

Abstract
There is increasing evidence that inflammation plays a role in the development of delayed cerebral vasospasm (DCV) after subarachnoid hemorrhage (SAH). Lipopolysaccharide (LPS) is an activator of the innate inflammatory system causes DCV in animal models. The effect of low-dose LPS has been shown to be protective in stroke models but has not been investigated in SAH. Two treatments were studied: 1) a single intraperitoneal (i.p.) dose of 0.6mg/kg injected 24 hours prior to SAH, and 2) four daily doses administered prior to SAH. DCV was determined by India ink angiography at day 6, behavioral testing was done in a different cohort of animals, and analysis of brain chemokine levels was accomplished by dot blot. Vessel caliber was improved compared to the SAH group in the single injection group (ldLPS ×4) (p<0.05). In the multiple injections group (ldLPS ×4), the vessel caliber was similar to SAH (p<0.05). ldLPS ×1 improved performance on the Barnes maze test whereas the ldLPS ×4 was worse (p<0.001). Brain levels of the inflammatory chemokine KC were decreased in the ldLPS ×1 and increased in the ldLPS ×4. Single injection low-dose LPS preconditioning is protective for DDAV whereas the multiple injection course exacerbates DDAV. This further supports that inflammation plays an important role in the development of DDAV and that modulating the inflammatory system may be a potential target for future therapies in SAH.
Keywords: Inflammation, Lipopolysaccharide, Tolerance, Delayed Cerebral Vasospasm, Subarachnoid Hemorrhage
Introduction
Delayed neurological deterioration associated with vasospasm (DDAV) in aneurismal subarachnoid hemorrhage (SAH) is the major cause of morbidity and mortality in patients who survive the initial bleeding episode(1). This vexing disease presents itself with spasm of proximal arteries and delayed neurological deficits 4-12 days after the aneurysm rupture. Although the term delayed cerebral ischemia (DCI) and delayed neurological ischemic deficits (DIND) have been used to describe the proposed cause of the injury, recent evidence suggests that mechanisms other than ischemia may be at play (2-4). A more appropriate term to describe the phenomenon may be “delayed deterioration associated with vasospasm (DDAV) ” to denote the uncertainty in the cause of the brain injury.
Our laboratory has focused on the role of early innate inflammation in both the vascular and cerebral manifestations of DDAV. Lipopolysaccharide A from E. coli (LPS) is a known signaling molecule of the innate immune system mediated through the TLR4 receptor on the neutrophil and endothelial cell surface. We have previously shown that the neutrophil percentage in the cerebrospinal fluid (CSF) early in the course of SAH can predict who will later develop DDAV(5). We have shown that myeloid cell depletion in a mouse model of DDAV ameliorates both the vascular and the behavioral effects(6). Previous animal work has shown that early administration of modulators of innate inflammation can alter the course of the disease(7-10). Direct administration of LPS into the CSF without SAH causes vasospasm(9) and systemic administration of LPS worsens DDAV after SAH in neutrophil dependent manner(11).
Data from stroke animal models show that low-dose LPS injection prevents inflammation and is neuroprotective(12-14). The mechanism of this protection has been proposed to be due to overexpression of genes coding for proteins in the TLR-signaling pathway(15). In this study, we find that systemic administration of low-dose LPS one day prior to SAH protects against DDAV but four daily sequential doses does not.
Materials and Methods
All experiments were conduct under the supervision of the Cleveland Clinic Institutional Animal Care and Use Committee (IACUC). Animals were randomized into six groups for the vasospasm study and four groups for the behavioral, immunohistochemistry, and chemokine studies, 1) Sham surgery (Sham), 2) SAH (SAH), 3) low-dose LPS administration one day prior to SAH (ldLPS ×1), and 4) low-dose LPS administration daily for 4 days prior to SAH (ldLPS ×4) for all the studies and the two controls groups 1) low-dose LPS administration one day prior to sham surgery (sham ldLPS ×1) and 2) low-dose LPS administration four days prior to sham surgery (sham ldLPS ×4) in the vessel diameter study. All surgeries were done by one investigator (SS) who randomly assigned animals to each of the three treatment groups. Analysis of the perfusion experiments and all behavioral tests were done by a different investigator (SKM) blinded to the surgical assignments.
Experimental SAH
We studied male C57BL6 mice (Jackson Labs, Maine) weighing 20-32 g, 10-12 week of age. Our murine model of SAH has been described(16). Briefly, mice were anesthetized and placed in a prone position. An incision was made in the midline of the neck, the atlanto-occipital membrane was punctured and a subarachnoid vein was transected. The bleeding was allowed to stop spontaneously after which the incision was closed. Saline injection sham surgery involved the same procedure except that the atlanto-occipital membrane was entered with a 30-gauge needle and 50 μl of saline was instilled. All animals that had surgery survived all the post hemorrhage testing.
LPS administration
0.6 mg/kg of LPS in 150 μl diluent (Sigma Aldrich, Saint Louis, MO) was injected into the peritoneal cavity of experimental animals either 24 hours prior SAH (ldLPS ×1) or daily for 4 days prior to SAH (ldLPS ×4). This dosage was increased 3-fold from previous studies of low dose LPS-induced tolerance in stroke(12, 15) due to the one day protocol in our study (unlike 2 day prior to stroke in published protocols).
India ink assessment of vessel caliber
Animals were anesthetized with pentobarbital (6 mg/100 g i.p.); trans-cardiac perfusion was performed with 20 ml 4% paraformaldehyde followed by 10 ml of warmed 5% India ink in gelatin. Animals were decapitated, and their brains were removed carefully preserving the vasculature. The circle of Willis vasculature was examined under the surgical microscope and relevant pictures captured (Leica, Wetzler, Germany) and analyzed with Adobe Photoshop CS2 (San Jose CA). The diameter of the middle cerebral artery (MCA) segment was measured 1 mm from the posterior wall of the carotid artery by a member of the research team not involved in the surgery and blinded to the intervention. All measurements were made on the 6th day after the hemorrhage based on previous work that shows that DCI occurs around the 6th post hemorrhage day in our model6. Ten mice were used for each of the four conditions (Sham, SAH, ldLPS ×1 and ldLPS ×4).
Behavioral testing
Animals in the behavioral study underwent three batteries of tests in groups of 10 mice. All tests were done at the same time of the day by a single handler (SKM) who was blinded to the treatment allocation. Groups of 5-10 animals randomized for treatment and blinded to the handler were tested together. This was repeated until 10 animals from each group were completed. On days 2 after the hemorrhage, the mice underwent rotarod testing to evaluate motor function and coordination. The 3rd day post hemorrhage, the mice underwent Y-maze testing to evaluate spatial working (immediate) memory. On day 2 and 3, the animals also had training for the Barnes maze. On days 4 through 9, the mice were tested in a Barnes maze to test spatial learning, short-term and long-term memory.
In the rotarod test, time on the rod and maximum speed of rotation (RPM) were measured. Animals were placed in one of 4 separated chambers with the spinning rod and a landing base. After a habituation for 30 seconds at low speed, the mice were tested with a continuously increasing rate of speed for 5 min or until the subject fell off the rod.
In the Y-maze, animals were placed in a random orientation in the center of a high-walled maze that has 3 equal-length passages in a Y configuration. The mice were recorded by video monitoring during 5 minutes of exploration and scored later. Percent alterations were calculated as the ratio of the number of times the animals chose to enter the three arms of the maze in succession to the total number of unique arms the animal chose to enter.
In the Barnes maze, the mice are placed on a round table with multiple open holes in the periphery one of which contains an under-mounted box that represents a safe haven for the mice. There are visual cues on the wall around the table for the animal to orient itself. Each animal had 2 days of training during which it was gently guided to the goal box. On subsequent days, animals were placed in the center of the table in a covered enclosure until ready for the trial. The cover was lifted and mice were allowed to find the goal box. The movements were recorded by video monitoring and scored later. The time it took to find the goal box (latency) was recorded.
Immunohistochemistry
The four groups of animals were tested using monoclonal anti-microglial cell antibody (Iba1) (Generous gift from Dr. Bruce Trapp) and flourochrome-conjugated monoclonal antibody against ICAM-1 (BioLegend, San Diego,CA)(which tests for endothelial activation). 30μm brain slices were incubated in primary antibody for 24 h using a free floating technique. Briefly, slices were microwaved for 2 min in citrate buffer followed by incubation in 500 μl of 3% goat serum and primary antibody at 20 °C overnight in a 24 well plate. Iba1 antibodies were incubated with a HRP-conjugated isotype-specific secondary antibody and resolved with 3,3′-diaminobenzidine (DAB).
Chemokine determination
Brain inflammatory chemokines levels were determined by Dot-Blot analysis (R&D Systems, Minneapolis, MN) 24 hours after surgery. Animals were anesthetized with pentobarbital (6 mg/100 g i.p.); trans-cardiac perfusion was performed with 20 ml 1% PBS. Animals were decapitated, and their brains were removed carefully and homogenized. After tissue protein extraction, the supernatant was incubated on the Dot Blot membrane. Three brain samples were pooled to increase concentrations of chemokines. Radioactive dots were visualized by radiography. Dot intensity was measured and normalized to a positive control and analyzed with Image J (NIH, Bethesda, MD).
Statistics
Statistical analysis was performed with the aid of Graphpad Prism 5.0 (Graphpad Software Inc. San Diego, CA). Multiple comparisons were assessed using 1-way ANOVA. Comparisons between individual groups in experiments where 1-way ANOVA was significantly different were assessed using Tukey’s Multiple Comparison Test. 2-way ANOVA was used to evaluate the effects of SAH over time. A p-value of 0.05 was considered significant in all comparisons.
Results
Vasospasm and behavioral assessment after SAH
Comparison of all four groups in the study for the vascular constriction shows that the groups are significantly different (ANOVA, F(5, 30)=10.56, p<0.0001). SAH animals had significant vasospasm at day 6 compared to sham control animals (Tukey’s Multiple Comparison Test, p<0.05) (Figure 1A). On Barnes maze behavioral testing, the four groups were significantly different in respect to time to find the goal box (2-way ANOVA, F(3,15)= 47.76, p<0.0001). SAH animals took a significantly longer time to find the goal box than the sham control animals (2-way ANOVA, F(1,5)=60.05, p<0.0001)(Figure 1B). The differences between the SAH and sham animals in both vasospasm and behavioral assessment confirm an earlier finding from our laboratory(6). There were no significant differences between the groups in regards to rotorod testing or Y-maze testing.
Figure 1.
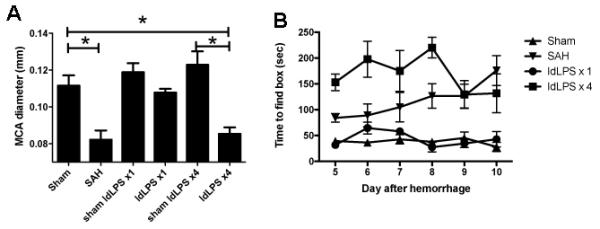
Single dose vs. multiple dose low-dose LPS schedule in SAH. A) MCA diameter six days after SAH shows that SAH and low dose LPS given 4 days prior to SAH (ldLPS ×4) have significantly smaller diameters indicating vasospasm (ANOVA, F(5,30)=10.56, p<0.0001). The low dose LPS administered once does not exhibit vasospasm compared to sham ldLPS ×1 (Tukey’s multiple comparisons test, p=NS). *signifies p<0.05 on Tukey’s multiple comparisons test for comparisons shown by bars. B) On Barnes maze test, animals in the ldLPS ×4 group and SAH had worsened times to find the goal box than the sham or ldLPS ×1 (2-way ANOVA, F(3,15)=47.76, p<0.0001). Together, these data show that a single dose of LPS prior to SAH prevents both the vasospasm and behavioral effects whereas the four-dose regimen prior to SAH is not protective.
Single dose preconditioning with LPS ameliorates both the vasospasm and the behavioral effects of SAH while multiple dose preconditioning does not
Animals in the ldLPS ×1 group did not show significant vasospasm (Tukey’s Multiple Comparison Test, ldLPS ×1 to SAH, p<0.05) or behavioral changes on Barnes maze (2-way ANOVA, ldLPS ×1 to SAH, F(1,5)= 28.36, p<0.0001) when compared to sham animals (Figures 1A and B). Conversely, animals in the ldLPS ×4 group had similar vasospasm (Tukey’s Multiple Comparison Test, ldLPS ×4 to SAH, NS) to animals with SAH. Interestingly, the behavioral changes on Barnes maze were more severe than the SAH animals (2-way ANOVA, ldLPS ×4 to SAH, F(1,5)= 7.898, p=0.0064). These results show that a single dose of LPS prior to SAH is protective of both vasospasm and the behavioral consequences whereas multiple doses appears to not be protective and makes the behavioral manifestations worse.
Immunohistochemical changes suggest that ldLPS ×1 decreases brain and vascular inflammation whereas ldLPS ×4 exacerbates inflammation
Immunohistochemical analysis of brain slices shows that animals in the ldLPS ×1 have decreased staining for I-CAM-1 on endothelial cells of the blood vessels suggesting that despite SAH, the endothelial cells are not activated, and ramified morphology of the microglia suggesting decreased microglial activation. By contrast, animals in the ldLPS ×2 group shows the opposite with increased staining of I-CAM-1 and ameboid morphology of the microglia suggesting inflammation (Figure 2). Animals in the Sham and SAH groups, the ICAM-1 staining and morphology of microglia predictably shows signs of inflammation in SAH group and no inflammation in the Sham group.
Figure 2.
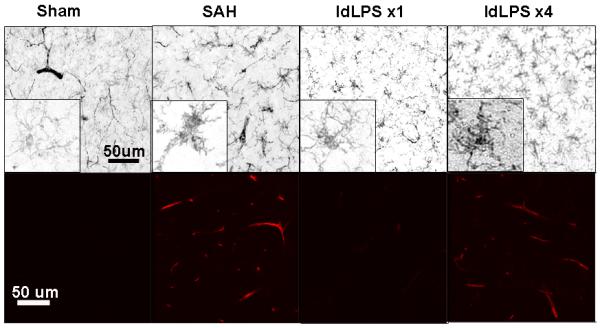
Immunohistochemical analysis of brain slices. Sham animals show ramified morphology of microglia (by Iba1 staining resolved with DAB) (above) and no vascular staining for ICAM-1 (below, confocal microscopy) suggesting no inflammation. SAH animals show ameboid morphology of microglia and increased vascular staining for ICAM-1. Animals in the single dose LPS course (ldLPS ×1) show a pattern similar to sham animals. Animals in the multiple dose course (ldLPS ×4) show a pattern similar to SAH animals. Non-microglial staining on Iba1-stained sections represents non-specific blood vessel staining.
The keratinocyte-derived chemokine (KC) is suppressed in ldLPS ×1 and increased in ldLPS ×4 animals after SAH
Mice do not express homologs to the human CXCL1 (Groα) and CXCL8 (IL-8) chemokines. Instead, they express the analogous KC chemokine that stimulates the CXCR2 receptor on neutrophils. We tested whole brain levels of KC in mice in the ldLPS ×1 and ldLPS ×4 groups. In order to develop adequate signal, 3 brains were pooled for each experiment. Each experiment represents pooled samples of three different animals. We find that compared to sham and SAH animals, animals in the ldLPS ×1 group had lower whole brain KC levels and ldLPS ×4 group had higher KC levels at both day 1 and day 6 after SAH. This supports the findings in the vasospasm, behavioral assessment and immunohistochemical analysis that a single dose of LPS decreases inflammation whereas the multiple-dose course increases inflammation. This further suggests that part of the mechanism of inflammation suppression is due to decreased signaling for neutrophils.
Discussion
Our study shows that a single dose of low dose LPS given prior to SAH prevents both the vascular and behavioral sequelae of DDAV. Interestingly, the same dose of LPS given four consecutive days prior to SAH has no effect on vasospasm and opposite effects on behavioral ability on Barnes maze tests. Immunohistochemical staining and chemokine analysis suggest that single dose LPS silences the inflammatory response to the blood whereas multiple doses increases the inflammatory signaling in the brain.
The mechanism by which low dose LPS pretreatment induces ‘tolerance’ in stroke has been investigated. In the case of LPS induced tolerance, the mechanism revolves around induction of IRF3 activation in the brain(15, 17). It is still not clear if systemic administration of LPS directly affects the brain parenchyma or works through mediators in the endothelium.
In our study, we found differential effects of LPS depending on the administration schedule. This suggest that a few possible mechanisms that require further research: 1) the endothelium mediates depression of brain inflammatory responses and has a summated effect based on LPS doses. That is to say, a single dose of LPS decreases endothelial propensity to signal inflammation and multiple doses summate to release inflammatory signaling molecules. Or 2) LPS crosses the blood brain barrier intact and signals inflammatory cells in the brain (microglia, astrocytes, etc.) to develop an inflammatory response.
The immunohistochemical studies presented here suggest that the endothelium is the gatekeeper of inflammation and that the brain follows the lead of the endothelium. This affords welcomed opportunities to interact with the endothelial lining without having to devise drug or biological therapies that cross the blood-brain barrier. We did not investigate activation of IRF3 or other TLR4 pathway mediators.
There are a number of limitations of our study. First, the severity of DDAV elicited by our model is mild compared to other models. It is possible that the mechanisms are different for mild versus severe DDAV. We feel that this mild form of DDAV is similar to what we see clinically in patients with good to medium grade SAH (Hunt and Hess 1-3). Second, our study does not address directly the molecular mechanism of tolerance. We felt that it was more important to determine the cells involved before entertaining specific molecular or cellular mechanisms.
The development of DDAV appears to be dependent on inflammation after SAH. How early this inflammation develops is still unknown but of critical importance for the development of meaningful therapies. In stroke models, increased inflammatory mediator levels are seen as early as three hours after the infarction(15, 18, 19). In stroke in humans, inflammation and edema develop over the span of 3-5 days. In SAH patients, DDAV begins later. It is possible that this is due to later activation of the inflammatory system making first day therapies realistic.
This study adds to our understanding of how inflammation in the brain of patients with SAH may lead to DDAV. It focuses the attention on the acute innate inflammatory response and confirms earlier findings that neutrophils are important in this process. Further work will need to focus on the molecular pathway that initiates this inflammatory response. This is the best hope for a viable treatment or preventative.
Figure 3.
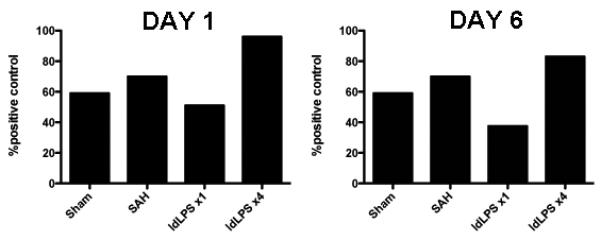
Level of KC in pooled brain samples. In both the Day 1 samples (left) and Day 6 samples (right), low dose LPS administered one time prior to SAH (ldLPS ×1) decreases the production of the neutrophil attractant chemokine KC whereas the four-dose regimen prior to SAH increases KC production.
Acknowledgements
The authors would like to thank Shu-mei Man for help with confocal imaging.
This work was supported by a grant from the NIH (K08-NS051350 JJP) and the Cleveland Clinic Cerebrovascular Center (Institutional Support JJP).
Footnotes
Financial Disclosure: The authors have no conflicts to report.
Contributor Information
Saksith Smithason, Department of Neurosurgery, Cleveland Clinic, Cleveland, OH, smithas@ccf.org.
Shari K. Moore, Neuroinflammation Research Center, Cleveland Clinic, Cleveland, OH, moores2@ccf.org.
J. Javier Provencio, Neuroinflammation Research Center and Cerebrovascular Center, Cleveland Clinic, Cleveland, OH.
References
- 1.van Gijn J, Rinkel GJ. Subarachnoid haemorrhage: diagnosis, causes and management. Brain. 2001;124(Pt 2):249–278. doi: 10.1093/brain/124.2.249. [DOI] [PubMed] [Google Scholar]
- 2.Etminan N, Vergouwen MD, Ilodigwe D, Macdonald RL. Effect of pharmaceutical treatment on vasospasm, delayed cerebral ischemia, and clinical outcome in patients with aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. J Cereb Blood Flow Metab. 2011 doi: 10.1038/jcbfm.2011.7. Translated from Eng. in Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stein SC, Levine JM, Nagpal S, LeRoux PD. Vasospasm as the sole cause of cerebral ischemia: how strong is the evidence? Neurosurgical Focus. 2006;21(3):E2. doi: 10.3171/foc.2006.21.3.2. [DOI] [PubMed] [Google Scholar]
- 4.Vergouwen MD, Ilodigwe D, Macdonald RL. Cerebral infarction after subarachnoid hemorrhage contributes to poor outcome by vasospasm-dependent and -independent effects. Stroke. 2011;42(4):924–929. doi: 10.1161/STROKEAHA.110.597914. Translated from eng. in eng. [DOI] [PubMed] [Google Scholar]
- 5.Provencio JJ, et al. CSF neutrophils are implicated in the development of vasospasm in subarachnoid hemorrhage. Neurocrit Care. 2010;12(2):244–251. doi: 10.1007/s12028-009-9308-7. Translated from eng. in eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Provencio JJ, Altay T, Smithason S, Moore SK, Ransohoff RM. Depletion of Ly6G/C(+) cells ameliorates delayed cerebral vasospasm in subarachnoid hemorrhage. J Neuroimmunol. 2010 doi: 10.1016/j.jneuroim.2010.10.016. Translated from Eng. in Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pradilla G, et al. Prevention of vasospasm by anti-CD11/CD18 monoclonal antibody therapy following subarachnoid hemorrhage in rabbits. J Neurosurg. 2004;101(1):88–92. doi: 10.3171/jns.2004.101.1.0088. Translated from eng. in eng. [DOI] [PubMed] [Google Scholar]
- 8.Zubkov AY, Tibbs RE, Aoki K, Zhang JH. Prevention of vasospasm in penetrating arteries with MAPK inhibitors in dog double-hemorrhage model. Surg Neurol. 2000;54(3):221–227. doi: 10.1016/s0090-3019(00)00290-1. discussion 227-228. [DOI] [PubMed] [Google Scholar]
- 9.Recinos PF, et al. Controlled release of lipopolysaccharide in the subarachnoid space of rabbits induces chronic vasospasm in the absence of blood. Surg Neurol. 2006;66(5):463–469. doi: 10.1016/j.surneu.2006.04.010. discussion 469. [DOI] [PubMed] [Google Scholar]
- 10.Zhou C, et al. Caspase inhibitors prevent endothelial apoptosis and cerebral vasospasm in dog model of experimental subarachnoid hemorrhage. Journal of Cerebral Blood Flow & Metabolism. 2004;24(4):419–431. doi: 10.1097/00004647-200404000-00007. [DOI] [PubMed] [Google Scholar]
- 11.Smithason S, Moore SK, Provencio JJ. Systemic administration of LPS worsens delayed deterioration associated with vasospasm after subarchnoid hemorrhage through a myeloid cell dependent mechanism. Neurocrit Care. 2011 doi: 10.1007/s12028-011-9651-3. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenzweig HL, et al. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke. 2004;35(11):2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. Translated from eng. in eng. [DOI] [PubMed] [Google Scholar]
- 13.Rosenzweig HL, et al. Endotoxin preconditioning protects against the cytotoxic effects of TNFalpha after stroke: a novel role for TNFalpha in LPS-ischemic tolerance. J Cereb Blood Flow Metab. 2007;27(10):1663–1674. doi: 10.1038/sj.jcbfm.9600464. Translated from eng. in eng. [DOI] [PubMed] [Google Scholar]
- 14.Tasaki K, et al. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Res. 1997;748(1-2):267–270. doi: 10.1016/s0006-8993(96)01383-2. Translated from eng. in eng. [DOI] [PubMed] [Google Scholar]
- 15.Stevens SL, et al. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. J Neurosci. 2011;31(23):8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011. Translated from eng. in eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Altay T, et al. A novel method for subarachnoid hemorrhage to induce vasospasm in mice. J Neurosci Methods. 2009;183(2):136–140. doi: 10.1016/j.jneumeth.2009.06.027. Translated from eng. in eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marsh B, et al. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. J Neurosci. 2009;29(31):9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. Translated from eng. in eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Broad A, Kirby JA, Jones DE. Toll-like receptor interactions: tolerance of MyD88-dependent cytokines but enhancement of MyD88-independent interferon-beta production. Immunology. 2007;120(1):103–111. doi: 10.1111/j.1365-2567.2006.02485.x. Translated from eng. in eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Broad A, Jones DE, Kirby JA. Toll-like receptor (TLR) response tolerance: a key physiological “damage limitation” effect and an important potential opportunity for therapy. Curr Med Chem. 2006;13(21):2487–2502. doi: 10.2174/092986706778201675. Translated from eng. in eng. [DOI] [PubMed] [Google Scholar]