

Abstract
Continuity of cycling cell lineages relies on the activities of undifferentiated stem cell-containing subpopulations. Transition to a differentiating state must occur periodically in a fraction of the population to supply mature cells, coincident with maintenance of the undifferentiated state in others to sustain a foundational stem cell pool. At present, molecular mechanisms regulating these activities are poorly defined for most cell lineages. Spermatogenesis is a model process that is supported by an undifferentiated spermatogonial population and transition to a differentiating state involves attained expression of the KIT receptor. We found that impaired function of the X chromosome-clustered microRNAs 221 and 222 (miR-221/222) in mouse undifferentiated spermatogonia induces transition from a KIT– to a KIT+ state and loss of stem cell capacity to regenerate spermatogenesis. Both Kit mRNA and KIT protein abundance are influenced by miR-221/222 function in spermatogonia. Growth factors that promote maintenance of undifferentiated spermatogonia upregulate miR-221/222 expression; whereas exposure to retinoic acid, an inducer of spermatogonial differentiation, downregulates miR-221/222 abundance. Furthermore, undifferentiated spermatogonia overexpressing miR-221/222 are resistant to retinoic acid-induced transition to a KIT+ state and are incapable of differentiation in vivo. These findings indicate that miR-221/222 plays a crucial role in maintaining the undifferentiated state of mammalian spermatogonia through repression of KIT expression.
Keywords: Differentiation, KIT, Spermatogonial stem cell, Undifferentiated spermatogonia, miR-221, miR-222, Mouse
INTRODUCTION
Male fertility requires continual spermatogenesis, a process dependent on activities of an undifferentiated spermatogonial population composed of spermatogonial stem cells (SSCs) and transient amplifying progenitor spermatogonia (de Rooij and Russell, 2000; Oatley and Brinster, 2012). Self-renewal of SSCs maintains a constant pool from which progenitors will arise and amplify in number before committing to a pathway of terminal differentiation. During steady-state conditions, spermatogenesis initiates when undifferentiated progenitor spermatogonia transition to a differentiating state, thereby committing to eventual formation of spermatozoa. In testes of mice, the timeframe of spermatogenesis is 35 days and constant cycling of the germ cell lineage relies on occurrence of the undifferentiated-to-differentiating spermatogonial transition at a defined interval (Oakberg, 1956; Clermont and Trott, 1969). To provide robustness, a majority of undifferentiated spermatogonia make the transition and only a small subpopulation remains in an undifferentiated state. At present, molecular mechanisms regulating transition from an undifferentiated to a differentiating state in mammalian spermatogonia are undefined.
In testes of mammals, a somatic cell population composed of Sertoli, Leydig and myoid cells supports germ cell activities. Signaling from a combination of growth factors secreted from these somatic cells, including glial cell line-derived neurotrophic factor (GDNF), fibroblast growth factor 2 (FGF2), and colony stimulating factor 1 (CSF-1), promotes maintenance and proliferation of the undifferentiated spermatogonial population, including self-renewal of SSCs (Meng et al., 2000; Kubota et al., 2004b; Oatley et al., 2009). In addition, signaling from retinoic acid (RA) induces spermatogonial transition from an undifferentiated to a differentiating state (Morales and Griswold, 1987; van Pelt and de Rooij, 1990b; Van Pelt and De Rooij, 1990a). Thus, undifferentiated spermatogonia are in an active state of mitosis while being primed to initiate differentiation upon activation of RA signaling.
A hallmark of the transition to a differentiating state in mammalian spermatogonia is the attainment of KIT receptor expression (Yoshinaga et al., 1991; de Rooij, 1998; Schrans-Stassen et al., 1999). Expression of KIT protein is absent in undifferentiated spermatogonia and becomes first detectable on the surface of differentiating spermatogonia and persists until the leptotene stage of spermatocyte development (Manova et al., 1990; Yoshinaga et al., 1991; Schrans-Stassen et al., 1999). In addition, expression of KIT on the surface of spermatogonia is inversely related to SSC capacity (Shinohara et al., 2000; Kubota et al., 2003). Furthermore, during neonatal testis development the undifferentiated spermatogonial population develops from transition of prospermatogonia (e.g. gonocytes) that are KIT+ (Yoshinaga et al., 1991). Thus, establishment of an undifferentiated state in spermatogonia coincides with suppression of KIT expression, which is maintained until activation of RA signaling induces transition to a differentiating state.
The expression profile for KIT in male germ cells (present in prospermatogonial precursors, suppressed in undifferentiated spermatogonia and subsequently induced upon transition to a differentiating state) suggests silencing by small RNAs. Mounting evidence indicates small non-coding RNAs, such as microRNAs (miRNAs), play essential roles in maintenance of stem and progenitor cell populations by regulating the stability and translation of mRNAs (Ghildiyal and Zamore, 2009). Importantly, recent studies with mice showed that miRNA biogenesis in germ cells is required for normal spermatogenesis (Maatouk et al., 2008; Korhonen et al., 2011; Romero et al., 2011; Liu et al., 2012). However, the role of individual miRNAs for maintenance of specific stages of germ cell development is largely unexplored. Studies of the hematopoietic progenitor population revealed the regulation of KIT protein translation by a cluster of miRNAs located on the X chromosome, miR-221 and miR-222 (Felli et al., 2005). Interestingly, miR-221 and miR-222 are transcribed from the same promoter as a single transcriptional unit, and overexpression has been detected in several cancer cell types, indicating a role in promotion of the undifferentiated state (Ciafrè et al., 2005; He et al., 2005; Galardi et al., 2007; Gillies and Lorimer, 2007; Igoucheva and Alexeev, 2009; Di Leva et al., 2010; Pineau et al., 2010). In the current study, we tested the hypothesis that miR-221/222 play an important role in regulating the undifferentiated state in spermatogonia. We found that miR-221/222 repress transition from a KIT– to a KIT+ state and influence maintenance of stem cell capacity.
MATERIALS AND METHODS
Animals
Male C57BL/6J and lacZ expressing B6;129S-Gt(ROSA)26Sor/J (designated Rosa) mice (The Jackson Laboratory; Bar Harbor, ME, USA) were used as germ cell donors. Recipients for SSC transplantation were F1 progeny of C57BL/6J × 129S1/svlmJ mice. All animal procedures were approved by the Washington State University Institutional Animal Care and Use Committee (IACUC).
Apoptosis and proliferation analysis
The percentage of apoptotic and mitotic cells in cultures of undifferentiated spermatogonia were determined using flow cytometric analysis (FCA) with the Guava Nexin Apoptosis Assay and Click-iT EdU Detection Kit (Invitrogen), respectively.
Isolation of THY1+ and KIT+ spermatogonia
Single-cell suspensions of testis cells were subjected to magnetic-activated cell sorting (MACS) using antibodies recognizing THY1 (Miltentyi Biotec, CD90.2 Microbeads) or KIT (Miltenyi Biotec, CD117 Microbeads) as described previously (Kubota et al., 2004a; Oatley and Brinster, 2006).
Primary cultures of THY1+ undifferentiated spermatogonia
Primary cultures of mouse undifferentiated spermatogonia were generated from isolated THY1+ testis cells as described previously (Kubota et al., 2004b; Oatley and Brinster, 2006) and maintained in mouse serum-free media (mSFM) (Kubota et al., 2004b) with supplementation of 20 ng/ml recombinant human GDNF (Peprotech; Rocky Hill, NJ, USA) and 1 ng/ml recombinant human FGF2 (BD Biosciences; San Jose, CA, USA).
RT-PCR analysis for Kit mRNA
RNA was isolated using Trizol Reagent (Invitrogen), and 0.5-1 μg of RNA was reverse transcribed using oligo (d)T priming and SuperScript 3 reverse transcriptase (Invitrogen). PCR reactions were performed with previously validated primers designed to recognize a 765-bp segment of Kit mRNA (Keller et al., 1993). All samples were also subjected to PCR with primers recognizing Gapdh mRNA: 5′-AACTTTGGCATTGTGGAAGGGCTC-3′ and 5′-TGGAAGAGTGGGAGTTGCTGTTGA-3′. PCR products were visualized using agarose gel electrophoresis.
Flow cytometric analysis of KIT+ cells
Cultured THY1+ undifferentiated spermatogonia were collected as single-cell suspensions by incubation in trypsin-EDTA solution and pelleted by centrifugation at 600 g for 7 minutes. Cells (1×106) were incubated with monoclonal rat anti-mouse KIT antibody conjugated to PE/Cy5 (1:100; Abcam, Cambridge, MA, USA) on ice for 30 minutes followed by washing in PBS and were analyzed using FCA (Guava Easy Cyte Plus Flow Cytometer, Millipore Corporation, Hayward, CA, USA). Control cells for gating were processed using identical conditions but with omission of primary antibody.
Single cell immunocytochemical staining
Cultured THY1+ undifferentiated spermatogonia were dissociated from STO feeder cells and single-cell suspensions generated by trypsin-EDTA digestion. Cells were adhered to poly-l-lysine-coated coverslips followed by fixation with 4% paraformaldehyde (PFA) and incubation in PBS with 0.1% Triton X-100 (PBS-T). Nonspecific antibody binding was blocked by incubation in 10% normal goat or donkey serum diluted in PBS-T followed by overnight incubation at 4°C with rat anti-KIT (1 μg/ml; Millipore), rabbit anti-PLZF (4 μg/ml; Santa Cruz Biotechnology), rabbit anti-SALL4 (1 μg/ml; Abcam) or guinea pig anti-SOHLH1 (1:200; gift from Dr A. Rajkovic, University of Pittsburgh) primary antibodies. Negative controls were incubated with normal rabbit or goat IgG in place of primary antibody. Alexa546-conjugated goat anti-rabbit IgG (Invitrogen), rhodamine-conjugated goat anti-guinea pig IgG (Santa Cruz Biotechnology) or Alexa546-conjugated donkey anti-rat IgG (Invitrogen) antibodies were used for secondary labeling. Coverslips were mounted onto glass slides with VectaShield medium containing DAPI and examined by fluorescence microscopy. Negative controls were used to set the background fluorescent intensity for each sample. The percentage of positive cells staining for KIT, PLZF (ZBTB16), SALL4 or SOHLH1 was determined by counting fluorescently labeled cells in ten random fields of view on each coverslip and dividing by the total number of cells in the same field (i.e. DAPI-stained nuclei).
miR-221 in situ hybridization analysis
Testes from adult mice were fixed in 4% PFA, embedded in paraffin and processed for in situ hybridization (ISH) as described previously (Nuovo et al., 2009). Briefly, cross-sections (5 μm) were adhered to glass slides, deparaffinized, rehydrated and incubated in 0.2 N HCl followed by washing in DEPC-treated water. Sections were treated with 1 μg/ml proteinase K followed by immersion in 0.2% glycine then 100% ethanol and were then air dried. Sections were then incubated overnight at 37°C with 200 nM digoxigenin (DIG)-labeled locked nucleic acid (LNA) miR-221 or scrambled probes (Exiqon, Woburn, MA, USA). The LNA-miR-221 probe has homology to both the immature and mature forms and therefore can hybridize with transcript in the nucleus and cytoplasm. On the next day, sections were washed and incubated with BCIP p-toluidine salt solution (Sigma) at 37°C and counterstained with Nuclear Fast Red. For fluorescent ISH staining, sections were incubated with an anti-DIG/fluorescein fab fragment antibody (1:500), and coverslips mounted with Prolong gold anti-fade reagent containing DAPI. Immunofluorescence co-staining in serial cross-section was conducted to localize PLZF protein and miR-221 in the same cells. Briefly, serial sections were boiled in citrate buffer (10 mM citric acid, 0.05% Tween-20, pH 6.0) for antigen retrieval. Nonspecific antibody binding was blocked by incubation with 10% normal donkey serum and sections were incubated with anti-PLZF antibody (1:200; Santa Cruz Biotechnology) overnight at 4°C, followed by washing in PBS and incubation with Alexa546 donkey anti-rabbit IgG (Invitrogen) secondary antibody.
miR-221 and miR-222 inhibitor and mimic treatments
Cultured THY1+ undifferentiated spermatogonia (1×105) were transfected with commercially available chemically modified antisense oligonucleotide inhibitors of miR-221 and miR-222 (100 nM each; mirVana, Life Technologies, product #4464084) or non-targeting control oligonucleotide (100 nM; Life Technologies, product #AM17010) for 24 hours. For miR-221/222 transient overexpression, cells (1×105) were transfected for 18 hours with 50 nM commercially available stability-enhanced miR-221 (product #C-310583-07-0005) and miR-222 (product #C-310584-07-0005) oligonucleotide mimics (MirIDIAN Mimic) or non-targeting negative control #1 mimic (product #IN-001005-01-05), all from Dharmacon (Chicago, IL, USA). All transfections were performed with Lipofectamine 2000 reagent (Invitrogen) in mSFM supplemented with GDNF and FGF2. On the next day, fresh media with growth factors was replaced or cells were collected by gentle pipetting, pelleted by centrifugation at 600 g for 7 minutes, washed in mSFM three times and processed for downstream analysis.
miR-221 and miR-222 lentiviral overexpression in cultured THY1+ undifferentiated spermatogonia
DNA sequences corresponding to pre-miR-221 and pre-miR-222 were amplified from genomic DNA using the primers: pre-miR-221, 5′-AAACGCGTTTTGCTATGATGCTCAGCTTCA-3′ and 5′-GGGCTAGCACCACCAATCGAATTATTTGAGG-3′; or pre-miR-222, 5′-AAACGCGTTTCCAAAACTGAATGAATGAATAATG-3′ and 5′-GGGCTAGCATTCAGAGCACAAGAAAAATATGC-3′. PCR products were sub-cloned into the pLV-EF1α-MCS-IRES-Puro lentiviral vector (Biosettia). Lentiviral particles were produced in HEK 293T cells as described previously (Kaucher et al., 2012). Single-cell suspensions of cultured Rosa THY1+ spermatogonia were transduced with lentiviral vectors using a multiplicity of infection (MOI) of five in the presence of polybrene (8 μg/ml) for 18 hours without STO feeder cells. Cells were then transferred onto STO feeder cells and allowed to recover for 24 hours followed by puromycin (1 μg/ml) treatment for 72 hours to select cells with stable incorporation of transgenes. Cells were allowed to recover for 24 hours prior to collection for transplantation analysis or extraction of RNA.
Quantitative RT-PCR analysis for miR-221, miR-222 and Kit
RNA was collected using Trizol reagent (Invitrogen). For miR-221/222 analysis, 10 μg of RNA was reverse transcribed using miR-221, miR-222 or sno202 (Snord68; endogenous control) specific primers (all from Applied Biosystems), and qPCR reactions performed with validated TaqMan probes for sno202, miR-221 and miR-222 using an ABI 7500 Fast Sequence Detection system. The primer annealing step of the reverse transcription reaction did not include heat denaturation. For Kit mRNA analysis, 1 μg of RNA was reverse transcribed using oligo (d)T priming and SuperScript 3 reverse transcriptase (Invitrogen) and qPCR reactions performed with validated TaqMan probes for Kit and the constitutively expressed gene ribosomal protein S2 (Rps2). CT values for miR-221, miR-222 or Kit in each sample were subtracted by corresponding CT values for sno202 or Rps2 to generate ΔCT values. Relative abundance of miR-221, miR-222 or Kit in each sample was determined using the formula: 2–ΔΔCT. For comparison between treatments, fold-differences were determined by dividing the relative abundance values for control and treated samples by the mean of the controls (Schmittgen and Livak, 2008).
Northern blot analysis
Small RNAs were isolated using the mirVana miRNA Isolation Kit (Invitrogen) and 2 μg separated in a 15% TBE-Urea gel by electrophoresis followed by transfer onto a nylon membrane and UV crosslinking. Blots were pre-hybridized in Ultra-Hyb Buffer (Ambion) followed by addition of 32P-labeled probe. Probes were synthesized for miR-221, miR-222 and U6 (Rnu6; loading control) with the MirVana MirNA Probe Synthesis Kit (Ambion) using α32P-labeled UTP (PerkinElmer, Waltham, MA, USA). Probe sequences were: miR-221, 5′-GAAACCCAGCAGAATGTAGCT-3′; miR-222, 5′-GAGACCCAGTAGCCAGATGTAGCT-3′; and U6, 5′-GCAGGGGCCATGCTAATCTTCTCTGTATCG-3′. Following overnight incubation, membranes were washed three times with North2South Hybridization Stringency Buffer (Thermo Scientific, Rockford, IL, USA) and exposed to BioMax film (Kodak, Rochester, NY, USA) for 1-72 hours at –80°C before developing.
SSC transplantation
Cultured Rosa THY1+ undifferentiated spermatogonia were collected as single-cell suspensions by digesting entire culture wells including STO feeders with trypsin-EDTA solution. Cells were diluted in mSFM at a concentration of 1×106 cells/ml and ∼10 μl of suspension was microinjected into seminiferous tubules of F1 recipient mice that had been pre-treated with busulfan (60 mg/kg body weight) 2 months previously to eliminate endogenous germ cells as described previously (Oatley and Brinster, 2006). Recipient testes were examined for colonies of donor-derived spermatogenesis 1 or 2 months after transplantation by incubation with X-gal to stain for β-galactosidase activity.
Western blot analysis
Testes and cultured spermatogonia were lysed in RIPA buffer and 25-50 μg of protein separated in NuPAGE 4-12% Bis-Tris gels followed by transfer onto nitrocellulose membranes and blocking in 5% nonfat dry milk. Membranes were incubated overnight at 4°C with antibodies against PLZF (Santa Cruz Biotechnology), SALL4 (Abcam), SOHLH1 (gift from Dr A. Rajkovic) and KIT (ACK2 clone; Millipore), all at 1:1000. The next day, blots were washed and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (Santa Cruz Biotechnology) at 1:5000. Detection was achieved using PicoWest Chemiluminescent Substrate (Thermo Scientific) and digital images captured with a FujiFilm LAS-4000 molecular imager. Blots were stripped with Restore Buffer (Thermo Scientific) and re-probed with an anti-TUBB1 (Abnova) antibody at 1:3000.
Statistical analysis
Data are presented as means±s.e.m. for three independent experiments. Differences between means were examined using the general linear model one-way ANOVA or t-test function of GraphPad Prism 5 (La Jolla, CA, USA) or SPSS statistical software (IBM Corporation). Multiple comparisons analysis was conducted using Tukey’s posthoc test. Differences between means were considered significant at P<0.05.
RESULTS
The Kit gene is transcribed in mouse undifferentiated spermatogonia but protein translation is suppressed
Investigating molecular pathways regulating undifferentiated spermatogonia is challenging using in vivo experimentation owing to rarity of the cells. Therefore, we utilized primary cultures of isolated THY1+ undifferentiated spermatogonia. Maintenance of these cells with STO feeder cell monolayers in serum-free media supplemented with the growth factors GDNF and FGF2 supports formation and long-term expansion of germ cell clumps (Fig. 1A) (Kubota et al., 2004b). These clumps represent the undifferentiated spermatogonial population in vivo consisting of SSCs that regenerate spermatogenesis after transplantation (Fig. 1B) and non-stem cell progenitor spermatogonia with nearly all cells (∼99%) being KIT– (Fig. 1C) (Oatley et al., 2010). Here, we found that treatment of cultured germ cell clumps with all-trans retinoic acid (AtRA), a bioactive form of RA, for 24 hours induced 44.8±2.9% (n=3 different cultures) of cells to become KIT+, indicating transition to a differentiating state (Fig. 1C). By comparison, only 0.6±0.1% (n=3 different cultures) and 1.2±0.2% (n=3 different cultures) of cells were KIT+ in untreated and vehicle (DMSO)-treated cultures, respectively (Fig. 1C). Based on immunocytochemical staining, 24.3±1.2% (n=3 different cultures) of the cells were found to be KIT+ after AtRA treatment, whereas only 1.3±0.3% (n=3 different cultures) of cells stained for KIT expression in vehicle-treated control cultures (Fig. 1D).
Fig. 1.

Undifferentiated characteristics and KIT phenotype of cultured mouse THY1+ spermatogonia. (A) Representative phase-contrast image of germ cell clumps (arrows) generated from isolated THY1+ spermatogonia. Scale bar: 100 μm. (B) Representative image of a recipient mouse testis 2 months after transplantation with cultured lacZ-expressing THY1+ spermatogonial clumps. Each blue segment is a colony of spermatogenesis derived from a single transplanted SSC. Scale bar: 2 mm. (C) Representative images of flow cytometric analysis for KIT+ cells in cultures of THY1+ spermatogonia receiving no treatment, DMSO for 24 hours or 0.5 μm AtRA for 24 hours. Percentages of cells determined to be KIT+ for each condition are indicated. (D) Representative images of fluorescence immunocytochemical staining for KIT (red) in cultures of THY1+ spermatogonia treated with DMSO or 0.5 μm AtRA for 24 hours. KIT-stained cells are indicated by arrows. DNA is stained with DAPI (blue). Scale bars: 100 μm. (E) RT-PCR analysis for Kit mRNA in freshly isolated THY1+ spermatogonia from testes of juvenile mice, cultured THY1+ spermatogonial clumps (Undiff Spg) and KIT+ differentiating spermatogonia (Diff Spg) isolated from testes of adult mice. –RT samples were not subjected to reverse transcription. Analysis of Gapdh mRNA was used as a loading control. (F) Representative images of western blot analysis for KIT protein in two different cultures of THY1+ undifferentiated spermatogonia. Protein lysates from adult mouse testes were used as a positive control and analysis of TUBB1 was used as a loading control. (G) Representative images of western blot analyses for SALL4, SOHLH1 and PLZF protein in two different cultures of THY1+ undifferentiated spermatogonia. Analysis of TUBB1 was used as a loading control. (H) Percentage of cultured THY1+ undifferentiated spermatogonia expressing PLZF, SALL4, SOHLH1 and KIT. Values were determined from immunocytochemical staining of single cell suspensions generated from germ cell clumps and are mean±s.e.m. of three different cultures.
Induction of a KIT+ state in cultured THY1+ undifferentiated spermatogonia could result from activation of Kit transcription or regulation of protein translation. Previous studies suggest that Kit mRNA is present in undifferentiated spermatogonia of mouse testes when KIT protein is absent (Schrans-Stassen et al., 1999; Oatley et al., 2006; Prabhu et al., 2006; Oatley et al., 2009). Here, we confirmed the presence of Kit mRNA in cultured THY1+ undifferentiated spermatogonia using RT-PCR analysis (Fig. 1E); however, KIT protein was not detectable using western blot analysis (Fig. 1F). Next, we examined whether proteins known to regulate Kit transcription including spermatogenesis and oogenesis HLH 1 (SOHLH1) (Suzuki et al., 2012), zinc finger and BTB domain containing 16 (ZBTB16 or PLZF) (Filipponi et al., 2007) and sal-like 4 (SALL4) (Hobbs et al., 2012) are expressed by cultured THY1+ undifferentiated spermatogonia. Western blot analysis revealed the presence of all these proteins in cultured THY1+ undifferentiated spermatogonial populations, which are primarily KIT– (Fig. 1G). Furthermore, based on immunocytochemical staining, 97.0±0.6%, 58.0±2.4% and 47.1±5.9% of cells in the cultured THY1+ undifferentiated spermatogonial population (n=3 different cultures) were found to express PLZF, SOHLH1 and SALL4, respectively (Fig. 1H; supplementary material Fig. S1). However, only 1.1±0.3% of the cells were KIT+ (Fig. 1H; supplementary material Fig. S1), indicating that a repressive mechanism exists to prevent protein translation and, therefore, transition to a KIT+ state.
Expression of miR-221/222 distinguishes KIT– undifferentiated and KIT+ differentiating spermatogonia
The finding that KIT expression is suppressed in cultured undifferentiated spermatogonia even in the presence of active Kit transcription suggests regulation by small RNAs. Studies of the hematopoietic progenitor cell population and several cancer cell types showed that Kit mRNA is a target of the X chromosome clustered miRNAs 221 and 222 (Ciafrè et al., 2005; Felli et al., 2005; He et al., 2005; Galardi et al., 2007; Gillies and Lorimer, 2007; Igoucheva and Alexeev, 2009; Di Leva et al., 2010; Pineau et al., 2010). Here, we used northern blot and in situ hybridization (ISH) analyses to profile expression of miR-221 and miR-222 in the undifferentiated spermatogonial population of mouse testes. Isolation of pure populations of undifferentiated spermatogonia is not possible utilizing currently available methods and the overall number of cells that can be collected using enrichment strategies is limiting for analyses requiring large amounts of sample. Therefore, we used primary cultures of THY1+ undifferentiated spermatogonia. Northern blot analysis revealed that mature transcripts for both miR-221 and miR-222 are present in these cultures, which are primarily KIT–, and are absent in KIT+ spermatogonia isolated from testes of adult mice (Fig. 2A). Next, we performed ISH analysis to localize miR-221/222 expression in cross-sections of testes from adult (2 months of age) C57BL/6 mice that contain all spermatogonial subtypes. Because the genome organization of miR-221 and miR-222 is such that the sequences are transcribed as a single pre-miRNA from the same promoter (Di Leva et al., 2010), we focused the analysis using a probe to miR-221 only. We observed staining for miR-221 expression within germ cells residing along the basement membrane of seminiferous tubules and with a morphological appearance of undifferentiated spermatogonia (Fig. 2B-D). Importantly, the miR-221-expressing spermatogonia also stained for expression of the undifferentiated spermatogonial protein PLZF (Fig. 2E-G). Taken together, these findings indicate that the undifferentiated spermatogonial population can be defined by expression of miR-221/222, whereas miR-221/222 is absent in differentiating spermatogonia.
Fig. 2.

Expression of miR-221/222 in mouse spermatogonia. (A) Representative images of northern blot analysis of miR-221 and miR-222 in cultures of THY1+ undifferentiated spermatogonia (Undiff Spg), KIT+ differentiating spermatogonia (Diff Spg) isolated from testes of adult mice, and mouse brain (+ control). Analysis of U6 snRNA was used as a loading control. (B-D) Representative images of ISH staining for miR-221 expression in cross-sections of testes from adult mice. DNA was stained with Nuclear Fast Red and arrows indicate spermatogonia stained for miR-221 expression. C shows a higher magnification image of the stained spermatogonia indicated by the arrow in B. D is negative control staining with a scrambled probe. Scale bars: in B,D, 100 μm; in C, 20 μm. (E-G) Representative images of co-fluorescence labeling for miR-221 (green) by ISH (E,F) and the undifferentiated spermatogonial protein PLZF (red) by immunostaining (G) in serial cross-sections of testes from adult mice. F shows a higher magnification image of the miR-221-stained spermatogonia indicated by the arrow in E. G is an overlay of image F with a serial cross-section stained for the undifferentiated spermatogonial marker PLZF. DAPI (blue) was used to stain DNA and arrows indicate undifferentiated spermatogonia staining for expression of miR-221 and PLZF. Scale bars: in E, 100 μm; in F,G, 20 μm.
Inhibition of miR-221/222 function in cultured spermatogonia impairs maintenance of the undifferentiated state
Core attributes of undifferentiated cell populations are the ability for proliferative amplification without commitment to terminal differentiation and the capacity for a portion of the cells to perform as stem cells for regenerating cell lineages. For mammalian male germlines, commitment to differentiation involves transition from a KIT– to a KIT+ state, and stem cells represent a portion of the undifferentiated spermatogonial population capable of regenerating continual spermatogenesis. To investigate whether miR-221/222 plays an important role in regulating the undifferentiated state, we treated primary cultures of THY1+ spermatogonia with a combination of synthetic inhibitors of miR-221 and miR-222. The miR inhibitors hybridize with mature forms of target miRNAs thereby preventing interaction of seed sequences with 3′ UTRs of mRNAs. After 24 hours of treatment, qPCR analysis revealed significant inhibition of both miR-221 and miR-222 in cells treated with the combination of miR-221/222 inhibitors compared with control cells (Fig. 3A). Next, we examined the effects of miR-221/222 inhibitor treatment on the KIT phenotype of cultured THY1+ undifferentiated spermatogonia. After 24 hours of treatment, qRT-PCR analysis revealed that Kit mRNA abundance was significantly (P<0.05) increased by 1.7±0.4-fold (n=3 different cultures) in cultures treated with miR-221/222 inhibitors compared with control cultures (Fig. 3B). Using flow cytometric analysis, we found that after 24 hours of treatment with miR-221/222 inhibitors, 10.8±1.1% of cells were KIT+ (n=3 different cultures), which was significantly (P<0.05) greater than the 0.7±0.2% KIT+ cells (n=3 different cultures) in control inhibitor-treated cultures (Fig. 3C,E). This finding was confirmed with immunocytochemical staining that showed 14.5±2.3% of cells were KIT+ (n=3 different cultures) in cultures treated with miR-221/222 inhibitors, which was significantly (P<0.05) greater than the 1.4±0.3% KIT+ cells (n=3 different cultures) in control inhibitor-treated cultures (Fig. 3D,E). Importantly, KIT protein was detectable using western blot analysis 24 hours after miR-221/222 inhibitor treatment but was not detectable in control inhibitor-treated cells (Fig. 3F). Moreover, the abundance of factors implicated as regulators of Kit transcription, including PLZF, SOHLH1 and SALL4, was not different in cultures treated with miR-221/222 inhibitors compared with non-targeting control inhibitor-treated cultures (Fig. 3G). Thus, the abundance of miR-221/222 influences the KIT phenotype of cultured spermatogonia irrespective of factors that influence Kit transcription.
Fig. 3.

Effects of miR-221/222 inhibition on maintenance of the undifferentiated state in cultured THY1+ undifferentiated spermatogonia. (A) qRT-PCR analysis to monitor miR-221 and miR-222 inhibition in cultured THY1+ undifferentiated spermatogonia 24 hours after treatment with a combination of synthetic miR-221/222 inhibitors or a scrambled non-targeting control inhibitor. (B) Quantitative comparison of Kit mRNA abundance in cultured THY1+ undifferentiated spermatogonia 24 hours after treatment with miR-221/222 inhibitors or a scrambled non-targeting control inhibitor. (C) Representative images of flow cytometric analysis for KIT+ cells in cultures of THY1+ undifferentiated spermatogonia treated with control or miR-221/222 inhibitors. Percentages of cells determined to be KIT+ for each treatment are indicated. (D) Representative images of fluorescence immunocytochemical staining for KIT+ cells in single cell suspensions of cultured THY1+ undifferentiated spermatogonia treated with control or miR-221/222 inhibitors. Arrows indicate KIT+ cells. Scale bars: 100 μm. (E) Quantitative comparison of KIT+ cells in cultures of THY1+ undifferentiated spermatogonia 24 hours after treatment with a combination of miR-221 and miR-222 inhibitors or a scrambled non-targeting control inhibitor using flow cytometric analysis (FCA) or immunocytochemical staining (ICC). (F) Representative images of western blot analysis of KIT protein expression in cultured THY1+ undifferentiated spermatogonia treated with a non-targeting control or a combination of miR-221/222 inhibitors for 24 hours. Protein lysate of testes from an adult mouse was used as a positive control and TUBB1 was used as a loading control. (G) Representative images of western blot analysis of PLZF, SALL4 and SOHLH1 protein abundance in cultured THY1+ undifferentiated spermatogonia 24 hours after treatment with a non-targeting control or a combination of miR-221/222 inhibitors. TUBB1 was used as a loading control. (H) Representative images of recipient mouse testes 2 months after transplantation with cultured Rosa lacZ-expressing THY1+ undifferentiated spermatogonia treated with a non-targeting control or a combination of miR-221/222 inhibitors for 3 days. Each blue segment is a colony of spermatogenesis derived from a single transplanted SSC. Scale bar: 2 mm. (I) Quantitative comparison of SSC numbers in cultures of lacZ-expressing THY1+ undifferentiated spermatogonia treated with a non-targeting control or a combination of miR-221/222 inhibitors for 3 days. SSC numbers were derived from β-galactosidase-stained colonies of donor-derived spermatogenesis in recipient testes and normalized to 105 cells injected. (J) Quantitative comparison of the number of cells recovered from THY1+ undifferentiated spermatogonial cultures prior to transplantation analysis at 3 days after treatment with a non-targeting control or a combination of miR-221/222 inhibitors. (K) Quantitative comparison of the percentage of apoptotic cells in cultures of THY1+ undifferentiated spermatogonia treated with a non-targeting control or a combination of miR-221/222 inhibitors for 2 days. All quantitative data are mean±s.e.m. of three different cultures. *P<0.05.
Next, we treated cultures of THY1+ undifferentiated spermatogonia derived from lacZ-expressing Rosa donor mice with miR-221/222 or control inhibitors for 3 days followed by transplantation into testes of recipient mice to measure stem cell content. Recipient testes were examined 2 months later for donor-derived colonies of spermatogenesis, which could be identified by staining for β-galactosidase activity and are indicative of stem cell capacity of the cultured cell population (Brinster and Avarbock, 1994; Brinster and Zimmermann, 1994). The average number of donor-derived colonies of spermatogenesis generated by miR-221/222 inhibitor-treated cultures was 49.0±11.2 colonies/105 cells injected (n=3 different cultures and 18 recipient testes) which was significantly (P<0.05) reduced by 59% compared with the 120.0±23.9 colonies/105 cells injected (n=3 different cultures and 18 recipient testes) produced by control inhibitor-treated cultures (Fig. 3H,I). However, the total number of THY1+ undifferentiated spermatogonia recovered from the culture wells was not different between control and miR-221/222 inhibitor-treated cultures (Fig. 3J). Furthermore, the percentage of cells with EdU incorporation into DNA, a measure of active mitosis, was not different between control and miR-221/222 inhibitor cultures 2 days after treatment (supplementary material Fig. S2). Moreover, after 2 days of treatment the percentage of apoptotic cells was not different between control and miR-221/222 inhibitor-treated cultures (Fig. 3K). Collectively, these findings suggest that miR-221/222 plays an important role in maintaining stem cell capacity of the undifferentiated spermatogonial population.
Expression of miR-221/222 in undifferentiated spermatogonia is regulated by extrinsic signals
Previous studies showed that signaling from the cytokines GDNF, FGF2 and CSF-1 promotes maintenance of undifferentiated spermatogonia, including self-renewal of SSCs (Meng et al., 2000; Kubota et al., 2004b; Oatley et al., 2009). However, transition from an undifferentiated to a differentiating state in spermatogonia is controlled by activation of RA signaling (Morales and Griswold, 1987; van Pelt and de Rooij, 1990b; Van Pelt and De Rooij, 1990a; Sugimoto et al., 2012). Here, we used qRT-PCR analysis to measure changes in the abundance of miR-221 and miR-222 in cultured THY1+ undifferentiated spermatogonia following individual exposure to extrinsic factors. The maintenance of these cells as a proliferative stem cell-containing population in vitro requires supplementation of culture media with GDNF and FGF2, and co-culture with feeder cells that secrete other factors including CSF-1 (Lim and Bodnar, 2002; Talbot et al., 2012). Therefore, we removed the germ cell clumps from feeder cell monolayers and subjected them to overnight (∼18 hours) withdrawal of exogenous growth factors before treating them individually with GDNF, FGF2 or CSF-1 for 8 hours. All three factors significantly (P<0.05) upregulated the abundance of miR-221 by >2-fold and miR-222 by >1.5-fold (n=3 different cultures) compared with control cells not treated with factors (Fig. 4A). Next, we used qRT-PCR and western blot analyses to examine the relationship between the abundance of miR-221 and miR-222 and KIT protein after exposure to AtRA. In cells treated with DMSO, KIT protein could not be detected but the expression was upregulated to detectable levels after exposure to AtRA for 24 hours (Fig. 4B). The expression of miR-221 and miR-222 was significantly (P<0.05) reduced after AtRA exposure for 24 hours to only 38.5±14.5% (n=3 different cultures) and 59.7±5.1% (n=3 different cultures) of that in DMSO-treated control cells, respectively (Fig. 4C). Taken together, these results indicate that signals promoting maintenance of the undifferentiated spermatogonial population induce expression of miR-221/222, whereas RA-regulated transition from a KIT– undifferentiated to a KIT+ differentiating state coincides with downregulation of miR-221/222 expression.
Fig. 4.
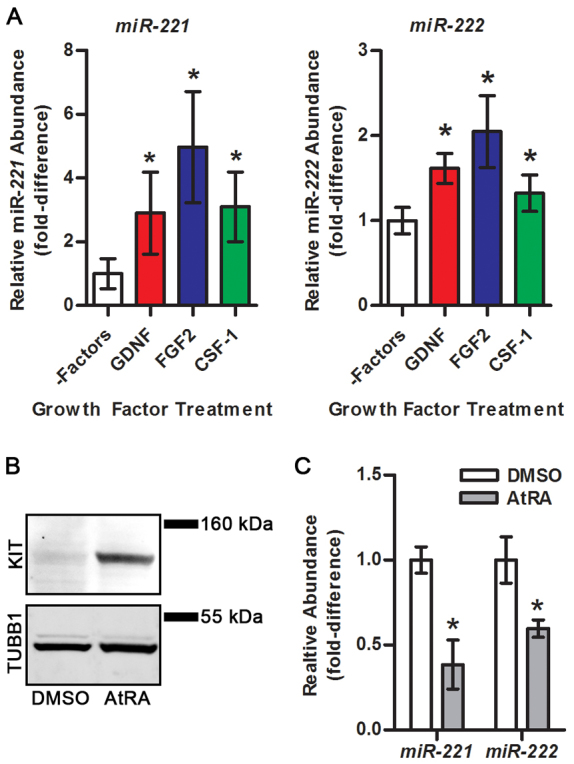
Effects of exposure to soluble factors on the expression of miR-221 and miR-222 in cultured THY1+ undifferentiated spermatogonia. (A) Quantitative comparison of miR-221 and miR-222 abundance in cultured THY1+ undifferentiated spermatogonia treated with GDNF, FGF2 or CSF-1 for 8 hours after overnight (18 hours) withdrawal from growth factors and feeder cells (–Factors) using qRT-PCR analysis. (B) Representative images of western blot analysis for KIT protein expression in cultured THY1+ undifferentiated spermatogonia after treatment with DMSO or 0.5 μM AtRA for 24 hours. TUBB1 was used as a loading control. (C) Quantitative comparison of miR-221 and miR-222 abundance in cultured THY1+ undifferentiated spermatogonia treated with DMSO or 0.5 μM AtRA for 24 hours using qRT-PCR analysis. All quantitative data are mean±s.e.m. of three different cultures. *P<0.05 compared with control (–Factors for A and DMSO for C).
Overexpression of miR-221/222 in undifferentiated spermatogonia inhibits the capacity for differentiation
Having found that inhibition of miR-221/222 function in cultured undifferentiated spermatogonia induces transition to a KIT+ state and loss of SSC capacity, we aimed to determine whether aberrant overexpression of miR-221/222 in spermatogonia could affect differentiation potential. First, we examined the impact of transient miR-221/222 overexpression on transition from a KIT– to a KIT+ state upon activation of RA signaling. To achieve this, cultured THY1+ undifferentiated spermatogonia were transfected with synthetic mimics of miR-221 and miR-222, which significantly (P<0.05) increased their abundance by ∼17-fold and ∼4-fold compared with cells treated with control mimics, respectively, as measured by qRT-PCR analysis (Fig. 5A). Using flow cytometric analysis, we found that the percentage of KIT+ cells after 24 hours exposure to AtRA was 13.7±2.6% (n=3 different cultures) in cultures treated with control mimics, which was significantly greater than the 2.4±0.7% KIT+ cells (n=3 different cultures) in vehicle (DMSO)-treated cultures (Fig. 5B). By comparison, only 7.9±2.9% of cells (n=3 different cultures) transfected with miR-221/222 mimics to induce transient overexpression were KIT+ after 24 hours exposure to AtRA, which represents a significant (P<0.05) reduction of 58% compared with control mimic-treated cells also exposed to AtRA (Fig. 5C). These results indicate that the abundance of miR-221/222 in undifferentiated spermatogonia influences the ability of RA signaling to induce transition to a differentiating state.
Fig. 5.

Impact of miR-221 and miR-222 overexpression in cultured THY1+ undifferentiated spermatogonia on differentiation capacity. (A) Quantitative comparison of miR-221 and miR-222 abundance in cultured THY1+ undifferentiated spermatogonia treated with a scrambled control or a combination of miR-221/222 mimics using qRT-PCR analysis. (B) Representative image of histogram overlays for flow cytometric analysis to determine the percentage of KIT+ cells in cultures of THY1+ undifferentiated spermatogonia treated with DMSO (vehicle control, gray fill), 0.5 μM AtRA+scrambled control mimics (red line) or 0.5 μM AtRA+combination of miR-221/222 mimics (blue line). (C) Quantitative comparison of the percentage of KIT+ cells in cultures of THY1+ undifferentiated spermatogonia treated with DMSO (gray), 0.5 μM AtRA+scrambled control mimics (red) or 0.5 μM+combination of miR-221/222 mimics (blue) using flow cytometric analysis. Data were obtained from scatter plots. (D) Quantitative comparison of miR-221 and miR-222 abundance in cultured THY1+ undifferentiated spermatogonia stably transduced with an empty Efα1 lentiviral expression vector (control), an Efα1-miR-221 lentiviral overexpression vector, or an Efα1-miR-222 lentiviral overexpression vector using qRT-PCR analysis. (E) Representative whole-mount images of dissected seminiferous tubules from testes of recipient mice 1 month after transplantation with control, miR-221-overexpressing, or miR-222-overexpressing cultured Rosa THY1+ undifferentiated spermatogonia. Robust colonies of differentiating donor germ cells were seen in seminiferous tubules transplanted with control cells. By contrast, short chains of spermatogonia with halted differentiation (arrows) were observed in tubules transplanted with miR-221- or miR-222-overexpressing cells. Insets are magnified views of representative germ cell chains. Scale bars: 1 mm (main panels) or 100 μm (insets). All quantitative data are mean±s.e.m. of three different cultures. *P<0.05.
Next, we examined the impact of constitutive miR-221/222 overexpression in undifferentiated spermatogonia on the capacity for differentiation in vivo. Cultured Rosa THY1+ undifferentiated spermatogonia were transduced with lentiviral vectors containing miR-221 or miR-222 coding sequences under control of the constitutively active human elongation factor 1α (Ef1α; EEF1A1) promoter. In comparison to control cells transduced with an empty vector, the abundance of miR-221 and miR-222 was 4.3-fold and 3.0-fold greater, respectively, in cells stably transduced with overexpression vectors (Fig. 5D). We transplanted cells stably expressing the transgenes into testes of recipient mice and examination 2 months later revealed that no spermatogenic colonies were derived from miR-221- or miR-222-overexpressing cells (n=4 testes examined), but colonies of donor spermatogenesis were derived from control cells (data not shown). This finding indicated that miR-221- and miR-222-overexpressing cells were either incapable of colonizing recipient testes or that the germ cells were lost over the 2 months in vivo period. Therefore, we examined testes at 1 month after transplantation and found that control cells derived expansive and densely packed colonies indicative of differentiating germ cells (Fig. 5E). In stark contrast, cells harboring overexpression of miR-221 or miR-222 generated small colonies consisting of short chains of spermatogonia only (n=8 testes and two different cultures) (Fig. 5E). In addition, the number of colonies derived from miR-221- or miR-222-overexpressing cells was lower compared with control cells containing the empty vector. Cells overexpressing miR-221 produced 5.3±2.7 colonies/105 cells transplanted and miR-222-overexpressing cells produced 4.1±3.2 colonies/105 cells transplanted, whereas control cells generated 44.0±18.9 colonies/105 cells transplanted. By contrast, the number of cells collected from culture wells prior to transplantation was not different between control (6.2±0.7×105 cells) and miR-221- (6.4±0.9×105 cells) or miR-222- (6.4±0.8×105 cells) overexpressing cells. Taken together, these findings indicate that constitutive expression of miR-221 or miR-222 in undifferentiated spermatogonia suppresses the ability to regenerate spermatogenesis following transplantation into recipient testes.
DISCUSSION
Expression of KIT in spermatogonia is a hallmark of the transition from an undifferentiated to a differentiating state, yet understanding of the regulatory molecular mechanisms is rudimentary. Recent studies with mice showed that SOHLH1 induces Kit transcription in a subset of spermatogonia and is required for formation of differentiating spermatogonia (Suzuki et al., 2012). Also, PLZF was shown to repress Kit transcription in hematopoietic progenitors (Spinello et al., 2009), immortalized cell lines and isolated mouse spermatogonia (Filipponi et al., 2007), and is required for maintenance of the undifferentiated spermatogonial population in mice (Buaas et al., 2004; Costoya et al., 2004). Furthermore, the transcription factor SALL4 was shown to antagonize the function of PLZF in spermatogonia in order to promote differentiation (Hobbs et al., 2012). Those findings indicate that SALL4 sequesters PLZF from the Kit promoter, thereby allowing for SOHLH1 to activate transcription in a subset of undifferentiated spermatogonia that are primed for transition to the differentiating state.
In the current study, we found that PLZF is expressed by most undifferentiated spermatogonia with SALL4 and SOHLH1 being present in a subset of the population. These PLZF+SALL4+SOHLH1+ cells are likely to possess active Kit transcription and are primed to differentiate; however, the cells lack KIT protein implying the existence of a post-transcriptional mechanism preventing the transition to a KIT+ differentiating state. In addition, we show that exposure to RA relieves the repression, thereby promoting upregulation of KIT protein abundance and transition to a KIT+ state. Importantly, we also found that inhibition of miR-221/222 function induced the transition to a KIT+ state irrespective of RA stimulation, and exposure to RA downregulates miR-221/222 abundance. Moreover, we found that Kit mRNA abundance is downregulated by miR-221/222 in primary cultures of undifferentiated spermatogonia, and previous studies showed that RA stimulation increases Kit mRNA abundance in cultured neonatal mouse testes, which are enriched for undifferentiated spermatogonia (Zhou et al., 2008). Taken together, these findings indicate that the machinery required for Kit transcription is present and active in at least a portion of the undifferentiated spermatogonial population, but the abundance of miR-221/222 controls conversion from a KIT– to a KIT+ state by repressing both mRNA stability and protein translation. We propose that in the undifferentiated spermatogonial state, miR-221/222 abundance is upregulated by niche growth factors including GDNF, FGF2 and CSF-1 until downregulated by activation of RA signaling, which subsequently promotes conversion to a KIT+ state (Fig. 6). It will be of interest in future studies to determine the mechanism by which RA regulates miR-221/222 expression given that an RA receptor (RAR) binding site has not been identified within the promoter region (Saumet et al., 2009).
Fig. 6.
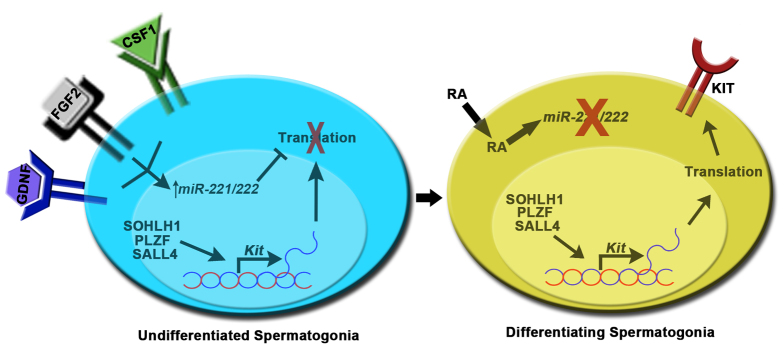
Model for the role of miR-221/222 in regulating spermatogonial fate. In the KIT– undifferentiated state, molecular machinery controlling Kit transcription is active but KIT protein is absent owing to translational repression by miR-221/222. Signaling from niche growth factors including GDNF, FGF2 and CSF-1 induce the expression of miR-221/222. At a defined interval within the seminiferous cycle, the activation of RA signaling leads to downregulation of miR-221/222 abundance which removes the suppression of Kit mRNA and promotes transition to a KIT+ differentiating state.
Continual spermatogenesis relies on a small subpopulation of undifferentiated spermatogonia that possess stem cell capacity and are often referred to as SSCs. Self-renewal by SSCs maintains a long-lasting foundational stem cell pool and progenitor spermatogonia arise from this pool to transiently amplify in number before committing to terminal differentiation. We found that transient inhibition of miR-221/222 results in decline of the stem cell pool in cultured undifferentiated spermatogonial populations. The self-renewal rate of stem cells in these cultures is ∼6 days (Kubota et al., 2004b); thus, the decline in stem cell numbers by 3 days after transient inhibition of miR-221/222 was unlikely to be a result of impaired proliferation. Indeed, the total number of THY1+ undifferentiated spermatogonia recovered from the cultures after 3 days was not different between treatments, indicating that proliferation was not greatly impacted by transient inhibition of miR-221/222 function. Furthermore, the percentage of apoptotic cells was unaffected by miR-221/222 inhibition implying that cell survival was not a major factor in the decline of stem cell capacity. An intriguing possibility is that upregulation of KIT expression, which occurred after miR-221/222 inhibition, allowed for activation of signaling from KIT ligand (KITL), thereby causing irreversible loss of stem cell potential. Maintenance of THY1+ undifferentiated spermatogonia as a proliferative stem cell-containing population in vitro requires co-culture with feeder cell monolayers and in this study we utilized STO feeder cells, which are known to secrete KITL (Lim and Bodnar, 2002; Talbot et al., 2012). These results indicate that the stem cell capacity of spermatogonia is linked to miR-221/222 abundance.
Overall, our studies add an important piece of information to our understanding of the molecular mechanism regulating maintenance of the undifferentiated state in mammalian spermatogonia. This process is essential for sustaining the germline in adulthood and is therefore required for male fertility. It is plausible to postulate that germ cell deficiency in expression of miR-221/222 will result in aging-related loss of the germline and could be an underlying cause of male infertility. Furthermore, the core molecular mechanisms regulating maintenance of an undifferentiated state are thought to be shared among tissue-specific stem cell populations (Arnold et al., 2011; Simons and Clevers, 2011). Thus, miR-221/222 might play an important role in maintenance of the undifferentiated stem cell pool of several cell lineages and could prove to be useful as a diagnostic marker for degenerative diseases and a therapeutic target for regenerative medicine.
Supplementary Material
Acknowledgments
We thank Dr A. Rajkovic (University of Pittsburgh) for generous donation of SOHLH1 antibody.
Footnotes
Funding
This work was supported by the National Institutes of Health [grant number HD061665 to J.M.O.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.087403/-/DC1
References
- Arnold K., Sarkar A., Yram M. A., Polo J. M., Bronson R., Sengupta S., Seandel M., Geijsen N., Hochedlinger K. (2011). Sox2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 9, 317–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster R. L., Avarbock M. R. (1994). Germline transmission of donor haplotype following spermatogonial transplantation. Proc. Natl. Acad. Sci. USA 91, 11303–11307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brinster R. L., Zimmermann J. W. (1994). Spermatogenesis following male germ-cell transplantation. Proc. Natl. Acad. Sci. USA 91, 11298–11302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buaas F. W., Kirsh A. L., Sharma M., McLean D. J., Morris J. L., Griswold M. D., de Rooij D. G., Braun R. E. (2004). Plzf is required in adult male germ cells for stem cell self-renewal. Nat. Genet. 36, 647–652 [DOI] [PubMed] [Google Scholar]
- Ciafrè S. A., Galardi S., Mangiola A., Ferracin M., Liu C. G., Sabatino G., Negrini M., Maira G., Croce C. M., Farace M. G. (2005). Extensive modulation of a set of microRNAs in primary glioblastoma. Biochem. Biophys. Res. Commun. 334, 1351–1358 [DOI] [PubMed] [Google Scholar]
- Clermont Y., Trott M. (1969). Duration of the cycle of the seminiferous epithelium in the mouse and hamster determined by means of 3H-thymidine and radioautography. Fertil. Steril. 20, 805–817 [DOI] [PubMed] [Google Scholar]
- Costoya J. A., Hobbs R. M., Barna M., Cattoretti G., Manova K., Sukhwani M., Orwig K. E., Wolgemuth D. J., Pandolfi P. P. (2004). Essential role of Plzf in maintenance of spermatogonial stem cells. Nat. Genet. 36, 653–659 [DOI] [PubMed] [Google Scholar]
- de Rooij D. G. (1998). Stem cells in the testis. Int. J. Exp. Pathol. 79, 67–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij D. G., Russell L. D. (2000). All you wanted to know about spermatogonia but were afraid to ask. J. Androl. 21, 776–798 [PubMed] [Google Scholar]
- Di Leva G., Gasparini P., Piovan C., Ngankeu A., Garofalo M., Taccioli C., Iorio M. V., Li M., Volinia S., Alder H., et al. (2010). MicroRNA cluster 221-222 and estrogen receptor alpha interactions in breast cancer. J. Natl. Cancer Inst. 102, 706–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felli N., Fontana L., Pelosi E., Botta R., Bonci D., Facchiano F., Liuzzi F., Lulli V., Morsilli O., Santoro S., et al. (2005). MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc. Natl. Acad. Sci. USA 102, 18081–18086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipponi D., Hobbs R. M., Ottolenghi S., Rossi P., Jannini E. A., Pandolfi P. P., Dolci S. (2007). Repression of kit expression by Plzf in germ cells. Mol. Cell. Biol. 27, 6770–6781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galardi S., Mercatelli N., Giorda E., Massalini S., Frajese G. V., Ciafrè S. A., Farace M. G. (2007). miR-221 and miR-222 expression affects the proliferation potential of human prostate carcinoma cell lines by targeting p27Kip1. J. Biol. Chem. 282, 23716–23724 [DOI] [PubMed] [Google Scholar]
- Ghildiyal M., Zamore P. D. (2009). Small silencing RNAs: an expanding universe. Nat. Rev. Genet. 10, 94–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillies J. K., Lorimer I. A. (2007). Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 6, 2005–2009 [DOI] [PubMed] [Google Scholar]
- He H., Jazdzewski K., Li W., Liyanarachchi S., Nagy R., Volinia S., Calin G. A., Liu C. G., Franssila K., Suster S., et al. (2005). The role of microRNA genes in papillary thyroid carcinoma. Proc. Natl. Acad. Sci. USA 102, 19075–19080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobbs R. M., Fagoonee S., Papa A., Webster K., Altruda F., Nishinakamura R., Chai L., Pandolfi P. P. (2012). Functional antagonism between Sall4 and Plzf defines germline progenitors. Cell Stem Cell 10, 284–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igoucheva O., Alexeev V. (2009). MicroRNA-dependent regulation of cKit in cutaneous melanoma. Biochem. Biophys. Res. Commun. 379, 790–794 [DOI] [PubMed] [Google Scholar]
- Kaucher A. V., Oatley M. J., Oatley J. M. (2012). NEUROG3 is a critical downstream effector for STAT3-regulated differentiation of mammalian stem and progenitor spermatogonia. Biol. Reprod. 86, 164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller G., Kennedy M., Papayannopoulou T., Wiles M. V. (1993). Hematopoietic commitment during embryonic stem cell differentiation in culture. Mol. Cell. Biol. 13, 473–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korhonen H. M., Meikar O., Yadav R. P., Papaioannou M. D., Romero Y., Da Ros M., Herrera P. L., Toppari J., Nef S., Kotaja N. (2011). Dicer is required for haploid male germ cell differentiation in mice. PLoS ONE 6, e24821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota H., Avarbock M. R., Brinster R. L. (2003). Spermatogonial stem cells share some, but not all, phenotypic and functional characteristics with other stem cells. Proc. Natl. Acad. Sci. USA 100, 6487–6492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota H., Avarbock M. R., Brinster R. L. (2004a). Culture conditions and single growth factors affect fate determination of mouse spermatogonial stem cells. Biol. Reprod. 71, 722–731 [DOI] [PubMed] [Google Scholar]
- Kubota H., Avarbock M. R., Brinster R. L. (2004b). Growth factors essential for self-renewal and expansion of mouse spermatogonial stem cells. Proc. Natl. Acad. Sci. USA 101, 16489–16494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J. W., Bodnar A. (2002). Proteome analysis of conditioned medium from mouse embryonic fibroblast feeder layers which support the growth of human embryonic stem cells. Proteomics 2, 1187–1203 [DOI] [PubMed] [Google Scholar]
- Liu D., Li L., Fu H., Li S., Li J. (2012). Inactivation of Dicer1 has a severe cumulative impact on the formation of mature germ cells in mouse testes. Biochem. Biophys. Res. Commun. 422, 114–120 [DOI] [PubMed] [Google Scholar]
- Maatouk D. M., Loveland K. L., McManus M. T., Moore K., Harfe B. D. (2008). Dicer1 is required for differentiation of the mouse male germline. Biol. Reprod. 79, 696–703 [DOI] [PubMed] [Google Scholar]
- Manova K., Nocka K., Besmer P., Bachvarova R. F. (1990). Gonadal expression of c-kit encoded at the W locus of the mouse. Development 110, 1057–1069 [DOI] [PubMed] [Google Scholar]
- Meng X., Lindahl M., Hyvönen M. E., Parvinen M., de Rooij D. G., Hess M. W., Raatikainen-Ahokas A., Sainio K., Rauvala H., Lakso M., et al. (2000). Regulation of cell fate decision of undifferentiated spermatogonia by GDNF. Science 287, 1489–1493 [DOI] [PubMed] [Google Scholar]
- Morales C., Griswold M. D. (1987). Retinol-induced stage synchronization in seminiferous tubules of the rat. Endocrinology 121, 432–434 [DOI] [PubMed] [Google Scholar]
- Nuovo G. J., Elton T. S., Nana-Sinkam P., Volinia S., Croce C. M., Schmittgen T. D. (2009). A methodology for the combined in situ analyses of the precursor and mature forms of microRNAs and correlation with their putative targets. Nat. Protoc. 4, 107–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakberg E. F. (1956). Duration of spermatogenesis in the mouse and timing of stages of the cycle of the seminiferous epithelium. Am. J. Anat. 99, 507–516 [DOI] [PubMed] [Google Scholar]
- Oatley J. M., Brinster R. L. (2006). Spermatogonial stem cells. MethodsEnzymol. 419, 259–282 [DOI] [PubMed] [Google Scholar]
- Oatley J. M., Brinster R. L. (2012). The germline stem cell niche unit in mammalian testes. Physiol. Rev. 92, 577–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oatley J. M., Avarbock M. R., Telaranta A. I., Fearon D. T., Brinster R. L. (2006). Identifying genes important for spermatogonial stem cell self-renewal and survival. Proc. Natl. Acad. Sci. USA 103, 9524–9529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oatley J. M., Oatley M. J., Avarbock M. R., Tobias J. W., Brinster R. L. (2009). Colony stimulating factor 1 is an extrinsic stimulator of mouse spermatogonial stem cell self-renewal. Development 136, 1191–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oatley J. M., Kaucher A. V., Avarbock M. R., Brinster R. L. (2010). Regulation of mouse spermatogonial stem cell differentiation by STAT3 signaling. Biol. Reprod. 83, 427–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pineau P., Volinia S., McJunkin K., Marchio A., Battiston C., Terris B., Mazzaferro V., Lowe S. W., Croce C. M., Dejean A. (2010). miR-221 overexpression contributes to liver tumorigenesis. Proc. Natl. Acad. Sci. USA 107, 264–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prabhu S. M., Meistrich M. L., McLaughlin E. A., Roman S. D., Warne S., Mendis S., Itman C., Loveland K. L. (2006). Expression of c-Kit receptor mRNA and protein in the developing, adult and irradiated rodent testis. Reproduction 131, 489–499 [DOI] [PubMed] [Google Scholar]
- Romero Y., Meikar O., Papaioannou M. D., Conne B., Grey C., Weier M., Pralong F., De Massy B., Kaessmann H., Vassalli J. D., et al. (2011). Dicer1 depletion in male germ cells leads to infertility due to cumulative meiotic and spermiogenic defects. PLoS ONE 6, e25241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saumet A., Vetter G., Bouttier M., Portales-Casamar E., Wasserman W. W., Maurin T., Mari B., Barbry P., Vallar L., Friederich E., et al. (2009). Transcriptional repression of microRNA genes by PML-RARA increases expression of key cancer proteins in acute promyelocytic leukemia. Blood 113, 412–421 [DOI] [PubMed] [Google Scholar]
- Schmittgen T. D., Livak K. J. (2008). Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3, 1101–1108 [DOI] [PubMed] [Google Scholar]
- Schrans-Stassen B. H., van de Kant H. J., de Rooij D. G., van Pelt A. M. (1999). Differential expression of c-kit in mouse undifferentiated and differentiating type A spermatogonia. Endocrinology 140, 5894–5900 [DOI] [PubMed] [Google Scholar]
- Shinohara T., Orwig K. E., Avarbock M. R., Brinster R. L. (2000). Spermatogonial stem cell enrichment by multiparameter selection of mouse testis cells. Proc. Natl. Acad. Sci. USA 97, 8346–8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons B. D., Clevers H. (2011). Strategies for homeostatic stem cell self-renewal in adult tissues. Cell 145, 851–862 [DOI] [PubMed] [Google Scholar]
- Spinello I., Quaranta M. T., Pasquini L., Pelosi E., Petrucci E., Pagliuca A., Castelli G., Mariani G., Diverio D., Foà R., et al. (2009). PLZF-mediated control on c-kit expression in CD34(+) cells and early erythropoiesis. Oncogene 28, 2276–2288 [DOI] [PubMed] [Google Scholar]
- Sugimoto R., Nabeshima Y., Yoshida S. (2012). Retinoic acid metabolism links the periodical differentiation of germ cells with the cycle of Sertoli cells in mouse seminiferous epithelium. Mech. Dev. 128, 610–624 [DOI] [PubMed] [Google Scholar]
- Suzuki H., Ahn H. W., Chu T., Bowden W., Gassei K., Orwig K., Rajkovic A. (2012). SOHLH1 and SOHLH2 coordinate spermatogonial differentiation. Dev. Biol. 361, 301–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot N. C., Sparks W. O., Powell A. M., Kahl S., Caperna T. J. (2012). Quantitative and semiquantitative immunoassay of growth factors and cytokines in the conditioned medium of STO and CF-1 mouse feeder cells. In Vitro Cell. Dev. Biol. Anim. 48, 1–11 [DOI] [PubMed] [Google Scholar]
- Van Pelt A. M., De Rooij D. G. (1990a). The origin of the synchronization of the seminiferous epithelium in vitamin A-deficient rats after vitamin A replacement. Biol. Reprod. 42, 677–682 [DOI] [PubMed] [Google Scholar]
- van Pelt A. M., de Rooij D. G. (1990b). Synchronization of the seminiferous epithelium after vitamin A replacement in vitamin A-deficient mice. Biol. Reprod. 43, 363–367 [DOI] [PubMed] [Google Scholar]
- Yoshinaga K., Nishikawa S., Ogawa M., Hayashi S., Kunisada T., Fujimoto T., Nishikawa S. (1991). Role of c-kit in mouse spermatogenesis: identification of spermatogonia as a specific site of c-kit expression and function. Development 113, 689–699 [DOI] [PubMed] [Google Scholar]
- Zhou Q., Li Y., Nie R., Friel P., Mitchell D., Evanoff R. M., Pouchnik D., Banasik B., McCarrey J. R., Small C., et al. (2008). Expression of stimulated by retinoic acid gene 8 (Stra8) and maturation of murine gonocytes and spermatogonia induced by retinoic acid in vitro. Biol. Reprod. 78, 537–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.