

Abstract
The role of different amygdala nuclei (neuroanatomical subdivisions) in processing Pavlovian conditioned fear has been studied extensively, but the function of the heterogeneous neuronal subtypes within these nuclei remains poorly understood. We used molecular genetic approaches to map the functional connectivity of a subpopulation of GABAergic neurons, located in the lateral subdivision of the central amygdala (CEl), which express protein kinase C-delta (PKCδ). Channelrhodopsin-2 assisted circuit mapping in amygdala slices and cell-specific viral tracing indicate that PKCδ+ neurons inhibit output neurons in the medial CE (CEm), and also make reciprocal inhibitory synapses with PKCδ− neurons in CEl. Electrical silencing of PKCδ+ neurons in vivo suggests that they correspond to physiologically identified units that are inhibited by the conditioned stimulus (CS), called CEloff units (Ciocchi et al, this issue). This correspondence, together with behavioral data, defines an inhibitory microcircuit in CEl that gates CEm output to control the level of conditioned freezing.
The amygdala is a medial temporal lobe region that plays a central role in the acquisition and expression of Pavlovian conditioned fear1-3. The amygdala contains multiple anatomically defined nuclei4. Associative learning largely occurs in the lateral nucleus (LA), while the central nucleus (CEA) is thought to control the expression of fear5,6. However CEA contains at least 3 subnuclei (CEm, CEl and capsular CE)3,4 and multiple neuronal subtypes7-10, the role of which is poorly understood. A full understanding of amygdala function requires its dissection at the cellular level.
Here we have used genetic methods to investigate the functional connectivity and behavioral relevance of a GABAergic subpopulation within CEl11-13, marked by expression of protein kinase C-δ (PKCδ). These neurons inhibit output neurons in CEm, and also make reciprocal inhibitory connections with PKCδ− neurons within CEl. Genetic silencing14,15 in conjunction with in vivo electrophysiology indicates that these neurons likely correspond to “CEloff” units identified by Ciocchi et al.16, that are inhibited by the conditioned stimulus (CS). Taken together, these data define the functional connectivity and behavioral relevance of an inhibitory microcircuit within CEl9,17, that gates output from CEm12.
Properties of PKCδ+ neurons in CEl
We sought stable markers for CEA subpopulations18,19, which could be used to genetically manipulate their activity in vivo20-22. PKCδ marks ∼50% of CEl GABAergic neurons (Fig. 1a-d and Table S1). These neurons are distinct from those expressing corticotropin releasing hormone9 (CRH; Fig. 1e-g) or dynorphin10 (Dyn; Fig. S1a-c), while ∼40% express enkephalin9 (Enk; Fig. 1h-j and Table S1) and ∼65% express the oxytocin receptor (OxtR; Fig. S1d-f and Table S1), which is implicated in inhibitory gating of CEm12.
Figure 1. Characterization of CEl PKCδ+neurons.
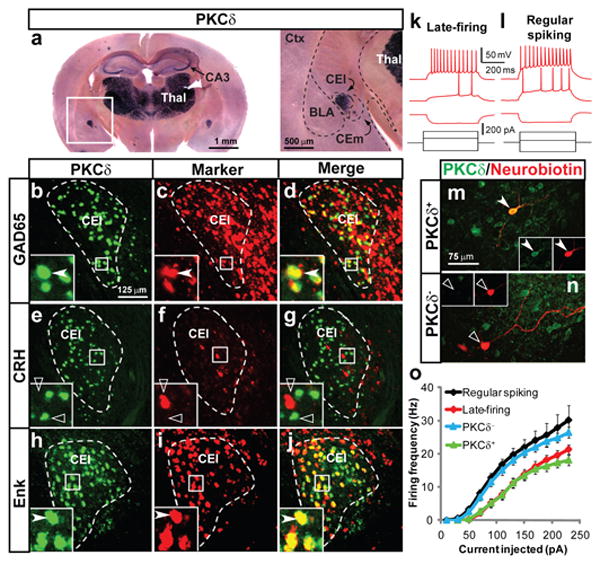
a, In situ hybridization (ISH) for PKCδ mRNA. CA3, hippocampus; Thal, thalamus. Boxed area at higher magnification on right. b-j, Double-label fluorescence ISH (dFISH) for PKCδ mRNA and the indicated markers. Insets, boxed areas. Filled and open arrowheads indicate doubly vs. singly labeled cells, respectively. k-l, Whole-cell patch clamp recordings in acute amygdala slices. Voltage changes (red) produced by respective current injections (black) are illustrated. Resting membrane potentials were adjusted to ∼ -65 mV. m, n, Neurobiotin-filled CEl neurons recorded in (k, l) after fluorescent streptavidin labeling and immunostaining for PKCδ. o, Stimulus-response (I/O) curves for neurons based on electrophysiological profile (late-firing, n = 22; regular spiking, n = 14), or PKCδ expression (PKCδ+, n = 14; PKCδ-, n = 12) in CEl. See also Tables S2-S4.
We examined the electrophysiological properties of PKCδ+ neurons in acute amygdala slices. Due to their low spontaneous activity, spiking was evoked by injection of depolarizing current. This analysis confirmed three types of neurons: late-firing, regular spiking, and low-threshold bursting neurons23 (Fig. 1k, l and Table S2). Neurobiotin fills and antibody staining, as well as recording from fluorescently labeled PKCδ+ neurons in transgenic mice (see below) indicated that most PKCδ+ neurons are late-firing (Fig. 1m, o and Table S3), while the PKCδ- population contains both regular spiking and late-firing units (Fig. 1n, o and Table S3). Thus, PKCδ+ neurons have relatively homogeneous electrophysiological properties (Table S4, P < 0.0001, Fisher's Exact Test).
Functional connectivity of PKCδ+ neurons
To gain genetic access to PKCδ+ neurons, transgenic mice were generated24 harboring a bacterial artificial chromosome (BAC) expressing Cre recombinase and the alpha subunit of a CFP-tagged version of the C. elegans glutamate-sensitive chloride channel GluCl25 (Fig. 2a), separated by an internal ribosome entry site (ires)26. Double-labeling for GluClα-CFP and PKCδ in these PKCδ:GluClα-ires-Cre mice, as well as crossing to Cre-dependent lacZ-expressing reporter mice, revealed neuron-specific expression of the transgene (Fig. 2j-m) that correctly recapitulated the pattern of endogenous PKCδ expression (Fig. 2b-e, f-i).
Figure 2. Transgenic targeting of PKCδ+neurons.
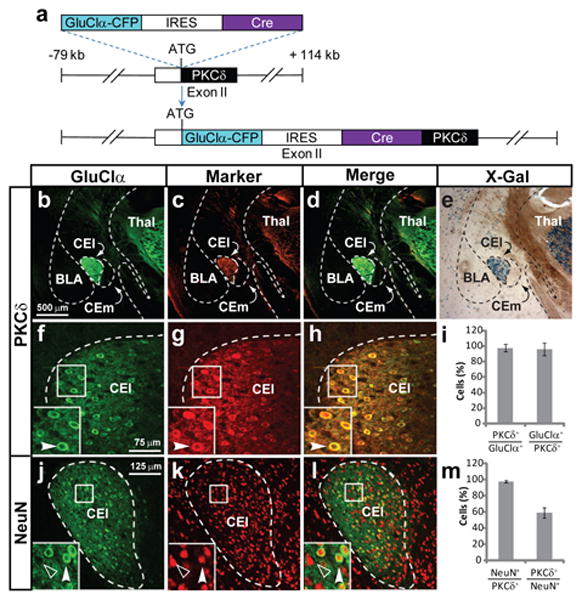
a, Design of PKCδ∷GluClα−CFP-iCre BAC transgene. b-d, f-h, j-l, Expression of transgene visualized by double-label immunofluorescent labeling (dIFL) for GFP and the indicated markers. i, m, Quantification of f-h and j-l, respectively. Values are mean ± SEM, n = 3. (e) X-gal staining of PKCδ∷GluClα-iCre; Rosa∷loxP-STOP-loxP-lacZ mice reveals β-galactosidase expression (blue) in correct locations. Scale bar in b applies to c-h and j-l.
CEl is known to contain GABAergic neurons that project to CEm7,12,27,28. We traced the anterograde projections of PKCδ+ neurons to CEm by injecting the CEl of the transgenic mice with a Cre-dependent adeno-associated virus (AAV) encoding humanized renilla GFP29 (hrGFP) (Fig. 3a-j). Recombination of injected AAVs was restricted to PKCδ+ neurons (Fig. S2f-j). hrGFP+ fibers derived from CEl projected to CEm (Fig. 3h, inset) (Fig. 3i, j), a result confirmed by retrograde tracing from CEm using cholera toxin B-subunit (CTB) (Fig. 3k, l). 60.9±5.7% (mean±S.E.M., n = 6) of CTB+ neurons in CEl were PKCδ+ (Fig. 3m-o, inset white arrowhead), while the remainder were PKCδ−.
Figure 3. CEl PKCδ+neurons directly inhibit CEm output neurons.
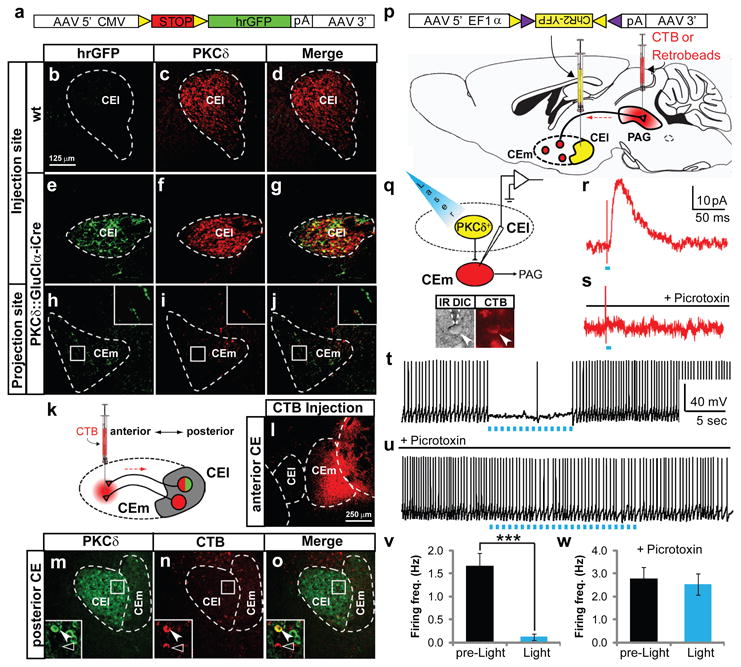
a-j, Anterograde axonal tracing using Cre-dependent hrGFP AAV29. k-o, Retrograde tracing from CEm using red fluorescent CTB. Scale bars for b-j, l-m, respectively. p-w, Optogenetic activation of CEl PKCδ+ neurons inhibits PAG projecting neurons in CEm. p, Schematic illustrating double-injection/slice recording experiments. q, CTB+ CEm cell (CTB, arrowhead) with patch electrode (IR DIC, dashed lines) is illustrated. r-v, Whole-cell voltage-clamp (Vhold = -40 mV) (r, s) or current clamp (t-v) recordings from a back-labeled CEm neuron. r,s, IPSC triggered by a 2 ms 473 nm laser pulse, with (s) or without (r) 100 μM picrotoxin. t-v, suppression of depolarizing current injection-evoked spiking by 473 nm laser pulses (2 ms, 15 Hz), with (u) or without (t) 100 μM picrotoxin. v, w, Quantification of data in t (n = 5 cells; * P < 0.001, t-test) and v (n = 3 cells; P = 0.75, t-test), respectively.
Freezing is controlled by projections from CEm to the peri-aqueductal gray (PAG)30-32, but CEm contains multiple neuronal subpopulations with different connectivities32. To determine whether PKCδ+ neurons make inhibitory synapses onto PAG-projecting CEm output neurons, we combined whole-cell patch clamp recording of retrogradely-labeled CEm output neurons, with Cre-dependent optogenetic activation33 of PKCδ+ neurons (Fig. 3p, q), in acute amygdala slices. Selective expression of channelrhodopsin-2 (ChR2)34 in PKCδ+ neurons was achieved by injecting a Cre-dependent AAV encoding ChR2-YFP35 into the CEl of PKCδ:GluClα-ires-Cre transgenic mice (Fig. S9a). Whole-cell patch-clamp recordings from ChR2-expressing PKCδ+ neurons (identified using native fluorescence for YFP (ChR2) and CFP (GluClα; Fig. S9b)) confirmed that spiking could be induced with ∼90% efficiency using 473 nm light flashes at 15 Hz (Fig. S9g). Retrograde labeling of CEm projection neurons in the same animals was achieved by injection of Alexa-555-conjugated CTB into the PAG (Fig. 3p), permitting their prospective identification using native fluorescence (Fig. 3q, CTB, arrowhead).
In slices prepared from dually injected animals, optogenetic activation of CEl PKCδ+ neurons elicited robust, picrotoxin-sensitive IPSCs in CEm output neurons (success rate 100%) (Fig. 3r, s; IPSC amplitude, 18.3±2.3 pA, n = 6 cells), with an average latency = 3.7±0.2 ms (range 2-5 ms, n = 30) and mean temporal jitter = 0.83±0.16 ms (n = 6), consistent with monosynaptic transmission36. It also suppressed current injection-evoked action potentials in CEm output neurons (Fig. 3t, v), in a picrotoxin-sensitive manner (Fig. 3u, w). Thus, CEl PKCδ+ neurons make inhibitory connections onto PAG-projecting CEm output neurons.
Light-activation of ChR2-expressing PKCδ+ neurons also evoked monosynaptic IPSCs, and suppressed current injection-evoked action potential firing, in CEl PKCδ− neurons (identified by the absence of CFP fluorescence), in a picrotoxin-sensitive manner (Fig. 4a-g). Therefore, PKCδ+ neurons also make local inhibitory connections within CEl onto PKCδ− neurons.
Figure 4. PKCδ+and PKCδ- make reciprocal inhibitory connections in CEl.
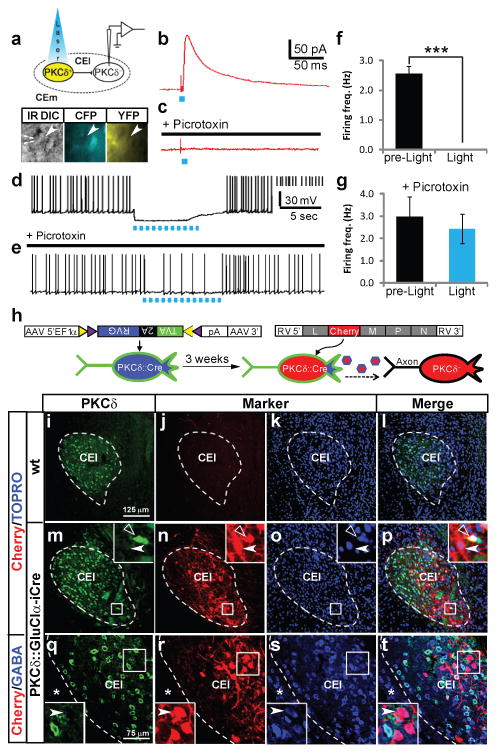
a, Schematic and micrograph from slice preparation. Arrowhead indicates PKCδ(CFP)minus; (YFP)- recorded cell in CEl. b-e, Whole-cell voltage clamp (Vhold = -40 mV) (b, c) or current clamp (d, e) recordings from PKCδ- CEl neuron showing light-triggered (d), picrotoxin-sensitive (c) IPSC, or picrotoxin-sensitive (e) suppression of depolarizing current injection-evoked spiking (d) by ChR2 activation (blue dots; 2 ms, 15 Hz). f, g, Quantification of data in d (f; n = 5 cells, *** P < 0.001, t-test) and e (g; n = 3 cells, P = 0.66, t-test), respectively. h, Schematic illustrating cell-specific Rabies virus infection. i-p, immunostaining for PKCδ(i,m), intrinsic mCherry fluorescence (j, n) and nuclear staining with TOPRO-3 (k, o) 3 days after RVΔG injection of transgenic (m-p) and wild-type (i-l) mice. Primary infected PKCδ+ neurons (m, n insets, open arrowheads) and retrogradely labeled PKCδ- cells (m, n insets, filled arrowheads) are indicated. q-t, Triple labeling for PKCδ (q) and GABA (s) and mCherry (r). mCherry-labeled PKCδ- cells are GABAergic (inset, arrowhead). Scale bars for f-m, n-q.
To determine whether, conversely, PKCδ+ neurons receive inhibitory input from PKCδ- neurons, we employed a cell-specific modification of a virally based mono-synaptic retrograde tracing method37. PKCδ∷GluClα-ires-Cre transgenic mice were first injected with a Cre-dependent AAV encoding both an avian receptor, TVA, and a complementing RV-G protein deleted from the rabies virus strain RVΔG (Fig. 4h, left)38. Three weeks later, animals were injected in the same site with Env(A)-pseudotyped RVΔG virus expressing the red fluorescent protein mCherry (Fig. 4h, right; see Methods). Since Env(A) directs infection exclusively to TVA-expressing cells37, this manipulation yields expression of mCherry in CEl PKCδ+ neurons in transgenic (Fig. 4m-p; inset, open arrowheads), but not wild-type (Fig. 4i, j) mice. Expression of G by AAV-infected PKCδ+ neurons permits trans-synaptic spread of the RVΔG virus to input neurons, but no further spread occurs37. Monosynaptic retrograde labeling of PKCδ− neurons occurred extensively in CEl (Fig. 4m, n, p; inset, filled arrowheads), and most labeled neurons were GABAergic (Fig. 4q-t; inset, arrowhead). These data suggest that PKCδ+ neurons receive inhibitory input from PKCδ- neurons.
PKCδ+ neurons correspond to CEloff units
Single-unit recording experiments in freely behaving mice have identified two types of CEl units with opposite responses to the CS16: units activated by the tone (“CElon” cells), and units inhibited by it (“CEloff”) cells (Fig. 5i-l). We wished to determine whether PKCδ+ neurons correspond to either of these classes of units. As the in vivo recordings employ extracellular electrodes16, the cells cannot be filled for antibody staining. Therefore, we examined the effect of reversibly silencing PKCδ+ neurons on the activity of CElon and CEloff units, using a mammalian codon-optimized form39 of the IVM-sensitive chloride channel GluCl from C. elegans14,15, mutated to eliminate glutamate sensitivity25. Functional expression of this ionotropic receptor requires two subunits: GluClα and GluClβ. To restrict expression of GluClαβ to CEl PKCδ+ neurons, we employed an intersectional strategy in which GluClα–CFP was expressed transgenically in all PKCδ+ neurons (Fig. 2b), while GluClβ-YFP25 was expressed in CEl from an AAV vector by stereotaxic injection (Fig. 5a; S2a-e).
Figure 5. IVM/GluCl-mediated suppression of CEl PKCδ+neuronal activity.
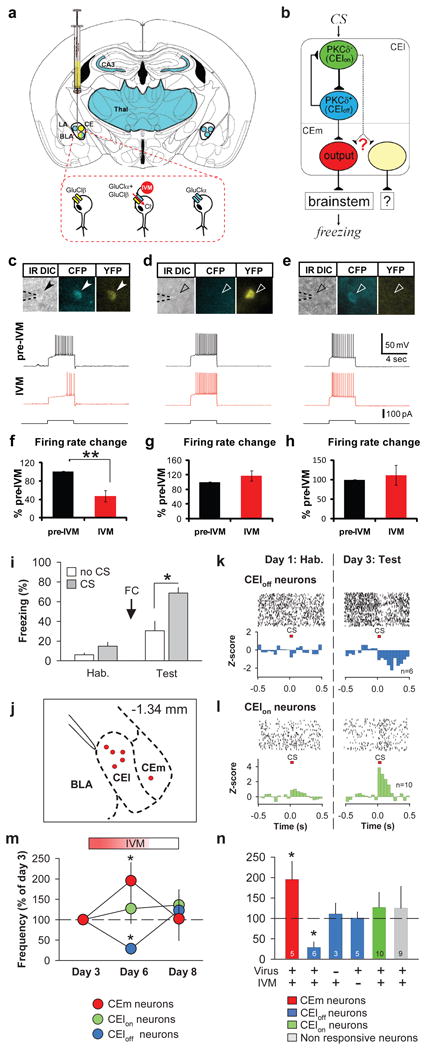
a, Strategy for selective expression of GluClαβ in CEl PKCδ+ neurons. Yellow, CMV-driven AAV∷GluClβ virus; blue, PKCδ∷GluClα-CFP transgene expression. b, Summary of inhibitory connections in CEA (see also Ciocchi et al16). c-e, Current injection (bottom trace)-evoked spiking (top, black trace) is suppressed by 20 nM IVM (middle, red trace) in PKCδ+ cells expressing GluClαβ (c), but not in cells expressing either GluClβ-YFP (d) or GluClα-CFP (e). f-h, Quantification of results in c-e, respectively (f, n = 4 cells, P = 0.005); g, n = 5 cells, P = 0.73; h, n = 4 cells, P = 0.66; paired t-tests). i-n, IVM/GluCl-mediated silencing of PKCδ+ neurons during chronic in vivo recording. i, Behavioral data for mice used in recording experiments. Hab, habituation; FC, fear conditioning. (n = 5) j, Coronal schematic showing recording sites in CEl and CEm (-1.34 mm posterior to bregma). BLA: basolateral amygdala. k, l Representative raster plots (upper) and normalized population peristimulus time histograms (lower) of CEloff (k; lower, n = 6 neurons from 3 mice) and CElon neurons (l; lower, n = 10 neurons from 4 mice) (P < 0.05). Confirmation of unit isolation is shown in Figure S11. m, Tonic activity of CEloff neurons (One-way ANOVA (F(2,15) = 4.845, P = 0.024) with post-hoc Bonferroni t-test (* P < 0.05), but not of CElon neurons (One-way ANOVA (F(2,27) = 0.391, P = 0.680)), is significantly (Day 6) and reversibly (Day 8) reduced by IVM (10 mg/kg, i.p.), while tonic activity of CEm neurons is increased (n = 5 units from 1 mouse; one-way Kruskal-Wallis ANOVA (H = 7.487, with 2 degrees of freedom, P = 0.024) with post-hoc Tukey's HSD (* P < 0.05)). n, Specificity controls for (m). IVM injection without virus infection (n = 3; P = 0.765); vehicle (DMSO) injection with AAV2∷GluClβ virus infection (n =5; P = 0.940). CElon neurons (n = 10; P = 0.497); CS-non responsive neurons (n = 9; P = 0.644). *P < 0.05 (paired t-tests with vs. without IVM in virus-injected mice).
We first confirmed silencing of PKCδ+ neurons using IVM/GluCl in acute amygdala slices from such mice. Neurons expressing GluClα and/or GluClβ could be prospectively identified by native CFP and YFP fluorescence, respectively (Fig. 5c-e). Bath application of IVM reduced the input resistance of cells expressing GluClαβ, but not of cells expressing either GluClα or GluClβ, from 337±41 to 277±36 MΩ (n = 4, P < 0.04, paired t-test). IVM also significantly suppressed current injection-evoked spiking in neurons co-expressing both subunits (Fig. 5c-h), as well as sub-threshold EPSPs evoked by bipolar electrode stimulation of LA (Figure S3; such bipolar stimulation did not evoke spiking of PKCδ+ neurons in our slice preparation).
We next examined the effect of silencing PKCδ+ neurons on CElon and CEloff unit activity16, in PKCδ∷GluClα-ires-Cre transgenic mice previously injected in CEl with the AAV∷GluClβ virus. CElon and CEloff units were first identified by CS presentation in conditioned animals, prior to IVM administration. The spontaneous spiking activity of these units was subsequently measured before, and 3-5 days after, IVM administration. The tonic activity of CEloff units was strongly suppressed following IVM treatment, while that of CElon units was unaffected (Fig. 5m, n). This effect of IVM reversed within 48 hrs after treatment (Fig. 5m), and was not observed in IVM-treated uninjected transgenic animals, or in virally injected animals not treated with IVM (Fig. 5n). Importantly, silencing PKCδ+ neurons by IVM/GluCl also increased CEm unit activity (Fig. 5m, n red symbols), consistent with our observation that PKCδ+ CEl neurons inhibit CEm output neurons (Fig. 3q-w). The simplest interpretation of these data is that CEloff units are PKCδ+ neurons, although this does not necessarily imply the converse.
Finally, we tested the effect of suppressing PKCδ+ neuronal activity on fear conditioning (see Methods). Because of the slow decay kinetics of IVM in vivo14, we initially investigated the effect of silencing during both training and testing. While IVM at the dose used (10 mg/kg) did not significantly affect freezing in wild-type animals (Fig. S4a), to avoid potential interactions between IVM treatment and viral infection, in most experiments single subunit-expressing control animals (transgenic or wild-type animals injected with GluClα or GluClβ virus, respectively) were also treated with IVM. These control groups were not significantly different from each other (Fig. S4b) and were pooled for statistical analysis.
In some experiments (n=5-8 animals/group), using a high-titer (1013/ml) preparation of AAV2:GluClβ14, a significantly higher level of freezing was observed in experimental animals than in controls; however in other such experiments using different virus preparations, no difference between groups was detected (Supplementary Footnote 1). Quantitative histological analysis (Fig. S10) revealed considerable variation in the level and bilaterality of GluClβ-YFP expression among individual animals (Fig. S5a-c). Therefore, data from multiple experiments (total n=54 animals/group) were pooled for statistical analysis. A Randomized Block ANOVA40 with 6 experimental and 6 control subjects assigned to each of 9 blocks based on infection rate (total n=108; Table 5 and Fig. S5d-f) indicated a significant block × group interaction during both the CS presentation (F(8, 90) = 2.298, P<0.05 by post-hoc Bonferroni t-test) and post-CS periods (F(8, 90) = 2.459, P < 0.05), but not the baseline (BL) period (F(8,90) = 1.41, P = 0.205). Freezing was significantly higher in the experimental group only in the block with the highest infection rate (Table I, Table S5 and Fig. S5e, f). Freezing in the experimental group was also significantly higher than controls (including GluClαβ animals tested without IVM; n=6) among animals expressing GluClbeta; bilaterally in CEA at a level above the median infection rate (Fig. S7). There was no significant decrease in baseline locomotor activity, or in the activity burst produced by the first US presentation during training (Fig. S8) in experimental animals, indicating that the increased freezing is not a consequence of either decreased locomotor activity, or increased US-sensitivity13. Freezing levels during the pre-training and pre-test baseline were unaffected by IVM in experimental animals (Table S5, Figs. S5 and S7 and data not shown).
Table I. Behavioral effect of silencing PKCδ+ neurons in CEl.
| Group (Block 9) | Inf. Ratea (%) | BL Freezing (%) | CS Freezing (%) | Post-CS freezing (%) |
|---|---|---|---|---|
| Control (GluClα or GluClβ + IVM) | 40.0±0.8 | 5.8±2.2 | 49.4±10.4 | 19.2±6.0 |
| Experimental (GluClαβ +IVM) | 40.6±3.5 | 2.6±1.5 (n. s.) | 91.1±4.7 (P < .05) | 75.3±8.4 (P < .001) |
The data are derived from the Randomized Block ANOVA in Table S5 (total n=108; 6 experimental and 6 control animals assigned to each of 9 blocks), and illustrate the block with the highest level of viral infection in CEl (aInfection Rate, % of PKCδ+ cells expressing GluClβ-YFP; see Fig. S10). The control group is pooled (Fig. S4b) single subunit-expressing mice (GluClα or GluClβalone) treated with IVM. Values represent the mean±S.E.M. n.s., not significantly different. P values are from post-hoc Bonferroni t-test. A significant linear component to the block × group interaction indicated that the difference between groups tended to increase with infection rate (F(1,90) = 22.98, P < 0.0001 for CS, F(1, 90) = 8.85, P < 0.006 for post-CS). See also Table S5 and Figure S5. Control GluClαβ-expressing mice not treated with IVM were significantly different from the experimental group among animals exhibiting bilateral AAV infection above the median rate (n=6-21; Fig. S7).
Discussion
We have used genetically based methods to investigate the functional connectivity of a subpopulation of CEl GABAergic neurons identified by expression of PKCδ. Our data suggest that these neurons participate in a recurrent inhibitory circuit within CEl that inhibits CEm output to brainstem centers that control freezing, consistent with earlier suggestions9,12. In the accompanying paper16, Ciocchi et al. identify two populations of CEl units in vivo with opposite responses to the CS. The data presented here strongly suggest that CEloff units are PKCδ+ neurons, linking molecular and physiological identity. Because PKCδ+ neurons pre-exist in untrained animals, this linkage suggests that CEloff and CElon units, which are robustly identifiable after conditioning (Fig. 5k, l), arise via plasticity-dependent changes in deterministic CEl subpopulations, rather than by stochastic selection from a population of initially homogeneous cells.
The link between PKCδ+ neurons and CEloff units is also consistent with complementary connectivity data in the two studies. For example, Ciocchi et al. have shown that in vivo, the activity of CEloff units is inversely correlated with the activity of CElon units, suggesting reciprocal inhibition16. Our channelrhodopsin-assisted circuit mapping36 and cell-specific monosynaptic retrograde viral tracing37 data directly demonstrate mutual inhibitory connections between PKCδ+ and PKCδ− neurons in CEl. Ciocchi et al. have also shown that CS exposure both inhibits CEloff unit spiking, and increases CEm unit activity16, with a short latency implying an inhibitory connection. Our data directly demonstrate inhibitory synapses from CEl PKCδ+ neurons onto brainstem-projecting CEm output neurons, and indicate that silencing the former increases the tonic activity of the latter in vivo. Thus, our direct mapping of PKCδ+ synaptic connectivity in amygdala slices is consistent with inferences of CEloff unit connectivity based on correlative in vivo recordings (Fig. 5b).
Consistent with these electrophysiological and connectional data, genetic silencing of PKCδ+ neurons yielded a statistically significant enhancement of conditional freezing (during both the CS-on and post-CS periods), among those animals exhibiting the highest level of AAV∷GluClβ infection in CEl. Nevertheless, these behavioral results should be interpreted with caution, given the variability in results between individual experiments (see Supplementary Footnote 1). Furthermore, while IVM treatment caused a decrease in CEloff tonic spiking activity in GluClαβ -expressing animals, how this effect influences CS-evoked freezing, which is correlated with phasic CS-response in CEloff unit activity16, is not yet clear.
While the CEl circuitry defined by this study and Ciocchi et al.16 is remarkably consistent, the behavioral results in the two studies differ in some respects. For example, muscimol injection into CEl evoked freezing in unconditioned animals16, while selective silencing of PKCδ+ neurons using IVM/GluCl did not. Furthermore, transient inhibition of CEl during training using muscimol attenuated fear conditioning16, while chronic silencing of PKCδ+ neurons during both training and testing did not. We have not yet examined the behavioral effects of silencing PKCδ+ neurons during training vs. testing, and this will be an important question for future studies. Differences in the consequences of muscimol inhibition of CEl vs. IVM/GluCl silencing of CEl PKCδ+ neurons may reflect differences in the cellular specificity, efficiency or time-course of the two types of manipulations (Supplementary Footnote 2).
The CEl/CEm micro-circuit revealed by these companion studies has interesting properties worthy of further investigation. For example, the mutual inhibitory connections between CEloff and CElon units could provide a positive-feedback loop to amplify CS-evoked activity in CElon units (Fig. 5b)41. These antagonistic connections could also potentially encode different states, through stable imbalances in tonic activity16, or different oscillatory regimes, that could influence the learning and/or expression of conditioned fear. A currently unresolved paradox is why, if CElon units (like CEloff units) send inhibitory projections to CEm16, activation of CElon units by the CS results in increased rather than decreased activity of CEm output neurons (see Supplementary Footnote 3). Resolution of this paradox will require distinguishing whether these two CEl populations target the same or different classes of CEm neurons32, and the relative strength of these connections. Genetically based synaptic tracing and functional manipulations of CElon units should clarify this issue, as well as provide direct tests of their causal role in conditioned freezing and other emotional behaviors. The ability to prospectively identify and selectively manipulate PKCδ+ and other molecularly defined CEl subpopulations42,43 should also open the way to investigating their roles in animal models of phobic or anxiety disorders, and in the mechanism of action of drugs used to treat such disorders44.
Methods Summary
Histochemical methods
Single color and double fluorescent ISHs were performed on fresh frozen sections, using DIG (Roche) and DNP (Perkin Elmer) labeled RNA probes. Immunofluorescence was carried out on cryosections of 4 % PFA perfused tissue following standard protocols.
Generation of PKCδ∷GluClα-ires-Cre transgenic mice
A GluClα-ires-Cre cassette was inserted into PKCδ BAC clone RP23-283B12 (CHORI). The modified BAC was injected into FVB mouse embryos (GENSAT). Transgenic founders were backcrossed (n > 5) to C57Bl6/N. These mice are available through GENSAT24.
Pharmacogenetic silencing in vitro and in vivo
PKCδ∷GluClα-ires-Cre transgenic or wild-type mice were injected with ∼109 particles of AAV∷GluClβ (or, in control experiments, AAV∷GluClα) into CE and allowed 4 weeks for recovery. For fear conditioning experiments, on day 1, mice were habituated and then injected with IVM (10 mg/kg (Phoenix)). On day 2, animals received tone/foot shock pairings in context A (Coulbourn, Med Associates). On day 3, (and for in vivo recordings also on days 6 and 8), animals were placed in context B and freezing was scored prior to (baseline), during and after CS presentations. For in vitro recordings, acute brain slices were prepared and superfused with 20 nM IVM in ACSF to induce pharmacogenetic silencing. Single-unit recordings were performed in freely behaving animals as described16.
Optogenetic circuit dissection
PKCδ∷GluClα-ires-Cre transgenic mice were injected with 109 particles of Cre-dependent ChR2 AAV into CE, and (in some animals) CTB into PAG for retrograde labeling. Four weeks later, neurons were light-stimulated, in acute brain slices, through a 200 μm optical fiber (Thorlabs) emitting 5-10 mW 473 nm laser light (Crystalaser).
Virus based transsynaptic tracing
PKCδ∷GluClα-ires-Cre transgenic mice were sequentially injected into CE with 104 particles of AAV encoding Cre-dependent TVA and rabies B19 glycoprotein (G). Three weeks later, animals were injected in the same site with 105 particles of RVΔG rabies virus pseudotyped with EnvA, and analyzed 1 week later. All animal experiments were conducted under protocols approved by the Caltech Institutional Animal Care and Use Committee (IACUC) and the Salk Institute Biosafety Committee.
Methods
Generation of PKCδ∷GluClα-iCre transgenic mice
The PKCδ∷GluClα-iCre targeting construct was assembled in pGEM-T Easy (Promega) by PCR-cloning nucleotides -425 to -1 (+1 corresponding to the PKCδ start codon, primer set 5′-ACACACCGCGGCGCGCCCTAAAGAGGCAGGAGGCATGTG and 5′-CCATGATGGAGCCTGGAGTGAG) and +4 to +561 (primer set 5′-TCTCTGCTAGCCCGGGACCCTTCCTGCGCATCTC and 5-TGTGTGGTCGACTTAATTAAACTAGTGACCTTTCCAGCCATCACGTG) of PKCδ genomic sequences 5′ and 3′ to the GluCl-α open reading frame (ORF)39, using Ksp I-blunt/Mlu NI and Nhe I-Sal I restriction sites, respectively. The resulting PKCδ∷GluClα cassette was then cloned into the pLD53 shuttle vector 45. An additional expression cassette containing the Encephalomyocarditis virus internal ribosome entry site (IRES) followed by an ORF coding for Cre-recombinase was cloned into the Nhe I site immediately downstream GluClα. This vector was then used to introduce the GluClα-iCre expression cassette into a bacterial artificial chromosome (BAC) clone containing the complete PKCδ gene (RP23-283B12) by Rec A-mediated homologous recombination in bacteria 45 (for final sequence see Supplementary Information). Homologous recombination was verified by PCR (5′ arm primer set 5′-AGACCAGGGTAGGAGTCGGTG and 5′-GATCAGGGAAGCGATGATCAG, 3′ arm primer set 5′-GAGACCAAGACCGAGTGGAA and 5′-CACAGGTTAGCCATGACCTG) and southern blotting (5′ arm probe primer set 5′-TGTTCATGGGGTTTCTCACAG and 5′-ACCGACTCCTACCCTGGTCAG, 3′ arm probe primer set 5′-AGGTCATGGCTAACCTGTGG and 5′-GGCAGAGAAGTCAGACTGGG). Transgenic mice were generated by pronuclear injection of CsCl prepared BAC DNA linearized with P-Sce I in FVB embryos 24, which yielded two independent transgenic lines with indistinguishable expression pattern. One of them was backcrossed for n>5 generations to C57Bl6/N. Germ line transmission and genotyping of transgenic offspring were traced by PCR on genomic tail DNA (primer set 5′- GCTACATCAAGGCCATCGAC and 5′- AACTCCAGCAGGACCATGTGATCG).
In situ hybridizations
ISH was performed on 20 μm fresh frozen brain sections.
Processing of sections, hybridization and probe detection for single color in situ hybridizations were carried out following standard protocols 19, with the following specifications: Probes were cloned from whole mouse brain cDNA library (Invitrogen; GAD65 primer set 5′-ATCTCCAACATGTATGCCATGCTCATTGCC and 5′-TTACAAATCTTGTCCGAGGCGTTCGA, CRH primer set 5′-AACGGAGTAAGGGCAGGAATGGAGACAGAG and 5′-GTTGCTGTGAGCTTGCTGAGCTAACTGCTCTGC, Enk primer set 5′-TAGGGTCCAAGCTCTCATTGAGGCACCCGG and 5′-GCTTCAGAACCGCATAAAGCCCCCGTAT, OxtR 5′-CTGGCCCACCAGGCCAGCCGCTGGGTGGTG and 5′-AATCCCCATCTCCTTGGGAATTTTAGGAT) transcribed from linearized templates in pTeasy (Promega) or pCR2.1 (Invitrogen) using Sp6 (9PIP108, Promega) or T7 (9PIP207, Promega) polymerase and Digoxigenin labeled nucleotide mix (11277073910, Roche), used at a final concentration of 1 μg/ml, and detected with Anti-Dig-HRP, Fab fragments (1-207-733, Roche).
Processing of sections and hybridization for double fluorescence ISH was carried out as above, with the following specifications: DIG and DNP-UTP (NEL555001, Perkin Elmer) probe pairs were used at a final concentration of 1 μg/ml each. For detection, sections were blocked first in 20 % sheep serum in 0.1M Tris-HCl, pH7.5, 0.15M NaCl, 0.05% Tween-20 and NEN-Blocking solution (Perkin Elmer) for 30 min, each. DIG labeled probe was detected with anti-DIG Horseradish peroxidase (POD) (1: 500, 2h), Biotin-Tyramide (1:100, 8 min, RT, Perkin Elmer), Vecta Stain Elite ABC Kit (Vector PK-6100) and Cy3-Tyramide (Cy3 Tyramide NEL704A, PerkinElmer, 1: 50, 1h). Horseradish peroxidase was inactivated by incubation in 3 % H2O2 in PBS (1 h), followed by heat denaturation in TE (5 min, 85 C). Sections were blocked again as above and DNP-labeled nucleotides were detected with anti-DNP Horseradish peroxidase (1:500, 2h), DNP-Tyramide, anti-DNP Horseradish peroxidase (1:500, 2h, all TSA DNP Kit, NEL747A, PerkinElmer) and Fluorescein-Tyramide (1: 100, NEL701, PerkinElmer). Sections were mounted in Fluoro Gel (17985-10, Electron Microscopy Sciences). All washing steps and incubations were carried out according to the respective manufacturers' recommendations. Sections were visualized on Zeiss Axioscope, and Leica TCS SP confocal, microscopes.
Immunofluorescent labelling
For IFL, mice were transcardially perfused with 4 % paraformaldehyde in PBS. Brains were removed and cryoprotected in 15 % sucrose (16 h, 4 C). 20-30 μm cryo-sections were air dried for 30 min, rehydrated in PBST (PBS + 0.1 % Trition-X-100). In some instances, immunoreactivity was increased by incubating the slides in 10 mM Sodium citrate, 0.05 % Tween-20, pH 6 for 10 min at 95 C. Non-specific binding was blocked with 1 % BSA in PBST for 30 min. Primary antibodies, diluted 1:300-1000 in blocking solution (mouse anti PKCδ, 610398 BD Biosciences; Guinea Pig anti Dynorphin, GP10110 Neuromics; rabbit anti GFP A11122, Invitrogen; rabbit anti hrGFP 240142, Stratagene; Mouse anti NeuN MAB377, Chemicon) were incubated at for 16 h at 4 °C. Standard secondary antibodies (Invitrogen) in blocking solution were incubated for 3 h at RT. Unbound primary and secondary antibody was each washed by incubating three times in PBST for 10 min. Sections were mounted in Fluoro Gel (17985-10, Electron Microscopy Sciences) and visualized on Zeiss Axioscope, and Leica TCS SP confocal, microscopes.
Animal Maintenance
Animals were group housed at 23 C with ad libitum access to food and water in a 13 h day, 11 h night cycle, day starting at 7 AM.
Histological analysis
All histological quantifications (Fig. 2i, m, S2e, j, S5, S7, S10 and Tables S1, S5) are based on at least three coronal sections spaced equidistantly along the rostro-caudal axis of CE.
The intrinsic fluorescence of GluClα−CFP, or GluClβ-YFP can be readily detected in live brain slices (Fig. 4a, 5c-e, S9a, b). In the perfusion fixed tissue used for all histological analysis, however, the fluorescent signal is substantially weaker. Therefore, GFP IFL directed against its C/YFP tag was used wherever possible for more accurate results (Fig. 1b-h, 1j-l, S2b-d, S5). However, this immunohistochemical method could not be used to quantify the extent of infection by AAV∷GluClβ-YFP in PKCδ∷GluClαCFP-iCre transgenic mice, because of cross-reactivity of the anti-GFP antibody with the transgene-encoded CFP. Therefore, the fraction of virally infected PKCδ+ neurons in PKCδ∷GluClαCFP-iCre transgenic brains was estimated by quantifying the number of cells expressing intrinsic YFP fluorescence (which could be spectrally distinguished from the endogenous CFP-fluorescence of the GluClα transgene). However, because this intrinsic fluorescence signal underestimates the extent of viral infection in perfusion-fixed tissue (see above), we constructed a standard curve using wild-type mice infected with AAV:GluClβ-YFP virus, in which the number of infected cells measured using native YFP fluorescence was plotted against the number of infected PKCδ+ cells measured using double-immunofluorescence staining with anti-GFP and anti-PKCδ antibodies (Fig. S10). This standard curve was then used to estimate the fraction of PKCδ+ cells that would have been detectable by direct immunofluorescence labeling for GFP, based on quantification of native YFP fluorescence. In the behavioral experiments in which wild-type and PKCδ∷GluClα- CFP-iCre transgenic animals were classified based on their infection rates (Table S5, Table I, Fig. S5, S7), this estimation method was applied to all experimental groups, including wild-type animals.
Electrophysiological slice recordings
Standard mouse brain slice preparation and whole cell recording were performed as described14. Briefly, 250 μm thick coronal sections were prepared with a vibratome (VT1000S, Leica), using ice-cold glycerol-based artificial cerebrospinal fluid (gACSF, in mM: 252 glycerol, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, and 11 glucose, oxygenated with 95%O2/5%CO2). Slices were allowed to recover for at least 1 hour at 32°C and kept at room temperature in standard artificial cerebrospinal fluid (ACSF, in mM: 126 NaCl, 1.6 KCl, 1.2 NaH2PO4, 1.2 MgCl2, 2.4 CaCl2, 18 NaHCO3, and 11 glucose, oxygenated with 95%O2/5%CO2). Cells expressing CFP or YFP were visualized by infrared DIC and fluorescence video microscopy (Olympus BX51). Whole-cell voltage and current recordings were performed at 30 °C with a MultiClamp 700B amplifier and Digidata 1440A (Molecular Devices, Sunnyvale, CA). The patch pipette with a resistance of 5-8 MΩ was filled with an intracellular solution containing (in mM): 135 Potassium gluconate, 5 EGTA, 0.5 CaCl2, 2 MgCl2, 10 HEPES, 2 Mg-ATP, and 0.1 GTP, PH 7.2, 280-300 mOsm. Data were sampled at 10 kHz, filtered at 3 kHz and analyzed with pCLAMP 10 software.
Electrophysiological characterization of PKCδ+ cells
The relation of genetic marker and electrophysiological type was analyzed using Fisher's exact test with the null hypothesis that PKCδ+ cells have the same proportion of late-firing neurons as do CEl neurons as a whole. We recorded 38 CEl neurons without knowing their genetic subtypes, 21 of which were late-firing neurons. 14 of the17 non late-firing neurons were regular-spiking, 3 low-threshold bursting cells (Table S2, S3). We also recorded 57 PKCδ+ neurons based on their CFP expression, and found 49 of them are late-firing neurons, 8 of them are non late-firing neurons (all 8 cells are regular-spiking cells; Table S4). Fisher's exact test revealed that PKCδ+ neurons have a relatively homogeneous electrophysiological property of late-firing neurons compared to CEl neurons as a whole.
Stereotaxic surgery
2-4 months old male mice were deeply anesthetized with either ketamine/xylazine or isoflurane, injected i. p. with 500 μl 20 % mannitol (Phoenix pharmaceuticals), and placed in a stereotaxic frame (Kopf). The skull was exposed and perforated using a stereotaxic mounted drill at the desired coordinates (Table S6). For post-operative care mice were injected i. p. with 2 mg/kg BW ketoprofen, and supplied with drinking water containing 80 mg/l Trimethoprim, 400 mg/l sulfamethoxazole and 200 mg/l Ibuprofen for 10 days.
Viral injections
Viral particles were delivered unilaterally by stereotaxic (see above) injection with through stereotaxic mounted motor driven 32 G 45 degree beveled steel cannulas (Micro 4 controller equipped with Hamilton system, World Precision Instruments) or glass capillaries (Micro4 controller equipped with Microject system, Word Precision Instruments) to 1-4 injection sites/hemisphere at flow rates of 50-100 nl/min.
Pharmacogenetic silencing in vitro
Mice were unilaterally injected with a total of 2 μl AAV GluClα or -β virus (serotype 2; 1-10 × 1012 particles/ml in PBS) into CE (see above). Four weeks after surgery, slices (see above) were perfused with 20 nM IVM in ACSF for 20 min to induce pharmacogenetic silencing.
Pharmacogenetic silencing in vivo
Mice were injected bilaterally with a total of 2 μl AAV GluClα or -β virus (serotypes 2, 5, 8; 1-10 × 1012 particles/ml in PBS) into CE (see above). Four weeks after surgery, on day 1, animals were habituated 20 min to context A and given single i. p. dose of 1 % IVM injectable solution (Phoenix) at 10 mg/kg BW, or vehicle. On day 2, animals were fear conditioned in context A connected to a shock scrambler (Coulbourn Instruments, Allentown, PA; Med Associates, St. Albans, VT). After 20 min mice were exposed to training trials of 85 dBA 2 kHz 20 s tone immediately followed by a 2 sec 0.5 mA foot shock with 198 sec inter-trial interval (ITI). To avoid masking of the consequences of the experimental manipulation by ceiling and floor effects, the number of training trials (3-6) was adjusted such that freezing levels averaged around 50 % in the genetic/viral control. During the training session, behavior was recorded with infrared sensors (Coulbourn Scientific) or video cameras (Med Associates). On day 3, mice were placed in context B, differentiated from context A by a modified wall, floor and ceiling. Before each trial, all surfaces were cleaned with water and disinfectant. After a 5 min baseline period, two test trials of a 30 s CS followed by a 1 min post-CS period were presented. Freezing was scored either manually by an observer blind to the experimental group of the animal (Coulbourn Scientific), or electronically (Med Associates). Freezing during the CS and post-CS periods are represented as bins of these two test trials. Baseline freezing was averaged over 3 min before the first CS presentation.
To investigate the relationship between freezing and the extent of viral infection (Fig. S5, a-c), freezing data were correlated with the fraction of PKCδ+ expressing GluClβ, estimated as described above using native fluorescence for YFP and the standard curve in Fig. S7.
To investigate differences in freezing between experimental and control groups, freezing in the experimental group was compared to genetic/viral control groups by 2-way ANOVA with infection rate as blocking variable (Table S5, Fig. S5, d-f).
Retrograde tracing with CTB
CTB (0.5 % in PBS; List) was delivered by stereotaxic (see above) iontophoresis with a positive-pulsed current of 5 μA for 2 min. The animals were sacrificed one week post-injection, and the brains were processed for IHC.
Optogenetic circuit dissection
Mice were unilaterally injected with a total of 2 μl or Cre-dependent AAV ChR2 virus (serotypes 2, 5; 1-10 × 1012 particles/ml in PBS) into CE (see above). Slices (see above) were stimulated with an optic fiber (200 μm core diameter, ThorLabs), coupled to a 473 nm laser (CrystaLaser) and mounted on a 3-D micromanipulator (MPC365, Sutter) with the fiber end positioned on the edge of CEl.
Cre-dependent monosynaptic tracing with Rabies virus
Cre-expressing cells were primed for subsequent infection and monosynaptic retrograde spread of EnvA-pseudotyped, glycoprotein gene-deleted rabies virus, by stereotaxic injection (see above) of 180 nl of Cre-dependent AAV expressing the avian receptor protein TVA and rabies B19 glycoprotein (AAV9-pEF1α-FLEX-GTB) into CE. The genomic structure of AAV9-pEF1α-FLEX-GTB is: L_ITR-EF1α Pro-Kozak-(FLEX cassette46 containing GFP-T2A-hTVA-E2A-hB19G)-STOP-WPRE-polyA-R_ITR. The start ATG has been deleted from all three genes, and transcription start is mediated by a Kozak sequence that precedes the FLEX cassette 47. Genes were linked together through the use of 2A elements, which allow for the expression of multiple genes under the control of a single promoter48,49. Both the TVA and B19G genes were codon-optimized for expression in mammalian cells. The virus was prepared through a crude lysate extraction of transfected 293T cells, and has a genomic titer of 108 particles/ml. Although reverse-complemented GFP is detectable in the AAV genome, Cre-expressing cells expressed undetectable levels of GFP, either through native fluorescence or antibody-amplified imaging, while TVA and B19G appear to express normally.
Three weeks later, mice were injected at the same site with 180 nL of glycoprotein gene-deleted rabies virus 50 that has been pseudotyped with the avian sarcoma leucosis virus glycoprotein EnvA 38. The glycoprotein gene has been replaced with mCherry. The resulting virus, (EnvA)SAD-dG-mCherry, is incapable of infecting mammalian neurons in the absence of its binding partner, TVA, and cannot spread retrogradely in the absence of the rabies B19 glycoprotein 37. The biological titer of 109 particles/ml was determined through infection of TVA-expressing 293T cells. One week after the second injection, the animals were sacrificed and the brains processed for IHC.
Pharmacogenetic silencing with in vivo electrophysiological recordings
Mice were injected bilaterally with a total of 2 μl AAV GluClα virus (serotypes 2; 1-10 × 1012 particles/ml in PBS) into CE (see above). Four weeks later, on day 1, mice were habituated to 4 CS presentations (total CS duration of 30 s, consisting of 50-ms pips repeated at 0.9 Hz, 2-ms rise and fall; pip frequency: 7.5 kHz, 80 dB sound pressure level) in context B. On day 2, mice were conditioned with 5 CS/US (0.6 mA, 1 s) pairings with ITIs of 20-180 s in context A (Coulbourn). On days 3, 6, 8, mice were exposed to 4 CS presentations in context B. During these periods, individual neurons were recorded extracellularly in freely behaving mice. Spikes of individual neurons were sorted by time-amplitude window discrimination and template matching as previously described51,52. Cluster quality was verified by quantifying the cluster separation52 (Fig. S8). Unit isolation was verified using auto- and cross-correlation histograms. Spike rasters and histograms were constructed by aligning sweeps relative to the CS onset, and CS-evoked responses were normalized to baseline activity using a z-score transformation. Detailed methodological information about in vivo single unit recordings of CE neurons is presented in Ciocchi et al..
Supplementary Material
Acknowledgments
We thank Nat Heinz and Xiaoching Gong (Rockefeller University/GENSAT) for generating BAC transgenic mice, Clifford Saper for providing the Cre-dependent hrGFP AAV construct, Cheng Xiao for training in slice electrophysiology and preliminary experiments, Henry Lester for advice on the GluCl system, Laurent van Tright for performing in situ hybridizations, Walter Lerchner for providing a CAG-driven GluClβ construct, Angela Chang for help with behavioral scoring, Monica Martinez for tail genotyping, Gaby Mosconi for laboratory management and Jennifer Alex, Ruben Bayon and Reyna Sauza for animal care. This work was supported by NIH grant 1 R01 MH085082-01A1 and by funds from the Caltech “Conscious Mouse” project. W.H. was supported by a fellowship of the Human Frontier Science Program and P.K. by the Jane Coffin Childs Memorial Fund for Medical Research. S.C. and A.L. were supported by the Novartis Research Foundation. D.J.A. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Author Contributions: W.H. initiated the project, generated BAC constructs, designed experiments, performed anatomical, viral injections and behavioral experiments, and wrote the manuscript; P.K. contributed to experimental design, performed viral injections, behavioral experiments, data analysis and interpretation; H.C. contributed to experimental design, performed viral injections and slice electrophysiology experiments, data analysis and interpretation; S.C. and A.L. designed performed and interpreted in vivo recording experiments (Fig. 5); N.W. and E.M.C. performed rabies virus injection experiments; R.P. performed supplementary behavioral experiments and M.S.F. contributed to their interpretation and to statistical analysis; J.B. and H.-W.D. performed supplementary stereotaxic viral injection experiments; K.D. provided Cre-dependent ChR2 constructs and advice; D.J.A. conceived the project, contributed to experimental design and interpretation and wrote the manuscript. P.K., H.C. and S.C. contributed equally. All authors discussed the results and commented on the manuscript.
Supplementary information is linked to the on-line version of the paper at www.nature.com/nature
References
- 1.Davis M, Walker DL, Myers KM. Role of the amygdala in fear extinction measured with potentiated startle. Ann N Y Acad Sci. 2003;985:218–232. doi: 10.1111/j.1749-6632.2003.tb07084.x. [DOI] [PubMed] [Google Scholar]
- 2.LeDoux JE. Emotion circuits in the brain. Annu Rev Neurosci. 2000;23:155–184. doi: 10.1146/annurev.neuro.23.1.155. [DOI] [PubMed] [Google Scholar]
- 3.Paré D, Quirk GJ, LeDoux JE. New vistas on amygdala networks in conditioned fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- 4.Pitkänen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20:517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- 5.Maren S, Quirk GJ. Neuronal signalling of fear memory. Nat Rev Neurosci. 2004;5:844–852. doi: 10.1038/nrn1535. [DOI] [PubMed] [Google Scholar]
- 6.Medina JF, Repa JC, Mauk MD, LeDoux JE. Parallels between cerebellum- and amygdala-dependent conditioning. Nat Rev Neurosci. 2002;3:122–131. doi: 10.1038/nrn728. [DOI] [PubMed] [Google Scholar]
- 7.Cassell MD, Freedman LJ, Shi C. The intrinsic organization of the central extended amygdala. Ann N Y Acad Sci. 1999;877:217–241. doi: 10.1111/j.1749-6632.1999.tb09270.x. [DOI] [PubMed] [Google Scholar]
- 8.Cassell MD, Gray TS, Kiss JZ. Neuronal architecture in the rat central nucleus of the amygdala: a cytological, hodological, and immunocytochemical study. J Comp Neurol. 1986;246:478–499. doi: 10.1002/cne.902460406. [DOI] [PubMed] [Google Scholar]
- 9.Day HEW, CUrran EJ, Watson SJ, Akil H. Distinct neurochemical populations in the rat central nucleus of the amygdala and bed nucleus of the stria terminalis: evidence for their selective activation by Interleukin-1β. J Comp Neurol. 1999;413:113–128. [PubMed] [Google Scholar]
- 10.Marchant NJ, Densmore VS, Osborne PB. Coexpression of prodynorphin and corticotrophin-releasing hormone in the rat central amygdala: evidence of two distinct endogenous opioid systems in the lateral division. J Comp Neurol. 2007;504:702–715. doi: 10.1002/cne.21464. [DOI] [PubMed] [Google Scholar]
- 11.Ehrlich I, et al. Amygdala inhibitory circuits and the control of fear memory. Neuron. 2009;62:757–771. doi: 10.1016/j.neuron.2009.05.026. [DOI] [PubMed] [Google Scholar]
- 12.Huber D, Veinante P, Stoop R. Vasopressin and oxytocin excite distinct neuronal populations in the central amygdala. Science. 2005;308:245–248. doi: 10.1126/science.1105636. [DOI] [PubMed] [Google Scholar]
- 13.Wilensky AE, Schafe GE, Kristensen MP, LeDoux JE. Rethinking the fear circuit: the central nucleus of the amygdala is required for the acquisition, consolidation, and expression of Pavlovian fear conditioning. J Neurosci. 2006;26:12387–12396. doi: 10.1523/JNEUROSCI.4316-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lerchner W, et al. Reversible silencing of neuronal excitability in behaving mice by a genetically targeted, ivermectin-gated Cl- channel. Neuron. 2007;54:35–49. doi: 10.1016/j.neuron.2007.02.030. [DOI] [PubMed] [Google Scholar]
- 15.Slimko EM, McKinney S, Anderson DJ, Davidson N, Lester HA. Selective electrical silencing of mammalian neurons in vitro by the use of invertebrate ligand-gated chloride channels. J Neurosci. 2002;22:7373–7379. doi: 10.1523/JNEUROSCI.22-17-07373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ciocchi S, et al. Encoding of conditioned fear in central amygdala inhibitory circuits. Nature. 2010 doi: 10.1038/nature09559. this issue, in press. [DOI] [PubMed] [Google Scholar]
- 17.Day HE, Nebel S, Sasse S, Campeau S. Inhibition of the central extended amygdala by loud noise and restraint stress. Eur J Neurosci. 2005;21:441–454. doi: 10.1111/j.1460-9568.2005.03865.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zirlinger M, Anderson D. Molecular dissection of the amygdala and its relevance to autism. Genes Brain Behav. 2003;2:282–294. doi: 10.1034/j.1601-183x.2003.00039.x. [DOI] [PubMed] [Google Scholar]
- 19.Zirlinger M, Kreiman G, Anderson DJ. Amygdala-enriched genes identified by microarray technology are restricted to specific amygdaloid subnuclei. Proc Natl Acad Sci U S A. 2001;98:5270–5275. doi: 10.1073/pnas.091094698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Callaway EM. A molecular and genetic arsenal for systems neuroscience. Trends Neurosci. 2005;28:196–201. doi: 10.1016/j.tins.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 21.Luo L, Callaway EM, Svoboda K. Genetic dissection of neural circuits. Neuron. 2008;57:634–660. doi: 10.1016/j.neuron.2008.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang F, Aravanis AM, Adamantidis A, de Lecea L, Deisseroth K. Circuit-breakers: optical technologies for probing neural signals and systems. Nat Rev Neurosci. 2007;8:577–581. doi: 10.1038/nrn2192. [DOI] [PubMed] [Google Scholar]
- 23.Chieng BC, Christie MJ, Osborne PB. Characterization of neurons in the rat central nucleus of the amygdala: cellular physiology, morphology, and opioid sensitivity. J Comp Neurol. 2006;497:910–927. doi: 10.1002/cne.21025. [DOI] [PubMed] [Google Scholar]
- 24.Gong S, et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 2003;425:917–925. doi: 10.1038/nature02033. [DOI] [PubMed] [Google Scholar]
- 25.Li P, Slimko EM, Lester HA. Selective elimination of glutamate activation and introduction of fluorescent proteins into a Caenorhabditis elegans chloride channel. FEBS Lett. 2002;528:77–82. doi: 10.1016/s0014-5793(02)03245-3. [DOI] [PubMed] [Google Scholar]
- 26.Wagstaff MJ, et al. Gene transfer using a disabled herpes virus vector containing the EMCV IRES allows multiple gene expression in vitro and in vivo. Gene therapy. 1998;5:1566–1570. doi: 10.1038/sj.gt.3300754. [DOI] [PubMed] [Google Scholar]
- 27.Veinante P, Freund-Mercier MJ. Branching patterns of central amygdaloid nucleus afferents in the rat: Single axon reconstructions. Ann N Y Acad Sci. 2003;985:552–553. [Google Scholar]
- 28.Sun N, Yi H, Cassell MD. Evidence for a GABAergic interface between cortical afferents and brainstem projection neurons in the rat central extended amygdala. J Comp Neurol. 1994;340:43–64. doi: 10.1002/cne.903400105. [DOI] [PubMed] [Google Scholar]
- 29.Gautron L, Lazarus M, Scott MM, Saper CB, Elmquist JK. Identifying the efferent projections of leptin-responsive neurons in the dorsomedial hypothalamus using a novel conditional tracing approach. J Comp Neurol. 2010;518:2090–2108. doi: 10.1002/cne.22323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.De Oca BM, De Cola JP, Maren S, Fanselow MS. Distinct regions of the periaqueductal gray are involved in the acquisition and expression of defensive responses. J Neurosci. 1998;18:3426–3432. doi: 10.1523/JNEUROSCI.18-09-03426.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JJ, Rison RA, Fanselow MS. Effects of amygdala, hippocampus, and periaqueductal grady lesions on short- and long-term contextual fear. Behav Neurosci. 1993;107:1093–1098. doi: 10.1037//0735-7044.107.6.1093. [DOI] [PubMed] [Google Scholar]
- 32.LeDoux JE, Iwata J, Cicchetti P, Reis DJ. Different projections of the central amygdaloid nucleus mediate autonomic and behavioral correlates of conditioned fear. J Neurosci. 1988;8:2517–2529. doi: 10.1523/JNEUROSCI.08-07-02517.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kravitz AV, et al. Regulation of parkinsonian motor behaviours by optogenetic control of basal ganglia circuitry. Nature. 2010;466:622–626. doi: 10.1038/nature09159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang F, et al. Multimodal fast optical interrogation of neural circuitry. Nature. 2007;446:633–639. doi: 10.1038/nature05744. [DOI] [PubMed] [Google Scholar]
- 35.Cardin JA, et al. Targeted optogenetic stimulation and recording of neurons in vivo using cell-type-specific expression of Channelrhodopsin-2. Nat Protoc. 2010;5:247–254. doi: 10.1038/nprot.2009.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Petreanu L, Huber D, Sobczyk A, Svoboda K. Channelrhodopsin-2-assisted circuit mapping of long-range callosal projections. Nat Neurosci. 2007;10:663–668. doi: 10.1038/nn1891. [DOI] [PubMed] [Google Scholar]
- 37.Wickersham IR, et al. Monosynaptic restriction of transsynaptic tracing from single, genetically targeted neurons. Neuron. 2007;53:639–647. doi: 10.1016/j.neuron.2007.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wickersham IR, Finke S, Conzelmann KK, Callaway EM. Retrograde neuronal tracing with a deletion-mutant rabies virus. Nat Methods. 2007;4:47–49. doi: 10.1038/NMETH999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Slimko EM, Lester HA. Codon optimization of Caenorhabditis elegans GluCl ion channel genes for mammalian cells dramatically improves expression levels. J Neurosci Methods. 2003;124:75–81. doi: 10.1016/s0165-0270(02)00362-x. [DOI] [PubMed] [Google Scholar]
- 40.Edwards AL. Experimental Design in Psychological Research. 4th edn. Holt, Reinhard & Winston; 1972. pp. 249–251. [Google Scholar]
- 41.Wickens JR, Arbuthnott GW, Shindou T. Simulation of GABA function in the basal ganglia: computational models of GABAergic mechanisms in basal ganglia function. Prog Brain Res. 2007;160:313–329. doi: 10.1016/S0079-6123(06)60018-6. [DOI] [PubMed] [Google Scholar]
- 42.Gozzi A, et al. A neural switch for active and passive fear. Neuron. 2010;67:656–666. doi: 10.1016/j.neuron.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 43.Tsetsenis T, Ma XH, Lo Iacono L, Beck SG, Gross C. Suppression of conditioning to ambiguous cues by pharmacogenetic inhibition of the dentate gyrus. Nat Neurosci. 2007;10:896–902. doi: 10.1038/nn1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ressler KJ, Mayberg HS. Targeting abnormal neural circuits in mood and anxiety disorders: from the laboratory to the clinic. Nat Neurosci. 2007;10:1116–1124. doi: 10.1038/nn1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gong S, Yang XW, Li C, Heintz N. Highly efficient modification of bacterial artificial chromosomes (BACs) using novel shuttle vectors containing the R6Kgamma origin of replication. Genome Res. 2002;12:1992–1998. doi: 10.1101/gr.476202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atasoy D, Aponte Y, Su HH, Sternson SM. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J Neurosci. 2008;28:7025–7030. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seidler B, et al. A Cre-loxP-based mouse model for conditional somatic gene expression and knockdown in vivo by using avian retroviral vectors. Proc Natl Acad Sci U S A. 2008;105:10137–10142. doi: 10.1073/pnas.0800487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Furler S, Paterna JC, Weibel M, Bueler H. Recombinant AAV vectors containing the foot and mouth disease virus 2A sequence confer efficient bicistronic gene expression in cultured cells and rat substantia nigra neurons. Gene Ther. 2001;8:864–873. doi: 10.1038/sj.gt.3301469. [DOI] [PubMed] [Google Scholar]
- 49.Szymczak AL, et al. Correction of multi-gene deficiency in vivo using a single ‘self-cleaving’ 2A peptide-based retroviral vector. Nat Biotechnol. 2004;22:589–594. doi: 10.1038/nbt957. [DOI] [PubMed] [Google Scholar]
- 50.Etessami R, et al. Spread and pathogenic characteristics of a G-deficient rabies virus recombinant: an in vitro and in vivo study. J Gen Virol. 2000;81:2147–2153. doi: 10.1099/0022-1317-81-9-2147. [DOI] [PubMed] [Google Scholar]
- 51.Herry C, et al. Switching on and off fear by distinct neuronal circuits. Nature. 2008;454:600–606. doi: 10.1038/nature07166. [DOI] [PubMed] [Google Scholar]
- 52.Nicolelis MA, et al. Chronic, multisite, multielectrode recordings in macaque monkeys. Proc Natl Acad Sci U S A. 2003;100:11041–11046. doi: 10.1073/pnas.1934665100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.