

Abstract
The clinical features and common molecular alterations of adenoid cystic carcinoma (ACC) are reviewed in this paper. ACC is an uncommon neoplasm that most frequently arises in salivary glands and related tissue in the head and neck region. ACC has distinct histologic features, with cribriform and tubular growth patterns of basaloid cells displaying a predominantly myoepithelial cellular phenotype. This neoplasm also has uncommon clinical features of rare regional lymph node metastasis and a prolonged but relentlessly progressive clinical course. Clinical outcome in ACC is correlated to histologic grade, which is correlated to the degree of aneuploidy and genetic alterations present in the tumor genomes. Recent studies have identified that the majority of ACC contain alterations of the MYB gene, usually resulting in a fusion gene product with the NFIB gene by a t(6;9) translocation event. The molecular consequences of this alteration are incompletely understood, as are secondary molecular alterations that contribute to the neoplastic phenotype of ACC.
Keywords: Adenoid cystic carcinoma, MYB, NFIB, Translocation
Adenoid cystic carcinoma (ACC) is a relatively uncommon tumor, with a reported incidence of 4.5 cases per million individuals [1]. The age range for diagnosis is wide, with tumors arising at almost equal incidence in any decade of adulthood. The salivary and lacrimal glands are among the most common sites at which this tumor arises. It is the most common malignancy to arise in the minor salivary glands, and is also one of the most common cancers of the parotid and sublingual salivary glands [2–6]. ACC also arises in glandular tissue of the nasal passages and tracheobronchial tree. ACC arises less frequently in sites outside of the head and neck, including the breast and vulva.
Histologically, ACC are composed of small basaloid epithelial tumor cells, with small to moderate amounts of cytoplasm. The nuclei tend not to be pleomorphic, and have small or inconspicuous nucleoli. The tumor is composed of cells that exhibit either luminal epithelial differentiation or myoepithelial differentiation, with myoepithelial differentiation predominating [7, 8]. Several growth patterns have been described for ACC (Fig. 1). The most common and classic pattern is characterized by tumor cells arranged in variably-sized nests of cells with distinct, punched-out spaces or pseudocysts, in a so-called cribriform pattern. The cribriform spaces contain hyaline material, variably eosinophilic or basophilic in color. Most carcinomas with such an appearance will secrete mucin; however, in ACC, the secreted substances are basement membrane constituents, including proteoglycans [9–11]. A second growth pattern, typically mixed with the cribriform pattern, is a tubular pattern where the tumor infiltrates in separate gland-like groups with single central lumens. The third growth pattern is a solid growth pattern, where tumor cells grow in sheets without lumen formation. This third pattern has been recognized as representing a higher grade of tumor. Szanto et al. [12] proposed a grading scheme for ACC, based on the degree of solid growth pattern, illustrated in Fig. 1. This grading scheme has reproducibly been associated with poorer prognosis in distinct patient cohorts [13–16]. That this grading scheme does indeed reflect a form of tumor progression is supported by cases where ACC tumors have displayed increasing solid growth and histologic atypia during the course of tumor recurrence, with increasingly aggressive clinical behavior [17]. ACC may also rarely undergo dedifferentiation, with a high grade anaplastic carcinoma component arising in a conventional ACC [18–20]. High-grade transformation is characterized by irregular, jagged infiltrating islands of tumor cells or large confluent areas of solid growth, and the background stroma displays a fibrocellular desmoplasic change, which is typically not seen in conventional ACC. The tumor cells display greater atypia, a higher mitotic rate, and usually have more cytoplasm than conventional ACC cases.
Fig. 1.

Histologic types and grades of ACC. a Cribriform pattern, with no solid growth component (Grade 1). b Tubular pattern, with no solid component (Grade 1). c Cribriform growth pattern, with areas of solid growth comprising less than 30 % of tumor (Grade 2). d Predominantly solid growth pattern (Grade 3). All figures H&E stain, original magnification ×40
The clinical course of conventional ACC is typically a slow but relentlessly progressive one. Although the majority of patients with ACC are alive at 5 years post diagnosis, the majority will go on to die of their disease [14, 21]. Although good local control is usually achieved by resection of the primary tumor, often accompanied by post-operative radiation therapy, late recurrence is common both regionally and at distant sites. Local recurrence is attributed in part due to the proclivity of ACC for perineural invasion [21–23]. The neurotropism also contributes to the infiltrative nature of this neoplasm, with deep penetration of vital structures of the craniofacial region along major nerve trunks [24]. Because of these clinical features, ACC has been described as “one of the most biologically destructive and unpredictable tumors of the head and neck” [25]. Unlike most carcinomas of the head and neck, ACC seldom metastasizes to regional lymph nodes [26]. Distant metastasis occurs in up to 40 % of cases, with the lungs being the most common site [26], but liver, kidney, bones and brain are also affected by metastatic disease. Due to the slow growth rate of this tumor, even patients with unresectable metastases may survive for many years before ultimately succumbing to their disease. ACC with high-grade transformation, however, show a markedly more aggressive clinical course, with shortened patient survival, and have a higher rate of lymph node metastasis than conventional ACC.
Several cytogenetic studies of ACC described a reciprocal t(6;9) translocation occurring in some tumors [27–30]. However it was not clear initially if this was a common finding in ACC, and the described breakpoints of the t(6;9) translocations did not appear to be consistent enough to map specific gene loci. This field of inquiry lay fallow until work from Goran Stenman’s laboratory was published in 2009 which described an analysis showing that the majority of ACC tumors harbor a t(6;9) translocation that joins the MYB and NFIB transcription factors in a fusion gene product [31]. The MYB gene appears to be the dominant contributor to the fusion product, with the DNA-binding and transcriptional regulatory portions of the MYB gene on chromosome 6 (6q22-q23) being fused to a small portion of the end of the NFIB gene on chromosome 9 (9p24.1). Though the molecular mechanisms are not entirely elucidated, this fusion gene appears to result in increased MYB transcriptional regulatory activity in the tumor cells. The levels of MYB gene product are generally elevated in ACC compared to most normal tissues, with the majority of ACC showing strong nuclear staining for MYB by immunohistochemistry (Fig. 2a).
Fig. 2.
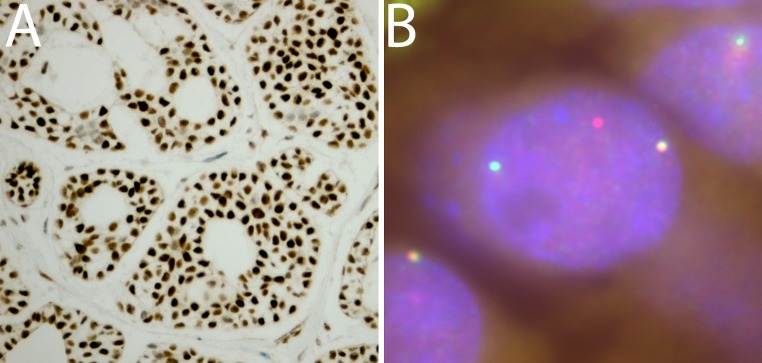
Examples of MYB alterations in ACC. a Immunohistochemical stain for MYB showing strong diffuse nuclear staining. Original magnification ×100. b Break-apart FISH analysis for MYB with 5′ green probe and 3′ red probe. The ACC tumor cell reveals one wild type MYB gene locus (yellow signal) and evidence of break-apart of the second MYB gene locus, with separate red and green signals. Original magnification ×1500
Though there is some disagreement in the literature regarding the percent of ACC tumors harboring this molecular alteration, most subsequent studies have shown that the majority of ACC have a disrupted MYB gene and/or evidence of a MYB-NFIB fusion RNA transcript [32–36]. One of the largest independent cohorts separate from the Stenman group came from a study at Stanford University, where 37 cases of ACC were studied by fluorescence in situ hybridization (FISH) of the MYB and NFIB loci, with 65 % of cases showing abnormalities of these loci [34]. An example of an ACC tumor displaying typical FISH findings for a broken-apart MYB gene locus is shown in Fig. 2b. One research group showed evidence that some ACC tumors that did not contain a translocated MYB gene had elevated levels of wild-type MYB transcript and protein through an unknown mechanism [32]. While, as previously noted, the majority of ACC will show strong MYB immunoreactivity, this stain cannot be used to exclude the diagnosis of ACC, as some tumors (up to a third of cases) that otherwise meet the histologic diagnosis of ACC may show weak or negative MYB staining [34, 36, 37]. MYB staining is also not specific for ACC, being observed in several other tumor types, including basaloid squamous cell carcinoma, an occasional histologic mimic of ACC [34, 36].
Thus it would appear that up-regulation of MYB transcriptional regulatory activity is a key component of tumorigenicity for the majority of, but not all, ACC tumors. Secondary genetic mechanisms are not well-understood, however. In attempts to identify genomic alterations that occur in ACC, there have been at least 6 studies of ACC cohorts employing the techniques of comparative genomic hybridization (CGH) and array comparative genomic hybridization (aCGH) which globally sample the copy number status of tumor genomes [38–43]. The results of these studies have been quite variable but areas of deletion that have been identified in two or more tumors in each of two or more of these studies include 1p (8–44 %), 5q (8–18 %), 6q (14–30 %), 9p (12–33 %), 9q (8–9 %), 12q (9–33 %), 13q (11–35 %), 14q (4–22 %), 17p (11–13 %), and Xp (8–9 %). Chromosomal areas of copy number gain that have been identified in two or more tumors in each of two or more of these studies include 1p (8–44 %), 8q (9–39 %), 11q (9–61 %), 16p (8–44 %), 16q (8–39 %), 17q 18–28 %), 19p (9–78 %), 22q (8–72 %). It is important to note that none of these studies segregated the ACC samples into MYB fusion positive and negative cases; hence, it is unknown if any of the described genetic abnormalities are associated positively or negatively with MYB gene locus translocation. It is also important to note that these techniques only identify changes in chromosomal copy number and will not detect reciprocal translocation events that are not associated with genetic deletion or amplification. Though there is some agreement on loci that show loss or low-level gain in a minority of ACC cases, evidence is generally lacking for specific genes that are targeted by these copy number variations identified by these studies, other than the deletions identified at 6q and 9p, which may be related to MYB-NFIB translocations events that were not perfectly reciprocal in genetic exchange. The studies are in agreement that very few high copy number gene amplification events were identified in any of the cohorts; hence, it does not appear that such a mechanism targets a specific gene in the majority of ACC.
When specific genes have been analyzed in ACC (outside of MYB), there has also been a distressing lack of consensus in the percent of ACC cases having mutations. For instance, analysis of the KIT gene has had studies leading to diametrically opposed conclusions as to the mutation rate in ACC [44–50]. However, at the current time, the consensus data does not identify any molecular alteration outside of the MYB gene that represents a common molecular mechanism in ACC. This is an active subject of research, and it is expected that planned and ongoing whole exome and whole genome sequencing studies of ACC cohorts will soon contribute to our knowledge in this area.
While secondary genetic hits remain to be elucidated in ACC, there does appear to be a consensus finding that the degree of genetic abnormality present in an ACC tumor is correlated with histologic grade. Analyses of chromosomal ploidy have shown that most classic non-solid ACC show a low degree of aneuploidy, much less than other types of carcinomas, but, ACC with solid histology show increased aneuploidy [51, 52]. Studies using CGH and aCGH show a correlation of the degree of genetic alterations (change in DNA copy number, chromosome number, genetic deletion) with the degree of solid growth pattern [39, 41, 42], consistent with the notion that ACCs with solid histology represent a later stage in tumor progression. One study of four ACC with high-grade transformation suggests that genomic amplification events are more common in transformed tumors than in conventional tumors, with identification of the MYC gene as perhaps a common mechanism for transformation [53].
Most carcinomas are thought to derive their abnormal genetic patterns from a process known as fusion-breakage-fusion, where unstable telomeres lead to a reiterative process of chromosomal fusion and breakage during subsequent mitotic cycles [54]. However, tumors that arise through a mechanism where a translocation event provides a primary driver mutation tend to have lower degrees of genetic abnormalities. A model is thus developing of the molecular pathogenesis of ACC. It would appear that most ACC begin as low-grade neoplasms, driven to neoplastic growth predominantly by genetic alteration of the MYB locus, with few if any secondary genetic hits. As these tumors continue to grow, eventually the tumors acquire secondary genetic hits, perhaps through a mechanism of telomere shortening and instability. Genetic instability is associated with increasing histologic grade and increased clinical aggressiveness, again presumably through involvement of other genetic loci that are involved in more typical and common forms of carcinoma. It may also be the case that these secondary mutations are diverse across the population of higher-grade ACC; hence, these tumors may not represent as uniform a genetic and biochemical population as low-grade ACC tumors.
In conclusion, recent findings indicate that up-regulation of the MYB proto-oncogene occurs in the majority of ACC, with translocation and gene fusion of the MYB and NFIB genes being the most common mechanism. The immediate future of research in this field of the molecular pathogenesis of ACC will be two-fold. One will be of diagnostic import, determining what combination of molecular assays (FISH, RT-PCR) is the most robust, sensitive and specific for identifying MYB/NFIB genetic alterations in the diagnosis and classification of ACC and related tumors. The second is determining the biochemical and biological effects of MYB activation in ACC tumor cells, in order to derive targeted therapies for this neoplasm, which is often highly-resistant to currently-available conventional therapies.
References
- 1.Bonaparte JP, Hart R, Trites J, Taylor MS. Incidence of adenoid cystic carcinoma in nova scotia: 30-year population-based epidemiologic study. J Otolaryngol Head Neck Surg. 2008;37:642–648. [PubMed] [Google Scholar]
- 2.Vander Poorten VL, Balm AJ, Hilgers FJ, Tan IB, Loftus-Coll BM, Keus RB, Hart AA. Prognostic factors for long term results of the treatment of patients with malignant submandibular gland tumors. Cancer. 1999;85:2255–2264. doi: 10.1002/(SICI)1097-0142(19990515)85:10<2255::AID-CNCR22>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 3.Spiro RH. Salivary neoplasms, overview of a 35 year experience with 2,807 patients. Head Neck Surg. 1986;8:177–184. doi: 10.1002/hed.2890080309. [DOI] [PubMed] [Google Scholar]
- 4.Isacsson G, Shear M. Intraoral salivary gland tumors: a retrospective study of 201 cases. J Oral Pathol. 1983;12:57–62. doi: 10.1111/j.1600-0714.1983.tb00316.x. [DOI] [PubMed] [Google Scholar]
- 5.Eveson JW, Cawson RA. Salivary gland tumours. A review of 2410 cases with particular reference to histological types, site, age and sex distribution. J Pathol. 1985;146:51–58. doi: 10.1002/path.1711460106. [DOI] [PubMed] [Google Scholar]
- 6.Font RL, Smith SL, Bryan RG. Malignant epithelial tumors of the lacrimal gland: a clinicopathologic study of 21 cases. Arch Ophthalmol. 1998;116:613–616. doi: 10.1001/archopht.116.5.613. [DOI] [PubMed] [Google Scholar]
- 7.Chen JC, Gnepp DR, Bedrossian CW. Adenoid cystic carcinoma of the salivary glands: an immunohistochemical analysis. Oral Surg, Oral Med Oral Pathol. 1988;65:316–326. doi: 10.1016/0030-4220(88)90116-8. [DOI] [PubMed] [Google Scholar]
- 8.Prasad AR, Savera AT, Gown AM, Zarbo RJ. The myoepithelial immunophenotype in 135 benign and malignant salivary gland tumors other than pleomorphic adenoma. Arch Pathol Lab Med. 1999;123:801–806. doi: 10.5858/1999-123-0801-TMIIBA. [DOI] [PubMed] [Google Scholar]
- 9.Cheng J, Saku T, Okabe H, Furthmayr H. Basement membranes in adenoid cystic carcinoma. An immunohistochemical study. Cancer. 1992;69:2631–2640. doi: 10.1002/1097-0142(19920601)69:11<2631::AID-CNCR2820691103>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 10.Frierson HF, Jr, El-Naggar AK, Welsh JB, Sapinoso LM, Su AI, Cheng J, Saku T, Moskaluk CA, Hampton GM. Large scale molecular analysis identifies genes with altered expression in salivary adenoid cystic carcinoma. Am J Pathol. 2002;161:1315–1323. doi: 10.1016/S0002-9440(10)64408-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kimura S, Cheng J, Ida H, Hao N, Fujimori Y, Saku T. Perlecan (heparan sulfate proteoglycan) gene expression reflected in the characteristic histological architecture of salivary adenoid cystic carcinoma. Virchows Arch. 2000;437:122–128. doi: 10.1007/s004280000209. [DOI] [PubMed] [Google Scholar]
- 12.Szanto PA, Luna MA, Tortoledo ME, White RA. Histologic grading of adenoid cystic carcinoma of the salivary glands. Cancer. 1984;54:1062–1069. doi: 10.1002/1097-0142(19840915)54:6<1062::AID-CNCR2820540622>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 13.Greiner TC, Robinson RA, Maves MD. Adenoid cystic carcinoma clinicopathologic study with flow cytometric analysis. Am J Clin Pathol. 1989;92:711–720. doi: 10.1093/ajcp/92.6.711. [DOI] [PubMed] [Google Scholar]
- 14.Lloyd S, Yu JB, Wilson LD, Decker RH. Determinants and patterns of survival in adenoid cystic carcinoma of the head and neck, including an analysis of adjuvant radiation therapy. Am J Clin Oncol. 2011;34:76–81. doi: 10.1097/COC.0b013e3181d26d45. [DOI] [PubMed] [Google Scholar]
- 15.Rutherford S, Yu Y, Rumpel CA, Frierson HF, Jr., Moskaluk CA: Chromosome 6 deletion and candidate tumor suppressor genes in adenoid cystic carcinoma. Cancer Lett. 2006;236:309–17. [DOI] [PubMed]
- 16.Zarbo RJ. Salivary gland neoplasia: a review for the practicing pathologist. Mod Pathol. 2002;15:298–323. doi: 10.1038/modpathol.3880525. [DOI] [PubMed] [Google Scholar]
- 17.Yamamoto Y, Saka T, Makimoto K, Takahashi H. Histological changes during progression of adenoid cystic carcinoma. J Laryngol Otol. 1992;106:1016–1020. doi: 10.1017/S0022215100121656. [DOI] [PubMed] [Google Scholar]
- 18.Seethala RR, Hunt JL, Baloch ZW, Livolsi VA, Leon Barnes E. Adenoid cystic carcinoma with high-grade transformation: a report of 11 cases and a review of the literature. Am J Surg Pathol. 2007;31:1683–1694. doi: 10.1097/PAS.0b013e3180dc928c. [DOI] [PubMed] [Google Scholar]
- 19.Cheuk W, Chan JKC, Ngan RKC. Dedifferentiation in adenoid cystic carcinoma of salivary gland: an uncommon complication associated with an accelerated clinical course. Am J Surg Pathol. 1999;23:465–472. doi: 10.1097/00000478-199904000-00012. [DOI] [PubMed] [Google Scholar]
- 20.Nagao T, Gaffey TA, Serizawa H, Sugano I, Ishida Y, Yamazaki K, Tokashiki R, Yoshida T, Minato H, Kay PA, Lewis JE. Dedifferentiated adenoid cystic carcinoma: a clinicopathologic study of 6 cases. Mod Pathol. 2003;16:1265–1272. doi: 10.1097/01.MP.0000097366.88165.08. [DOI] [PubMed] [Google Scholar]
- 21.Fordice J, Kershaw C, el-Naggar A, Goepfert H. Adenoid cystic carcinoma of the head and neck. Predictors of morbidity and mortality. Arch Otolaryngol Head Neck Surg. 1999;125:149–152. doi: 10.1001/archotol.125.2.149. [DOI] [PubMed] [Google Scholar]
- 22.Mendenhall WM, Morris CG, Amdur RJ, Werning JW, Hinerman RW, Villaret DB. Radiotherapy alone or combined with surgery for adenoid cystic carcinoma of the head and neck. Head Neck. 2004;26:154–162. doi: 10.1002/hed.10380. [DOI] [PubMed] [Google Scholar]
- 23.Chen AM, Bucci MK, Weinberg V, Garcia J, Quivey JM, Schechter NR, Phillips TL, Fu KK, Eisele DW. Adenoid cystic carcinoma of the head and neck treated by surgery with or without postoperative radiation therapy: prognostic features of recurrence. Int J Radiat Oncol Biol Phys. 2006;66:152–159. doi: 10.1016/j.ijrobp.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 24.Barrett AW, Speight PM. Perineural invasion in adenoid cystic carcinoma of the salivary glands: anatomical influences on oncological management. Curr Cancer Ther Rev. 2011;7:78–82. doi: 10.2174/157339411794474119. [DOI] [Google Scholar]
- 25.Conley J, Dingman DL. Adenoid cystic carcinoma in the head and neck (cylindroma) Arch Otolaryngol. 1974;100:81–90. doi: 10.1001/archotol.1974.00780040087001. [DOI] [PubMed] [Google Scholar]
- 26.Spiro RH. Distant metastasis in adenoid cystic carcinoma of salivary origin. Am J Surg. 1997;174:495–498. doi: 10.1016/S0002-9610(97)00153-0. [DOI] [PubMed] [Google Scholar]
- 27.Nordkvist A, Mark J, Gustavsson H, Bang G, Stenman G. Nonrandom chromosome rearrangements in adenoid cystic carcinoma of the salivary glands. Genes Chromosom Cancer. 1994;10:115–121. doi: 10.1002/gcc.2870100206. [DOI] [PubMed] [Google Scholar]
- 28.Jin Y, Mertens F, Limon J, Mandahl N, Wennerberg J, Dictor M, Heim S, Mitelman F. Characteristic karyotypic features in lacrimal and salivary gland carcinomas. Br J Cancer. 1994;70:42–47. doi: 10.1038/bjc.1994.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jin C, Martins C, Jin Y, Wiegant J, Wennerberg J, Dictor M, Gisselsson D, Strombeck B, Fonseca I, Mitelman F, Tanke HJ, Hoglund M, Mertens F. Characterization of chromosome aberrations in salivary gland tumors by FISH, including multicolor COBRA-FISH. Genes Chromosom Cancer. 2001;30:161–167. doi: 10.1002/1098-2264(2000)9999:9999<::AID-GCC1077>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 30.Higashi K, Jin Y, Johansson M, Heim S, Mandahl N, Biorklund A, Wennerberg J, Hambraeus G, Johansson L, Mitelman F. Rearrangement of 9p13 as the primary chromosomal aberration in adenoid cystic carcinoma of the respiratory tract. Genes Chromosom Cancer. 1991;3:21–23. doi: 10.1002/gcc.2870030105. [DOI] [PubMed] [Google Scholar]
- 31.Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci USA. 2009;106:18740–18744. doi: 10.1073/pnas.0909114106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mitani Y, Li J, Rao PH, Zhao YJ, Bell D, Lippman SM, Weber RS, Caulin C, El-Naggar AK. Comprehensive analysis of the MYB-NFIB gene fusion in salivary adenoid cystic carcinoma: Incidence, variability, and clinicopathologic significance. Clin Cancer Res. 2010;16:4722–4731. doi: 10.1158/1078-0432.CCR-10-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitani Y, Rao PH, Futreal PA, Roberts DB, Stephens PJ, Zhao YJ, Zhang L, Mitani M, Weber RS, Lippman SM, Caulin C, El-Naggar AK. Novel chromosomal rearrangements and break points at the t(6;9) in salivary adenoid cystic carcinoma: association with MYB-NFIB chimeric fusion, MYB expression, and clinical outcome. Clin Cancer Res. 2011;17:7003–7014. doi: 10.1158/1078-0432.CCR-11-1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.West RB, Kong C, Clarke N, Gilks T, Lipsick JS, Cao H, Kwok S, Montgomery KD, Varma S, Le QT. MYB expression and translocation in adenoid cystic carcinomas and other salivary gland tumors with clinicopathologic correlation. Am J Surg Pathol. 2011;35:92–99. doi: 10.1097/PAS.0b013e3182002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moskaluk CA, Baras AS, Mancuso SA, Fan H, Davidson RJ, Dirks DC, Golden WL, Frierson HF., Jr Development and characterization of xenograft model systems for adenoid cystic carcinoma. Lab Invest. 2011;91:1480–1490. doi: 10.1038/labinvest.2011.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brill LB, 2nd, Kanner WA, Fehr A, Andren Y, Moskaluk CA, Loning T, Stenman G, Frierson HF, Jr.: Analysis of MYB expression and MYB-NFIB gene fusions in adenoid cystic carcinoma and other salivary neoplasms. Mod Pathol. 2011;24:1169–76. [DOI] [PubMed]
- 37.Bell D, Roberts D, Karpowicz M, Hanna EY, Weber RS, El-Naggar AK. Clinical significance of Myb protein and downstream target genes in salivary adenoid cystic carcinoma. Cancer Biol Ther. 2011;12:569–573. doi: 10.4161/cbt.12.7.17008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bernheim A, Toujani S, Saulnier P, Robert T, Casiraghi O, Validire P, Temam S, Menard P, Dessen P, Fouret P. High-resolution array comparative genomic hybridization analysis of human bronchial and salivary adenoid cystic carcinoma. Lab Invest. 2008;88:464–473. doi: 10.1038/labinvest.2008.18. [DOI] [PubMed] [Google Scholar]
- 39.Rao PH, Roberts D, Zhao YJ, Bell D, Harris CP, Weber RS, El-Naggar AK. Deletion of 1p32-p36 is the most frequent genetic change and poor prognostic marker in adenoid cystic carcinoma of the salivary glands. Clin Cancer Res: Off J Am Assoc Cancer Res. 2008;14:5181–5187. doi: 10.1158/1078-0432.CCR-08-0158. [DOI] [PubMed] [Google Scholar]
- 40.Vekony H, Ylstra B, Wilting SM, Meijer GA, van de Wiel MA, Leemans CR, van der Waal I, Bloemena E. DNA copy number gains at loci of growth factors and their receptors in salivary gland adenoid cystic carcinoma. Clin Cancer Res. 2007;13:3133–3139. doi: 10.1158/1078-0432.CCR-06-2555. [DOI] [PubMed] [Google Scholar]
- 41.El-Rifai W, Rutherford S, Knuutila S, Frierson HF, Jr, Moskaluk CA. Novel DNA copy number losses in chromosome 12q12–q13 in adenoid cystic carcinoma. Neoplasia. 2001;3:173–178. doi: 10.1038/sj.neo.7900158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu Y, Baras AS, Shirasuna K, Frierson HF, Jr, Moskaluk CA. Concurrent loss of heterozygosity and copy number analysis in adenoid cystic carcinoma by SNP genotyping arrays. Lab Invest. 2007;87:430–439. doi: 10.1038/labinvest.3700536. [DOI] [PubMed] [Google Scholar]
- 43.Freier K, Flechtenmacher C, Walch A, Ohl S, Devens F, Burke B, Hassfeld S, Lichter P, Joos S, Hofele C. Copy number gains on 22q13 in adenoid cystic carcinoma of the salivary gland revealed by comparative genomic hybridization and tissue microarray analysis. Cancer Genet Cytogenet. 2005;159:89–95. doi: 10.1016/j.cancergencyto.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 44.Moskaluk CA, Frierson HF, Jr., El-Naggar AK, Futreal PA: C-kit gene mutations in adenoid cystic carcinoma are rare, Mod Pathol 2010, 23:905–906; Author reply 906–907. [DOI] [PubMed]
- 45.Aubry MC, Heinrich MC, Molina J, Lewis JE, Yang P, Cassivi SD, Corless CL. Primary adenoid cystic carcinoma of the lung: absence of KIT mutations. Cancer. 2007;110:2507–2510. doi: 10.1002/cncr.23075. [DOI] [PubMed] [Google Scholar]
- 46.Holst VA, Marshall CE, Moskaluk CA, Frierson HF., Jr KIT protein expression and analysis of c-kit gene mutation in adenoid cystic carcinoma. Mod Pathol. 1999;12:956–960. [PubMed] [Google Scholar]
- 47.Jeng YM, Lin CY, Hsu HC. Expression of the c-kit protein is associated with certain subtypes of salivary gland carcinoma. Cancer Lett. 2000;154:107–111. doi: 10.1016/S0304-3835(00)00387-6. [DOI] [PubMed] [Google Scholar]
- 48.Lin CH, Yen RF, Jeng YM, Tzen CY, Hsu C, Hong RL. Unexpected rapid progression of metastatic adenoid cystic carcinoma during treatment with imatinib mesylate. Head Neck. 2005;27:1022–1027. doi: 10.1002/hed.20274. [DOI] [PubMed] [Google Scholar]
- 49.Sorensen KB, Godballe C, de Stricker K, Krogdahl A. Parotid carcinoma: expression of kit protein and epidermal growth factor receptor. J Oral Pathol Med. 2006;35:286–291. doi: 10.1111/j.1600-0714.2006.00421.x. [DOI] [PubMed] [Google Scholar]
- 50.Vila L, Liu H, Al-Quran SZ, Coco DP, Dong HJ, Liu C: Identification of c-kit gene mutations in primary adenoid cystic carcinoma of the salivary gland. Mod Pathol. 2009;22:1296–302. [DOI] [PMC free article] [PubMed]
- 51.Saka T, Yamamoto Y, Takahashi H. Comparative cytofluorometric DNA analysis of pleomorphic adenoma and adenoid cystic carcinoma of the salivary glands. Virchows Arch B Cell Pathol Incl Mol Pathol. 1991;61:255–261. doi: 10.1007/BF02890426. [DOI] [PubMed] [Google Scholar]
- 52.Franzen G, Nordgard S, Boysen M, Larsen PL, Halvorsen TB, Clausen OP. DNA content in adenoid cystic carcinomas. Head Neck. 1995;17:49–55. doi: 10.1002/hed.2880170111. [DOI] [PubMed] [Google Scholar]
- 53.Seethala RR, Cieply K, Barnes EL, Dacic S. Progressive genetic alterations of adenoid cystic carcinoma with high-grade transformation. Arch Pathol Lab Med. 2011;135:123–130. doi: 10.5858/2010-0048-OAR.1. [DOI] [PubMed] [Google Scholar]
- 54.Londono-Vallejo JA. Telomere length heterogeneity and chromosome instability. Cancer Lett. 2004;212:135–144. doi: 10.1016/j.canlet.2004.05.008. [DOI] [PubMed] [Google Scholar]