

Abstract
This article reviews the clinical approach taken towards identification of the cause of hearing loss in children. A brief overview of the universal newborn hearing screening program is presented. Discussion is then focused on clinical elements of the diagnostic process with emphasis on the importance of the history, physical examination, and audiologic testing. The utility and appropriateness of additional diagnostic testing is considered, particularly with regards to the incorporation of diagnostic radiologic imaging and genetic testing. In the course of these discussions, the genetic and non- genetic causes of pediatric hearing loss are reviewed. Finally, the implications of a definitive identification of hearing loss etiology are considered.
Hearing loss is the fourth most common developmental disorder in the United States, and deafness is the most common sensory disorder. In the United States, the incidence of congenital hearing loss based on universal neonatal screening programs is estimated to be 1.1 per 1,000 with a range of 0.22–3.61 per 1,000 between individual states [1]. Indigent patients are at a higher risk of neonatal hearing loss than the average US population [2].
Generally, it is estimated that 50% of cases of congenital hearing loss are genetic in nature, 25% are acquired, and the remaining 25% are idiopathic. Of genetic causes for congenital hearing loss, approximately 30% are syndromic, and 70% are nonsyndromic [3]. Genetic causes can also be subdivided by inheritance pattern; approximately 77% of cases are autosomal recessive, 22% are autosomal dominant, 1% are X-linked, and <1% are passed via mitochondrial inheritance from the mother [4].
Prelingual deafness is particularly worrisome as it can engender many other disabilities. The ability to hear during the early years of life is critical for the development of speech, language, and cognition. Hearing impairment is also associated with a reduction in visual reception and fine motor skills as the child ages [5]. Early identification and intervention can prevent severe psychosocial, educational, and linguistic repercussions [6, 7]. The prevalence of congenital hearing loss is greater than twice that of all other diseases and syndromes routinely screened at birth combined [8]. Hence, universal newborn hearing screening programs have been implemented in most states across the country.
Universal Newborn Hearing Screening
As recently as 1988, the Commission on Education of the Deaf estimated that in the United States, congenital hearing loss was diagnosed in children at an average age of 2.5–3 years, a point in time significantly after the onset of the critical period for speech and language development [9]. At that time, the only children screened for hearing loss were those with significant risk factors as defined by the Joint Committee on Infant Hearing (JCIH) [10]. This directed method of screening missed cases of nonsyndromic sensorineural hearing loss (SNHL) and only identified about half of the children who had hearing loss [11]. In 1993, via a consensus statement, the National Institute of Health (NIH) advocated early detection of hearing loss not only in high- risk infants but universally in all infants before 3 months of age [12]. Presently, the JCIH endorses universal screening within a state- run system of early hearing detection and intervention (EHDI). This program has three goals: (1) to identify children with hearing loss by one month of age, (2) to formally diagnose children with hearing loss by three months, and (3) to intervene by 6 months [13].
Initial screening in healthy infants is typically performed using otoacoustic emissions (OAE) testing. A passing OAE result signifies proper functioning of the outer hair cells and the endocochlear potential within the cochlea and is usually interpreted as hearing sensitivity of 30 dB or better. If a child fails an OAE test, an auditory brainstem response (ABR) test is performed. Failing both elements of the hearing screening results in a referral to an audiologist for formal evaluation by 3 months of age.
Typically, ABR abnormalities are associated with a lack of OAE response. In some cases, however, a child will fail an ABR despite passing their OAE testing. This finding suggests auditory neuropathy/dysynchrony, a condition whereby the outer hair cells of the cochlea respond appropriately to auditory input but either the inner hair cells are unable to transduce the signals to the auditory nerve or there is dysfunction of the auditory nerve/brainstem pathways. Since auditory neuropathy/dysynchrony is particularly common in infants within the neonatal intensive care unit (NICU), it is recommended that all infants within the NICU undergo hearing screening using ABR alone [14].
The Colorado hearing screening program has reported that 98% of newborns in the state were screened for hearing loss between 2002–2004 [15]. While the screening rate is high, the remaining percentage of unscreened newborns still represents more than 200,000 children. Therefore, even during regularly scheduled well- child visits, the physician should be vigilant in monitoring the achievement of speech and language developmental milestones in an effort to detect previously missed or progressive hearing loss. The American Speech-Language-Hearing Association summarizes key speech and language developmental milestones on their website [16].
Diagnosing the Etiology of Hearing Loss
Presently, no formal consensus exists regarding which specific diagnostic tests should be ordered for pediatric hearing loss, but all otolaryngologists agree that a careful and complete history and physical examination along with the audiometric data are critical first steps towards diagnosing the cause of a child’s hearing loss.
History
The important elements of the history are summarized in table 1. Non-genetic causes of hearing loss can often be revealed during history taking. These acquired causes can be classified into several general categories: (1) infection, (2) prematurity/NICU admission, and (3) miscellaneous.
Table 1.
Elements of history taking for pediatric hearing loss
| Prenatal |
| Maternal infections |
| Toxoplasmosis, rubella, cytomegalovirus, herpes simplex, syphilis |
| Maternal medications |
| Aminoglycosides, quinine, cloroquine, thalidomide |
| Maternal illnesses |
| Diabetes |
| Perinatal |
| Prematurity |
| Low birth weight |
| Birth hypoxia (low APGAR scores) |
| Hyperbilirubinemia |
| Sepsis |
| NICU admission |
| Ototoxic medications |
| Postnatal |
| Viral illnesses (mumps, measles, chicken pox) |
| Bacterial meningitis |
| Recurrent or persistent otitis media with effusion |
| Head trauma |
| Noise trauma |
| Neurodegenerative disorders |
| Speech and language developmental milestones |
| Family history |
| First and second-degree relatives with hearing loss |
| Common origin from ethnically isolated areas |
| Consanguinity |
Infection once comprised the majority of acquired causes of hearing loss. Prenatally, any of the common maternal infections (toxoplasmosis, rubella, cytomegalovirus (CMV), herpes simplex, and syphilis) can manifest as hearing loss. Postnatally, viral illnesses and bacterial meningitis are causes of hearing loss. Advancements in prenatal and neonatal care and the institution of universal immunization programs have decreased the incidence of hearing loss from infectious etiology [17]. In particular, vaccination programs have nearly eliminated congenital rubella and H. influenza type B as significant causes of hearing loss [18, 19]. On the other hand, CMV infection is the most common infectious cause of congenital hearing loss and may be responsible for 12–25% of pediatric hearing loss [20].
As survival rates of premature infants have increased, hearing loss related to prematurity and NICU admissions have become more prevalent. It is not always apparent how prematurity or NICU care directly leads to hearing loss, but children in the NICU typically share one or more of the following traits: birth weight <1,500 g, low APGAR scores (0– 3 at 5 min, 0– 6 at 10 min), hypoxia re-quiringrespiratorysupport, hyperbilirubinemiaat levels requiring exchange transfusion, sepsis, and exposure to ototoxic medications such as amino-glycoside antibiotics or diuretics [21]. Most often, the hearing loss in these cases is sensorineural and permanent. There is some evidence, however, that correction of hyperbilirubinemia may improve hearing [22].
Other miscellaneous acquired causes of hearing loss that may be suspected after taking a history include chronic otitis media with effusion, congenital stapes fixation, ossicular chain malformation, congenital aural atresia, noise exposure, and head trauma.
Genetic causes of hearing loss are less easily identified by history, but occasionally other elements of a syndromic hearing loss may by identified. These include a history of visual impairment, kidney disease, or syncope spells. Attention to achievement of speech and language developmental milestones may reveal whether the hearing loss was present at birth or later in onset, and whether the hearing loss is progressive in nature.
Non-syndromic genetic hearing loss is far more difficult to identify by history alone. If hearing loss can be adequately identified in first and second degree relatives, constructing a pedigree may identify a pattern of inheritance. Also, children from consanguineous marriages or from family backgrounds arising from small, ethnically homogeneous areas such as Ashkenazi Jews or Japanese are more likely to suffer from a genetic form of hearing loss [23].
Physical Examination
The physical examination typically performed by an otolaryngologist is in actuality quite limited. Assessments are made of head size and symmetry, jaw size and symmetry, facial movement and symmetry, and external and middle ear morphology. These elements of the physical examination can often reveal pathology associated with acquired causes of hearing loss, particularly those that result in a conductive hearing loss. Examples include middle ear effusion, microtia, external canal atresia, cleft lip or palate, and craniofacial anomalies such as microcephaly, craniosynostosis, micrognathia, or facial asymmetry.
In other cases of congenital hearing loss, associated physical anomalies, particularly in the head and neck region, may be subtle or even nonexistent. Congenital hearing loss can result from one of more than 400 syndromes and be associated with defects in virtually any organ system (reviewed by Toriello et al. [24]). Collaboration between specialists therefore is essential to identify a syndrome.
Audiologic Studies
In neonates, an ABR can give accurate hearing thresholds between 1 and 4 kHz [25]. Auditory steady-state evoked potentials (ASSR) can measure auditory thresholds at levels higher than that often available clinically for ABR [26]. In older children, a behavioral audiogram can be obtained by means of visual reinforcement or conditioned play audiometry. Acquired hearing loss and syndromic genetic hearing loss can be associated with conductive, sensorineural, or mixed forms of hearing loss, but non-syndromic genetic hearing loss is almost always sensorineural. The shape of the audiogram can be used for audiometric profiling, a method that pairs a given frequency response curve with the most likely causative gene mutations [27].
Ancillary Studies
While a history, physical exam, and audiologic evaluation are mandatory in the workup of pediatric hearing loss, the decision to pursue further diagnostic testing is controversial. Some clinicians order an exhaustive array of tests to aid in diagnosis. Others select a few specific tests that may be of higher yield given particular findings on history or physical exam. Despite several studies to determine the usefulness of various sets of diagnostic tests, no consensus exists regarding the appropriate test battery for pediatric hearing loss [28]. Table 2 lists commonly ordered ancillary diagnostic tests.
Table 2.
Commonly ordered ancillary studies or consultations in the diagnostic workup for congenital pediatric hearing loss
| Study or consultation | Associated etiology for hearing loss |
|---|---|
| ANA, ESR, RF, anticardiolipin, immunoglobulins, complement studies | autoimmune disease |
| CBC | thallasemia, Sickle cell anemia |
| Platelet count | Fechtner syndrome, Epstein syndrome |
| BUN, creatinine | Alport syndrome* |
| Glucose | Alstrom syndrome*, diabetes mellitus |
| Urinalysis | Alport syndrome, metabolic disorders |
| Antibody titers | toxoplasmosis, rubella, CMV, herpes simplex |
| FT4, T3, TSH, perchlorate test | Pendred syndrome*, cretinism |
| RPR, FT-ABS | syphilis |
| ECG | Jervell Lange-Nielsen syndrome |
| CT Temporal Bone | temporal bone anomalies |
| MRI Brain and IAC | cochlear nerve hypoplasia/aplasia, CPA mass, brain lesions |
| Renal ultrasound | branchio-oto-renal syndrome |
| Connexin 26/30 genetic testing | connexin 26/30 mutation |
| Genetics consultation | any genetic etiology |
| Ophthalmology consultation | Usher syndrome*, Alport syndrome, Cogan’s syndrome, Norrie disease, Stickler syndrome |
Note that certain phenotypic features, such as goiter in Pendred syndrome and retinitis pigmentosa in Usher syndrome develop with age and would not be expected to be seen in an infant, thus a normal result would not exclude such a diagnosis in an infant or young child.
Imaging Studies
Diagnostic imaging is the most useful ancillary test in determining the cause of pediatric hearing loss. Diagnostic imaging typically consists of computed tomography (CT) of the temporal bone, magnetic resonance imaging (MRI) of the brain and internal auditory canal (IAC), or both. Studies have shown that 27–39% of children with hearing loss who undergo diagnostic imaging will demonstrate an anatomic abnormality that may explain the hearing loss [30, 31]. The most common temporal bone abnormality is an enlarged vestibular aqueduct (EVA). This may or may not be associated with a Mondini malformation in the cochlea and is suggestive of Pendred or Branchio-Oto-Renal syndromes. Other temporal bone anomalies include lateral semicircular canal dysplasia, small IACs, hypoplastic cochleas, and otic capsule lucencies [29]. Figure 1 demonstrates a variety of temporal bone malformations encountered in children with congenital hearing loss.
Fig. 1.
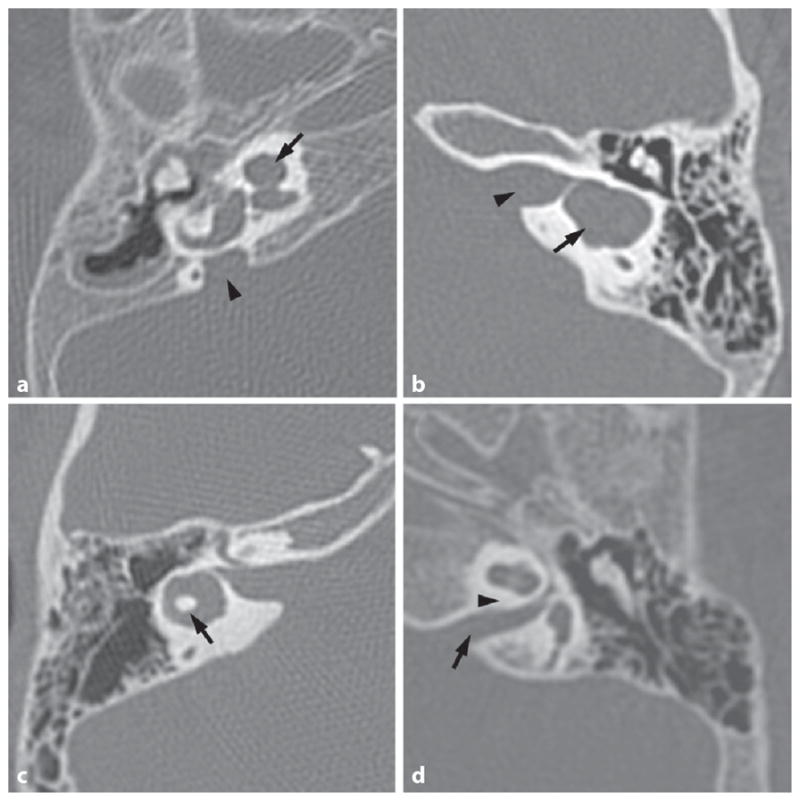
A variety of CT temporal bone images showing anomalies encountered in children with congenital hearing loss. a Axial CT of a right temporal bone with a Mondini malformation of the cochlea (arrow) and an enlarged vestibular aqueduct (arrowhead). b Axial CT of a left temporal bone with a common cavity malformation (arrow) and bony separation of common cavity from the IAC (arrowhead). c Axial CT of a right temporal bone with lateral semicircular canal dysplasia – the bone island in the center of the canal (arrow) is too small. d Axial CT of a left temporal bone with a narrow IAC (arrow) and no cochlear aperture for the auditory nerve to enter the cochlea (expected location is marked by arrowhead).
In many cases, temporal bone anomalies may not be pathognomonic for particular syndromes but may predict clinical course or surgical outcomes. For instance, children with EVA may show progressive sensorineural hearing loss, particularly after mild head trauma [30]. Children with abnormal connections between otic capsule structures and the middle ear may be prone to hearing loss, vestibular symptoms, or facial paralysis during occurrences of otitis media. Abnormal connections between the cerebrospinal fluid (CSF) space and inner ear structures such as EVA or absent bone at the modiolus may predispose patients to CSF gushers during and CSF leaks after certain ear surgeries [31]. These patients may also be more prone to meningitis. For patients who have had a history of meningitis, imaging may reveal labyrinthitis ossificans, a condition that complicates placement of a cochlear implant [32].
Renal ultrasound is a commonly performed diagnostic imaging procedure in the workup of pediatric hearing loss [33]. A renal ultrasound is ordered in suspected cases of BOR syndrome looking for developmental renal anomalies.
Laboratory Studies
Many studies suggest that performing a standard battery of laboratory tests is not particularly useful in identifying the etiology for hearing loss. Rather than blanketing all deaf children with multiple blood tests, it has been recommended that specific laboratory tests be ordered on the basis of the patient’s history, physical examination, and audiogram [28]. Laboratory tests that are typically ordered to evaluate pediatric hearing loss include complete blood count, serum chemistry panels, thyroid function tests, antibody titers for pre- or perinatal pathogens, autoimmune serologies, and urinalysis. Abnormal laboratory findings for autoimmune serologies occur nearly 25% of the time but are almost never helpful in determining the cause of the hearing loss [28].
Electrocardiography (ECG)
Electrocardiography has a diagnostic yield akin to laboratory testing but deserves mention because of the dire consequences of a missed diagnosis of Jervell and Lange-Nielsen syndrome. This syndrome, which includes deafness and prolonged QT intervals, may initially present as sudden death. An ECG may be obtained in any child with hearing loss to evaluate for this possibility, but would be particularly relevant in a child with a history of syncopal episodes or a family history of sudden infant death syndrome.
Consultations
Hearing loss is disproportionately associated with abnormalities of ocular structures. Therefore, most children with congenital hearing loss are also referred to an ophthalmologist, who can identify subtle ocular abnormalities such as retinitis pigmentosa that may indicate a particular etiology for the hearing loss. Children with nonsyndromic congenital hearing loss can experience decreased visual reception skills as they age [5]. Therefore, at the very least, the child’s visual acuity can be evaluated and optimized in order to minimize his or her sensory disabilities and maximize the potential for normal development.
Determining a possible syndromic etiology for hearing loss sometimes requires a familiarity with the constellation of physical findings beyond that of an otolaryngologist. Thus the expertise of a clinical geneticist can be helpful for making a clinical diagnosis of syndromic hearing loss and for pedigree analysis for non-syndromic hearing loss.
Genetic Testing
As our understanding of the molecular basis of hearing improves, a significant percentage of hearing loss that is presently idiopathic will likely be found to have a genetic basis. Non-syndromic hearing loss, which lacks associated physical findings, has typically been a diagnosis of exclusion. In the last 5 years, however, rapid escalation in the numbers of genes found to be responsible for non-syndromic hearing loss has made a definitive diagnosis more likely [34].
The finding in 1997 that up to 50% of autosomal recessive non-syndromic hearing loss in some populations may be due to mutations in the GJB2 gene that encodes Connexin 26 significantly improved the prospects of genetic testing for hearing loss [35]. Aside from Connexin 26, however, no consensus exists regarding which additional genes should be considered as part of routine testing. In an excellent review of the genetic approach toward diagnosing pediatric hearing loss, Rehm [36] proposes an algorithm that accounts for patterns of inheritance, timing of hearing loss, audiogram profile, and associated clinical findings in recommending specific genes to be tested. This algorithm will evolve as new genes are discovered, functions of existing gene products are revealed, and the prevalences of mutations in known genes are defined. On the other hand, cost reductions and advances in gene testing technology are likely to soon render obsolete any need for algorithmic testing.
Implications of Diagnosis
Every patient or family has different experiences with hearing loss, different attitudes towards genetic testing, and different expectations regarding the outcomes of genetic testing. Therefore, it is imperative to consult a genetic counselor that has experience in dealing with perceptions or misperceptions of genetic testing and managing expectations from testing results. In many instances, finding a genetic basis does not substantially change the patient’s treatment or prognosis, at present. Thus, some patients may consider genetic testing to be wasted effort; others, however, may appreciate an identifiable reason for their hearing loss. Parents who are considering having more children might benefit from knowing the inheritance pattern of hearing loss in their family and the likelihood that additional children will be deaf. In other instances, finding a genetic basis for hearing loss may indeed significantly impact a patient’s prognosis and management. For example, the diagnosis of Usher syndrome and its associated imminent blindness will direct a patient to concentrate on developing verbal rather than visual means of communication. A finding of mutations in the 12SrRNA gene allows a patient or patient’s family to recognize his or her susceptibility to aminoglycoside- induced hearing loss and to avoid aminoglycoside antibiotics use if possible. In a child with otoferlin mutations, auditory neuropathy/dyssynchrony would be established as the cause of hearing loss and in appropriate circumstances, rehabilitation might be more successful with cochlear implantation rather than hearing aids [37].
Conclusions
Significant advancements have been made in identifying genes important for hearing and understanding of the molecular function of these gene products. For neonatal hearing loss that is acquired or genetic hearing loss that is part of a syndrome, a thoughtful integration of the clinical history, the physical examination, and the audiologic testing may yield a diagnosis of etiology. Genetic testing offers the best chance for determining an etiology for non-syndromic hearing loss. An algorithm integrating our understanding of gene function with clinical findings can be used to select candidate genes that are most likely to be mutated. As more hearing genes are identified and their functions are elucidated, algorithms for genetic testing will become more refined and directed. Concomitantly, improvements in technology will allow for faster more cost-effective genetic testing.
References
- 1.Mehra S, Eavey RD, Keamy DG. The epidemiology of hearing impairment in the United States: newborns, children, and adolescents. Otolaryngol Head Neck Surg. 2009;140:461–472. doi: 10.1016/j.otohns.2008.12.022. [DOI] [PubMed] [Google Scholar]
- 2.Oghalai JS, et al. Neonatal hearing loss in the indigent. Laryngoscope. 2002;112:281–286. doi: 10.1097/00005537-200202000-00015. [DOI] [PubMed] [Google Scholar]
- 3.Hone SW, Smith RJH. Medical evaluation of pediatric hearing loss: laboratory, radiographic, and genetic testing. Otolaryngol Clin N Am. 2002;35:751– 764. doi: 10.1016/s0030-6665(02)00048-8. [DOI] [PubMed] [Google Scholar]
- 4.Morton NE. Genetic epidemiology of hearing impairment. Ann NY Acad Sci. 1991;630:16–31. doi: 10.1111/j.1749-6632.1991.tb19572.x. [DOI] [PubMed] [Google Scholar]
- 5.Pierson SK, Caudle SE, Krull KR, et al. Cognition in children with sensorineural hearing loss: etiologic considerations. Laryngoscope. 2007;117:1661–1665. doi: 10.1097/MLG.0b013e3180ca7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robinshaw HM. Early intervention for hearing impairment: differences in the timing of communicative and linguistic development. Br J Audiol. 1995;29:315–334. doi: 10.3109/03005369509076750. [DOI] [PubMed] [Google Scholar]
- 7.Yoshinaga-Itano C, et al. Language of early- and later-identified children with hearing loss. Pediatrics. 1998;102:1161– 1171. doi: 10.1542/peds.102.5.1161. [DOI] [PubMed] [Google Scholar]
- 8.Finitzo T, Crumley WG. The role of the pediatrician in hearing loss: from detection to connection. Pediatr Clin North Am. 1999;46:15–34. doi: 10.1016/s0031-3955(05)70078-x. [DOI] [PubMed] [Google Scholar]
- 9.Commission on Education of the Deaf. Education of the Deaf. Washington: 1988. Toward Equality. [Google Scholar]
- 10.Joint Committee on Infant Hearing: 1990 position statement. Am Speech/Language Hearing Assoc. 1991;33(suppl 5):3–6. [PubMed] [Google Scholar]
- 11.Mauk GW, White KR, Mortensen LB, et al. The effectiveness of screening programs based on high-risk characteristics in early identification of hearing impairment. Ear Hear. 1991;12:312–319. doi: 10.1097/00003446-199110000-00003. [DOI] [PubMed] [Google Scholar]
- 12.National Institutes of Health. NIH Consensus Development Conference Statement. Bethesda: National Institutes of Health; 1993. Early Identification of Hearing Impairment in Infants and Young Children; pp. 1–24. [PubMed] [Google Scholar]
- 13.Joint Committee on Infant Hearing: 2007 position statement. Pediatrics. 2007;120:898–921. [Google Scholar]
- 14.Cristobal R, Oghalai JS. Hearing loss in children with very low birth weight: current review of epidemiology and pathophysiology. Arch Dis Child Fetal Neonatal Ed. 2008;93:f462–f468. doi: 10.1136/adc.2007.124214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Christensen M, Thomson V, Letson GW. Evaluating the reach of universal newborn hearing screening in Colorado. Am J Prev Med. 2008;359:594–597. doi: 10.1016/j.amepre.2008.09.007. [DOI] [PubMed] [Google Scholar]
- 16.http://www.asha.org/public/speech/development/chart.htm.
- 17.Catlin FI. Prevention of hearing impairment from infection and ototoxic drugs. Arch Otolaryngol. 1985;111:377–384. doi: 10.1001/archotol.1985.00800080063007. [DOI] [PubMed] [Google Scholar]
- 18.Centers for Disease Control and Prevention. Progress toward elimination of rubella and congenital rubella syndrome – the Americas, 2003–2008. Morb Mortal Wkly Rep. 2008;57:1176– 1179. [PubMed] [Google Scholar]
- 19.Peltola H. Worldwide Haemophilus influenzae type B diseases at the beginning of the 21st century: global analysis of the disease burden 25 years after the use of the polysaccharide vaccine and a decade after the advent of conjugates. Clin Microbiol Rev. 2000;13:302–317. doi: 10.1128/cmr.13.2.302-317.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fowler KB, Boppana SB. Congenital cytomegalovirus (CMV) infection and hearing deficit. J Clin Virol. 2006;35:226– 231. doi: 10.1016/j.jcv.2005.09.016. [DOI] [PubMed] [Google Scholar]
- 21.Bamiou DE, Macardle B, Bitner-Glindzicz M, et al. Aetiological investigations of hearing loss in childhood: a review. Clin Otolaryngol. 2000;25:98– 106. doi: 10.1046/j.1365-2273.2000.00346.x. [DOI] [PubMed] [Google Scholar]
- 22.Kuriyama M, Tomiwa K, Konishi Y, et al. Improvement in auditory brainstem response of hyperbilirubinemic infants after exchange transfusions. Pediatr Neurol. 1986;2:127–132. doi: 10.1016/0887-8994(86)90002-0. [DOI] [PubMed] [Google Scholar]
- 23.Basil A. Childhood sensorineural hearing loss in consanguineous marriages. J Audiol Med. 1994;3:151–159. [Google Scholar]
- 24.Toriello HV, Reardon W, Gorlin RJ. Hereditary Hearing Loss and Its Syndromes. 2. Oxford: Oxford University Press; 1995. [Google Scholar]
- 25.Tomaski SM, Grundfast KM. A stepwise approach to the diagnosis and treatment of hereditary hearing loss. Pediatr Clin N Am. 1999;46:35–47. doi: 10.1016/s0031-3955(05)70079-1. [DOI] [PubMed] [Google Scholar]
- 26.Oghalai JS, Tonini R, Rasmus J, et al. Intra-operative monitoring of cochlear function during cochlear implantation. Cochlear Implants Int. 2009;10:1–18. doi: 10.1002/cii.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hildebrand MS, DeLuca AP, Taylor KR, et al. A contemporary review of Audiogene audioprofiling: a machine-based candidate gene prediction tool for autosomal dominant nonsyndromic hearing loss. Laryngoscope. 2009;119:2211–2215. doi: 10.1002/lary.20664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mafong DD, Shin EJ, Lalwani AK. Use of laboratory evaluation and radiologic imaging in the diagnostic evaluation of children with sensorineural hearing loss. Laryngoscope. 2002;112:1–7. doi: 10.1097/00005537-200201000-00001. [DOI] [PubMed] [Google Scholar]
- 29.Antonelli PJ, Varela AE, Mancuso AA. Diagnostic yield of high-resolution computed tomography for pediatric sensorineural hearing loss. Laryngoscope. 1999;109:1642–1647. doi: 10.1097/00005537-199910000-00018. [DOI] [PubMed] [Google Scholar]
- 30.Madden C, Halsted M, Benton C, et al. Enlarged vestibular aqueduct syndrome in the pediatric population. Oto Neurotol. 2003;24:625–632. doi: 10.1097/00129492-200307000-00016. [DOI] [PubMed] [Google Scholar]
- 31.Luntz M, Balkany T, Hodges AV, et al. Cochlear implants in children with congenital inner ear malformation. Arch Otolaryngol Head Neck Surg. 1997;123:974–977. doi: 10.1001/archotol.1997.01900090090013. [DOI] [PubMed] [Google Scholar]
- 32.Philipon D, Bergeron F, Ferron P, et al. Cochlear implantation in postmeningitic deafness. Oto Neurotol. 2010;31:83–87. doi: 10.1097/mao.0b013e3181c2a02d. [DOI] [PubMed] [Google Scholar]
- 33.Wang RY, Dawn LE, Ruder RO, et al. Sydromic ear anomalies and renal ultrasounds. Pediatrics. 2001;108:E32. doi: 10.1542/peds.108.2.e32. [DOI] [PubMed] [Google Scholar]
- 34.Hilgert N, Smith RJH, Van Camp G. Function and expression pattern of non-syndromic deafness genes. Curr Mol Med. 2009;9:546–564. doi: 10.2174/156652409788488775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kelsell DP, Dunlop J, Stevens HP, et al. Connexin 26 mutationsin hereditary non-syndromic sensorineural deafness. Nature. 1997;387:80–83. doi: 10.1038/387080a0. [DOI] [PubMed] [Google Scholar]
- 36.Rehm HL. A genetic approach to the child with sensorineural hearing loss. Semin Perinatol. 2005;29:173–181. doi: 10.1053/j.semperi.2004.12.002. [DOI] [PubMed] [Google Scholar]
- 37.Berlin CI, Hood LJ, Morlet T, et al. Multi-site diagnosis and management of 260 patients with auditory neuropathy/dys-synchrony (auditory neuropathy spectrum disorder) Int J Audiol. 2010;49:30–43. doi: 10.3109/14992020903160892. [DOI] [PubMed] [Google Scholar]