

Abstract
Alveolar soft part sarcoma (ASPS) constitutes a rare soft tissue malignant neoplasm comprising less than 1 % of all soft tissue sarcomas. ASPS demonstrates a strong predilection for adolescents and young adults, with a female predominance reported. The head and neck region is the most commonly affected region in pediatric patients with the tongue and orbit affected most commonly. Herein we present the clinical, radiographic, histopathologic, immunohistochemical and molecular features of two examples of ASPS affecting the oral cavity of 4 and 13 year-old boys, along with a focused review of the literature on intraoral ASPS in pediatric patients.
Keywords: Alveolar soft sarcoma, Children, Oral cavity, Tongue, Pathology, Immunohistochemistry, Molecular, ASPSCR1/TFE3 fusion, Management
Introduction
Alveolar soft part sarcoma (ASPS) constitutes a rare malignant soft tissue neoplasm comprising 0.5–0.9 % of all soft tissue sarcomas [1]. Christopherson et al. [2] were first to coin the term and although ASPS may occur at any age, it demonstrates a strong predilection for adolescents and young adults [3] between 15 and 35 years of age [1]. ASPS arising in infants and children younger than 15 years is considered rare [1, 4]. Although most ASPS occur in the extremities, tumors occurring in childhood and infancy have a predilection for the head and neck region, especially the orbit and tongue [1, 5–8].
Herein we present the clinical, radiographic, histopathologic, immunohistochemical and molecular features of ASPS occurring in the tongue of a 4 year old boy and the buccal mucosa of a 13 year old boy, along with a literature review with emphasis on pediatric intraoral ASPS cases.
Materials and Methods
Immunohistochemical evaluation was performed using antibodies against pan cytokeratin (Dako; monoclonal; AE1/AE3), desmin (Ventana; monoclonal; DE-R-11 and/or Dako, D33), S100 protein (Ventana; polyclonal), HMB45 (Dako, monoclonal, HMB-45), Melan-A (Dako, monoclonal, A103), neuron specific enolase (NSE) (Ventana; BBS/NC/V1-H14), synaptophysin (Ventana, polyclonal), chromogranin (Ventana, monoclonal, LK2H10), CD68 (Ventana; KP1), MYOD1 (Dako; monoclonal, DAS), vimentin (Ventana, monoclonal, V9), smooth muscle actin (Leica, monoclonal, asm-1), smooth muscle myosin heavy chain (Dako, monoclonal, SMMS-1), TFE3 transcription factor (Cell Marque; monoclonal; MRQ-37). All immunohistochemical stains were performed on a Ventana NexES automated immunohistochemistry system (Ventana Medical Systems, Tucson, AZ) according to the manufacturers’ instructions.
To detect a ASPSCR1/TFE3 fusion, reverse-transcription polymerase chain reaction (RT-PCR) was performed on total RNA extracted from paraffin-embedded tumor tissue (RecoverAllTM Total Nucleic Acid Isolation Kit; Ambion Diagnostics). The SuperScript III First-Strand Synthesis System by Invitrogen was utilized followed by end point PCR amplification, using previously reported primers [9] and appropriate controls. The thermocycling amplification conditions consisted of 1 cycle at 95 °C for 10 min, 54 °C for 1 min, 72 °C for 1 min, 15 cycles at 95 °C for 30 s, 54 °C for 1 min, 72 °C for 1 min, 21 cycles at 95 °C for 30 s, 54 °C for 1 min, 72 °C for 2 min and finally 1 cycle at 95 °C for 30 s, 54 °C for 1 min and 72 °C for 7 min. PCR products were run on a 2 % agarose gel to visualize the presence of a fusion product using ethidium bromide staining. Five millilitre of the final amplification product was sequenced with the corresponding primers in the forward and reverse directions, using the ABI PRISM Dye Terminator Cycle Sequencing Analyzer. The electropherograms were edited and analyzed by utilizing the Chromos software. DNA sequence breakpoint analysis was performed using BLAST sequence similarity searches (National Center for Biotechnology Information database).
A review of the English literature based on a MEDLINE search from 1966 to 2012 was performed and all cases of ASPS involving the oral cavity specifically were included in the review (Table 1). However, cases were excluded if the lesion arose primarily in the soft tissues of the upper neck, or involved the sinonasal tract at the time of presentation. Clinical series were selected if critical information about oral cavity tumors was included, but excluded if the information was too generalized. Duplicate reports were excluded from further evaluation. No foreign language articles were included.
Table 1.
Reported pediatric cases of oral cavity ASPS
| No | Author | Years | Age (years) | Sex | Location | Tumor size (cm) | Follow-up (years) | Clinical course |
|---|---|---|---|---|---|---|---|---|
| 1 | Christopherson [2] | 1952 | 12 | F | Base of tongue | 5 | 8 | NED |
| 2 | Caldwell [16] | 1956 | 4 | F | Base of tongue | 4 | 1 | DOD/M |
| 3 | Vakil [17] | 1963 | 10 | F | Posterior tongue | N/R | 0.5 | DOD/R |
| 4 | Buchanan [18] | 1975 | 17 | F | Infratemporal fossa | 6 | 4 | DOD/M |
| 5 | Olson [19] | 1976 | 5 | F | Tongue, NOS | 3 | 15 | NED |
| 6 | Ramanjaneyulu [20] | 1977 | 12 | F | Base of tongue | 4 | 3 | NED |
| 7 | Spector [21] | 1979 | 17 | F | Base of tongue | 4.8 | 3 | DOD/M |
| 8 | Master [22] | 1979 | 4 | F | Tongue, NOS | N/R | 15 | DOD/M |
| 9 | King [23] | 1983 | 5 | F | Tongue, NOS | N/R | 7 | NED |
| 10 | Chaudhry [24] | 1984 | 3.3 | F | Dorsal tongue | 2 | N/R | NP |
| 11 | Komori [25] | 1984 | 11 | F | Base of tongue | 2.5 | 5 | NED |
| 12 | Sawyer [26] | 1985 | 16 | F | Tongue, NOS | N/R | 0.6 | NED |
| 13 | Simmons [5] | 1989 | 1.6 | F | Dorsal tongue | 1.5 | 2.8 | NED |
| 14 | Cetik [27] | 1989 | 13 | F | Tongue, NOS | 1.5 | 1.5 | NED |
| 15 | Matsuno [28] | 1990 | 6 | M | Tongue, NOS | 3 | 2.3 | AWD/M |
| 16 | Marker [29] | 1995 | 17 | F | Mandibular angle | 4.5 | 19 | NED |
| 17 | Hunter [7] | 1998 | 3 | F | Tongue, NOS | N/R | 4 | N/R |
| 18 | Bentley [30] | 1999 | 5 | F | Base of tongue | 2.2 | 2 | NED |
| 19 | Castle [31] | 1999 | 3 | M | Tongue, NOS | 0.8 | 4 | NED |
| 20 | Yoshida [32] | 2000 | 2 | F | Dorsal tongue | 2 | 7.2 | NED |
| 21 | Noussios [33] | 2002 | 3 | M | Tongue, NOS | 3.3 | 3.5 | NED |
| 22 | Richards [34] | 2003 | 16 | M | Mandibular angle | N/R | 1.3 | NED |
| 23 | Inci [35] | 2004 | 17 | NP | Tongue, NOS | N/R | 3 | NED |
| 24 | Fanburg-Smith [8] | 2004 | 3 | F | Dorsal tongue | 2.5 | 11 | NED |
| 25 | Fanburg-Smith [8] | 2004 | 3 | F | Lateral border of tongue | N/R | 10 | NED |
| 26 | Fanburg-Smith [8] | 2004 | 5 | M | Base of tongue | N/R | 11 | NED |
| 27 | Fanburg-Smith [8] | 2004 | 5 | F | Midportion of tongue | 1 | 22 | NED |
| 28 | Fanburg-Smith [8] | 2004 | 5 | F | Dorsal tongue | N/R | N/R | N/R |
| 29 | Fanburg-Smith [8] | 2004 | 6 | M | Tongue, NOS | N/R | N/R | N/R |
| 30 | Fanburg-Smith [8] | 2004 | 6 | M | Dorsal tongue | 2.5 | 28 | NED |
| 31 | Fanburg-Smith [8] | 2004 | 7 | M | Ventral tongue | 1.3 | 27 | NED |
| 32 | Fanburg-Smith [8] | 2004 | 7 | M | Dorsal tongue | 2.5 | 16 | NED |
| 33 | Fanburg-Smith [8] | 2004 | 17 | M | Posterior tongue | 2.5 | 25 | NED |
| 34 | do Nascimento [6] | 2005 | 13 | F | Tongue, NOS | 3 | 5 | NED |
| 35 | Kim [36] | 2005 | 16 | M | Tongue, NOS | N/R | 0.5 | NED |
| 36 | Kim [36] | 2005 | 4 | F | Tongue, NOS | N/R | 0.7 | NED |
| 37 | Correia-Silva [37] | 2006 | 17 | F | Tongue, NOS | 2 | 1 | NED |
| 38 | Rodriguez-Velasco [4] | 2008 | 2 | F | Tongue, NOS | 1.5 | 2.8 | NED |
| 39 | Kumar [38] | 2010 | 7 | M | Tongue, NOS | 2.5 | 0.9 | NED |
| 40 | Present case #1 | 2012 | 4 | M | Dorsal tongue | 2 | 0.6 | NED |
| 41 | Present case #2 | 2012 | 13 | M | Buccal mucosa | 3.2 | 1.0 | NED |
N/R not reported, NED no evidence of disease, DOD dead of disease, R recurrent, M metastatic, AWD alive with disease
Case Reports
Case 1
A previously healthy 4 year-old boy was referred for evaluation of an asymptomatic mass of the dorsal tongue. The lesion was discovered serendipitously by the child’s parents. The patient’s medical history was unremarkable. Clinical examination revealed a firm, raised, well circumscribed, nontender, bluish mass located in the left dorsal side of the tongue, at the junction of the anterior two thirds and posterior third. The tumor measured approximately 2 cm in greatest dimension. No lingual motor disturbances were present. There were no enlarged lymph nodes in the neck, supraclavicular or axillary areas.
With a clinical differential diagnosis of a benign mesenchymal neoplasm, such as hemangioma, lymphangioma and rhabdomyoma, the patient underwent computed tomography which disclosed a 1.6 cm × 1.4 cm × 1.2 cm well-defined hyperdense lesion in the left superior aspect of the oral tongue, elevating the superior lingual edge (Fig. 1a, b). An excisional biopsy was performed under general anesthesia.
Fig. 1.
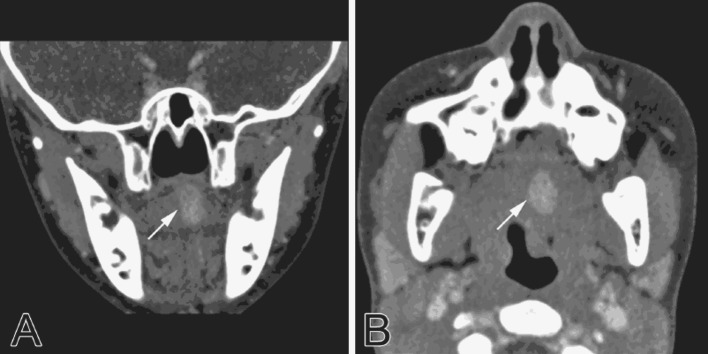
Computed tomography findings. a Coronal view depicted well delineated hyperdense nodular mass (arrow) on the left aspect of the tongue dorsum. b Axial view of the mass (arrow)
Histopathologic examination at low magnification revealed an infiltrating lesion with solid nodular growth pattern extending and partially infiltrating the subjacent skeletal muscle (Fig. 2, left). At medium magnification, the neoplastic cells were arranged in uniform sheets separated by thin septa of vascularized fibrous connective tissue, with focal organoid nested architecture, but no alveolar pattern. The tumor cells were round or polygonal and were characterized by one or two ovoid vesicular nuclei with numerous prominent nucleoli and low mitotic rate (4 per 10 HPFs). The cytoplasm varied from intensely eosinophilic to finely granular (Fig. 3b) or clear (Fig. 3c). Crystalloid PAS-positive, diastase resistant cytoplasmic inclusions were present focally.
Fig. 2.

Low power appearance of the tumor. Left note the lobular architecture of the proliferation of neoplastic cells. The tumor cell sheets are separated by thick fibrous septae. Right note lobules of proliferating neoplastic cells infiltrating the skeletal muscle supporting the buccal mucosa and separated by delicate fibrous septae
Fig. 3.

Histologic features of the tumor. a Low power appearance showing, in this portion of the tumor, thick fibrous septae separating sheets of neoplastic cells with no appreciable “alveolar” pattern. b Tumor cells with small, ovoid nuclei and granular cytoplasm, simulating granular cell tumor. c Tumor cells with large nuclei with open chromatin and prominent nucleoli, ample clear to faintly eosinophilic cytoplasm, and intracytoplasmic granular eosinophilic material. d PAS stain after diastase digestion showing positivity of the cytoplasmic granules
The neoplastic cells demonstrated diffuse intense nuclear positivity for TFE3 and diffuse moderate to intense cytoplasmic positivity for NSE. Focal CD68 cytoplasmic staining was also observed (Fig. 4d). There was no staining for cytokeratin AE1/AE3, desmin, or S-100 protein.
Fig. 4.

Immunohistochemical findings. a TFE-3 showing nuclear staining. b Diffuse cytoplasmic staining with NSE. cCytoplasmic staining with MYOD1. d Focal cytoplasmic staining with CD68
The TFE3 nuclear staining suggested translocation involving the TFE3 gene. RT-PCR demonstrated the presence of an ASPSCR1/TFE3 fusion transcript, that commonly results from the t(X;17) translocation, and is characteristic of alveolar soft part sarcoma (Fig. 5).
Fig. 5.

RT-PCR findings: identification of the ASPSCR1(ASPL)/TFE3 fusion transcript product in the tumor tissue
The initial biopsy material featured involvement of the superior, medial and lateral margins by neoplastic nests thus necessitating re-excision. No residual neoplasm was identified in the subsequent wider local excision and the patient’s postoperative course was uneventful. Postoperative imaging studies, including computed tomography of the thorax, abdomen, and pelvis, and MRI of the brain, showed no metastatic lesions. The patient has remained in good health and is disease-free 7 months after the initial diagnosis.
Case 2
A 13 year-old boy noted a rapidly enlarging mass of the left posterior buccal mucosa, at the occlusal line, about 2 months after a well-child examination. The patient stated that the lesion “popped” and bled. Computed tomography (CT) scans showed a mass medial to the angle of the left mandible measuring 3.2 cm in great dimension. No abnormalities were found on CT of the abdomen and pelvis. A 1.5-cm level IIA lymph node was palpable on physical examination. A biopsy was performed.
Microscopic examination revealed a lobulated soft tissue tumor below an intact oral squamous mucosa (Fig. 2, right) composed of large, round to polygonal cells with eosinophilic to clear cytoplasm, mild nuclear pleomorphism, prominent nucleoli and 5 mitoses per 20 HPFs. The tumor cells were arranged in nests, sheets and rare acinar-like structures (Fig. 3a), separated by a delicate meshwork of capillaries. There was no necrosis. Eosinophilic granular material was present in the cytoplasm of some of the tumor cells and crystals were highlighted with PAS, with (Fig. 3d) and without diastase, and phosphotungstic acid hematoxylin (PTAH). The tumor involved minor salivary glands and infiltrated skeletal muscle but was not connected to the surface epithelium. There was also perineural and angiolymphatic invasion. The bone was not involved, but the initial biopsy had positive margins.
Immunohistochemical stains revealed strong diffuse nuclear staining for TFE3 transcription factor (Fig. 4a), diffuse cytoplasmic NSE (Fig. 4b) and MYOD1 cytoplasmic positivity (Fig. 4c), focal weak or patchy desmin positivity, selective cytoplasmic staining of various intensity with CD68 and no staining for cytokeratin AE1/AE3, vimentin, smooth muscle actin, smooth muscle myosin heavy chain, melanocytic markers S-100, HMB45, Melan-A, and neuroendocrine markers synaptophysin and chromogranin.
RT-PCR demonstrated the presence of an ASPSCR1/TFE3 fusion transcript, quite characteristic of ASPS.
Based on the initial biopsy showing positive margins, a wide local re-excision accompanied by an ipsilateral modified radical neck dissection was performed, with residual tumor present, but negative lymph nodes. The patient received adjuvant chemotherapy consisting of ifosfamide and doxorubicin hydrochloride (COG ARST 0332 trial). The patient has remained in good health and is disease-free 12 months after the initial diagnosis.
Discussion
ASPS is an uncommon malignant soft tissue neoplasm, most commonly involving the lower extremities [1]. Lieberman et al. [10] reviewed the clinical and histopathologic features of 102 cases of ASPS. In their study the median age at diagnosis was 22 years (range 2–71 years) and the female to male ratio 1.5:1, with a higher F:M ratio in childhood and adolescence. ASPS arises in association with skeletal muscle or musculofascial planes, a fact that explains the strong predilection of this tumor for the thighs, buttocks and abdominal or chest walls [10]. The neoplasm is often indolent and may therefore be present for years before medical attention is sought [11]. Even then treatment may be delayed as the highly vascularized mass of ASPS is clinically misinterpreted as a benign or developmental lesion such as a hemangioma, lymphangioma or arteriovenous malformation [4].
Despite being a slow-growing tumor with an asymptomatic course, ASPS demonstrates a disproportionate frequency of distant metastases even at the early stages of disease [12]. In fact, data reviewed by Enzinger and Weiss [13] revealed high metastatic potential of the tumor, with 13/36 patients (36 %) presenting early metastases before the detection of the primary. The frequency of metastasis at presentation increases with the patient’s age at diagnosis [10]. Metastases occur most commonly in the lungs, followed by bones and the brain [11, 13]. ASPS has the strongest predilection for brain metastasis of all sarcomas [13].
The 5-year-survival rate of ASPS in children, adolescents and adults younger than 25 years is reportedly 83 % [11]. However, because of the propensity to late metastasis, survival rates decline steadily. In the large series of adults and children reported by Lieberman et al., [10] patients without metastases at presentation had survival rates of 77 % at 2 years, 60 % at 5 years, 38 % at 10 years and 15 % at 20 years. In that study, the local recurrence rate after adequate excision of the primary tumor was 16.7 % [10]. Tumor size and age at diagnosis were identified as significant prognostic factors, with primary tumors measuring less than 5 cm and those occurring in patients in the first and second decades of life demonstrating a more favorable clinical course [10, 11].
ASPS arising in the oral cavity are exceedingly rare in the general population [8]. Data from the files of the Armed Forces Institute of Pathology (AFIP) between 1970 and 2003 showed that only 13 out of 266 cases of ASPS (approximately 5 %) occurred in the tongue [4, 8]. However, in infants and children, 25 % of head and neck ASPS involve the tongue [14], this frequency increasing to 70 % when the orbits are excluded [15]. Table 1 summarizes all reported cases of intraoral ASPS in the pediatric population (0–18 years) [2, 4–8, 16–38], totaling forty-one cases, including the present series. There were 26 females and 14 males affected, with a median age at diagnosis of 8.4 years, ranging from 1.6 to 17 years. In one case the gender of the patient was not provided [35]. The most common intraoral location was the tongue accounting for 90.2 % (37/41) of the cases, followed by the mandibular angle (4.8 %, 2/40), the buccal mucosa (2.4 %, 1/41) and infratemporal fossa (2.4 %, 1/40). Among the lingual cases of ASPS, the dorsal surface and the base of the tongue were affected in 21.6 % (8/37) and 18.9 % (7/37) of the cases, respectively. In 17/37 cases, the specific location of the tumor within the tongue was not provided. The tumor size was reported in 28 cases and ranged from 0.8 to 6 cm (mean, 2.7 cm). The median period of follow-up after complete surgical treatment with or without supplementary chemotherapy and/or radiation therapy was 7.2 years (range 0.5–28 years). The majority of patients (31/41, 76 %) were alive with no evidence of disease during the provided follow-up period. One patient was alive with metastases 2.3 years after the initial diagnosis, and 5/41 (12.2 %) had died of disseminated ASPS with distant pulmonary, cerebral or skeletal metastases or from a recurrence of the primary tumor. In 4/41 patients, information on the clinical course was unavailable.
Compared to ASPS of the extremities, oral ASPS demonstrates distinct clinicopathologic characteristics [4, 8]. First, the median age of patients with extremity ASPS is around 20 years, while oral ASPS affects a younger population, with a reported median age of 5 years [8]. Secondly, the reported cases of oral ASPS show a higher female predominance than ASPS of the extremities [4, 7, 8]. The size of the tumors affecting the tongue is smaller (median, 2.5 cm) than that of ASPS involving other parts of the body [4]. The smaller size of the tumors and shorter interval to diagnosis may account for the overall better prognosis of oral ASPS occurring in children and adolescents [4, 8].
Histologically, ASPS is characterized by aggregates of large polygonal or round mesenchymal cells with abundant eosinophilic, granular or clear cytoplasm [1–5, 7–9]. The cells contain one or more vesicular nuclei with several prominent nucleoli, mild pleomorphism, relatively few mitoses and rare atypical mitoses. The neoplastic population is characterized by multinodular growth and mainly exhibits an organoid, nested architecture, with neoplastic cells separated by multiple variably thick septae of vascularized dense connective tissue. The loss of intercellular cohesion, with neoplastic cells adhering only to the fibrovascular septae, results in a “pseudo”-alveolar pattern [1–4, 6–8, 10, 12]. However, this alveolar architecture is not encountered in all cases [7, 8, 23, 28, 31]. As in one of the case presented herein, a solid, “non-alveolar” pattern was present, with the cells arranged in compact sheets and cords without the characteristic dyshesion [8]. Based on the observation that ASPS tumors in very young patients (≤3 years old) exhibit a solid morphology while lesions in older patients (>5 years old) acquire an organoid pattern, Fanburg-Smith et al. [8] have suggested that the neoplasm’s architecture may be an age-related characteristic. Specifically, they proposed that the “immature” neoplastic cells form solid nests that become progressively dyshesive. A consistent finding in ASPS of the tongue is vascular invasion which has not been correlated with distant metastases or worse prognosis [8].
ASPS are consistently negative for vimentin, cytokeratins, and epithelial membrane antigen, neuroendocrine markers (synaptophysin and chromogranin), and melanocytic markers. Desmin, S100 protein and NSE stains are occasionally positive. There may be cytoplasmic but not nuclear reaction with MYOD1.
The pathognomonic histologic feature of ASPS, first described by Masson [39], is the presence of granules and rod-like or rhomboid-shaped crystalline inclusions within the cytoplasm of the tumor cells. These crystalline inclusions are found in 25–100 % of cases [8, 39], both in primary and metastatic ASPS. PAS positive diastase resistant granules may represent precursors to the rod shaped crystals. The cell surface and cytoplasm in the region of these crystals stains for MCT1 and CD147 [1, 14, 15]. MCT1, the monocarboxylate transporter 1, is found predominantly in cardiac and skeletal muscle, and catalyzes the rapid transport of monocarboxylates from the rough endoplasmic reticulum across plasma membranes. CD147 functions, in part, as its chaperone protein [1, 40].
The histogenesis of ASPS remains obscure [1, 5, 8]. Historic designations of this neoplasm as malignant granular cell myoblastoma, malignant granular cell tumor and malignant nonchromaffin paraganglioma [41] reflect some of the proposed histogenetic derivations from neural or Schwann cell, paraganglia, and skeletal muscle [12]. In an effort to demonstrate a myogenic derivation of ASPS, the characteristic crystalloids encountered in ASPS have been interpreted as being composed of Z-band muscle substances [42].
Recently, based on the fact that ASPS show t(X;17) (p11;q25) involving the TFE3 gene, commercially available antibodies against the internal region of the TFE3 gene have been used to confirm the diagnosis of ASPS [43]. The antibody against TFE3 shows variably strong nuclear positivity in the majority of ASPS. However, TFE3 is not specific for this tumor, as it may stain the nuclei of granular cell tumor [44], paraganglioma, translocation related renal cell carcinomas and adrenocortical carcinoma [45, 46]. If there is doubt about the diagnosis based on clinical and histological features, RT-PCR for the ASPSCR1/TFE3 fusion transcript is a more specific method that can be performed on frozen or paraffin-embedded tissue [44, 47].
The der 17 t(X;17)(p11;q25)[48, 49] leads to rearrangement and fusion of ASPL and TFE3 genes. Two variants of ASPSCR1/TFE3 gene fusion have been reported. Both encode a chimeric protein consisting of the ASPL N-terminal region fused to TFE3 basic helix-loop-helix and leucine zipper DNA-binding domains. The expression of ASPSCR1/TFE3 chimeric protein is considered to cause transcriptional deregulation and has been implicated as a possible tumorigenic mechanism. Detection of t(X;17) or ASPL-TFE3 fusion transcripts are highly specific and sensitive markers for rendering the diagnosis of ASPS [44, 50]. A study comparing immunohistochemistry for TFE3 and CD147 to RT-PCR for ASPSCR1/TFE3 fusion transcript suggests that identification of the ASPSCR1/TFE3 fusion transcript is the most sensitive marker for diagnosing ASPS, surpassing the accuracy of immunohistochemical stain for TFE3 [44]. Of crucial importance to this method is the extraction of adequate RNA from the paraffin embedded samples. If this is not possible, fluorescence in situ hybridization (FISH) assay may be applied alternatively for the identification of TFE3 fusion gene [45, 51]. A similar but reciprocal t(X;17) (p11;q25) translocation and ASPSCR1/TFE3 fusion transcripts have been identified in a variant of renal cell carcinoma arising in pediatric and young adult patients [51, 52]. Specifically, in our patients, imaging evaluation failed to identify any kidney lesions.
Depending on the predominant architectural pattern of the tumor (typical alveolar or solid, sheet-like), the differential diagnosis of ASPS encompasses a variety of benign and malignant entities. Generally and traditionally, the differential diagnosis includes rhabdomyoma, granular cell tumor, PEComas, and, more remotely, clear cell sarcoma, renal cell carcinoma, extrarenal rhabdoid tumor, rhabdoid tumor of the kidney, hibernoma, other adipocytic tumors, and paraganglioma [3, 5, 14, 41]. Adult rhabdomyoma can be distinguished from ASPS by its large eosinophilic cells with small nuclei, the “spider-like” appearance of the cytoplasm [1, 5, 40] and its characteristic diffuse and strong cytoplasmic positivity for desmin and nuclear positivity for MYOD1 and myf4. In contrast, ASPS may exhibit variable cytoplasmic positivity for MYOD1 [53]. Both ASPS and granular cell tumor show granular eosinophilic cytoplasm and round to polygonal cells. However, while the nuclei of benign granular cell tumors are uniform, without atypia or prominent nucleoli, ASPS shows nuclear atypia and prominent nucleoli. Granular cell tumors are intensely and diffusely positive with S100 protein, while ASPS cells are usually S100 protein negative. PEComas are rare neoplasms characterized by a proliferation of epithelioid or spindle cells with a perivascular distribution and dual myomelanocytic differentiation, resulting in positivity for both melanocytic markers such as HMB45, Melan-A and microphthalmia transcription factor, and muscle markers including SMA, muscle-specific actin and calponin [54]. ASPS are consistently negative for melanocytic markers, which also helps their differentiation from clear cell sarcomas, which are positive for HMB45 as well as S100 protein [55].
The differential diagnosis from metastatic renal cell carcinoma is based on the absence of CD10, EMA, RCC, PAX2 and CA9 expression in ASPS. Extrarenal rhabdoid tumor and rhabdoid tumor of the kidney are malignant neoplasms with frequent polyphenotypic expression of vimentin, keratins, EMA, CD99, synaptophysin and NSE. Apart from the immunohistochemical differences separating extrarenal rhabdoid tumor and ASPS, identification of mutations and homozygous deletions of the SMARCB1 (INI1) gene or absence of nuclear staining for INI1 are commonly encountered in rhabdoid tumors, but absent in ASPS [53]. Sometimes ASPS may acquire clear cytoplasmic features and mimic lipomatous neoplasms including hibernoma, lipoblastoma or liposarcoma, but these are usually positive for S100 protein and show specific morphologic differences such as lipoblasts, distinguishing them from ASPS [1]. Paraganglioma shows a characteristic nested “zellballen” architecture [47], and demonstrates positivity for neuroendocrine markers, while S100 protein highlights the supporting sustentacular cells.
Surgical excision of the tumor with negative margins is considered the treatment of choice [5, 9, 18]. The benefit of adjuvant therapy in ASPS is controversial but radiotherapy and/or chemotherapy may be administered, and radiotherapy is particularly applied in cases of inadequate surgery [9]. According to Lieberman et al., radio- and chemotherapy had no statistically significant impact on the development of metastases [5]. ASPS prognosis is based mainly on clinical features such as location of the primary lesion, size of the tumor, and age of patient [1, 3, 5, 18]. ASPS can demonstrate an early metastasizing potential, sometimes prior to detection of the primary tumor, but can also metastasize decades after the initial diagnosis [1, 3–9, 15–20]. Hence, complete local excision, adjuvant therapy if clinically indicated, and long-term patient follow-up are advised for all cases of ASPS. Chemotherapeutic regimens used for the treatment of other soft tissue sarcomas generally lack efficacy in ASPS.
Treatments targeting the c-Met receptor (which is an ASPSCR1/TFE3 transcriptional target) with its downstream effectors, AKT and ERK (which are activated in ASPS) with tyrosine kinase receptor inhibitors, novel c-Met inhibitors, and AKT inhibitors, as well as anti-angiogenic drugs targeting the VEGF pathway have shown encouraging preliminary results.
Acknowledgments
The authors are indebted to Mr. Jonathan Henriksen (University of Minnesota) for his superb assistance with the illustrations.
References
- 1.Ordonez N, Ladanyi M. Alveolar soft part sarcoma. In: Fletcher C, Unni K, Mertens F, editors. World Health Organization classification of tumours, pathology and genetics, tumours of soft tissue and bone. Lyon: IARC Press; 2002. [Google Scholar]
- 2.Christopherson WM, Foote FW, Jr, Stewart FW. Alveolar soft-part sarcomas; structurally characteristic tumors of uncertain histogenesis. Cancer. 1952;5:100–111. doi: 10.1002/1097-0142(195201)5:1<100::AID-CNCR2820050112>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 3.Folpe AL, Deyrup AT. Alveolar soft-part sarcoma: a review and update. J Clin Pathol. 2006;59:1127–1132. doi: 10.1136/jcp.2005.031120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rodriguez-Velasco A, Ferman-Cano F, Cerecedo-Diaz F. Rare tumor of the tongue in a child: alveolar soft part sarcoma. Pediatr Dev Pathol. 2009;12:147–151. doi: 10.2350/07-07-0317.1. [DOI] [PubMed] [Google Scholar]
- 5.Simmons WB, Haggerty HS, Ngan B, Anonsen CK. Alveolar soft part sarcoma of the head and neck. A disease of children and young adults. Int J Pediatr Otorhinolaryngol. 1989;17:139–153. doi: 10.1016/0165-5876(89)90089-X. [DOI] [PubMed] [Google Scholar]
- 6.do Nascimento Souza KC, Faria PR, Costa IM, Duriguetto AF, Jr, Loyola AM. Oral alveolar soft-part sarcoma: review of literature and case report with immunohistochemistry study for prognostic markers. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:64–70. doi: 10.1016/j.tripleo.2004.07.002. [DOI] [PubMed] [Google Scholar]
- 7.Hunter BC, Devaney KO, Ferlito A, Rinaldo A. Alveolar soft part sarcoma of the head and neck region. Ann Otol Rhinol Laryngol. 1998;107:810–814. doi: 10.1177/000348949810700914. [DOI] [PubMed] [Google Scholar]
- 8.Fanburg-Smith JC, Miettinen M, Folpe AL, Weiss SW, Childers EL. Lingual alveolar soft part sarcoma; 14 cases: novel clinical and morphological observations. Histopathology. 2004;45:526–537. doi: 10.1111/j.1365-2559.2004.01966.x. [DOI] [PubMed] [Google Scholar]
- 9.Lazar AJ, Das P, Tuvin D, Korchin B, Zhu Q, Jin Z, Warneke CL, Zhang PS, Hernandez V, Lopez-Terrada D, Pisters PW, Pollock RE, Lev D. Angiogenesis-promoting gene patterns in alveolar soft part sarcoma. Clin Cancer Res. 2007;13:7314–7321. doi: 10.1158/1078-0432.CCR-07-0174. [DOI] [PubMed] [Google Scholar]
- 10.Lieberman PH, Brennan MF, Kimmel M, Erlandson RA, Garin-Chesa P, Flehinger BY. Alveolar soft-part sarcoma. A clinico-pathologic study of half a century. Cancer. 1989;63:1–13. doi: 10.1002/1097-0142(19890101)63:1<1::AID-CNCR2820630102>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 11.Kayton ML, Meyers P, Wexler LH, Gerald WL, Laquaglia MP. Clinical presentation, treatment, and outcome of alveolar soft part sarcoma in children, adolescents, and young adults. J Pediatr Surg. 2006;41:187–193. doi: 10.1016/j.jpedsurg.2005.10.023. [DOI] [PubMed] [Google Scholar]
- 12.Batsakis JG. Alveolar soft-part sarcoma. Ann Otol Rhinol Laryngol. 1988;97:328–329. doi: 10.1177/000348948809700325. [DOI] [PubMed] [Google Scholar]
- 13.Enzinger FM, Weiss SW. Soft tissue tumors. St. Louis: Mosby. Ix; 1983. p. 840. [Google Scholar]
- 14.Weiss SW. Alveolar soft part sarcoma: are we at the end or just the beginning of our quest? Am J Pathol. 2002;160:1197–1199. doi: 10.1016/S0002-9440(10)62545-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pappo AS, Parham DM, Cain A, Luo X, Bowman LC, Furman WL, Rao BN, Pratt CB. Alveolar soft part sarcoma in children and adolescents: clinical features and outcome of 11 patients. Med Pediatr Oncol. 1996;26:81–84. doi: 10.1002/(SICI)1096-911X(199602)26:2<81::AID-MPO2>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 16.Caldwell JB, Fadell EJ, Hughes KW. Alveolar soft-part sarcoma of the tongue; report of case. J Oral Surg. 1956;14:342–345. [PubMed] [Google Scholar]
- 17.Vakil VV, Sirsat MV. The natural history of alveolar soft-part sarcomas. Indian J Pathol Bacteriol. 1963;6:19–25. [PubMed] [Google Scholar]
- 18.Buchanan G. Two rare tumours involving the infratemporal fossa: alveolar soft part sarcoma and haemangiopericytoma. J Laryngol Otol. 1975;89:375–389. doi: 10.1017/S0022215100080506. [DOI] [PubMed] [Google Scholar]
- 19.Olson RA, Perkins KD. Alveolar soft-part sarcoma in the oral cavity: report of a case. J Oral Surg. 1976;34:73–76. [PubMed] [Google Scholar]
- 20.Ramanjaneyulu P. Residents page. Pathology quiz case 2. Arch Otolaryngol. 1977;103:438–440. doi: 10.1001/archotol.1977.00780240096019. [DOI] [Google Scholar]
- 21.Spector RA, Travis LW, Smith J. Alveolar soft part sarcoma of the head and neck. Laryngoscope. 1979;89:1301–1306. doi: 10.1002/lary.1979.89.8.1301. [DOI] [PubMed] [Google Scholar]
- 22.Master K, Berkmen YM. Pulmonary metastases 15 years after removal of an alveolar soft-part sarcoma of the tongue. Rev Interam Radiol. 1979;4:43–45. [PubMed] [Google Scholar]
- 23.King VV, Fee WE., Jr Alveolar soft part sarcoma of the tongue. Am J Otolaryngol. 1983;4:363–366. doi: 10.1016/S0196-0709(83)80023-4. [DOI] [PubMed] [Google Scholar]
- 24.Chaudhry AP, Lin CC, Lai S, Yamane G. Alveolar soft part sarcoma of the tongue in a female neonate. J Oral Med. 1984;39:2–7. [PubMed] [Google Scholar]
- 25.Komori A, Takeda Y, Kakiichi T. Alveolar soft-part sarcoma of the tongue. Report of a case with electron microscopic study. Oral Surg Oral Med Oral Pathol. 1984;57:532–539. doi: 10.1016/0030-4220(84)90313-X. [DOI] [PubMed] [Google Scholar]
- 26.Sawyer DR, Ajagbe HA, Abiose BO, Daramola JO. Alveolar soft-part sarcoma of the oral cavity. Report of a case. J Oral Med. 1985;40:139–141. [PubMed] [Google Scholar]
- 27.Cetik F, Ozsahinoglu C, Kivanc F, Secinti E. Alveolar soft part sarcoma of the tongue. J Laryngol Otol. 1989;103:952–954. doi: 10.1017/S002221510011059X. [DOI] [PubMed] [Google Scholar]
- 28.Matsuno Y, Mukai K, Itabashi M, Yamauchi Y, Hirota T, Nakajima T, Shimosato Y. Alveolar soft part sarcoma. A clinicopathologic and immunohistochemical study of 12 cases. Acta Pathol Jpn. 1990;40:199–205. doi: 10.1111/j.1440-1827.1990.tb03323.x. [DOI] [PubMed] [Google Scholar]
- 29.Marker P, Jensen ML, Siemssen SJ. Alveolar soft-part sarcoma of the oral cavity: report of a case and review of the literature. J Oral Maxillofac Surg. 1995;53:1203–1208. doi: 10.1016/0278-2391(95)90636-3. [DOI] [PubMed] [Google Scholar]
- 30.Bentley RP, Wake MJ, Raafat F. Alveolar soft part sarcoma of the tongue. Br J Oral Maxillofac Surg. 1999;37:451–454. doi: 10.1054/bjom.1999.0116. [DOI] [PubMed] [Google Scholar]
- 31.Castle JT, Goode RK. Alveolar soft part sarcoma of the tongue: report of an unusual pattern in a child. Ann Diagn Pathol. 1999;3:315–317. doi: 10.1016/S1092-9134(99)80029-X. [DOI] [PubMed] [Google Scholar]
- 32.Yoshida K, Kurauchi J, Shirasawa H, Kosugi I. Alveolar soft part sarcoma of the tongue. Report of a case. Int J Oral Maxillofac Surg. 2000;29:370–372. doi: 10.1016/S0901-5027(00)80055-7. [DOI] [PubMed] [Google Scholar]
- 33.Noussios G, Chouridis P, Petropoulos I, Karagiannidis K, Kontzoglou G. Alveolar soft part sarcoma of the tongue in a 3-year-old boy: a case report. J Med Case Reports. 2010;4:130. doi: 10.1186/1752-1947-4-130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richards SV, Welch DC, Burkey BB, Bayles SW. Alveolar soft part sarcoma of the mandible. Otolaryngol Head Neck Surg. 2003;128:148–150. doi: 10.1067/mhn.2003.17. [DOI] [PubMed] [Google Scholar]
- 35.Inci E, Korkut N, Erem M, Kalekoglu N. Alveolar soft tissue sarcoma. Hno. 2004;52:145–149. doi: 10.1007/s00106-003-0872-4. [DOI] [PubMed] [Google Scholar]
- 36.Kim HS, Lee HK, Weon YC, Kim HJ. Alveolar soft-part sarcoma of the head and neck: clinical and imaging features in five cases. AJNR Am J Neuroradiol. 2005;26:1331–1335. [PMC free article] [PubMed] [Google Scholar]
- 37.Correia-Silva JF, Duarte ECB, Lacerda JCT, De Sousa SCOM, Mesquita RA, Gomez RS. Alveolar soft part sarcoma of the tongue. Oral Oncol Extra. 2006;42:241–243. doi: 10.1016/j.ooe.2006.02.001. [DOI] [Google Scholar]
- 38.Kumar M, Patne S, Vishwanath A, Hasan Z. Lingual alveolar soft part sarcoma in a child managed successfully with surgery and chemotherapy. Indian J Cancer. 2010;47:234–235. doi: 10.4103/0019-509X.63023. [DOI] [PubMed] [Google Scholar]
- 39.Masson P. Tumeurs Humaines: Histologie, Diagnostics Et Techniques. Paris: Libraire Maloine; 1956. [Google Scholar]
- 40.Ladanyi M, Antonescu CR, Drobnjak M, Baren A, Lui MY, Golde DW, Cordon-Cardo C. The precrystalline cytoplasmic granules of alveolar soft part sarcoma contain monocarboxylate transporter 1 and CD147. Am J Pathol. 2002;160:1215–1221. doi: 10.1016/S0002-9440(10)62548-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Welsh RA, Bray DM, III, Shipkey FH, Meyer AT. Histogenesis of alveolar soft part sarcoma. Cancer. 1972;29:191–204. doi: 10.1002/1097-0142(197201)29:1<191::AID-CNCR2820290129>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 42.Mukai M, Iri H, Nakajima T, Hirose S, Torikata C, Kageyama K, Ueno N, Murakami K. Alveolar soft-part sarcoma. A review on its histogenesis and further studies based on electron microscopy, immunohistochemistry, and biochemistry. Am J Surg Pathol. 1983;7:679–689. doi: 10.1097/00000478-198310000-00008. [DOI] [PubMed] [Google Scholar]
- 43.Argani P, Antonescu CR, Illei PB, Lui MY, Timmons CF, Newbury R, Reuter VE, Garvin AJ, Perez-Atayde AR, Fletcher JA, Beckwith JB, Bridge JA, Ladanyi M. Primary renal neoplasms with the ASPL-TFE3 gene fusion of alveolar soft part sarcoma: a distinctive tumor entity previously included among renal cell carcinomas of children and adolescents. Am J Pathol. 2001;159:179–192. doi: 10.1016/S0002-9440(10)61684-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsuji K, Ishikawa Y, Imamura T. Technique for differentiating alveolar soft part sarcoma from other tumors in paraffin-embedded tissue: comparison of immunohistochemistry for TFE3 and CD147 and of reverse transcription polymerase chain reaction for ASPSCR1-TFE3 fusion transcript. Hum Pathol. 2012;43:356–363. doi: 10.1016/j.humpath.2011.05.004. [DOI] [PubMed] [Google Scholar]
- 45.Rekhi B, Ingle A, Agarwal M, Puri A, Laskar S, Jambhekar NA. Alveolar soft part sarcoma ‘revisited’: clinicopathological review of 47 cases from a tertiary cancer referral centre, including immunohistochemical expression of TFE3 in 22 cases and 21 other tumours. Pathology. 2012;44:11–17. doi: 10.1097/PAT.0b013e32834d7ba4. [DOI] [PubMed] [Google Scholar]
- 46.Williams A, Bartle G, Sumathi VP, Meis JM, Mangham DC, Grimer RJ, Kindblom LG. Detection of ASPL/TFE3 fusion transcripts and the TFE3 antigen in formalin-fixed, paraffin-embedded tissue in a series of 18 cases of alveolar soft part sarcoma: useful diagnostic tools in cases with unusual histological features. Virchows Arch. 2011;458:291–300. doi: 10.1007/s00428-010-1039-9. [DOI] [PubMed] [Google Scholar]
- 47.Pang LJ, Chang B, Zou H, Qi Y, Jiang JF, Li HA, Hu WH, Chen YZ, Liu CX, Zhang WJ, Li F. Alveolar soft part sarcoma: a bimarker diagnostic strategy using TFE3 immunoassay and ASPL-TFE3 fusion transcripts in paraffin-embedded tumor tissues. Diagn Mol Pathol. 2008;17:245–252. doi: 10.1097/PDM.0b013e31815d68d7. [DOI] [PubMed] [Google Scholar]
- 48.Joyama S, Ueda T, Shimizu K, Kudawara I, Mano M, Funai H, Takemura K, Yoshikawa H. Chromosome rearrangement at 17q25 and Xp11.2 in alveolar soft-part sarcoma: a case report and review of the literature. Cancer. 1999;86:1246–1250. doi: 10.1002/(SICI)1097-0142(19991001)86:7<1246::AID-CNCR20>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 49.Ladanyi M, Lui MY, Antonescu CR, Krause-Boehm A, Meindl A, Argani P, Healey JH, Ueda T, Yoshikawa H, Meloni-Ehrig A, Sorensen PH, Mertens F, Mandahl N, Van Den Berghe H, Sciot R, Dal Cin P, Bridge J. The Der(17)T(X;17)(P11;Q25) of human alveolar soft part sarcoma fuses the TFE3 transcription factor gene to ASPL, a novel gene at 17q25. Oncogene. 2001;20:48–57. doi: 10.1038/sj.onc.1204074. [DOI] [PubMed] [Google Scholar]
- 50.Zhong M, De Angelo P, Osborne L, Keane-Tarchichi M, Goldfischer M, Edelmann L, Yang Y, Linehan WM, Merino MJ, Aisner S, Hameed M. Dual-color, break-apart FISH assay on paraffin-embedded tissues as an adjunct to diagnosis of Xp11 translocation renal cell carcinoma and alveolar soft part sarcoma. Am J Surg Pathol. 2010;34:757–766. doi: 10.1097/PAS.0b013e3181dd577e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sidhar SK, Clark J, Gill S, Hamoudi R, Crew AJ, Gwilliam R, Ross M, Linehan WM, Birdsall S, Shipley J, Cooper CS. The T(X;1)(P11.2;Q21.2) translocation in papillary renal cell carcinoma fuses a novel gene PRCC to the TFE3 transcription factor gene. Hum Mol Genet. 1996;5:1333–1338. doi: 10.1093/hmg/5.9.1333. [DOI] [PubMed] [Google Scholar]
- 52.Kuroda N, Mikami S, Pan CC, Cohen RJ, Hes O, Michal M, Nagashima Y, Tanaka Y, Inoue K, Shuin T, Lee GH. Review of renal carcinoma associated with Xp11.2 translocations/TFE3 gene fusions with focus on pathobiological aspect. Histol Histopathol. 2012;27:133–140. doi: 10.14670/HH-27.133. [DOI] [PubMed] [Google Scholar]
- 53.Gomez JA, Amin MB, Ro JY, Linden MD, Lee MW, Zarbo RJ. Immunohistochemical profile of myogenin and myod1 does not support skeletal muscle lineage in alveolar soft part sarcoma. Arch Pathol Lab Med. 1999;123:503–507. doi: 10.5858/1999-123-0503-IPOMAM. [DOI] [PubMed] [Google Scholar]
- 54.Koutlas IG, Pambuccian SE, Jessurun J, Manivel JC, Gopalakrishnan R. Perivascular epithelioid cell tumor of the oral mucosa. Arch Pathol Lab Med. 2005;129:690–693. doi: 10.5858/2005-129-0690-PECTOT. [DOI] [PubMed] [Google Scholar]
- 55.Garcia De Marcos JA, Del Castillo-Pardo De Vera JL, Poblet E, Galdeano-Arenas M, Arroyo-Rodriguez S, Calderon-Polanco J, De Alava E. Clear cell sarcoma of the temporal region: case report, review of the literature, and genetic analysis. J Oral Maxillofac Surg. 2009;67:910–914. doi: 10.1016/j.joms.2008.06.092. [DOI] [PubMed] [Google Scholar]