed] [Google Scholar]
- 52.Zhen L, King AA, Xiao Y, Chanock SJ, Orkin SH, Dinauer MC. Gene targeting of X chromosome-linked chronic granulomatous disease locus in a human myeloid leukemia cell line and rescue by expression of recombinant gp91phox. Proc Natl Acad Sci U S A. 1993;90:9832–9836. doi: 10.1073/pnas.90.21.9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu X, Rottkamp CA, Hartzler A, Sun Z, Takeda A, Boux H, Shimohama S, Perry G, Smith MA. Activation of MKK6, an upstream activator of p38, in Alzheimer’s disease. J Neurochem. 2001;79:311–318. doi: 10.1046/j.1471-4159.2001.00597.x. [DOI] [PubMed] [Google Scholar]
- 54.Zimmerman MC, Dunlay RP, Lazartigues E, Zhang Y, Sharma RV, Engelhardt JF, Davisson RL. Requirement for Rac1-dependent NADPH oxidase in the cardiovascular and dipsogenic actions of angiotensin II in the brain. Circ Res. 2004;95:532–539. doi: 10.1161/01.RES.0000139957.22530.b9. [DOI] [PubMed] [Google Scholar]