

Abstract
Alkyl chloroformates induced indirect trapping of the retroconjugate addition reaction intermediate involved in the epimerization of lobeline is described. This strategy was applied to the conversion of (−)-lobeline to (−)-sedamine in high overall yield.
(−)-Lobeline (1a), a lipophilic alkaloidal constituent of Lobelia inflata LINN., has a long history of therapeutic usage ranging from emetic and respiratory stimulant to tobacco smoking cessation agent.1 Although lobeline has been generally accepted to act as a nicotinic receptor agonist, recent studies suggest its pharmacological action is more complex than previously thought. Studies in our laboratories have revealed a novel mechanism of action, i.e., inhibition of dopamine uptake and promotion of dopamine release from storage vesicles within dopaminergic presynaptic terminals, via an interaction with the tetrabenazine binding site on the vesicular monoamine transporter (VMAT-2).2 As part of a drug discovery program aimed at the development of therapeutic agents for treating central nervous system disorders, we were interested in the preparation of a series of structural analogues of lobeline. A literature search indicates that relatively few analogues of lobeline have been prepared for subsequent structure-activity relationship studies to define the lobeline pharmacophore.3 Moreover, most of the analogues of lobeline that appear in the literature were either defunctionalized or simplified lobeline analogues. 3
As reported in the literature,4,5 (−)-lobeline (1a) free base underwent a slow epimerization at C2 to afford a mixture of cis/trans lobeline (1a and 1b),6,7 which is believed to be the result of a self (base)-catalyzed equilibration via a transient retroconjugate addition reaction intermediate 2 (Scheme 1). With respect to our interest in the synthesis of novel lobeline analogues, this retroconjugate addition reaction was considered to be a key step. However, no spectroscopic evidence (UV, IR, NMR) for the existence of 2 has thus far been obtained, presumably because of its very short lifetime and/or its presence in very low concentration in the equilibrium mixture. Nevertheless, the occurrence of a retroconjugate addition reaction can be confirmed by trapping intermediate 2, which requires prevention of 2 from undergoing the intramolecular conjugate addition reaction by either blocking the enone system or blocking the resultant secondary amine. Our first attempts to trap the enone system in 2 met with failure: adding nucleophiles (dimethylamine or piperidine) to a methanol solution of 1a resulted in unstable enamine products of 1a; reducing 1a by Pd/C hydrogenation or NaBH3CN treatment gave solely the ketone reduced product of 1a; and oxidation of 1a by OsO4/NaIO4 afforded a product mixture that could not be characterized. Thus, we turned to the second strategy of trapping the secondary amine of 2 by preventing it from reacting as a nucleophile. We first treated 1a with excess HCl in MeOH (or CHCl3) at room temperature or at reflux.8 Unexpectedly, intermediate was not trapped, nor did epimerization occur (confirmed by NMR spectroscopic analysis). To further investigate this reaction, 9 a mixture of 1a/1b was used under the same conditions; in contrast to the literature reports,4 no change in the mixture was observed after treatment with HCl.10 Second, we treated 1a with excess Ac2O (with or without THF, or CH3CN) and NaOAc (or pyridine) at room temperature, or at 60 °C.11 However, at room temperature, 1a was completely transformed into the hydroxyl O-acetate ester. On increasing the reaction temperature to 60 °C, a di-O-acetyl derivative was obtained, due to additional O-acetylation of the enol tautomer of the keto moiety. No ring opening product was formed during these reactions. On the other hand, treatment of 1a with methyl chloroformate, benzyl chloroformate, or trichloroethyl chloroformate (TrocCl) furnished the corresponding ring opened product 4, which was likely formed via intermediate 2.12 Since alkyl chloroformates are well-known fragmentation reagents,13,14 compound 4 might also be generated via the quaternary acylammonium salt 3. To simplify the reaction, the hydroxyl group of 1a was protected as the tert-butyldimethylsilyl ether 5, which was epimerized, as expected, to form a cis/trans mixture in a ratio of about 1:1 (Scheme 2). Compound 5 was then treated with TrocCl15 in the presence of K2CO316 and the reaction proceeded cleanly to afford the open ring product 6 in almost quantitative yield (in most cases this was based on recovered material). 17 Further investigation of the reaction indicated that conversion rates from 5 to 6 were highly dependent on the type of organic solvent used in the reaction.18 The best result was obtained when the reaction was carried out in CH3CN19 for 48 h with 5 equiv of TrocCl in the presence of K2CO3 as base;20 under these conditions, compound 5 was converted to 6 in quantitative yield. It should be noted that the geometry of the double bond in compound 6 is exclusively Z and is exclusively Z in compounds 4. This result suggests the “trapped” intermediate is likely generated from chloroformate-induced fragmentation, rather than from the initial retroconjugate addition reaction followed by N-acylation (Figure 1). As depicted in Figure 1, among the possible intermediate configurations involved in the retroconjugate addition reaction (i.e., A-F), B (2,6-trans), C (2,6-cis), and E (2,6-cis/trans) are favored ones which will afford an E double bond product. On the other hand, in the case of the alkyl chloroformate induced fragmentation mechanism (i.e., A′-F′), due to the steric hindrance between the acylammonium group and the keto side chain, configurations A′ (2,6-cis), D′ (2,6-trans), and F′ (2,6-cis/trans) become favored ones, and will afford a Z double bond product.
SCHEME 1.

SCHEME 2.

FIGURE 1.

Possible intermediate configurations during the retroconjugate addition reaction (A-F) and alkyl chloroformate induced ring opening reaction (A′-F′).
The utility of compound 6 was explored as a novel synthon in the synthesis of the natural product (−)-sedamine (11) (Scheme 2).21 Thus, 6 was treated with OsO4/NaIO4 22 to afford the double bond cleaved product 7, which was not fully characterized. Without further purification, the unstable aldehyde 7 was immediately treated with NaBH4 to afford the primary alcohol 8 in 70% yield from 6. It is worth mentioning that, in coordination with the procedure reported by Lebreton et al.5 for the synthesis of lobeline, 7 could be employed as a useful starting material for the preparation of a variety of substituted aromatic analogues of 1a. Bromination of 8 with PPh3/CBr4 afforded the corresponding bromide 9, which was then treated with zinc dust in acetic acid to remove the Troc protecting group. The resulting acetate salt was treated with aqueous K2CO3 during the workup to release the free amine, which then underwent simultaneous cyclization to form the TBS protected (−)-sedamine 10. Deprotection of 10 via the method of Lebreton et al.5 afforded (−)-sedamine 11 in quantitative yield. Characterization data (IR, NMR, MS, melting point, and optical rotation) for 11 are in good agreement with those reported in the literature.21
In summary, the retroconjugate addition reaction intermediate of lobeline and 5 were indirectly trapped by alkyl chloroformate induced fragmentation, and the “trapped” intermediate 6 was utilized as a key intermediate in the conversion of (−)-lobeline to (−)-sedamine. Consequently, (−)-sedamine was prepared in 7 steps from 1a in 56% overall yield, a considerable improvement in both yield and number of steps compared to the procedure of Lebreton et al.5 Moreover, this “trapping” strategy can also be applied to the preparation of substituted aromatic analogues of the biologically active lobeline molecule.2
Experimental Section
2-{(6S)-6-[(2S)-2-(tert-Butyldimethylsilanoxy)-2-phenylethyl]-1-methylpiperidin-(2R)-2-yl}-1-phenylethanone (5)
To a solution of (−)-lobeline (1a) (2.02 g, 6.00 mmol), Et3N (1.25 mL, 9.00 mmol), and DMAP (55 mg, 0.45 mmol) in CH2Cl2 at 0 °C was added TBDMSCl (1.36 g, 9.00 mmol). After being stirred for 1 h at 0 °C and then overnight at room temperature, the reaction mixture was diluted with EtOAc (100 mL), washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (CHCl3/CH3OH 30:1) to give 5 (2.47 g, 91%) as a viscous yellow oil: IR (neat) ν 3061, 3030, 2930, 2886, 2856, 2799, 1685, 1463, 1449, 1361, 1253, 1082, 836, 776, 698 cm−1; 1H NMR δ (two epimers in a ratio of about 1:1) −0.23 (s, 3H × 0.5), −0.22 (s, 3H × 0.5), −0.01 (s, 3H × 0.5), 0.02 (s, 3H × 0.5), 0.86 (s, 9H × 0.5), 0.89 (s, 9H × 0.5), 1.28-1.75 (m, 7H), 1.93-2.10 (m, 1H), 2.21 (s, 3H × 0.5), 2.33 (s, 3H × 0.5), 2.46 (m, 0.5H), 2.86-3.00 (m, 1.5 H), 3.09-3.38 (m, 2H), 4.60-4.68 (m, 1H), 7.13-7.38 (m, 5H), 7.40-7.62 (m, 3H), 7.90-8.01 (m, 2H) ppm; 13C NMR δ − 4.7, −4.6, −4.2, −4.1, 18.4, 19.8, 24.7, 26.1, 26.5, 27.0, 27.4, 28.5, 31.9, 38.9, 40.5, 40.9, 44.7, 46.0, 54.8, 54.9, 59.8, 60.2, 72.5, 72.8, 126.0, 126.1, 126.9, 127.1, 128.0, 128.16, 128.19, 128.3, 128.7, 128.8, 133.1, 137.2, 137.3, 145.7, 145.8, 199.3, 199.4 ppm; MS (EI) m/z 451 (M+), 436, 332, 216, 98, 96 (100), 82; HRMS calcd for C28H41NO2Si m/z 451.2901, found 451.2901.
{(1S)-1-[(2S)-2-(tert-Butyldimethylsilanoxy)-2-phenylethyl]-7-oxo-7-phenylhept-5(Z)-enyl}methylcarbamic Acid 2,2,2-Trichloroethyl Ester (6)
To a suspension of 5 (1.04 g, 2.30 mmol) and K2CO3 (1.59 g, 11.50 mmol) in CH3CN (40 mL) was added TrocCl (2.44 g, 11.50 mmol), and the mixture was stirred at room temperature for 48 h and then filtered through Celite. The filtrate was concentrated, the residue was dissolved in CHCl3 (100 mL), and the organic liquors were washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (hexanes/EtOAc 20:1) to give 6 (1.42 g, quantitative yield) as a viscous colorless oil: [α]22D-25.0 (c 0.50, CHCl3); IR (neat) ν 3063, 3031, 2945, 2930, 2857, 1714, 1673, 1621, 1449, 1406, 1331, 1253, 1137, 837, 775, 701 cm−1; 1H NMR δ (two rotamers in a ratio of about 6:4) −0.22 (s, 3H × 0.4), −0.20 (3H, 3H × 0.6), 0.00 (s, 3H), 0.86 (s, 9H × 0.4), 0.87 (s, 9H × 0.6), 1.37-1.60 (m, 4H), 1.70-1.88 (m, 1H), 1.98-2.10 (m, 1H), 2.15-2.38 (m, 2H), 2.77 (s, 3H × 0.4), 2.78 (s, 3H × 0.6), 4.06-4.22 (m, 1H), 4.43 (A of AB, 1H × 0.4), 4.59 (t, J = 6.3 Hz, 1H × 0.6), 4.62 (t, J = 6.3 Hz, 1H × 0.4), 4.68-4.86 (A′B′, 2H × 0.6), 5.00 (B of AB, 1H × 0.4), 6.80 (d, J = 6.6 Hz, 1H × 0.4), 6.85 (d, J = 6.6 Hz, 1H × 0.6), 6.95 (dd, J = 13.2, 6.6 Hz, 1H × 0.6), 7.01 (dd, J = 13.2, 6.6 Hz, 1H × 0.4), 7.17-7.38 (m, 5H), 7.41-7.60 (m, 3H), 7.85-7.94 (m, 2H) ppm; 13C NMR δ − 4.70, −4.66, −4.3, 18.4, 24.8, 25.1, 26.1, 28.7, 31.8, 32.0, 32.6, 32.8, 44.2, 52.9, 72.7, 72.8, 75.1, 95.8, 96.2, 126.20, 126.24, 126.27, 126.34, 127.4, 127.6, 128.2, 128.4, 128.6, 132.8, 137.9, 144.4, 144.7, 149.1, 149.2, 154.67, 154.70, 190.7 ppm; MS (EI) m/z 568/570/572 (M+ − tBu), 462/464/466, 259, 233 (100), 221, 73; HRMS calcd for C27H3335Cl3NO4Si (M+ − tBu) m/z 568.1239, found 568.1221; HRMS calcd for C27H3335Cl237ClNO4Si (M+ − tBu) m/z 570.1209, found 570.1186.
{(1S)-1-[(2S)-2-(tert-Butyldimethylsilanoxy)-2-phenylethyl]-5-hydroxypentyl}methylcarbamic Acid 2,2,2-Trichloroethyl Ester (8)
To a solution of 6 (1.16 g, 1.89 mmol) in dioxane/H2O (3:1) was added OsO4 (10 mL, 1 mg/mL in dioxane/H2O 1:1) at room temperature. The reaction mixture was stirred for 10 min and then NaIO4 (1.21 g, 5.68 mmol) was added in portions over a period of 15 min. The resulting suspension was stirred for a further 4 h and then filtered through a Celite pad and the filter cake was rinsed with CHCl3. The aqueous phase was extracted with CHCl3 and the combined organic portions were washed with saturated Na2S2O3, dried over anhydrous Na2-SO4, filtered, and concentrated under reduced pressure. The resulting aldehyde 7 was dissolved in 20 mL of EtOH. NaBH4 (140 mg, 3.70 mmol) was added in portions at 0 °C. The mixture was stirred for 2 h, and then quenched with acetone. Organic solvent was evaporated under reduced pressure, the resulting residue was suspended in water (20 mL) and extracted with CHCl3 (3 × 30 mL), and the combined organic liquors were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (hexanes/EtOAc 6:1) to give 8 (701 mg, 70%) as a colorless oil: [α]22D −35.4 (c 0.59, CHCl3); IR (neat) ν 3433, 3030, 2940, 2931, 2858, 1715, 1472, 1456, 1407, 1331, 1254, 1138, 1092, 1064, 837, 777, 701 cm−1; 1H NMR δ (two rotamers in a ratio of about 6:4) −0.22 (s, 3H × 0.4), −0.20 (3H, 3H × 0.6), 0.01 (s, 3H), 0.87 (s, 9H × 0.4), 0.88 (s, 9H × 0.6), 1.20-1.64 (m, 7H), 1.65-1.89 (m, 1H), 1.95-2.10 (m, 1H), 2.15-2.38 (m, 2H), 2.77 (s, 3H × 0.4), 2.78 (s, 3H × 0.6), 3.57 (dd, J = 13.5, 6.6 Hz, 2H), 4.00-4.20 (m, 1H), 4.44 (A of AB, 1H × 0.4), 4.59 (t, J = 6.6 Hz, 1H × 0.6), 4.62 (t, J = 6.6 Hz, 1H × 0.4), 4.68-4.84 (A′B′, 2H× 0.6), 5.00 (B of AB, 1H × 0.4), 7.14-7.40 (m, 5H) ppm; 13C NMR δ − 4.70, −4.66, −4.3, 18.4, 22.4, 22.8, 26.1, 28.6, 32.0, 32.2, 32.6, 32.8, 44.2, 44.3, 53.1, 62.9, 63.0, 72.7, 72.8, 75.1, 95.8, 96.2, 126.21, 126.24, 127.4, 127.5, 128.2, 128.3, 144.5, 144.8, 154.7, 154.8 ppm; MS (EI) m/z 468/470/472 (M+ − tBu) (100), 454/456/462, 364/366/368, 259, 233, 221, 73; HRMS calcd for C19H2935Cl3NO4Si (M+ − tBu) m/z 468.0926, found 468.0919; HRMS calcd for C27H3335Cl237ClNO4Si (M+ − tBu) m/z 470.0896, found 470.0889.
{(1S)-1-[(2S)-2-(tert-Butyldimethylsilanoxy)-2-phenylethyl]-5-bromopentyl}methylcarbamic Acid 2,2,2-Trichloroethyl Ester (9)
To a solution of 8 (570 mg, 1.08 mmol) and CBr4 (464 mg, 1.40 mmol) in CH2Cl2 (10 mL) was added dropwise at 0 °C a solution of PPh3 (386 mg, 1.47 mmol) in CH2Cl2 (5 mL). The mixture was stirred at 0 °C for 1 h and poured into hexanes/EtOAc (4:1) (60 mL). The resulting suspension was filtered through a short silica gel column with hexanes/EtOAc (4:1). The combined filtrates were concentrated under reduced pressure. The crude product was purified by column chromatography (hexanes/EtOAc 20:1) to give 9 (595 mg, 93%) as a colorless oil: [α]22D −28.4 (c 0.89, CHCl3); IR (neat) ν 3031, 2945, 2930, 2886, 2857, 1717, 1457, 1252, 1142, 1092, 837, 777, 701 cm−1; 1H NMR δ (two rotamers in a ratio of about 6:4) −0.22 (s, 3H × 0.4), −0.20 (3H, 3H × 0.6), 0.01 (s, 3H), 0.87 (s, 9H × 0.4), 0.88 (s, 9H × 0.6), 1.25-1.60 (m, 4H), 1.68-1.95 (m, 3H), 1.98-2.10 (m, 1H), 2.77 (s, 3H × 0.4), 2.78 (s, 3H × 0.6), 3.29-3.38 (m, 2H), 4.06-4.20 (m, 1H), 4.44 (A of AB, 1H × 0.4), 4.59 (t, J = 6.6 Hz, 1H × 0.6), 4.62 (t, J = 6.6 Hz, 1H × 0.4), 4.67-4.86 (A′B′, 2H × 0.6), 5.00 (B of AB, 1H × 0.4), 7.19-7.40 (m, 5H) ppm; 13C NMR δ −4.7, −4.6, −4.2, 18.4, 24.8, 25.1, 26.1, 28.7, 31.4, 31.5, 32.5, 32.8, 33.7, 33.9, 44.2, 44.3, 53.0, 72.7, 72.8, 75.1, 95.8, 96.2, 126.2, 127.4, 127.6, 128.3, 128.4, 144.5, 144.8, 154.7 ppm; MS (EI) m/z 530/532/534/536 (M+ − tBu), 456/458/460/462 (100), 352/354/356/358, 318, 221, 98; HRMS calcd for C19H28-BrCl3NO3Si (M − tBu, A + 2 ion) m/z 532.0058, found 532.0024; HRMS calcd for C19H28BrCl3NO3Si (M − tBu, A + 4 ion) m/z 534.0034, found 534.0027.
(S)-2-[1-Methylpiperidin-(2S)-2-yl]-1-phenylethanol (10)
A suspension of 9 (440 mg, 0.75 mmol) and Zn dust (0.8 g) in HOAc/CH2Cl2 (3/1) (12 mL) was stirred vigorously at room temperature for 4 h. The mixture was filtered, and the filtrate was concentrated under reduced pressure, basified with a saturated K2CO3 solution (15 mL), and extracted with CHCl3 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (CHCl3/MeOH/NH4OH 30:1:0.2) to give 10 (238 mg, 96%) as a colorless oil: [α]22D −87.6 (c 0.93, CHCl3); IR (neat) ν 3029, 2931, 2857, 2780, 1463, 1254, 1005, 836, 773, 700 cm−1; 1H NMR δ −0.25 (s, 3H), 0.02 (s, 3H), 0.89 (s, 9H), 1.23-1.78 (m, 6H), 1.90 (m, 1H), 2.10-2.40 (m, 3H), 2.31 (s, 3H), 2.86 (dt, J = 12.0, 3.3 Hz, 1H), 4.70 (dd, J = 9.6, 3.0 Hz, 1H), 7.19-7.39 (m, 5H) ppm; 13C NMR δ −4.7, −4.1, 18.4, 23.6, 25.5, 26.1, 30.5, 42.8, 43.1, 56.5, 60.6, 72.5, 126.0, 127.2, 128.2, 145.6 ppm; MS (EI) m/z 333 (M+), 318, 276, 98 (100), 73; HRMS calcd for C30H35-NOSi m/z 333.2482, found 333.2493.
(−)-Sedamine (11)
To a solution of 10 (185 mg, 0.55 mmol) in EtOH (10 mL) was added concentrated HCl (0.1 mL). After 3 h at 55-60 °C, the reaction mixture was basified with saturated aqueous K2CO3 solution (10 mL). The aqueous layer was extracted with CHCl3 (3 × 20 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude product was purified by column chromatography (CHCl3/MeOH/NH4OH 10:1:0.2) to give 11 (124 mg, quantitative yield) as a white solid: mp 58-59 °C (lit.5 mp 58-60 °C); [α]22D −89.4 (c 1.0, EtOH) [lit.5 mp −87.1 (c 0.93, EtOH)]; IR (KBr) ν 3362, 3060, 3028, 2935, 2855, 2795, 1450, 1061, 701 cm−1; 1H NMR δ 1.25-1.82 (m, 7H), 2.12 (ddd, J = 14.4, 10.5, 9.6 Hz, 1H), 2.49 (s, 3H), 2.55 (m, 1H), 2.85 (m, 1H), 3.06 (m, 1H), 4.89 (dd, J = 10.8, 2.7 Hz, 1H), 7.20-7.42 (m, 10H) ppm; 13C NMR δ 20.7, 22.6, 26.0, 40.0, 40.2, 51.4, 61.1, 74.9, 125.6, 127.1, 128.3, 145.7 ppm; MS (EI) m/z 219 (M+), 129, 112, 104, 98 (100), 77, 70; HRMS calcd for C14H21NO m/z 219.1618, found 219.1614.
Supplementary Material
Acknowledgment
This research was supported by NIH grants DA 00399 and DA 13519.
Footnotes
Supporting Information Available: General experimental methods and NMR spectra of all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Millspaugh CF. American medicinal plants: an illustrated and descriptive guide to plants indigenous to and naturalized in the United States which are used in medicine. New York: Dover; 1974. Lobelia inflate; pp. 385–388. [Google Scholar]
- 2.Dwoskin LP, Crooks PA. Biochem. Pharmacol. 2002;63:89–98. doi: 10.1016/s0006-2952(01)00899-1. [DOI] [PubMed] [Google Scholar]
- 3.(a) Miller DK, Crooks PA, Zheng G, Grinevich VP, Norrholm S, Dwoskin LP. J. Pharm. Exp. Ther. 2004;310:1035–1045. doi: 10.1124/jpet.104.068098. [DOI] [PubMed] [Google Scholar]; (b) Flammia D, Malgorzata D, Damaj MI, Martin B, Glennon RA. J. Med. Chem. 1999;42:3726–3731. doi: 10.1021/jm990286m. [DOI] [PubMed] [Google Scholar]; (c) Terry AV, Jr, Williamson R, Gattu M, Beach JW, McCurdy CR, Sparks JA, Pauly JR. Neuropharmacology. 1998;37:93–102. doi: 10.1016/s0028-3908(97)00142-1. [DOI] [PubMed] [Google Scholar]
- 4.Compère D, Marazano C, Das BC. J. Org. Chem. 1999;64:4528–4532. [Google Scholar]
- 5.Felpin FX, Lebreton J. J. Org. Chem. 2002;67:9192–9199. doi: 10.1021/jo020501y. [DOI] [PubMed] [Google Scholar]
- 6.In an NMR study we observed that the equilibration was reached at a ratio of 46:54 cis:trans after 10 days at room temperature at a concentration of 2% (w/w) in CDCl3. We found that epimerization was faster in solvents of higher polarity, i.e. CD3OD > CD3CN > CD3-COCD3 > CDCl3.
- 7.The retroconjugate addition reaction of 1a free base occurred in various organic solutions; however, in the solid state, no epimerization of 1a was observed even after storing for several years at room temperature. For a recent example of a retroconjugate addition reaction in the solid state, see: Tan K, Alvarez R, Nour M, Cavé C, Chiaroni A, Riche C, d’Angelo J. Tetrahedron Lett. 2001;42:5021–5023.
- 8.For an example of the trapping of a retroconjugate addition reaction intermediate with HCl, see: Vázquez E, Galindo A, Gnecco D, Bernès S, Terán JL, Enríquez RG. Tetrahedron: Asymmetry. 2001;12:3209–3211.
- 9.Reference 4 reported that a 1a/1b mixture could be converted to the single epimer 1a by treating the mixture with HCl, which probably indicates that the HCl salt of the cis isomer of lobeline (1a·HCl) is a thermodynamically stable species.
- 10.These studies indicate that lobeline does not undergo a retroconjugate addition reaction under acidic conditions.
- 11.For an example of the trapping of a retroconjugate addition reaction intermediate with Ac2O/NaOAc, see: Doll MKH, Guggisberg A, Hesse M. Helv. Chim. Acta. 1996;79:973–981.
- 12.Although compounds 4 were each difficult to obtain in pure form from the complex reaction mixture, 1H NMR clearly showed signals of the double bond in these products, indicating the formation of the ringopen products.
- 13.For examples, see: Hobson JD, McCluskey JG. J. Chem. Soc. 1967:2015–2017. Toth JE, Fuchs PL. J. Org. Chem. 1986;51:2594–2596. Kim G, Chu-Moyer MY, Danishefsky SJ. J. Am. Chem. Soc. 1990;112:2003–2005.
-
14.In our hands, compound I was converted to ring opening compound II under similar conditions:
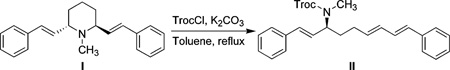
- 15.The use of methyl chloroformate or benzyl chloroformate also afforded similar results; however, using AcCl under the same conditions resulted in no reaction.
- 16.The use of NaHCO3 or Et3N also gave similar results; however, the use of stronger bases such as NaOH or t-BuOK gave low conversion rates, probably because these strong bases destroyed the chloroformates during the reaction.
- 17.Among a number of solvents that were in the reaction, three solvents, i.e. THF, acetone, and CH3CN, were investigated quantitatively, the others were estimated from TLC data.
- 18.Conversion rates were determined through a parallel reaction manner, using TLC to roughly estimate the amount of the product and the starting material. The following rank order was obtained: CH3-CN > EtOAc > dioxane > acetone > THF > toluene > hexanes > CHCl3. MeOH and DMF gave no reaction.
- 19.It was found not necessary to use anhydrous CH3CN; however, excessive water will afford a low conversion rate.
- 20.The reaction time was shortened when the molar ratio of TrocCl was increased or the temperature of the reaction was increased; however, the latter caused a decrease in yield.
- 21.Numerous syntheses of sedamine, either in racemic form or in enantiotopic form, have been reported; see: Angoli M, Barilli A, Lesma G, Passarella D, Riva S, Silvani A, Danieli B. J. Org. Chem. 2003;68:9525–9527. doi: 10.1021/jo035215g. and references therein; also see refs 4 and 5.
- 22.(a) Pappo R, Allen DS, Lemieux RU, Johnson WS. J. Org. Chem. 1956;21:478–479. [Google Scholar]; (b) Demuth M, Ritterskamp P, Weigt E, Schaffner K. J. Am. Chem. Soc. 1986;108:4149–4154. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.