

Abstract
A unique design paradigm to form core–shell particles based on interfacial radical polymerization is described. The interfacial initiation system is comprised of an enzymatic reaction between glucose and glucose oxidase (GOx) to generate hydrogen peroxide, which, in the presence of iron (Fe2+), generates hydroxyl radicals that initiate polymerization. Shell formation on prefabricated polymeric cores is achieved by localizing the initiation reaction to the interface of the core and a surrounding aqueous monomer formulation into which it is immersed. The interfacially confined initiation reaction is accomplished by incorporating one or more of the initiating species in the particle core and the remainder of the complementary initiating components in the surrounding media such that interactions and the resulting initiation reaction occur at the interface. This work is focused on engineering the reaction behavior and mass transport processes to promote interfacially confined polymerization, controlling the rate of shell formation, and manipulating the structure of the core–shell particle. Specifically, incorporating GOx in the precursor solution used to fabricate cores ranging from 100 to 200 μm, and the remainder of the complementary initiating components and monomer in the bulk solution prior to interfacial polymerization yielded shells whose average thickness was 20 μm after 4 min of immersion and at a bulk iron concentration of 12.5 mM. When the locations of glucose and GOx are interchanged, the average thickness of the shell was 15 or 100 μm for bulk iron concentrations of 45 and 12.5 mM, respectively. The initial locations of glucose and GOx also determine the degree of interpenetration of the core and the shell. Specifically, for a bulk iron concentration of 45 mM, the thickness of the interpenetrating layer averaged 12 μm when GOx was initially within the core, whereas no interpenetrating layer was observed when glucose was incorporated in the core. The polymeric shell formed by this technique is also demonstrated to be self-supporting following core degradation. This behavior is accomplished by fabricating the particle core hydrogel from monomers possessing degradable groups that can be irreversibly cleaved by light exposure following shell formation. When the coated particle was exposed to light, the shell remained intact while the core degraded as evidenced by a dramatic change in diffusion coefficient of fluorescent beads immobilized within the core.
Keywords: core−shell particles, interfacial radical polymerization, stimuli-responsive microparticles
Introduction
Microparticles composed of a distinct core and shell are useful in a variety of applications such as drug delivery,1−4 tissue engineering,1,5 catalysis,6,7 autonomic healing,8 and pigments,1 among others. The shell can function to isolate the core from harsh external environments, including pH, physiological, or biochemical stresses; serve as a barrier membrane that controls the release of encapsulated molecules; or provide a means to achieve integration of multiple components with spatial separation. A number of techniques have been reported in the literature that are capable of fabricating core–shell architectures such as layer-by-layer self-assembly of polyelectrolytes,9 magnetic coatings,10 selective withdrawal coatings,11 and interfacial polymerization.12,13 Here, we demonstrate a unique design paradigm for the fabrication of core–shell objects in the submillimeter range based on interfacial polymerization initiated by radical generating reactions. The initiation system consists of the specific reaction between glucose and glucose oxidase to generate hydrogen peroxide (H2O2), which, in the presence of Fe2+, forms hydroxyl radicals capable of efficiently initiating (meth)acrylate monomer chain polymerization (Figure 1c). By confining one of the initiating species in a hydrogel core and the remaining initiating components and monomer in a coating solution, the formation of a shell or coating is accomplished (Figure 1a,b).
Figure 1.
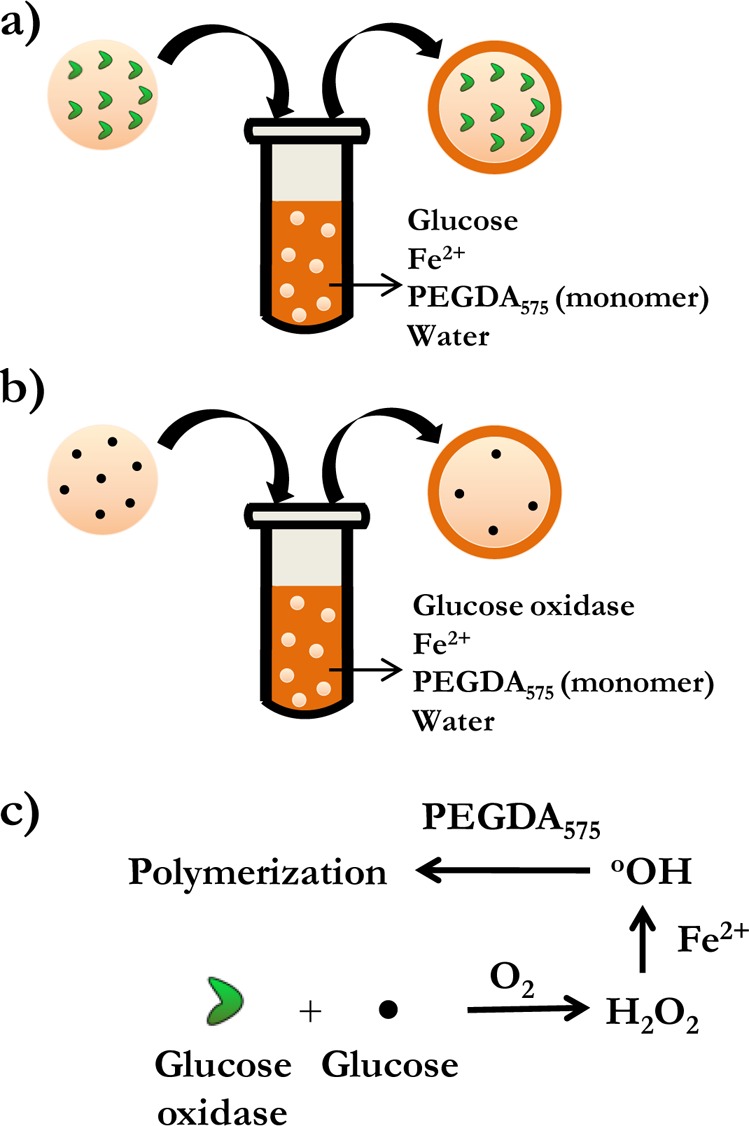
Schematic depicting the spatial organization of the initiating components in the hydrogel core and the bulk media prior to interfacial polymerization. (a) Glucose oxidase is incorporated in the core and (b) glucose is incorporated in the core. The complementary initiating species are in the coating solution. The simplified reaction mechanism underpinning the interfacial polymerization is shown in panel c.
The mild conditions of temperature and pressure employed, as well as the use of aqueous monomer solutions, render this technique suitable for encapsulating biological moieties. Unlike other surface-initiated and interfacial radical polymerizations that immobilize radicals or radical generating species at the surface or interface12,14 prior to polymerization, this technique uses a simpler approach to achieve interfacially confined initiating reactions. Here, the reaction is accomplished by mixing the initiating species into the precursor solutions used to fabricate the core and shell. Additionally, the coating that forms the shell is ideally conformal as a result of the isotropic mass transport properties of the core, which could enable cores with complex geometrical shapes to be coated uniformly. A particular implication of this approach is the ability to form liquid cores encapsulated in a shell of an unconventional geometry. This outcome is accomplished by dissolution of the core after the formation of the shell which is difficult to achieve when liquid cores are used directly as templates for the fabrication of the shell.
Johnson et al.15 have demonstrated the formation of conformal coatings on 3D hydrogels of up to several millimeters in dimension and Hume et al.23 used this approach to modify hydrogels with biological moieties to control dendritic cell activation. However, enabling interfacially confined polymerization on hydrogel cores in the submillimeter range requires a highly reactive surrounding phase that prevents the rapid diffusion of the initiating species into the bulk media, which ultimately leads to unconfined bulk polymerization. In addition, the significantly smaller size of the hydrogel makes it difficult to physically remove these cores from the coating solution immediately after the desired thickness of the shell has formed. This challenge necessitates the establishment of a self-limiting reaction or stimulus-responsive termination of polymerization. A portion of this manuscript is focused on engineering the reaction behavior and mass transport conditions to promote interfacially confined shell formation and enable control over the rate of shell growth and structure of the core–shell particle. A subsequent portion of the work demonstrates the ability of the shell to be self-supporting in the absence of a core with a defined geometry. This behavior is accomplished by degrading an initially coated solid core to form a liquid core encapsulated within the shell. The transition of the core from a gelled to a liquid state is achieved by fabricating the hydrogel core from monomers that degrade upon exposure to light.16
Experimental Section
Materials
Glucose oxidase (GOx) from Aspergillus niger, glucose, acrylamide/bisacrylamide (40%, 19:1), sorbitan monooleate (Span 80), poly(ethylene glycol) sorbitan monooleate (Tween 80), poly(ethylene glycol) diacrylate (PEGDA) (MW 575), ammonium persulfate, N,N,N′,N′-tetramethylethylenediamine (TEMED), triethanolamine, and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) used in this work were all obtained from Sigma Aldrich. Acryloxyethyl thiocarbamoyl rhodamine B (Rhodamine B acrylate) was obtained from Polysciences, and 2-(N-morpholino)ethanesulfonic acid (MES) buffer pH 4.5 was obtained from Teknova. Yellow-green fluorescent particles 20 nm and 2 μm in diameter (excitation/emission, 505/515 nm) were obtained from Invitrogen.
Microparticle Fabrication
The nonphotodegradable hydrogel microparticles used in this work were synthesized by an inverse suspension polymerization described as follows: An aqueous phase (0.5 mL) consisting of the monomer was suspended in an organic phase (5 mL) that was purged with nitrogen for 5 min. The aqueous phase was formulated by dissolving in an acrylamide/bisacrylamide solution (0.5 mL) the following components: ammonium persulfate (28 mg/mL), rhodamine B acrylate (0.005 wt %), and glucose oxidase (14 mg/mL) or glucose (0.1 M) depending on whether glucose or glucose oxidase is incorporated in the coating solution, respectively. The organic phase consisted of Span 80 (200 μL) and Tween 80 (35 μL) and hexane to make up the organic phase (5 mL). The two phases were then emulsified by sonication for 10 s after which polymerization was initiated by the addition of TEMED (10 μL) followed by magnetic stirring for 10 min. Following this procedure, the supernatant was carefully removed, and the remaining microparticles that settled at the bottom were resuspended in hexane (water) when glucose (glucose oxidase) was present in the microparticles. Since glucose is negligibly soluble in hexane, most of it remains in the water-swollen microparticles. The glucose oxidase (GOx) is trapped in the microparticles due to the smaller mesh size of the network compared to the hydrodynamic radius of GOx and, therefore, does not diffuse into the bulk media when the microparticles are resuspended in water.
Photodegradable Microparticle Fabrication
The monomers used in fabrication of photodegradable cores were poly(ethylene glycol) diphotodegradable-acrylate (PEGdiPDA; Mn = 4070 g/mol), synthesized as described earlier16,17 and poly(ethylene glycol) tetrathiol (PEG4SH; Mn 5000 g/mol) synthesized as described earlier.18 Photodegradable microparticles were prepared via inverse suspension polymerization, in which, PEGdiPDA was copolymerized with PEG4SH via base-catalyzed Michael addition in an aqueous phase that was suspended in an organic phase as previously reported.24 The organic phase comprised hexane (5 mL) containing Span 80 and Tween 80 (150 mg of a 3:1 ratio by weight respectively). The aqueous phase (0.25 mL) consisted of triethanolamine (300 mM) at pH 8.0 with PEGdiPDA (6.2 wt %), PEG4SH (3.8 wt %), and glucose (0.1 M). The polymerization reaction was allowed to proceed overnight. After the polymerization was completed, the suspension was centrifuged (Eppendorf Centrifuge model 5702) at 1000 rcf for 10 min, and the supernatant was decanted. The microparticles were then washed twice with hexanes and recovered with the same centrifugation conditions.
Interfacial Polymerization and Particle Characterization
Formation of the shell by interfacial polymerization was accomplished by injecting the coating solution (2 mL) into microparticle suspension (50 μL). The aqueous coating solution consisted of PEGDA575 (15 wt %), either glucose (0.1 M) or glucose oxidase (3 μM) depending on whether GOx or glucose is present in the microparticle respectively, iron(II) sulfate at the desired concentration, MES buffer pH 4.5 (10 mM), and either rhodamine B acrylate (0.005 wt %) or yellow-green fluorescent particles (0.01 vol%). The ensuing interfacial polymerization was allowed to continue for the desired time after which an aqueous solution (5 mL) containing TEMPO (6 mM) was added to arrest the polymerization. The supernatant was carefully removed, and the remaining particles were resuspended in deionized water before characterization. The fluorescence images of the coated particles were obtained by confocal microscopy using a Zeiss Pascal LSM 5 confocal microscope. The gels were excited with a 488 nm argon ion laser (543 nm helium neon laser) and fluorescence was monitored from 492 to 557 nm (547–680 nm).
Results and Discussion
Reaction Engineering at the Interface
The spatial organization of the initiating components can be achieved in two ways as represented in panels a and b in Figure 1 that differ in the initial location of the initiating components, namely glucose or glucose oxidase, prior to interfacial polymerization. The difference in the location of the enzyme and glucose has significant implications in the ability to control the thickness and properties of the shell, the interpenetrating layer, and the immersion time required to accomplish shell formation with minimal bulk polymerization. These are discussed in the following sections.
GOx Is Located in the Core and Glucose in the Bulk Solution
In this design, glucose oxidase (GOx) is incorporated in the core precursor solution while glucose, Fe2+, and the monomer, poly(ethylene glycol) diacrylate (MW 575) are included in the immersion phase (Figure 1a). Glucose oxidase is a bulky molecule (hydrodynamic radius, rh ≈ 43 Å) compared to the size of glucose (rh ≈ 3.5 Å). Therefore, GOx is effectively confined within the core on account of the smaller mesh size (<20 Å) of the polyacrylamide network, but glucose in the surrounding phase can diffuse into the core to initiate the reaction that generates hydrogen peroxide (H2O2). Interfacial polymerization occurs when the generated H2O2, whose locus of formation is always within the core, diffuses to the interface of the dynamically densifying shell and the surrounding media to react with the surrounding Fe2+ and generate hydroxyl radicals. This interplay of reaction behavior and the diffusion process results in shell growth in a self-limiting manner. The self-limiting growth results from H2O2, generated within the core, diffusing to the moving interface between the shell and the bulk monomer solution. In order to sustain shell growth, H2O2 must diffuse through the shell layer while avoiding reaction with Fe2+ that is diffusing into the shell to generate radicals at the shell-bulk interface.21,22 Otherwise, hydroxyl radical production occurs within the shell instead of near the desired interface, thereby contributing negligibly to shell growth.
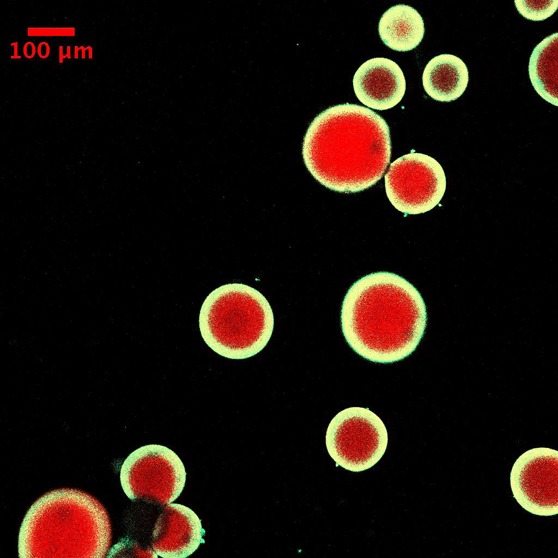
The resistance to diffusion of H2O2 increases with thickness and therefore decreases the likelihood of mass transport of initiating species into the bulk media. This approach results in minimal bulk polymerization when the microparticles are immersed in the coating solution for relatively long time periods. Figure 2 shows a representative fluorescent image of core–shell microparticles fabricated using this design at the end of 4 min of immersion. Another implication of hydrogen peroxide being generated in the core is that the polymerization of monomer diffusing into the core forms an interpenetrating network. This results because of persistent generation of hydroxyl radicals within the core due to the redox reaction between H2O2 and Fe2+ that has diffused into the core. Figure 3a provides evidence to support this hypothesis. In these experiments the core–shell particles were fabricated at two conditions, which differ in the fluorescent molecules that are used to render the shell fluorescent. In the first condition rhodamine B acrylate was used as the comonomer for the shell formulation, thereby allowing diffusion of the dye into the core enabled by the smaller size of this dye relative to the network mesh size. In the second condition fluorescent nanoparticles, more than 1 order of magnitude larger than the mesh size of the core, were incorporated in the shell formulation. Therefore, when rhodamine B acrylate is used, both the portion of the shell outside the core as well as the interpenetrating layer formed are observed in the fluorescent images. However, the interpenetrating layer is not observed when fluorescent nanoparticles are used; thereby enabling calculation of the interpenetrating layer thickness. Ensemble averaged thicknesses of the interpenetrating layer indicated that the thickness of the layer was ∼12 μm (Figure 3c).
Figure 2.
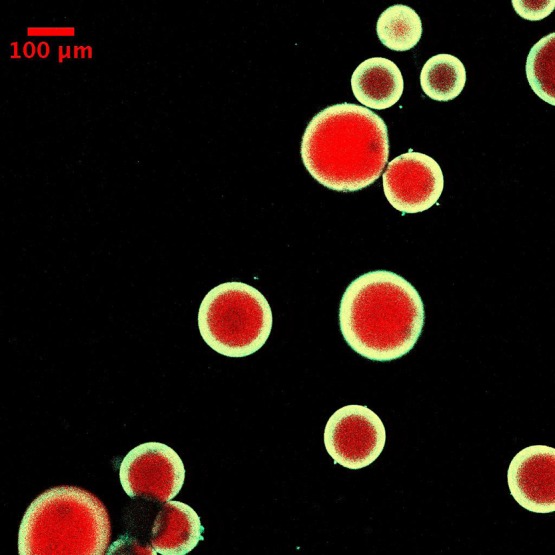
Representative fluorescence image of coated hydrogel cores when glucose oxidase is incorporated in the core. The concentration of GOx in the precursor solution used to fabricate the core was 14 mg/mL. The condition used for coating solution was 0.1 M glucose, 12.5 mM Fe2+, 0.01 vol. % yellow-green nanoparticles, 10 mM MES buffer pH 4.5, and 15 wt % PEGDA575. Immersion time was 4 min.
Figure 3.

Representative fluorescence images of coated hydrogel cores when rhodamine B acrylate was used to render the shell fluorescent (left) and yellow-green nanoparticles were incorporated in the shell to render the shell fluorescent (right). The initiating species incorporated in the core prior to interfacial polymerization was (a) GOx. The conditions used for the coating solution were 0.1 M glucose, 45 mM Fe2, 0.01 vol. % yellow-green nanoparticles (right), 0.005 wt % rhodamine B acrylate (left), 10 mM MES buffer pH 4.5, and 15 wt % PEGDA575. Immersion time was 4 min. (b) Glucose is within the core. The conditions used for the coating solution were 3 μM GOx, 45 mM Fe2, 0.01 vol. % yellow-green nanoparticles (right), 0.005 wt % rhodamine B acrylate (left), 10 mM MES buffer pH 4.5, and 15 wt % PEGDA575. Immersion time was 30 s. The thickness of the shell in each of the cases is shown in panel c.
Glucose Is Located in the Core and GOx in the Bulk Solution
In this design, the initial locations of glucose and GOx are interchanged such that glucose is located in the core and GOx in the bulk solution. The GOx cannot diffuse from the bulk media into the core, whereas glucose can rapidly diffuse into the bulk media to initiate the generation of hydrogen peroxide. This confinement of H2O2 generation into the bulk decreases the distances that H2O2 needs to diffuse to reach the nonstationary interface and initiate polymerization. The rapid diffusion of glucose into the bulk media and the resulting delocalization of the initiation reaction into the bulk solution enable the formation of thicker shells. Figure 4a shows a representative fluorescence image of the coated microparticles for the same bulk iron concentration used earlier (Figure 3), but at the end of 30 s. The average thickness of the shell was calculated to be ∼90 μm (Figure 4c). The significantly lower polymerization time employed decreases the likelihood of bulk polymerization resulting from the rapid diffusion of glucose into the bulk media. The polymerization was stopped after 30 s by injecting a high concentration of TEMPO, a powerful radical inhibitor, into the reacting mixture. Further, the thickness of the shell can be controlled by changing the bulk iron concentration, which acts to influence the degree of confinement of the initiation reaction. Previous work on the enzyme-mediated redox reaction mechanism has established that using higher concentrations of Fe2+ promotes localized polymerization by confining the redox reaction between H2O2 and Fe2+ to the interface as a result of enabling a highly reactive surrounding phase.21,22 Therefore, the concentration of iron was increased by a factor of ∼4 to enable inhibition in the bulk. However, the higher initiation rates adjacent to the interface, compared to the bulk media, allows interfacial polymerization to occur. This strategy resulted in the formation of shells ∼20 μm thick after 30 s of immersion as shown in Figure 4b and quantified in Figure 4c. Additionally, this design approach eliminated the formation of any observable interpenetrating networks (Figure 3b). Since the locus of hydrogen peroxide generation is within the bulk media, the likelihood of radicals being generated within the core is decreased, resulting in negligible polymerization of the monomer that diffuses into the core. The result is a distinct core and shell polymer network.
Figure 4.
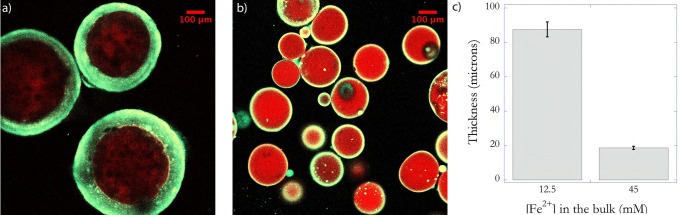
Representative fluorescence images of the coated hydrogel cores when glucose is incorporated in the core for two concentrations of iron (Fe2+) in the precursor solution used to form the shell (a) 12.5 and (b) 45 mM. Higher iron concentrations limit the shell’s growth resulting in a thinner shell (c). Glucose concentration in the precursor solution used to fabricate the core was 0.1 M. The conditions used for the coating solution were 3 μM GOx, 0.01 vol. % yellow-green nanoparticles, 10 mM MES buffer pH 4.5, and 15 wt % PEGDA575. Immersion time is 30 s.
Self-Supporting Shells: Formation of Core–Shell Particles with Liquid Cores
The ability of the shell to be self-supporting in the absence of a core with a well-defined geometry is demonstrated by using cores that possess photodegradable groups in the polymer network, as templates for shell formation. Irreversible, photoinduced cleavage of o-nitrobenzyl ether (NBE) moieties in the PEGdiPDA structure,24 initiates the transition from a gelled state to a solution state and hence the formation of a liquid core (Figure 5). To form a shell around the photodegradable cores, glucose was incorporated into the core and glucose oxidase in the bulk coating formulation prior to conducting interfacial polymerization. This strategy was chosen to increase the likelihood of formation of a distinct shell, thereby conserving the photodegradable properties of the hydrogel core. The fluorescent images of a core–shell particle prior to and after photodegradation are shown in Figure 6a,b, respectively. Before degradation (Figure 6a), the core can be distinctly seen, due to the higher background fluorescence resulting from the autofluorescence of photodegradable moieties, most notably of the interface of the core microsphere. The quantitative variation of fluorescence intensity, shown in Figure 6c highlights this idea. However, upon exposure to light, the photodegradable groups are cleaved and the resulting transition of the core from a cross-linked polymer to a degraded state (Figure 6b) decreased the fluorescence intensity (Figure 6c).
Figure 5.
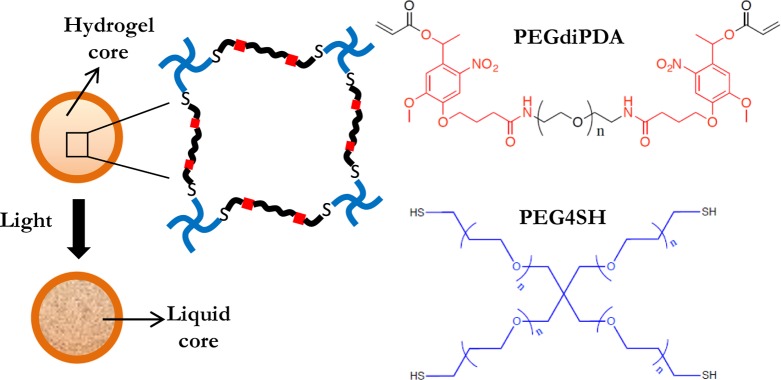
Schematic depicting photodegradation of the coated hydrogel core that possesses degradable groups in the polymer network. The core is fabricated by copolymerization of poly(ethylene glycol) diphotodegradable-acrylate (PEGdiPDA) and poly(ethylene glycol) tetrathiol (PEG4SH). The o-nitrobenzyl ether moieties (NBE) in the PEGdiPDA structure absorb strongly at 365 nm, and the resulting irreversible cleavage causes degradation of the cross-links and, ultimately, the formation a liquid core.
Figure 6.

Representative fluorescence images of coated degradable hydrogel cores before (a) and after (b) exposing to light. The concentration of glucose in the precursor solution used to fabricate the core was 0.1 M. The conditions used for forming the shell were 3 μM GOx, 45 mM Fe2+, 0.005 wt % rhodamine B acrylate, 10 mM MES buffer pH 4.5, and 15 wt % PEGDA575. Immersion time was 30 s. The core was exposed to light of wavelength 365 nm at an intensity of 20 mW/cm2 for 2 min. The normalized fluorescence intensity as a function of distance from the center of the particle before and after degradation (c). The degradation of the core is indicated by the drop in fluorescence intensity at the core–shell interface.
To provide direct evidence of the formation of a liquid core, the mass transport properties of the core were monitored before and after degradation. Monitoring was accomplished by incorporating fluorescent beads (2 μm in diameter) into the core formulation and using single particle tracking methodology20 to correlate the trajectories of the beads to the diffusion coefficient in the core. Figure 7 compares the average diffusion coefficient of the beads in the core before and after photodegradation. The diffusion coefficient before degradation is statistically insignificant from zero, whereas the value after photodegradation takes a finite value indicating the significantly increased mobility of the beads resulting from the photodegradation-induced gel to sol transition of the core. Besides demonstrating the ability to form free-standing structures, this investigation also highlights the potential toward constructing photoresponsive controlled release architectures. The conditions can be designed so that the rapid light-induced production of cleavage products is subsequently released into the surrounding environment at a rate that is determined by the properties of the shell.
Figure 7.
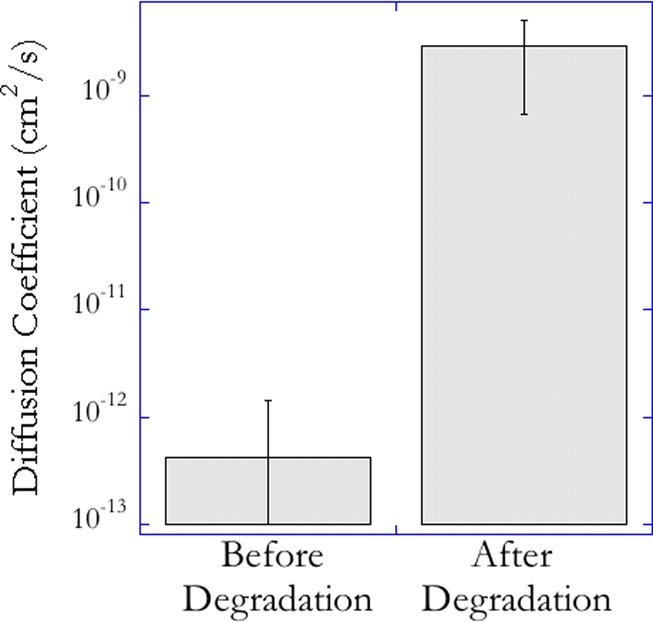
Average diffusion coefficient of fluorescent beads, 2 μm in diameter, incorporated into the core, prior to (left) and after photodegradation (right). These values were calculated by tracking the 2D trajectory of the beads before and after photodegradation and using a random walk model to correlate the mean square displacement of the beads to the diffusion coefficient.
Conclusions
Reaction engineering of the glucose oxidase-mediated redox reaction to enable core–shell formation by interfacial polymerization was described. The initial sequestration of glucose or glucose oxidase in the core is an important factor that controls the interfacial polymerization rate and the ultimate structure of the core–shell particle. Incorporating GOx in the core precursor solution and the remainder of the complementary initiating species in the bulk media prior to polymerization results in thinner shells and self-limiting shell growth. This is because the locus of hydrogen peroxide generation is always within the confines of the core resulting in a decreased likelihood of H2O2 diffusion to the nonstationary interface with increasing thickness to sustain shell growth. The presence of GOx in the core also leads to less distinct core and shell phases with a greater degree of interpenetration of the two caused by the necessity for glucose to diffuse into the core to form hydrogen peroxide. Switching the initial locations of glucose and GOx leads to rapid formation of thicker shells due to the delocalization of the hydrogen peroxide generation into the bulk solution resulting from diffusion of glucose into the bulk media. This switch also results in distinct core and shell phases as a result of negligible radical generation within the confines of the core. The polymeric shell formed by this technique is self-supporting when the initially solid core is photodegraded to form a liquid. This process for particle coating is robust and highly controllable. It could readily be used to coat particles formed by a variety of processes and core materials. This approach would enable the core particles to be more monodisperse, to have reversible degradation, and to have various material properties and particle sizes, all of which are broader than the core particle materials and techniques used here to demonstrate the coating process.
Acknowledgments
This work has been supported by the National Institutes of Health (Grant R21 EB 012188) and the National Science Foundation (DMR 1006711)
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Langer R. Acc. Chem. Res. 2000, 33(2), 94–101. [DOI] [PubMed] [Google Scholar]
- Caruso F. Adv. Mater. 2001, 13(1), 11–22. [Google Scholar]
- Freiberg S.; Zhu X. X. Int. J. Pharm. 2004, 282(1–2), 1–18. [DOI] [PubMed] [Google Scholar]
- Zhu Y.; Shi J.; Shen W.; Dong X.; Feng J.; Ruan M.; Li Y. Angew. Chem., Int. Ed. 2005, 44(32), 5083–5087. [DOI] [PubMed] [Google Scholar]
- Shenoy D. B.; Antipov A. A.; Sukhorukov G. B.; Möhwald H. Biomacromolecules 2003, 4(2), 265–272. [DOI] [PubMed] [Google Scholar]
- Bäumler H.; Georgieva R. Biomacromolecules 2010, 11(6), 1480–1487. [DOI] [PubMed] [Google Scholar]
- Zhu H. G.; McShane M. J. Langmuir 2005, 21(1), 424–430. [DOI] [PubMed] [Google Scholar]
- White S. R.; Sottos N. R.; Geubelle P. H.; Moore J. S.; Kessler M. R.; Sriram S. R.; Brown E. N.; Viswanathan S. Nature 2001, 409(6822), 794–797. [DOI] [PubMed] [Google Scholar]
- Chen W.; McCarthy T. J. Macromolecules 1997, 30(1), 78–86. [Google Scholar]
- Tsai S. S. H.; Wexler J. S.; Wan J. D.; Stone H. A. Appl. Phys. Lett. 2011, 99, 15. [Google Scholar]
- Cohen I.; Li H.; Hougland J. L.; Mrksich M.; Nagel S. R. Science 2001, 292(5515), 265–267. [DOI] [PubMed] [Google Scholar]
- Scott C.; Wu D.; Ho C. C.; Co C. C. J. Am. Chem. Soc. 2005, 127(12), 4160–4161. [DOI] [PubMed] [Google Scholar]
- Yow H. N.; Routh A. F. Soft Matter 2006, 2(11), 940–949. [DOI] [PubMed] [Google Scholar]
- Luo Y.; Gu H. Macromol. Rapid Commun. 2006, 27(1), 21–25. [Google Scholar]
- Johnson L. M.; DeForest C. A.; Pendurti A.; Anseth K. S.; Bowman C. N. ACS Appl. Mater. Interfaces 2010, 2(7), 1963–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloxin A. M.; Tibbitt M. W.; Anseth K. S. Nat. Protoc. 2010, 5(12), 1867–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloxin A. M.; Kasko A. M.; Salinas C. N.; Anseth K. S. Science 2009, 324(5923), 59–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbanks B. D.; Singh S. P.; Bowman C. N.; Anseth K. S. Macromolecules 2011, 44(8), 2444–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson L. M.; Fairbanks B. D.; Anseth K. S.; Bowman C. N. Biomacromolecules 2009, 10(11), 3114–3121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H.; Sheetz M. P.; Elson E. L. Biophys. J. 1991, 60(4), 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy R.; Bowman C. N. Biomaterials 2012, 33(29), 6909–6914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shenoy R.; Bowman C. N. Macromol. Theory Simul. 2013, 10.1002/mats.201200062. [DOI] [Google Scholar]
- Hume P. S.; Bowman C. N.; Anseth K. S. Biomaterials 2011, 32(26), 6204–6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbitt M. W.; Han B. W.; Kloxin A. M.; Anseth K. S. J. Biomed. Mater. Res., Part A 2012, 100A(7), 1647–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]