

Abstract
Bone marrow mesenchymal stem cells (bMSCs) are multipotent and preferred for cell therapy. However, the content of bMSCs is very low. To propagate a large number of primary bMSCs rapidly has become a prerequisite for bMSC study and application. Different methods of isolating and culturing bMSC were used and compared among groups: bMSCs of group A are isolated using direct adherence method and cultured by conventional medium changing; of group B are isolated using direct adherence method and cultured by low volume medium changing; of group C are isolated using density gradient centrifugation and cultured by conventional medium changing; of group D are isolated using density gradient centrifugation and cultured by low volume medium changing. The average population doubling time (PDT), average generation time and the cumulative cell doubling level were calculated for every group. bMSCs cultured with complete medium containing 10, 11 and 15 % FBS were allocated into group a, b and c separatedly. Cell numbers were counted everyday under a microscope, the population doubling level curve was plotted and PDT was calculated. The growth curve of bMSC in group a, b and c was made. Both density gradient centrifugation and direct adherence methods obtained relatively pure bMSCs. A larger quantity of primary bMSCs were obtained by direct adherence. bMSC proliferation was faster when cultured via the low volume medium changing method at a serum concentration of 11 % than the other methods. Isolating bMSC by direct adherence and culturing by low volume medium changing at a serum concentration of 11 % is preferential for bMSC propagation.
Keywords: Bone marrow mesenchymal stem cells, Cell culture, Serum concentration
bMSCs are a population of self-renewing and multipotent cells that can differentiate into adipocytes, osteocytes, chondrocytes, cardiomyocytes, etc. (Pittenger et al. 1999; Theise et al. 2000; Sanchez-Ramos et al. 2002; Orlic et al. 2001; In’t Anker et al. 2003, 2004; Erices et al. 2000; Zuk et al. 2002) They can be acquired conveniently and amplified in vitro. (Kim 2008) Self-transplantation of bMSCs for treatment has no problem of ethics and may arise the body immunologic tolerance by depressing T lymphocytes proliferation (Pelttari et al. 2008; Xu et al. 2004; Aggarwal and Pittenger 2005; Pittenger and Martin 2004), which has received more and more attention in the field of regenerative medicine (Mclntosh and Bartholomew 2000).
MSCs have been successfully isolated from samples (such as placenta, liver, blood, bone marrow, etc.) of many species including human, pig, rabbit, rat and other animals (Tse et al. 2003; Nicola et al. 2002; Le Blanc et al. 2003; Bartholomew et al. 2002). Rats are most common used experimental animals because it is easy and economical to keep them. Many researchers isolate bMSCs from rats bone marrow and amplify them. However it is difficult to acquire high numbers bMSCs in a short time because of their low content in the bone marrow and many things affecting their growth and proliferation, such as extracellular matrix (ECM), cytokines, mechanical stimulus and other chemical and physical factors (Jäger et al. 2005; Cowles et al. 2000; Wilson et al. 2003; Illario et al. 2005; Vellon et al. 2006; Kobayashi-Sakamoto et al. 2008; Conget and Minguell 1999; Larsen et al. 2006; Chang and Karin 2001; Giancotti and Ruoslahti 1999; Cácamo-Orive et al. 2008; Levy et al. 2008; Bǒcker et al. 2008; Runguang et al. 2007; Liedert et al. 2006; Ko and McCulloch 2001; Li et al. 2004; Simmons et al. 2003; Sekiya et al. 2004). It is a big problem for scientists to find a simple and practicable way to get more bMSCs within a shorter time.
Up to now, people are still looking for some ideal methods of isolating and culturing bMSCs, through which researcher might easily get the greatest amount of bMSCs and have them amplified rapidly. Our study aimed to find such an efficient and easy-to-practice method in order to provide convenience for research on bMSCs.
Materials and method
Rat bMSCs isolation using density gradient centrifugation with Percoll separation medium
Healthy 3–4 week specific pathogen free (SPF) grade Wistar rats weighing 100–120 g were sacrificed by cervical dislocation, soaked in 75 % ethanol for 10 min and then the femur and tibia were isolated under sterile condition. Samples were immersed in low glucose Dulbecco’s Modified Eagle’s Medium (L-DMEM, Hyclone, Logan, UT, USA) supplemented with 2 mmol/L l-glutamine (GIBCO, Grand Island, NY, USA), 100 U/mL penicillin (Tianjin Pharmaceuticals, Tianjin, China) and 100 μg/mL streptomycin (Tianjin Pharmaceuticals). Joint capsules at the ends of the diaphysis were removed without isolating epiphysis and the diaphysis was then divided. A disposable aseptic syringe was used to draw antibiotic supplemented L-DMEM medium and to repeatedly wash the bone marrow cavity to collect cells in a sterile petri dish. The obtained cell suspension was added onto the Percoll separation solution whose density sas 1.073 g/mL, and centrifuged at 400×g for 30 min at 26 °C. The cotton-like cells at the interface were collected and rinsed once in L-DMEM containing 10 % fetal bovine serum (FBS, GIBCO). Cells were further resuspended in complete medium (90 % L-DMEM, 10 % FBS, 100 μ/mL penicillin and 100 μg/mL streptomycin) and transferred to a 25 cm2 plastic culture flask for incubation at 37 °C in a 5 % CO2 supplemented incubator. Cells isolated from one rat were cultured in one flask.
Rat bMSCs isolation using direct adherence
Healthy 3–4 week SPF grade Wistar rats weighing 100–120 g were sacrificed by cervical dislocation, soaked in 75 % ethanol for 10 min and then the femur and tibia were isolated under sterile condition. Samples were immersed in L-DMEM, 100 U/mL penicillin and 100 μg/mL streptomycin. Joint capsules at the ends of the diaphysis were removed without isolating epiphysis and the diaphysis was then divided. A disposable aseptic syringe was used to draw antibiotic supplemented L-DMEM medium and to repeatedly wash the bone marrow cavity to collect cells in a sterile petri dish. The obtained cell suspension was centrifuged at 250×g for 5 min at 26 °C and rinsed once in L-DMEM containing 10 % FBS. Cells were further resuspended in complete medium (90 % L-DMEM, 10 % FBS, 100 μ/mL penicillin and 100 μg/mL streptomycin) and transferred to a 25 cm2 plastic culture flask for incubation at 37 °C in a 5 % CO2 supplemented incubator. Cells isolated from one rat were cultured in one flask.
bMSCs culture using various medium volume changes
One half of the culture medium volume was changed 24 h after obtaining primary cells. The culture medium of cells obtained using Percoll gradient centrifugation was changed every 3–4 days. For direct adherence isolated cells, the culture medium was changed 24 h after the initial medium change for the first time, and then every 3–4 days. Some of bMSCs isolated using Percoll gradient centrifugation and direct adherence methods (3 culture flasks for each, selected randomly) were cultured via low volume changing method. Another group (3 culture flasks for each, selected randomly) were routinely cultured with conventional method. Low medium changing method was that low volume of medium, namely 0.3–0.5 mL of the original medium, was left in the original flask after discarding the original medium and then adding fresh complete medium. Conventional medium changing was to discard all the original medium and then add fresh complete medium. The rats were randomly allocated into four groups, different methods of MSC isolation and culture were used and compared among every group: group A isolated using direct adherence and cultured with conventional medium changing; group B isolated using direct adherence and cultured with low volume medium changing; group C obtained using density gradient centrifugation and cultured with conventional medium changing; group D obtained using density gradient centrifugation and cultured with low volume medium changing.
Subculture
bMSCs were subcultured at 70–80 % confluence. Medium was discarded, bMSCs were gently rinsed and then 2–3 mL trypsin preheated to 37 °C was added for 3–5 min, followed by addition of 3–5 mL of complete medium to terminate digestion. The cell suspension was transferred into a 15 mL tube and centrifuged at 250×g for 5 min at 26 °C. The supernatant was discarded. bMSCs were resuspended in complete medium and subcultured at a 1:2 split ratio. Passage 1 cells were labeled as P1, passage 2 and 3 as P2 and P3 respectively, and so on.
Screening tests at various serum concentrations
bMSCs, which were isolated using direct adherence and cultured by low volume medium changing, were cultured with complete medium containing 10, 11 and 15 % FBS and allocated into groups a, b and c separatedly with three flasks of cells in each group. The cell number was counted everyday under a microscope in duplicate. We randomly selected five visual fields for every cell culture flask, counted the bMSC number for every visual field and calculated the mean. Then we plotted the PDL curve and calculated the PDT.
PDL and time calculation
We counted the numbers of MSCs of groups A, B, C and D, calculated the PDL and PDT, and then drew the PDL curves for the 4 group cells as above. Differences in the proliferation capacity of cells isolated using various methods and culture conditions were compared and calculated as follows:
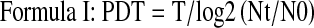 |
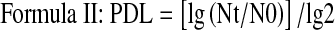 |
(N: cell number, T: incubation time 0–t, t: time t, and 0: initial time.
Growth curve
Passage 1 bMSCs of groups a, b and c were plated in 96-well culture plates at 1 × 104 cells/well in three wells of six culture plates each. One plate was taken out for OD (optical density) meaturement every 2 days until day twelve. MTT (3-(4,5-Dimethylthiazol--2-yl)-2,5-diphenyltetrazolium bromide) was added at a final concentration of 5 mg/mL. Four hours later, the medium was removed and DMSO (dimethyl sulphoxide) was added in. The results were read with an ELISA reader at 490 nm.
Immunocytochemical identification of cell surface antigens CD45 and CD90
P3 bMSCs were digested and seeded on coverslips in a six well culture dish at 8 × 103 cells/cm2 (coverslips were autoclaved, dried and coated with sterile poly-l-lysine). When bMSCs reached 80–90 % confluence, culture medium was discarded and the dish was rinsed three times in phosphate balanced solution (PBS) for 3 min each. bMSCs were fixed in paraformaldehyde, rinsed in PBS as described and blocked with 5 % bovine serum albumin (BSA) for 30 min. The blocking solution was removed and the primary antibody (mouse anti-rat CD45/CD90, 1:100, Becton, Dickinson and Co, Franklin Lakes, NJ, USA) was added without rinsing, followed by incubation at 4 °C overnight. Samples were rinsed as described, the secondary antibody (biotinylated rabbit anti-mouse IgG, Becton, Dickinson and Co) was added and incubated at 37 °C for 30 min. Samples were rinsed three times in PBS for 5 min each, one drop of Strept Avidin–Biotin Complex (SABC, Wuhan Boster Biological Engineering Co., Ltd, Wuhan, China) was added to the coverslips and then incubated at room temperature for 1 h and samples were rinsed in PBS as previously described, and then diaminobenxidine (DAB, Wuhan Boster Biological Engineering Co., Ltd) was added for color development over 8–10 min. Samples were rinsed in distilled water counterstained with hematoxylin. Samples were dehydrated in 1 % acid alcohol and rinsed in running water, followed by a secondary dehydration in 75 % ethanol and absolute ethyl alcohol for 2 min, and then clarified with xylene twice for 5 min each. Samples were mounted with coverslips and observed under a light microscope.
Flow cytometric detection of cell surface antigens CD45, CD90, CD14, CD34, CD73 and CD105
Digested P3 bMSCs in an optimal growth state were rinsed in PBS and resuspended in 0.5 mL PBS. Mouse anti-rat CD45- fluorescein isothiocyanate (FITC, Becton, Dickinson and Co, 3 μL), CD90- phycoerythrin (PE, Becton, Dickinson and Co, 3 μL), CD14-FITC (BD Biosciences, San Jose, CA, USA, 3μL), CD73-PE (BD Biosciences, 3 μL), CD105-FITC (Peprotech, Rocky Hill, NJ, USA, 3 μL) and CD34-FITC (Santa Cruz Biotechnology, Santa Cruz, USA, 20 μL) monoclonal antibodies were added separately, followed by 30 min incubation in the dark at 4 °C. Labeled bMSCs were rinsed in PBS, centrifuged at 200×g for 5 min and resuspended in PBS. Labeled bMSCs were then analyzed using a flow cytometer. Unstained cells were used as the control.
MSC-derived adipocyte identification
P3 bMSCs were digested and seeded on poly-l-lysine-coated coverslips in a six well culture dish at 8 × 103 cells/cm2. After adhering, bMSCs were differentiated into adipocytes using adipogenesis induction medium [DMEM/F12(Hyclone), 0.1 mmol/L 3-isobutyl-1-methylxanthine (IBMX, Tianjin Pharmaceuticals), 10 mg/L insulin (Tianjin Pharmaceuticals), 0.1 mmol/L indometacin (Tianjin Pharmaceuticals), 1μmol/L dexamethasone (Sigma, St. Louis, MO, USA), 10 % FBS]. After 2 weeks, coverslips from induction and control groups were rinsed three times in PBS, fixed in 4 % paraformaldehyde for 15 min, rinsed three times in PBS again and gently rinsed in 60 % isopropanol. Samples were then stained with an appropriate amount of oil red-O (Sigma) for 30 min, rinsed three times in distilled water, mounted with glycerol and observed under a microscope (Sekiya et al. 2004).
MSC-derived osteoblast identification
P3 bMSCs were digested and seeded as described above. After adhering, MSCs were differentiated into osteoblasts using osteogenesis induction medium [DMEM/F12, 0.01 μmol/L dexamethasone,10 mmol/L β-Na glycerophosphate (Sigma), 50 μmol/L ascorbic acid C (Tianjin Pharmaceuticals), 10 % FBS] for 5 days. After 2 weeks, coverslips from induction and control groups were rinsed twice in PBS, fixed in 95 % ethanol for 10 min and washed three times with distilled water. Samples were then treated with 0.1 % alizarin red (Sigma)-Tris–HCl (Tianjin Pharmaceuticals) (pH = 8.3) at 37 °C for 30 min, rinsed with distilled water and observed under a microscope (Sheehan and Hrapchak 1980).
Results
Isolation and culture of primary bMSCs
After 24-hour culture, primary cells adhered and most of the round cells with strong refractivity were erythrocyte. After adhering, bMSCs were rod-shaped or spindle-shaped. When the medium was changed, the majority of non-adherent cells, such as red cells, were eliminated and adherent cells gradually proliferated. A proportion of the cells extended to become spindle-shaped and others were predominantly short rod-like, triangular or stelliform. After 1–5 days, adherent mitotic cells developed into dispersed cell clones of unequal size. Cells extended to become long spindle-shaped with 1–2 cell nuclei, circular central nuclei or one to several nucleoli, and the cytoplasm became clear with high refractivity with progressive medium changes. After 2–6 days, clones enlarged in a fish-stock-like growth state and gradually fused with other clones. A small proportion of heterologous adherent cells grew to the final stage of primary culture. One passage could be subcultured after 4–10 days and another after a further 4–10 days. Primary cells became round when treated with trypsin. Three to four h after subculture the majority of cells adhered, and after 3–4 days cells were confluent, spindle-shaped and grew in a whorl-like state.
Light microscopy showed that group A (Fig. 1A) and Group B (Fig. 1B) cells were spindle-shaped or polygonal 24 h after seeding with an average initial population density of 200 cells/cm2, and group C (Fig. 1C) and group D (Fig. 1D) cells after seeding with an average initial population density of 50 cells/cm2. After 6 days of subculture, group A cells generated colonies and grew in a whorl-like state until 80 % confluency (Fig. 1A1). Group B cells after 4 days of subculture generated colonies and grew in a whorl-like state until 90 % confluency (Fig. 1B1). And group C cells after 10 days of subculture locally generated colonies without growing in a whorl-like state (Fig. 1C1). Group D cells after 8 days of subculture locally generated colonies and grew in a homodromous rather than whorl-like state (Fig. 1D1). Figure 1E shows evaluation of various isolation methods and culture conditions on the growth rate of rat bMSCs. The PDL of group B cells (isolated using direct adherence combined with low volume medium changing) was significantly higher than that of other groups and group B cells proliferated rapidly (P < 0.05). Cell proliferation in groups was as follows: B > A > D > C.
Fig. 1.

A: group A cells after 24 h; B: group B cells after 24 h; C: group C cells after 24 h; D: group D cells after 24 h; A1: group A cells after 6 days of subculture; B1: group B cells after 4 days of subculture; C1: group C cells after 10 days of subculture; D1: group D cells after 8 days of subculture; E shows evaluation of various isolation methods and culture conditions on the growth rate of rat bMSCs
Average PDT for P5 group A cells was 36.6 ± 0.9 h, average generation time was 6 days, and the cumulative cell doubling level was 39.6; average PDT for P5 group B cells was 23.5 ± 1.1 h, average generation time was 4 days and the cumulative cell doubling level was 40.1; average PDT for P5 group C cells was 49.8 ± 1.2 h, average generation time was 10 days and the cumulative cell doubling level was 36.7; average PDT for P5 group D cells was 48.0 ± 0.8 h, average generation time was 8 days and the cumulative cell doubling level was 38.7 (Table 1).
Table 1.
shows average PDT and the cumulative cell doubling level of every group cells untill the P5
| Average PDT for P5 | Average generation time (days) | The cumulative cell doubing level | |
|---|---|---|---|
| Group A | 36.6 ± 0.9 h | 6 | 39.6 |
| Group B | 23.5 ± 1.1 h | 4 | 40.1 |
| Group C | 49.8 ± 1.2 h | 10 | 36.7 |
| Group D | 48.0 ± 0.8 h | 8 | 38.7 |
Screening tests at different serum concentrations
Light microscopy shows group a (Fig. 2a) and b (Fig. 2b) cells were spindle-shaped or polygonal and irregularly arranged 24 h after seeding with an average initial population density density of 200 cells/cm2, and more mitotic cells were observed in group b cells. Group c cells (Fig. 2c) were also spindle-shaped or polygonal 24 h after seeding with an average initial population density of 200 cells/cm2 and more polygonal cells were observed. Group a cells did not grow in a whorl-like state but reached 80 % confluency with colonies after 3 days (Fig. 2a1). Group b cells grew in a whorl-like state and reached 90 % confluency after 3 days (Fig. 2b1). No obvious colonies were observed among group c cells after 3 days, although more cells were observed in the mitotic phase of proliferation (Fig. 2c1). Figure 2d shows evaluation of various serum concentrations for rat bMSC growth. The proliferation velocity of cells cultured in 11 % FBS was significantly higher than that of the other groups (P < 0.05). We may conclude that the proliferation velocity of cells in groups are in the following order: b > a > c.
Fig. 2.

A–C show MSC of group a, group b and group c after 24 h of culturing separately. And a1, b1 and c1 show MSC of group a (10% FBS), group b (11% FBS) and group c (15% FBS) after 3 days of culturing separately. d shows evaluation of various serum concentrations for rat bMSC growth. The proliferation velocity of cells cultured in 11 % FBS was significantly higher than that of other groups (P < 0.05)
Average PDT for P5 group a cells was 23.5 ± 1.1 h, average generation time was 4 days and cumulative cell doubling level was 40.1; average PDT for P5 group b cells was 20.5 ± 0.9 h, average generation time was 3.5 days and cumulative cell doubling level was 46; average PDT for P5 group c cells was 30.7 ± 1.3 h, average generation time was 7 days and cumulative cell doubling level was 39.7 (Table 2). We found that cells cultured with 11 % FBS (group b) showed a higher proliferation rate (Fig. 2d).
Table 2.
shows average PDT and the cumulative cell doubling level of cells of group a, b and c
| Average PDT for P5 | Average generation time (days) | The cumulative cell doubling level | |
|---|---|---|---|
| Group a | 23.5 ± 1.1 h | 4 | 40.1 |
| Group b | 20.5 ± 0.9 h | 3.5 | 46 |
| Group c | 30.7 ± 1.3 h | 7 | 39.7 |
Growth test of MSCs of group a, b and c (MTT) showed that cells in group a had the highest proliferation rate (P < 0.05, Fig. 3).
Fig. 3.

Comparison of proliferation rate of MSCs of group a, group b and group c. MSCs of group b showed a higher growth rate when compared to the proliferation rate of the other two groups
Identification of MSCs
Immunocytochemical identification of cell surface antigens CD45 and CD90. Immunochemical identification of cell surface antigens CD45 (–) (Fig. 4a) and CD90 (+) (Fig. 4b).
-
Detection of cell surface antigens CD45, CD90, CD14, CD105, CD34 and CD73 of MSC using flow cytometry.
Phenotypic cell surface marker analysis of P3 bMSCs using flow cytometry found that bMSCs highly expressed CD90 (98.4 %), CD73 (97.6 %) and CD105 (98.5 %) rather than CD45 (0.38 %), CD14 (0.45 %) and CD34 (0.37 %) (Fig. 5).
-
Induced bMSC differentiation into adipocytes.
bMSCs were differentiated into adipocytes. After 2 weeks, cell morphology changed, lipid droplets emerged and oil red-O staining was positive (Fig. 4c).
-
Induced bMSC differentiation into osteoblasts.
bMSCs were differentiated into osteoblasts. After 2 weeks, mineralized nodules emerged and alizarin red staining was positive (Fig. 4d).
Fig. 4.

Immunohistochemical staining for surface antigens CD45 and CD90 on rat bMSCs: a MSC marker CD45 (–) and DAB staining (200×); b MSC marker CD90 (+) and DAB staining (200×). c lipid droplets emerged after adipogenesis induction of MSCs and oil red-O staining was positive. d Mineralized nodules emerged after osteogenesis induction of MSCs and alizarin red staining was positive
Fig. 5.

Detection of cell surface antigens CD45, CD90, CD14, CD105, CD34 and CD73 of MSC using flow cytometry
Discussion
MSCs proliferate in abundance under in vitro culture conditions while maintaining a highly undifferentiated state. MSCs can be easily isolated, cultured and propagated in vitro. Additionally, because bone marrow sources are rich, bMSCs can be conveniently and safely collected (Sotiropoulou et al. 2006; Satija et al. 2007) with minimal invasiveness. Thus, MSCs have great prospects in cell replacement therapy and tissue engineering. Because the human bMSC percentage is small, ~1/104–l05 mononuclear cells, and declines with age, such a small number of bMSCs can barely meet the needs of cellular engineering. Therefore, it is particularly important to establish an easy and feasible method for bMSC isolation, purification and propagation.
Common methods for isolating MSCs include whole marrow direct adherence and density gradient centrifugation. Whole marrow direct adherence refers to regular culture medium changing to remove non-adherent cells based on the stem cell adherence characteristic to achieve MSC purification. Density gradient centrifugation refers to the extraction of mononuclear cells for adherent culture according to the proportion of mononuclear bone marrow cells. With an in-depth understanding of MSC surface antigens; immunoassays, flow cytometry and immunobead methods have been applied to isolate and purify MSC surface antigens. However, cells isolated using flow cytometry or immunobead methods proliferate slowly. In addition to the high cost and technical difficulties, the extensive application of these methods is reasonably limited. In 1976, Friedenstein et al. (1976) found that adherent bone marrow mononuclear cells could differentiate into osteoblasts, chondroblasts, adipocytes and sarcoblasts under certain induction conditions in a plastic culture dish. Also, these cells maintained their multipotenial differentiation after 20–30 passages. This finding has played an important role in research, which not only confirms the existence of bMSCs, but also established a simple and feasible method for in vitro MSC isolation and culture. These methods have been widely applied in the actual practice.
The bone marrow stromal cell system consists of a variety of cell populations that can be assigned based on their morphological characteristics to reticular cells, adipocytes, adipocyte precursors, smooth muscle-like and fibroblast-like cells, and endothelioid and epithelioid cells. Bone marrow hematopoietic stem cells, red blood cells and white blood cells grow in a suspended state in vitro while MSCs grow adherently, therefore suspended cells can be eliminated by changing the culture medium to obtain adherently growing MSCs. bMSC isolation and culture methods vary among laboratories. In this study, bMSCs isolated using direct adherence and density gradient centrifugation methods were compared. Direct adherence is simple and convenient, obtains more MSCs than density gradient centrifugation and has an appropriate cell density for growth in culture flasks. In addition, bMSC purity increases after medium changes. Direct adherence can be regarded as the preferential method for MSC isolation and is supported by previously reported studies (Rouger et al. 2007; Yang and Yang 2008).
The percentage of bMSCs is small, however MSC differentiation and proliferation capacities are strong and MSCs can maintain a comparatively high vitality during in vitro culture. MSC growth in vitro is affected by a variety of factors such as oxygen concentration, culture medium, serum concentration and soluble growth factors. MSCs can grow more vigorously under appropriate hypoxic conditions (2 % oxygen), which is closer to their physiological state. However, MSC growth is limited under relatively hyperoxic in vitro conditions and oxygen concentration control is inconvenient for culturing large numbers of MSCs. In addition, growth factors are often expensive and optimal culture medium for MSCs has been finalized, so this study focused on various serum concentrations for MSC culture which is easier to control. Serum is beneficial for MSC propagation; however, serum can also induce premature differentiation. Moreover, serum composition such as cytokine content is not yet fully understood and various serum concentrations may not all be conducive for MSC growth. In this study, an optimal serum concentration was chosen from three concentrations commonly used, with the purpose of benefiting the majority of MSC researchers. Nonetheless, the optimal condition for MSC growth is still unknown. Due to limitations, this study remains inconclusive until a wider range of screening analyses and other related research methods are available.
MSCs undergo stagnant, logarithmic growth and plateau phases under in vitro culture conditions. We confirmed that the percentage of bMSCs is comparatively small; however, bMSC proliferation is very strong. In this experiment, normal MSCs were obtained using simple and convenient methods of isolation and culture, providing easy and reliable cell sources for experimentation and clinical research.
Studies suggest that (Woodbury et al. 2000, 2002; Zannettino et al. 2007; Gang et al. 2007; Pittenger et al. 1999; Romanou et al. 2005) bMSCs are positive for surface antigens CD105 (SH2), CD73 (SH3, SH4), STRO-1, actin, CD29, CD44, CD90, CD106, CD120a, CD124, CD166 and HLA-ABC, while hematopoietic lineage markers CD14, CD34 and CD45 are negative. SH2 and SH3 are relatively specific MSC antigens, however monoclonal antibodies directed against these antigens are rare. Therefore, SH2 and SH3 cannot be used for routine bMSC detection. Currently, MSC identification is mainly based on morphology and functional characteristics (Cheng 2004; Reger 2008). Because CD90 is expressed on bMSCs and absent on hematopoietic stem cells, and CD45 is absent on bMSCs and positive on hematopoietic stem cells, this indicates that cultivated cells are bMSCs rather than hematopoietic stem cells. According to minimum standards put forward by the International Society for Cellular Therapy (ISCT), multipotent MSCs from various origins should meet the following criteria: 1) adherence to a plastic flask; 2) expression of surface antigens CD73, CD90, CD105 with >95 % expression, while hematopoietic cell markers CD45, CD34, CD14 and HLA-DR are negative with ≤2 % expression; 3) ability to differentiate into osteoblasts, adipocytes and cartilage cells in vitro (Dominici et al. 2006). bMSCs isolated and propagated using the various methods in this study, meet these requirements.
P1 bMSCs propagate rapidly, however due to insufficient purity are inappropriate for transplantation. Moreover, P8 bMSCs display an obvious aging trend and are considered inappropriate for transplantation. P3–5 cells have high purity and are available at sufficient. Thus, P3–5 cells are an appropriate selection for MSC transplantation.
Cell growth is affected by cell interactions, and secreted cytokines can promote growth. MSCs secrete interleukin 6-8 (IL6–8), stem cell factor (SCF) and other cytokines of great importance for maintaining growth. Thus, to minimize damage from the microenvironment, culture methods using low and common medium volume changes were compared, proving that low medium changes yield a greater proportion of MSCs.
Conclusion
We may conclude that more MSCs can be gained through the method of direct adherence compared with the method of density gradient centrifugation, and the researcher will get pure bMSCs after medium changing and subculture. MSCs cultured by low medium changing method grow and proliferate significantly faster than those cultured by conventional changing method. 11 % FBS in the medium is the most efficient concentration among these three different concentrations of FBS used for MSC culture, which are 11, 10, 15 %. Generally speaking, the usage of direct adherence method during MSC isolation, low medium changing and 11% FBS in medium is optimal in MSC culture by now. This simple and practical technique will facilitate the study of MSC, both for examing their biological properties as well as their therapeutic potential in clinical practice.
Acknowledgments
Authors received fund from Liaoning province high school program (2008s248).
References
- Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell response. Blood. 2005;105(4):1815–1822. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- Bartholomew A, Sturgeon C, Siatska M, et al. Mesenchymal stem cues suppress lyruphocyte proliferation in vitro and prolong skin graft Surrlval in vivo. Exp Hematol. 2002;30(1):42–48. doi: 10.1016/S0301-472X(01)00769-X. [DOI] [PubMed] [Google Scholar]
- Bǒcker W, Docheva D, Prall WC, et al. IKK-2 is required for TNF-alpha-induced invasion and proliferation of human mesenchymal stem cells. J Mol Med. 2008;86(10):1183–1192. doi: 10.1007/s00109-008-0378-3. [DOI] [PubMed] [Google Scholar]
- Cárcamo-Orive I, Tejados N, Delgado J, et al. ERK2 protein regulates the proliferation of human mesenchymal stem cells without affecting their mobilization and differentiation potential. Exp Cell Res. 2008;314(8):1777–1788. doi: 10.1016/j.yexcr.2008.01.020. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410(6824):37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Cheng Z, Zhou D, Song S, et al. Oriented induction of umbilical cord blood and bone marrow mesenchymal stem cells into osteoblasts and comparison of the osteogenic activity. Chin J Clin Rehabil. 2004;35(8):7973–7975. [Google Scholar]
- Conget PA, Minguell JJ. Phenotypical and functional properties of human bone marrow mesenchymal progenitor cells. J Cell Physiol. 1999;181(1):67–73. doi: 10.1002/(SICI)1097-4652(199910)181:1<67::AID-JCP7>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Cowles EA, Brailey LL, Gronowicz GA. Integrin-mediated signaling regulates AP-1 transcription factors and proliferation in osteoblasts. J Biomed Mater Res. 2000;52(4):725–737. doi: 10.1002/1097-4636(20001215)52:4<725::AID-JBM18>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- Dominici M, Le Blanc K, Mueller I, et al. Minimal criteria for defining multipotent mesenchymal stromal cells. The internatlonal society for cellular therapy Position statement. Cytotherapy. 2006;8:315–317. doi: 10.1080/14653240600855905. [DOI] [PubMed] [Google Scholar]
- Erices A, Conget P, Minguell JJ. Mesenchymal progenitor cells in human umbilical cord blood. Br J Haematol. 2000;109(1):235–242. doi: 10.1046/j.1365-2141.2000.01986.x. [DOI] [PubMed] [Google Scholar]
- Friedenstein AJ, Gorskaja JF, Kulagina NN (1976) Fibroblast precursors in normal and irradiated mouse hematopoietic organs. Exp Hematol 4:267–274 [PubMed]
- Gang EJ, Bosnakovski D, Figueiredo CA, et al. SSEA-4 identifies mesenchymal stem cells from bone marrow. Blood. 2007;109(4):1743–1751. doi: 10.1182/blood-2005-11-010504. [DOI] [PubMed] [Google Scholar]
- Giancotti FG, Ruoslahti E. Integrin signaling. Science. 1999;285(5430):1028–1032. doi: 10.1126/science.285.5430.1028. [DOI] [PubMed] [Google Scholar]
- Illario M, Cavallo AL, Monaco S, et al. Fibronectin-induced proliferation in thyroid cells is mediated by αvβ3 integrin through Ras/Raf-1/MEK/ERK and calcium/CaMKII signals. Clin Endocrinol Metab. 2005;90(5):2865–2873. doi: 10.1210/jc.2004-1520. [DOI] [PubMed] [Google Scholar]
- In’t Anker PS, Scherjon SA, Kleijburg-van der Keur C, et al. Amniotic fluid as a novel source of mesenchymal stem cells for therapeutic transplantation. Blood. 2003;102(4):1548–1549. doi: 10.1182/blood-2003-04-1291. [DOI] [PubMed] [Google Scholar]
- In’t Anker PS, Scherjon SA, Kleijburg-Van Der Keur C, et al. Isolation of mesenchymal stem cells of fetal or maternal origin from human placenta. Stem Cells. 2004;22(7):1338–1345. doi: 10.1634/stemcells.2004-0058. [DOI] [PubMed] [Google Scholar]
- Jäger M, Feser T, Denck H, et al. Proliferation and osteogenic differentiation of mesenchymal stem cells cultured onto three different polymers in vitro. Ann Biomed Eng. 2005;33(10):1319–1332. doi: 10.1007/s10439-005-5889-2. [DOI] [PubMed] [Google Scholar]
- Kim SM, Jung JU, Ryu JS, et al. Effects of gangliosides on the differentiation of human mesenchymal stem cells into osteoblasts by modulating epidermal growth factor receptors. Biochem Biophys Res Commun. 2008;371(4):866–871. doi: 10.1016/j.bbrc.2008.04.162. [DOI] [PubMed] [Google Scholar]
- Ko KS, McCulloch CA. Intercellular mechanotransduction: cellular circuits that coordinate tissue responses to mechanical loading. Biochem Biophys Res Commun. 2001;285(5):1077–1083. doi: 10.1006/bbrc.2001.5177. [DOI] [PubMed] [Google Scholar]
- Kobayashi-Sakamoto M, Isogai E, Hirose K, et al. Role of alphav integrin in osteoprotegerin-induced endothelial cell migration and proliferation. Microvasc Res. 2008;76(3):139–144. doi: 10.1016/j.mvr.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Larsen M, Artym VV, Green JA, et al. The matrix reorganized: extracellular matrix remodeling and integrin signaling. Curr Opin Cell Biol. 2006;18(5):463–471. doi: 10.1016/j.ceb.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Le Blanc K, Tammik L, Sundberg B, Haynesworth SE, Ringdén O (2003) Mesenchymal stem cells inhabit and stimulate mixed lymphocyte cultures and mitogenic responses independently of the major histocompatibility complex. Scand J Immumol 57:11–20 [DOI] [PubMed]
- Levy O, Dvir T, Tsur-Gang O, et al. Signal transducer and activator of transcription 3-A key molecular switch for human mesenchymal stem cell proliferation. Int J Biochem Cell Biol. 2008;40(11):2606–2618. doi: 10.1016/j.biocel.2008.05.010. [DOI] [PubMed] [Google Scholar]
- Li YJ, Batra NN, You L, et al. Oscillatory fluid flow affects human marrow stromal cell proliferation and differentiation. J Orthop Res. 2004;22(6):1283–1289. doi: 10.1016/j.orthres.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Liedert A, Kaspar D, Blakytny R, et al. Signal transduction pathways involved in mechanotransduction in bone cells. Biochem Biophys Res Commun. 2006;349(1):1–5. doi: 10.1016/j.bbrc.2006.07.214. [DOI] [PubMed] [Google Scholar]
- Mclntosh KR, Bartholomew A. Stromal cell modulation of the immune system: a potential role for mesenchymal stem cell. Graft. 2000;3:324–328. [Google Scholar]
- Nicola DM, Carlo-Stella C, Magni M, et al. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood. 2002;99:3838–3843. doi: 10.1182/blood.V99.10.3838. [DOI] [PubMed] [Google Scholar]
- Orlic D, Kajstura J, Chimenti S, Jakoniuk I, Anderson SM, Li B, Pickel J, McKay R, Nadal-Ginard B, Bodine DM, Leri A, Anversa P. Bone marrow cells regenerate infarcted myocardium. Nature. 2001;410:701–705. doi: 10.1038/35070587. [DOI] [PubMed] [Google Scholar]
- Pelttari K, Steck E, Richter W. The use of mesenchymal stem cells for chondrogenesis. Injury. 2008;39(suppl 1):S58–S65. doi: 10.1016/j.injury.2008.01.038. [DOI] [PubMed] [Google Scholar]
- Pittenger MF, Martin BJ. Mesenchymal stem cells and their potential as cardiac therapeutics. Circ Res. 2004;95(1):9–20. doi: 10.1161/01.RES.0000135902.99383.6f. [DOI] [PubMed] [Google Scholar]
- Pittenger MF, Mackay AM, Beck SC, et al. Multilineage potential of adult human mesenchymal stem cells. Science. 1999;284(5411):143–147. doi: 10.1126/science.284.5411.143. [DOI] [PubMed] [Google Scholar]
- Reger RL, Tucker AH, Wolfe MR. Differentiation and characterization of human MSCs. Methods Mol Biol. 2008;449:93–107. doi: 10.1007/978-1-60327-169-1_7. [DOI] [PubMed] [Google Scholar]
- Romanov YA, Darevskaya AN, Merzlikina NV, et al. Mesenchymal stem cells from human bone marrow and adipose tissue: isolation, characterization, and differentiation potentialities. Bull Exp Biol Med. 2005;140(1):138–143. doi: 10.1007/s10517-005-0430-z. [DOI] [PubMed] [Google Scholar]
- Rouqer K, Fornasari B, Armenqol V, et al. Progenitor cell isolation from muscle derived cells based on adhesion properties. J Histochem Cytochem. 2007;55(6):607–618. doi: 10.1369/jhc.6A6954.2007. [DOI] [PubMed] [Google Scholar]
- Runguang L, Jingfan S, Mingfa W. Infection of mechanics on mesenchymal stem cells in vitro. J Clin Rehabil Tissue Eng Res. 2007;11(3):551–554. [Google Scholar]
- Sanchez-Ramos JR. Neural cells derived from adult bone marrow and umbilical cord blood. J Neurosci Res. 2002;69(6):880–893. doi: 10.1002/jnr.10337. [DOI] [PubMed] [Google Scholar]
- Satija NK, Gurudutta GU, Sharma S, et al. Mesenchymal stem cells: molecular targets for tissue engineering. Stem Cells Dev. 2007;16(1):7–23. doi: 10.1089/scd.2006.9998. [DOI] [PubMed] [Google Scholar]
- Sekiya I, Larson BL, Vuoristo JT, Cui J-G, Prockop DJ. Adipogenic differentiation of human adult stem cells from bone marrow stroma (MSCs) J Bone Miner Res. 2004;19(2):256–264. doi: 10.1359/JBMR.0301220. [DOI] [PubMed] [Google Scholar]
- Sheehan DC, Hrapchak BB. Theory and practice of histotechnology. 2. St. Louis, Mo: Mosby; 1980. [Google Scholar]
- Simmons CA, Matlis S, Thornton AJ, et al. Cyclic strain enhances matrix mineralization by adult human mesenchymal stem cells via the extracellular signal-regulated kinase (ERK1/2) signaling pathway. J Biomech. 2003;36(8):1087–1096. doi: 10.1016/S0021-9290(03)00110-6. [DOI] [PubMed] [Google Scholar]
- Sotiropoulou PA, Perez SA, Salagianni M, et al. Characterization of the optimal culture conditions for clinical scale production of human mesenchymal stem cells. Stem Cells. 2006;24(2):462–471. doi: 10.1634/stemcells.2004-0331. [DOI] [PubMed] [Google Scholar]
- Theise ND, Nimmakayalu M, Gardner R, Illei PB, Morgan G, Eperman L, Henegariu O, Krause DS. Liver from bone marrow in humans. Hepatology. 2000;32:11–16. doi: 10.1053/jhep.2000.9124. [DOI] [PubMed] [Google Scholar]
- Tse WT, Pendleton JD, Beyer WM, et al. Suppression of allogeneic T cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. 2003;75(3):389–397. doi: 10.1097/01.TP.0000045055.63901.A9. [DOI] [PubMed] [Google Scholar]
- Vellon L, Menendez JA, Lupu R. A bidirectional “alpha(v)beta(3) integrin-ERK1/ERK2 MAPK” connection regulates the proliferation of breast cancer cells. Mol Carcinog. 2006;45(10):795–804. doi: 10.1002/mc.20242. [DOI] [PubMed] [Google Scholar]
- Wilson SH, Ljubimov AV, Morla AO, et al. Fibronectin fragments promote human retinal endothelial cell adhesion and proliferation and ERK activation through α5β1 integrin and PI 3-kinase. Invest Ophthalmol Vis Sci. 2003;44(4):1704–1715. doi: 10.1167/iovs.02-0773. [DOI] [PubMed] [Google Scholar]
- Woodbury D, Schwarz EJ, Prockop DJ, et al. Adult rat and human bone marrow stromal cells differentiate into neurons. J Neurosci Res. 2000;61(4):364–370. doi: 10.1002/1097-4547(20000815)61:4<364::AID-JNR2>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- Woodbury D, Reynolds K, Black IB. Adult bone marrow stromal stem cells express germ Line, ectodermal, endodermal, and mesodermal genes prior to neurogenesis. J Neurosci Res. 2002;69(6):908–917. doi: 10.1002/jnr.10365. [DOI] [PubMed] [Google Scholar]
- Xu W, Zhang X, Qian H, et al. Mesenchymal stem cells from adult human bone marrow differentiate into a cardiomyocyte phenotype in vitro. Exp Biol Med. 2004;229(7):623–631. doi: 10.1177/153537020422900706. [DOI] [PubMed] [Google Scholar]
- Yang F, Yang N. Comparison of two methods of in vitro isolation of adult bone marrow mesenchymal stem cells. J Clin Rehabil Tissue Eng Res. 2008;12(3):473–476. [Google Scholar]
- Zannettino AC, Paton S, Kortesidis A, et al. Human mulipotential mesenchymal/stromal stem cells are derived from a discrete subpopulation of STRO-1bright/CD34/CD45(-)/glycoPhorin-A-bone marrow cells. Haematoloqica. 2007;92(12):1707–1708. doi: 10.3324/haematol.11691. [DOI] [PubMed] [Google Scholar]
- Zuk PA, Zhu M, Ashjian P, et al. Human adipose tissue is a source of multipotent stem cells. Mol Biol Cell. 2002;13(12):4279–4295. doi: 10.1091/mbc.E02-02-0105. [DOI] [PMC free article] [PubMed] [Google Scholar]