

Abstract
Bone morphogenetic proteins (BMPs) have unexpectedly diverse activities establishing different aspects of dorsal neural circuitry in the developing spinal cord. Our recent studies have shown that, in addition to spatially orienting dorsal commissural (dI1) axons, BMPs supply ‘temporal’ information to commissural axons to specify their rate of growth. This information ensures that commissural axons reach subsequent signals at particular times during development. However, it remains unresolved how commissural neurons specifically decode this activity of BMPs to result in their extending axons at a specific speed through the dorsal spinal cord. We have addressed this question by examining whether either of the type I BMP receptors (Bmpr), BmprIa and BmprIb, have a role controlling the rate of commissural axon growth. BmprIa and BmprIb exhibit a common function specifying the identity of dorsal cell fate in the spinal cord, whereas BmprIb alone mediates the ability of BMPs to orient axons. Here, we show that BmprIb, and not BmprIa, is additionally required to control the rate of commissural axon extension. We have also determined the intracellular effector by which BmprIb regulates commissural axon growth. We show that BmprIb has a novel role modulating the activity of the actin-severing protein cofilin. These studies reveal the mechanistic differences used by distinct components of the canonical Bmpr complex to mediate the diverse activities of the BMPs.
Keywords: Axon guidance, Axon outgrowth, Bone morphogenetic proteins, Cofilin, Commissural axons, Spinal cord, Chick, Mouse
INTRODUCTION
As axons grow, they encounter multiple directional cues in the constantly changing environment of the developing embryo (Dickson, 2002). To interpret this guidance information correctly, axons must modulate their speed to reach these cues at the right time in development. What mechanism controls this process? Our studies have suggested that, in addition to directing axon orientation, guidance signals also provide ‘temporal’ information by regulating actin polymerization in the growth cone to control the rate of axon growth (Phan et al., 2010). We have been examining this mechanism using the trajectory of commissural axons within the developing spinal cord as a model system. Commissural (dI1) neurons arise in response to a putative gradient of bone morphogenetic proteins (BMPs) secreted from the roof plate (RP) at the dorsal midline (Lee and Jessell, 1999). They subsequently extend axons away from the RP to project towards the floor plate (FP) at the ventral midline. Our studies have identified that BMPs can both orient commissural axon growth away from the RP (Augsburger et al., 1999) and regulate their rate of extension through the dorsal spinal cord (Phan et al., 2010). Thus, BMPs direct remarkably diverse cellular processes for commissural neurons depending on their stage of development.
How are these disparate activities translated by commissural neurons to result in the correct outcome at the correct time? In the canonical signaling pathway, BMPs activate a heterodimeric complex of type I and type II BMP receptors (Bmprs) to result in the phosphorylation of the receptor-activated Smads, a complex of transcriptional activators (Heldin et al., 1997). However, the regulation of cytoskeletal changes downstream of BMP signaling has been linked to the Lim domain kinase 1 (Limk1)-cofilin pathway. In this pathway, Limk1 is ‘primed’ by binding to the tail of BmprII and then activated after BMP binding (Foletta et al., 2003; Lee-Hoeflich et al., 2004). Limk1 then phosphorylates, and thereby inactivates, cofilin (Arber et al., 1998). Active cofilin depolymerizes actin and tends to stimulate neurite outgrowth (Meberg and Bamburg, 2000). The balance between the activation states of Limk1-cofilin is crucial for many axon behaviors, including turning away from chemorepellents (Wen et al., 2007) and regulating their speed of growth (Endo et al., 2007; Phan et al., 2010).
However, the mechanism by which a commissural neuron discriminates between the canonical Smad and non-canonical Limk1-cofilin signaling pathway remains unclear. Our studies have suggested that this choice depends on which type I Bmprs are present in the cell during development (Yamauchi et al., 2008). The specification of commissural cell fate is a redundant function of both type I Bmprs, BmprIa and BmprIb (Timmer et al., 2002; Wine-Lee et al., 2004). By contrast, only BmprIb is required in commissural neurons to reorient their axons away from the RP (Yamauchi et al., 2008). Here, we show that BmprIb, but not BmprIa, is also required for the ability of BMPs to control the rate of commissural axon outgrowth, lending further support to a model in which BmprIb uniquely functions to mediate the ability of BMPs to act as axon guidance signals.
What is the mechanistic basis for the distinct activities of BmprIb? Our previous studies have implicated BmprII in the regulation of commissural axon growth rate, by acting as a scaffold for Limk1 (Phan et al., 2010). Does this interaction further require a particular complement of the Bmpr complex, to ensure the specific regulation of axon outgrowth rate by BMPs? Or is the activation of Limk1 non-specific, such that Limk1 is constitutively activated in the cytosol after dissociating from BmprII? Here, we provide evidence for the former model by showing that BmprIb, but not BmprIa, is required to regulate cofilin activity. The activity of BmprIb inversely affects cofilin activity and can be blocked by an isoform of cofilin that cannot be regulated by Limk1. Taken together, these studies demonstrate for the first time that the activities of Bmpr1a and BmprIb can diverge through the use of distinct second messengers. The presence of BmprIb confers commissural neurons with the ability to regulate Limk1-cofilin activity and thereby control the speed at which they extend axons.
MATERIALS AND METHODS
Immunohistochemistry
Antibody staining was performed on 30 μm-thick cryosectioned tissues, dissociated neurons or whole-mount tissues as described previously (Augsburger et al., 1999). Fluorescence images were collected on Carl Zeiss LSM510 confocal and Axiovert 200M microscopes. Images were processed using Adobe Photoshop CS4. The intensity of phospho (p)-cofilin staining was quantified using identical settings on the confocal microscope to image sections and dissociated neurons from control and mutant Bmpr littermates that underwent immunohistochemistry on the same slide.
Antibodies against the following proteins were used: mouse: neuronal class III β-tubulin at 1:2000 (Tuj1; Covance), Tag1 at 1:6 (4D7) (Dodd et al., 1988), green fluorescent protein (GFP) at 1:2000 (3E6, Invitrogen), Cre at 1:1000 (Covance), HA at 1:1000 (Covance), His at 1:1000 (Covance), Myc at 1:10 (9E10) (Evan et al., 1985); rabbit: yellow fluorescent protein (YFP) recognized by αGFP at 1:2000 (Invitrogen), phosphorylated (p)-cofilin at 1:100 (Santa Cruz Biotechnology), panLh2 (Lhx2/9) at 1:1000 (L1) (Liem et al., 1997); sheep: GFP at 1:2000 (Biogenesis); guinea pig: Olig2 at 1:20,000 (Skaggs et al., 2011). Cyanine3-, cyanine5- or FITC-coupled secondary antibodies were obtained from Jackson ImmunoResearch.
In ovo electroporation and expression constructs
Hamilton and Hamburger (HH) stage 13-15 (Hamburger and Hamilton, 1992) White Leghorn chick embryos (AA Laboratory Eggs) were electroporated and processed as previously described (Yamauchi et al., 2008). Math1 (Atoh1) enhancer expression constructs were generated as previously described (Phan et al., 2010). Plasmid constructs were electroporated in the following amounts: Math1::fGfp, 0.1 μg/μl; Math1::caBmprIb-HA-IRES-fGfp, 1.5 μg/μl; Math1::wtcofilin-myc, 1.0 μg/μl; and Math1::cofilinS3A-his, 1.0 μg/μl.
For HH stage 18, the number of commissural neurites was calculated as a percentage of the GFP+ Lhx2/9+ cells per cryosection. The length of GFP+ axons, from cell body to growth cone, was quantified using NIH Image J. For HH stages 22/23 and 27/28, axon outgrowth was quantified as previously described (Fig. 1L) (Yamauchi et al., 2008). At HH stage 27/28, the Lhx2/9+ cell bodies have started their migration into deeper layers of the dorsal spinal cord, thus there are more axons at the intermediate (INT) level compared with the mid-dorsal (MD) boundary. All statistical analyses used a one-tailed Student’s t-test.
Fig. 1.

Constitutive activation of BmprIb persistently delays commissural axon outgrowth. (A-K,M) Constructs encoding farnesylated (f) GFP (control, A,B,E,F,I) or constitutively active (ca) BmprIb-HA-IRES-fGFP (experimental, C,D,G,H,J) were ectopically expressed under the control of the Math enhancer by in ovo electroporation at either Hamburger-Hamilton (HH) stages 13/14 (A-D) or 14/15 (E-J). Chicken embryos were harvested at HH stages 18 (A-D), 22/23 (E-H) or 27/28 (I,J). Transverse sections of the spinal cord were examined for the extent of fGFP+ (green) and HA+ (blue) axon outgrowth and number of Lhx2/9+ commissural neurons (red). The distribution of HA-tagged BmprIb was indistinguishable from fGFP expressed from the IRES cassette (inset panel, H). (A-D,K,M) Control and experimental Lhx2/9+ neurons are born in similar numbers and initiate axon growth at the same time. Over 80% of Lhx2/9+ neurons electroporated with fGFP (n=69 sections, three embryos) or caBmprIb (n=68 sections, four embryos) have initiated axon growth (probability of similarity between the control and experimental conditions, P>0.18, Student’s t-test). However, the caBmprIb-IRES-fGFP+ axons (n=43 sections, five embryos) have extended significantly less (P<0.0036) than the GFP+ axons (n=49 sections, three embryos). (E-J,K) caBmprIb+ commissural axons continue to grow more slowly than fGFP+ axons. By HH stage 27/28, 87% of commissural neurons had extended GFP+ axons, 90% of these axons subsequently reached the FP (n=132 sections, six embryos). Similarly, 85% of caBmprIb-IRES-fGFP+ neurons (n=56 sections, four embryos) extended axons (P>0.38). However, progressively fewer of these axons reach the different boundaries in the spinal cord, such that only 70% of these axons have reached the FP (P<1.9×10–3). (L) The extent of commissural axon outgrowth was quantified by determining whether axons had crossed one of four arbitrary boundaries: mid-dorsal (MD), intermediate (INT), mid-ventral (MV) or the FP. (N) Control axons extending through HH stage 20/21 dorsal spinal cords have a velocity of 13.5±0.6 μm/hour (n=11 neurons, two embryos) whereas the caBmprIb+ axons grow significantly slower (P<0.002) with a velocity of 9.8±0.8 μm/hour (n=13 neurons, three embryos). Error bars represent s.e.m. Scale bar: in A-D, 40 μm; in E-H, 50 μm; in I,J, 70 μm.
Generation and analysis of mutant mice
BmprIb mice were inbred in a mixed genetic background (129/Sv/C57/B6), whereas all other mice were inbred in an identical background (129/Sv). Mice were genotyped by PCR as previously described: BmprIaflox (Mishina et al., 2002), BmprIb (Yi et al., 2000), Math1::tauGfp (Imondi et al., 2007), Math1::Cre (Matei et al., 2005), Rosa26R::Yfp (Srinivas et al., 2001). In all cases, only relevant littermates were used as controls: BmprIa experiments: BmprIa+/+±Math1::Cre; BmprIb experiments: BmprIb+/+.
To assess dI1 cell fate, the number of Olig2+ and Cre recombinase+ (BmprIa) or Lhx2/9+ (BmprIb) cells were counted in sections from embryonic day (E) 10.5 control and mutant embryos. Data were plotted as the number of Olig2+ cells versus either Cre+ or Lhx2/9+ cells per section and a logarithm trend line was fitted to each data set using Microsoft Excel 2008 for Mac.
Whole-mount fillet preparations were dissected from the upper brachial to lower thoracic region of E10.5 spinal cords from the surrounding mesoderm in dispase-free medium. Spinal cords were cut along the FP at the ventral midline, embedded in collagen (BD Biosciences), fixed and immunostained. GFP+ axons were measured from cell body to growth cone and binned according to number of dI1 cells, as assessed by Cre+ or Lhx2/9+ cells, per 100 μm hemi-segment to normalize for developmental age. Math1::Cre and Lhx2/9 are present in the same population of neurons (Yamauchi et al., 2008), but Math1::Cre is expressed earlier than Lhx2/9, thus more Cre+ cells are present per 100 μm hemi-segment at the earliest stages of commissural axiogenesis compared with Lhx2/9+.
Time-lapse imaging of fillet preparations
Time-lapse imaging of chicken embryos was performed as described previously (Phan et al., 2010).
For time-lapse imaging of mouse embryos, fillet preparations of E11.5 lumbar spinal cords were dissected as described above and embedded in collagen in glass-bottom dishes (MatTek). Explants were incubated in OptiMEM (Gibco) and 1×penicillin/streptomycin/glutamine (PSG) (Gibco) for 1 hour prior to imaging in a 37°C, 5% CO2 cell culture incubator. The remaining embryos were kept in Hibernate E media (Gibco) at 4°C in the dark until ready for dissection. Time-lapse live imaging was performed at 37°C using an enclosed Zeiss Axiovert 200M inverted microscope fitted with an XL S1 incubator, P Lab-Tek S1 heating insert, S temperature module and a XL S heating unit (Pecon). Images were collected every 5 minutes for 4-8 hours in 7 μm z-slice intervals producing 49-84 μm-thick z-stack images. Images were captured with a Zeiss HRm camera and Apotome without optical sectioning.
Axon growth rates were quantified in a region 100-250 μm from the RP, the closest region to the RP where there was no interfering fluorescence from the GFP+ cell bodies. Retracting axons were not included in the quantification; however, there is no significant difference in the frequency or rate of retracting axons between control and mutant spinal cords. The growth of axons in the z-plane was compensated for by measuring the depth to which axons grew within the fillet using projected z-stack side views. Movies were exported as AVI files at a rate of 30 minutes per second, converted to Mov files and processed in Final Cut Pro 7.
Dissociated cell culture
The dorsal third of brachial and thoracic E11.5 mouse spinal cords were dissected and dissociated as previously described (Moore and Kennedy, 2008). Dissociated neurons from each embryo were cultured in parallel on poly-d-lysine-coated german glass coverslips (Bellco Glass) in Neurobasal medium containing 10% fetal bovine serum and 1×PSG. For the experiments measuring axon length, neurons were grown for 24 hours in serum and then for 24 hours without serum. Similar differences in length were observed with or without BMP7, suggesting that there is an endogenous source of BMPs in the cultures. For experiments measuring pcofilin levels, neurons were grown for 20 hours in serum, then in serum-free OptiMEM medium containing 1×PSG for 4 hours. Neurons were stimulated with 25 ng/ml recombinant human BMP7 (R&D Systems) or an equal volume of 0.05% bovine serum albumin (BSA) for 5 minutes, fixed in 4% paraformaldehyde (J.T.Baker) and 0.5% glutaraldehyde (Sigma-Aldrich), and then processed for immunostaining.
RESULTS
Constitutive activation of BmprIb persistently slows commissural axon outgrowth
The RP-resident BMPs control the rate at which commissural axons extend through the dorsal spinal cord (Phan et al., 2010). What is the nature of the signaling complex that transduces this activity? Our studies have shown that BmprIb regulates the orientation of commissural axons (Yamauchi et al., 2008): these studies also revealed that expressing a constitutively active (ca) form of BmprIb in chicken commissural neurons resulted in these neurons extending their axons apparently more slowly through the developing spinal cord. By Hamilton and Hamburger (HH) stage 22/23, chicken commissural axons expressing caBmprIb and farnesylated green fluorescent protein (fGFP) under the control of the Math1 enhancer (Helms et al., 2000) were significantly shorter in length than control GFP+ axons (Fig. 1E-H; Fig. 6I) (Yamauchi et al., 2008). However, it remained unresolved whether this difference in axon length resulted from a delay in commissural axon initiation or if axon outgrowth was rather stalled or slowed.
Fig. 6.

Cofilin overexpression rescues the axon growth delay seen after constitutive activation of BmprIb. (A-H) Constructs encoding wild-type (wt) cofilin-myc (A-D) or cofilinS3A-his (E-H) were overexpressed in the spinal cord under the control of the Math1 enhancer in combination with either Math1::fGfp (A,B,E,F) or Math1::caBmprIb-HA-IRES-fGfp (C,D,G,H). Chicken embryos were in ovo electroporated at HH stage 14/15 and harvested at HH stage 22/23. Transverse sections of the spinal cord were examined for the extent of GFP+ axon outgrowth (green) and number of Lhx2/9+ commissural neurons (red). (C-H) The presence of wtcofilin-myc in Math1+ neurons does not rescue the delay in axon outgrowth caused by caBmprIb expression. By contrast, the presence of cofilinS3A-his restores the axon growth to a similar extent as observed in the Math1::fGfp control. Yellow arrowheads indicate the extent of axon outgrowth. Scale bar: 30 μm. (I) About 20% of commissural neurons electroporated with Math1::fGfp extended axons to the FP (n=200 sections, 11 embryos). Electroporation of Math1::wtcofilin (n=48 sections, four embryos) or Math1::cofilinS3A (n=54 sections, four embryos) results in ∼40% of GFP+ axons reaching the FP (compared with Math1::fGfp control: wtcofilin, P<2.4×10–7; cofilinS3A P<2.6×10–12, Student’s t-test). Less than 10% of caBmprIb+ Math1+ neurons (compared with Math1::fGfp control, P<9.4×10–10; n=122 sections, six embryos) and <5% of caBmprIb+ wtcofilin+ Math1+ neurons extend axons to the FP (compared with Math1::caBmprIb, P<0.0013; n=121 sections, five embryos). By contrast, co-electroporation of cofilinS3A and caBmprIb rescued the caBmprIb axon outgrowth phenotype to similar levels to that seen in the Math1::fGfp control (P>0.13; n=54 sections, four embryos). Error bars represent s.e.m. (J) Model for the regulation of cofilin activity by the BMP signaling pathway. The activity of Limk1 is ‘primed’ by binding to a site on the tail of BmprII. Upon BMP binding, Limk1 is released into the cytosol in a phosphorylated, activated form, that inactivates cofilin. These studies suggest that BmprIb is required for the activation of Limk1 (dotted arrow).
To determine whether the constitutive activation of BmprIb results in a specific delay in commissural axon initiation, chicken embryos were in ovo electroporated with Math1::fGfp or Math1::caBmprIb-IRES-fGfp expression constructs at HH stage 13/14. The embryos were permitted to develop to HH stage 18 (Fig. 1A-D), the stage at which commissural axon extension begins (Phan et al., 2010). The introduction of caBmprIb does not result in any alterations in the specification of dorsal commissural neuron fate: Lhx2/9+ dl1 commissural neurons (Liem et al., 1997) are present in similar numbers on the electroporated and non-electroporated sides of spinal cords (supplementary material Fig. S1). Moreover, there was no significant difference in the number of control and experimental Lhx2/9+ neurons initiating axiogenesis (Fig. 1K). However, the experimental GFP+ commissural axons were found to be ∼40% shorter on average than the control GFP+ axons (Fig. 1M). Thus, the presence of caBmprIb does not delay axon initiation, but rather results in the immediate slowing of axon outgrowth.
To examine whether the defect observed at HH stage 22/23 resulted from stalled or slowed axon growth, embryos were allowed to develop to HH stage 27/28 to determine the extent of axon outgrowth from caBmprIb+ commissural neurons ∼1 day later in development. Axon outgrowth was quantified by counting the number of Lhx2/9+ neurons that extended GFP+ axons past four arbitrary boundaries in the spinal cord: mid-dorsal (MD), intermediate (INT), mid-ventral (MV) and floor plate (FP) (Fig. 1L) (Yamauchi et al., 2008). By HH stage 27/28, the control commissural axons have largely completed their trajectories through the transverse plane of the spinal cord: of the Lhx2/9+ GFP+ neurons that extended axons to the MD level, 90% of these axons subsequently reached the FP (Fig. 1I,K). Although a similar number of Lhx2/9+ caBmprIb-IRES-GFP+ neurons extend axons beyond the MD boundary (Fig. 1J), progressively fewer axons reached the subsequent levels compared with controls (Fig. 1K). However, the observed decrease in commissural axon outgrowth was far less than that seen in HH stage 22/23 spinal cords (Fig. 1H; Fig. 6I) (Yamauchi et al., 2008), suggesting that elevating BMP signaling slowed, rather than stalled, axon outgrowth. Strongly supporting this model, imaging live axons growing in elecroporated tissue explants in vitro demonstrated that the caBmprIb-IRES-GFP+ axons had a ∼30% slower average velocity than control GFP+ axons as they grew through the dorsal spinal cord (Fig. 1N; supplementary material Movie 1). This decreased rate of growth is consistent with the shorter length of axons extending from caBmprIb-IRES-GFP+ axons (Fig. 1M; Fig. 6I).
Taken together, these studies demonstrate that the constitutive activation of BmprIb in dI1 neurons results in persistent reduction in the speed of commissural axons growing through the dorsal spinal cord.
Commissural neurons deficient in BmprIb, but not BmprIa, extend longer axons in vitro
If elevating BMP signaling in commissural neurons slows axon growth, then, conversely, lowering BMP signaling should accelerate the rate of axon outgrowth. Moreover, our previous studies have also suggested that BmprIb specifically transduces the ability of BMPs to regulate commissural axon orientation. The other type I Bmpr, BmprIa, did not have a significant effect on commissural axon outgrowth when misexpressed in chicken spinal cords (Yamauchi et al., 2008). Thus, if BmprIb is the critical receptor that mediates the ability of BMPs to control the rate of axon outgrowth, then commissural axon growth should be accelerated in the absence of BmprIb, but not in the absence of BmprIa.
To address this question, we examined mice mutant for either BmprIa or BmprIb in combination with a genetically encoded Math1::tauGfp reporter, which expresses GFP in the dI1 population of dorsal commissural axons (Imondi et al., 2007; Hazen et al., 2012). Although BmprIb loss-of-function mutants are viable (Yi et al., 2000; Yi et al., 2001), the loss of BmprIa is lethal (Mishina et al., 1995). Therefore, we took a conditional approach to inactivate BmprIa, crossing a floxed allele of BmprIa (Mishina et al., 2002) to transgenic mice expressing Cre recombinase under control of the Math1 enhancer (Matei et al., 2005). To check that Cre recombination was active in commissural neurons, we crossed mice from the Math1::Cre line to a Rosa26R(lox-stop-lox)::Yfp reporter strain (Srinivas et al., 2001). At E10.5 and E11.5, Cre protein was observed in Lhx2/9+ commissural neurons (supplementary material Fig. S2B,F) (Yamauchi et al., 2008) and YFP was present both in the cell bodies and Tag1 (Cntn2)+ axons of commissural neurons (supplementary material Fig. S2C,D,G,H, arrows).
As a first approach to examining whether the loss of BmprIa or BmprIb alters commissural axon outgrowth, we assessed the length of GFP+ axons extending from dissociated control (Math1::Cre; Math1::tauGfp; BmprIa+/+ or Math1::tauGfp; BmprIb+/+) and mutant (Math1::Cre; Math1::tauGfp; BmprIaflox/flox or Math1::tauGfp; BmprIb–/–) commissural neurons (Fig. 2A). BmprIa mutant commissural axons were similar in length to those from littermate control commissural neurons (Fig. 2B,C,F). By contrast, BmprIb–/– neurons extended axons that were almost 20% longer on average than the controls (Fig. 2D-F). Thus, the absence of BmprIb, but not BmprIa, results in increased commissural axon growth in vitro supporting the hypothesis that a specific activity of BmprIb is to slow the rate of commissural axon extension.
Fig. 2.

Dissociated BmprIb–/– neurons extend longer axons than controls in vitro. (A) Schematic of the experimental procedure for dissociated neuron cultures. Dissociated neurons from the dorsal third of brachial and thoracic E11.5 mouse spinal cords (dissected as indicated by yellow dotted lines) were analyzed after 48 hours growth (see Materials and methods for details). (B-F) Commissural axons were detected using antibodies against type III β-tubulin (Tuj1, red in B-E) which label all neuronal processes (Geisert and Frankfurter, 1989) and a genetically encoded reporter, Math1::tauGfp (Imondi et al., 2007) (green in B-E). Tuj1+ GFP+ axon outgrowth was comparable (P>0.048, Student’s t-test) in BmprIa control (Math1::Cre;BmprIa+/+; n=354 neurons, eight embryos) and mutant (Math1::Cre;BmprIaflox/flox, n=80 neurons, four embryos) littermates. By contrast, the BmprIb–/– Tuj1+ GFP+ axons extended almost 20% further (P<1.2×10–6; n=287 neurons, three embryos) than those from control littermates (n=327 neurons, three embryos). Error bars represent s.e.m. Scale bar: 20 μm.
Commissural axons deficient in BmprIb, but not BmprIa, show accelerated growth in vivo
In a second approach to examine the effect of functionally inactivating the type I Bmprs on the growth rate of commissural axons, we compared the length of commissural axons growing in vivo in E10.5 type I Bmpr control and mutant spinal cords. To avoid complications that could arise from even minor developmental heterogeneities between embryos, we sought to use the number of either Lhx2/9+ or Cre+ commissural neurons as an indicator of developmental age, rather than the axial level of the spinal cord. This strategy requires that the removal of either type I Bmpr has no effect on the number of commissural neurons. To assess this possibility, we correlated a BMP-independent indicator of spinal cord development (Liem et al., 2000), the number of Olig2+ motor neuron progenitors (pMNs) (Novitch et al., 2001), with the extent of either Lhx2/9+ or Cre+ commissural neuron development in E10.5 control (supplementary material Fig. S3A,B) or mutant (supplementary material Fig. S3C,D) spinal cords. The number of Olig2+ pMNs declined in a similar manner with respect to Cre+ neurons in BmprIa control and mutant spinal cords (supplementary material Fig. S3E), and Lhx2/9+ neurons in BmprIb control and mutant spinal cords (supplementary material Fig. S3F). This observation strongly suggests that the specification of commissural cell fate is normal in the type I Bmpr mutant mice and that the numbers of commissural neurons can be used as an accurate indicator of the stage of spinal cord development.
To assess the extent of commissural axon outgrowth, longitudinal ‘fillet’ preparations of the spinal cord were dissected from E10.5 type I Bmpr control and mutant spinal cords (Fig. 3A). At E10.5, commissural neurons are in the process of extending axons away from the RP (Augsburger et al., 1999) and towards the FP (Tessier-Lavigne et al., 1988; Placzek et al., 1990), such that the length of the commissural trajectories markedly increases along the caudal-to-rostral axis of the spinal cord. Similar to our observations using in vitro cultures, the length of the GFP+ commissural axons growing in vivo were indistinguishable in BmprIa mutant and control fillets (Fig. 3B-G,N, brackets). By contrast, the GFP+ commissural axons extending in the caudal regions of BmprIb–/– fillets were on average 10-20% longer than those in control fillets (Fig. 3H-M,O, brackets). However, although this trend persisted, the difference in commissural axon length was not statistically significant at more rostral levels of the spinal cord, most likely because of the challenge of scoring GFP+ commissural axon length against the background staining of Math1::tauGfp in the motor column (Hazen et al., 2012).
Fig. 3.

Commissural axons extend more rapidly in vivo in the absence of BmprIb. (A) Explants were taken from the brachial and thoracic levels of E10.5 mouse spinal cord. The boxed region represents the orientation of the images in panels B-M. (B-M) Commissural axons were detected using antibodies against Tag1 (blue in B,E,H,K) and a genetically encoded reporter, Math1::tauGfp (green in A,B,E,H,K). Antibodies against Cre (red in B,E) or Lhx2/9 (red in H,K) were used to normalize the stage of commissural neuron development. The extent of commissural axon outgrowth was equivalent in the BmprIa control (Math1::Cre; Math1::tauGfp; Bmpr1a+/+; bracket in C) and mutant (Math1::Cre; Math1::tauGfp; Bmpr1aflox/flox; bracket in F) fillets. By contrast, commissural axons extended further in the BmprIb–/– fillets (bracket in I) compared with control fillets (bracket in L). (N) There was no significant difference between axon growth within the BmprIa mutant and control fillets at three stages of Cre+ commissural neuron development: 20-39 Cre+ neurons (P>0.30, Student’s t-test; control: n=61 axons, eight embryos, mutant: n=63 axons, three embryos); 40-59 Cre+ neurons (P>0.45; control: n=196 axons, mutant: n=210 axons); and 60-79 Cre+ neurons (P>0.04, control: n=440 axons, mutant: n=240 axons). (O) By contrast, the BmprIb-deficient axons extended up to 18.5% further than the control axons: 1-19 Lhx2/9+ neurons (P<0.0091, control: n=159 axons, four embryos, mutant: n=262 axons, six embryos); 20-39 Lhx2/9+ neurons (P<0.0014, control: n=387 axons, mutant: n=520 axons); and 40-59 Lhx2/9+ neurons (P>0.20, control: n=85 axons, mutant: n=566 axons). Error bars represent s.e.m. Scale bar: 50 μm.
To examine whether the difference in axon length observed in BmprIb mutants results from commissural axons growing at an accelerated rate, we directly measured the velocity of commissural axon growth in the dorsal spinal cord using time-lapse imaging. Supporting our previous results that BmprIa does not transduce the ability of the BMPs to regulate the rate of commissural axon outgrowth, BmprIa mutant commissural axons grew at 13±0.5 μm/hour on average, a statistically identical velocity to that of littermate control commissural axons (Fig. 4C). By contrast, commissural axons in BmprIb–/– fillets grew at 17±0.8 μm/hour, an average velocity 3 μm/hour faster than the littermate controls (Fig. 4A-C; supplementary material Movie 2). This 20% average increase in velocity is in good agreement with the 18.5% increase in commissural axon length seen at caudal levels in BmprIb mutants (Fig. 3O).
Fig. 4.
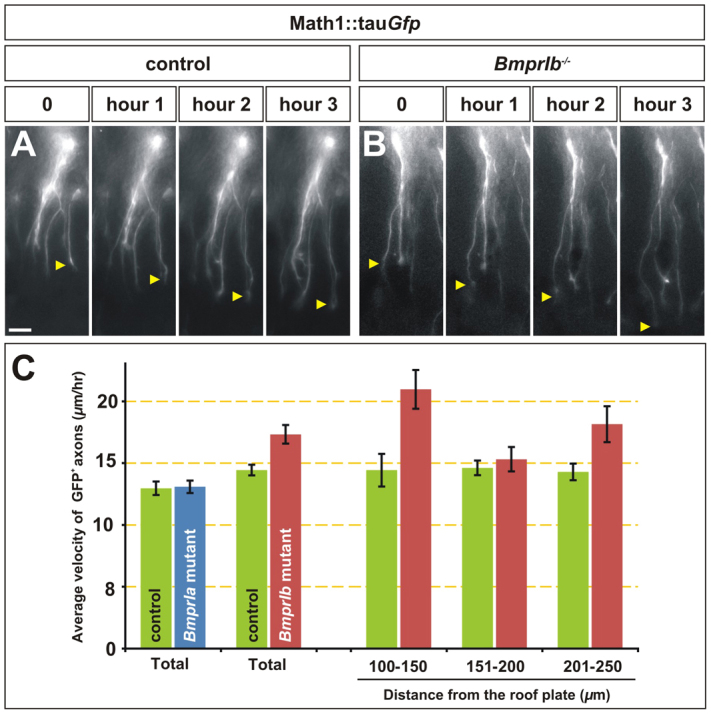
BmprIb–/– axons have accelerated growth rates in the mouse dorsal spinal cord. (A-C) The growth rate of GFP+ commissural axons was monitored using time-lapse imaging in longitudinal preparations of lumbar spinal cord taken from E11.5 BmprIa control (Math1::Cre; Math1::tauGfp; Bmpr1a+/+) mutant (Math1::Cre; Math1::tauGfp; Bmpr1aflox/flox) or BmprIb control (Math1::tauGfp; Bmpr1b+/+) and mutant (Math1::tauGfp; Bmpr1b–/–) littermates. (A,B) Stills taken from a BmprIb control (A) and mutant (B) spinal cord (see also supplementary material Movie 2). The arrows indicate the progress of an advancing growth cone. Scale bar: 20 μm. (C) There was no statistical difference in the rate of growth of GFP+ axons in BmprIa mutants and controls (P>0.43, Student’s t-test; control, n=53 neurons, five embryos; mutant, n=48 neurons, four embryos). By contrast, BmprIb mutant commissural axons grew on average 20% faster than their respective littermate controls (P<0.00017; control, n=107 neurons, seven embryos; mutant, n=67 neurons, five embryos). Moreover, the control GFP+ axons grew consistently at about 15 μm/hour, whereas the speed of the BmprIb–/– GFP+ axons varied significantly as follows: 100-150 μm from the RP, 45% faster than control axons (P<0.0018; control, n=37 measurements; mutant, n=52 measurements); 150-200 μm from the RP, no difference (P>0.25; control, n=154; mutant, n=108); 200-250 μm from the RP, 25% faster than control axons (P<0.012; control, n=171; mutant, n=34). Error bars represent s.e.m.
We further examined how the rate of commissural axon outgrowth changed as the axons extended through specific 50 μm regions of the dorsal spinal cord, starting at a region 100-250 μm from the RP. Control and BmprIa mutant commissural axons grew at a rate of 11-15 μm/hour throughout the interval examined, showing no statistical differences in the speed within each 50 μm region (Fig. 4C; data not shown). However, the BmprIb–/– commissural axons showed striking variations in their velocities. They grew ∼45% faster than control axons within the 50 μm interval closest to the RP, at an average rate of 21±1.6 μm/hour (Fig. 4C). The commissural axons slowed in the next 50 μm interval, dropping to an average velocity similar to control (Fig. 4C), and then grew ∼25% faster than control in the next 50 μm interval, at a rate of 18±1.5 μm/hour (Fig. 4C). Thus, the BmprIb mutant axons showed a much wider range in the rate at which they grew, compared with either control or BmprIa mutant axons.
Taken together, these studies show that the loss of BmprIb, and not BmprIa, results in accelerated axon outgrowth in vivo, suggesting that the BMPs slow commissural axon outgrowth by specifically activating BmprIb.
BmprIb is required to downregulate cofilin activity
What is the mechanism by which BmprIb controls the rate of commissural axon outgrowth? Our recent studies have suggested that BMPs alter the balance between the activation states of Limk1 and cofilin to regulate the speed of commissural axon extension (Phan et al., 2010). In particular, BMP7 stimulation of commissural neurons resulted in the activation of Limk1, which in turn phosphorylated, and thereby inactivated, cofilin (Phan et al., 2010). In light of these previous results, we examined whether the altered rates of axon outgrowth, observed after modulating BmprIb activity, were a consequence of BmprIb regulating the activity of Limk1-cofilin in commissural neurons. This hypothesis predicts that decreased BmprIb activity will result in a decrease in the level of phosphorylated inactive cofilin.
To assess whether BmprIa or BmprIb is required for the BMP-mediated inactivation of cofilin, we examined the distribution of phosphorylated (p)-cofilin in transverse sections taken from E10.5 BmprIa and BmprIb control and mutant embryos. Whereas the loss of BmprIa had no effect on the intensity of pcofilin staining in the dorsal spinal cord (Fig. 5A,B,E), there was a 20% decrease in the level of pcofilin in the BmprIb mutants (Fig. 5C-E), suggesting that only BmprIb regulates cofilin activity. To determine whether BmprI-deficient neurons responded appropriately to BMP stimulation, we briefly treated cultures of dissociated commissural neurons taken from E11.5 control or mutant mouse spinal cords with either BMP7 recombinant protein or a vehicle control. The loss of BmprIa had no effect on the response of commissural neurons to BMP7 (Fig. 5N; data not shown). By contrast, there was no increase in the level of pcofilin after BMP7 stimulation of BmprIb–/– commissural neurons (Fig. 5J-N) whereas pcofilin was significantly increased in the BmprIb+/+ commissural neuron cultures treated with BMP7 (Fig. 5H,I,N). Thus, BmprIb is required to transduce the ability of BMP7 to phosphorylate cofilin.
Fig. 5.

BmprIb is required to regulate cofilin activity in response to BMP stimulation. (A-E) E10.5 control [Math1::cre (A) or BmprIb+/+ (C)], or mutant [Math1::Cre; BmprIaflox/flox (B) or BmprIb–/– (D)], mouse brachial spinal cords labeled with antibodies against phospho (p)-cofilin (red and gray) and Tag1, which labels commissural neurons (green). The intensity of dorsal pcofilin staining was similar (P>0.12, Student’s t-test) in BmprIa mutant (n=200 sections, three embryos) and control littermates (n=144 sections, four embryos). By contrast, pcofilin staining was 20% lower (P<2.4×10–4) in BmprIb–/– embryos (n=195 sections, five embryos) compared with control littermates (n=212 sections, five embryos). (F-N) Dissociated E11.5 mouse commissural neurons from BmprIa and BmprIb control or mutant littermates were stimulated in parallel with either vehicle (mock stimulation) or BMP7 recombinant protein and then labeled with antibodies against pcofilin (red or gray, F-M), GFP to distinguish the Math1+ commissural neurons (green, F,H,J,L) and type III β-tubulin (blue, Tuj1, F,H,J,L). Control (n=141 neurons, five embryos) and mutant (n=291 neurons, eight embryos) BmprIa neurons behave equivalently in response to BMP7 or mock stimulation (N and data not shown, P>0.05). By contrast, there was >50% decrease in pcofilin activation in the BmprIb–/– neurons (L,M; n=50 neurons, three embryos) compared with the controls (H,I; P<2.57×10–7; n=39 neurons, three embryos), such that the level of pcofilin was similar in BMP7-treated (M) and mock-treated (G,K) cultures. Error bars represent s.e.m. Scale bars: in A-D, 30 μm; in F-M, 20 μm.
A non-phosphorylatable form of cofilin rescues the growth defects observed after constitutively activating BmprIb
To examine further whether BmprIb regulates cofilin activity, we assessed whether increasing the amount of cofilin was sufficient to rescue the delay in axon outgrowth observed after constitutively activating BmprIb. Moreover, we wanted to determine whether BmprIb works through Limk1 to alter the activation state of cofilin. Towards these goals, we assessed the effect on commissural axon outgrowth of two forms of cofilin: wild-type (wt) cofilin, which can be regulated by Limk1, and cofilinS3A, an analog that cannot be phosphorylated by Limk1 (Arber et al., 1998). Our previous studies have shown that overexpressing wt-cofilin in chicken commissural neurons results in their extending significantly faster growing axons than neurons electroporated with the Math1::fGfp control construct alone (Phan et al., 2010). We confirmed these studies (Fig. 6A,B,I), and also observed that cofilinS3A+ commissural axons grow at rates comparable to wt-cofilin+ commissural axons (Fig. 6E,F,I). Thus, increasing the amount of free cofilin can, by itself, enhance axon outgrowth.
However, these two forms of cofilin do not have equivalent abilities to rescue the BmprIb-mediated axon outgrowth defect. By HH stage 22/23, ∼20% of control GFP+ commissural neurons have extended axons to the FP (Fig. 6I). By contrast, only 10% of the caBmprIb+ commissural axons have reached the FP (Fig. 6I). Co-expression of Math1::cofilinS3A with Math1::caBmprIb-IRES-fGfp in commissural neurons rescues this defect: 20% of co-electroporated neurons now project axons to the FP (Fig. 6G-I). Thus, cofilinS3A can bypass the upstream regulatory events mediated by caBmprIb, thereby restoring normal axon growth at the FP. However, concomitantly increasing the level of wt-cofilin and caBmprIb further exacerbates the commissural axon growth defect. The rate of axon extension declined even further, such that <5% of the commissural axons reached the FP (Fig. 6C,D,I). This result strongly suggests a direct regulatory link between BmprIb, Limk1 and cofilin (Fig. 6J): caBmprIb acting through Limk1 can further slow the rate of commissural axon growth by inactivating both the wt-cofilin and the endogenous cofilin, thereby stabilizing actin in its filamentous form.
DISCUSSION
Modulating the activity of BmprIb alters the rate of commissural axon growth
In addition to the guidance cues that provide directional information to orient axons (Butler and Tear, 2007), axons also appear to be directed by extrinsic guidance cues to grow at a particular speed. Such ‘temporal’ information can control the rate and/or time at which directional cues are interpreted and is thus an important mechanism to ensure that axonal circuits develop in synchrony with the rest of the developing embryo. We had previously shown that the BMP RP chemorepellent provides temporal cues to commissural neurons (Phan et al., 2010), controlling the rate at which they extend axons through the dorsal spinal cord. These studies had implicated the type II BMP receptor (BmprII) as an essential factor that mediates the ability of BMPs to control the speed at which commissural axons grow. Here, we show that the other component of the canonical Bmpr complex, BmprIb, is also required to establish the correct rate of commissural axon growth. BmprIa is not required for temporal control, further demonstrating the specificity of the action of type I Bmprs.
Our previous time-lapse imaging studies had found that commissural axons in chicken embryos normally extend at ∼13 μm/hour (Phan et al., 2010). However, when BMP signaling was decreased in commissural neurons using a truncated form of BmprII, their axons now grew at ∼18 μm/hour. We see remarkably similar changes in the velocity of commissural axons deficient for BmprIb. Control mouse axons grow at ∼14 μm/hour whereas BmprIb–/– neurons extend axons at ∼17 μm/hour. Moreover, control axons grew at a relatively constant speed through the dorsal spinal cord. BmprIb–/– axons show far more variability: accelerating and decelerating from 15 to 21 μm/hour depending on their distance from the RP. It remains unresolved whether the BmprIb–/– axons change their speed consistently as they grow, or if this variability rather reflects a role for BMP signaling in stabilizing growth rates through the dorsal spinal cord. Supporting this latter possibility, we had predicted that commissural axons might accelerate while growing down the presumptive gradient of BMPs in the dorsal spinal cord (Phan et al., 2010). However, this model is not the case in rodents. Rather, commissural axons normally grow at the same average rate through the dorsal spinal cord, suggesting that the BMP chemorepellent establishes the speed of growth from the outset of axiogenesis. That the rate of axon outgrowth becomes more variable in the absence of BmprIb signaling further suggests that BMPs might also function in a long-range manner to keep the rate of growth constant.
BmprIb regulates the balance between the activities of Limk1-cofilin
The regulation of Limk1-cofilin activity is important for balancing commissural axon growth and guidance decisions (Phan et al., 2010). However, it remains unclear how the Limk1-cofilin pathway is regulated by BMPs. Other groups have implicated a regulatory interaction between BmprII and Limk1, in which binding to the intracellular tail of BmprII ‘primes’ Limk1 for activation (Foletta et al., 2003; Lee-Hoeflich et al., 2004). However, the molecular events that lead to Limk1 being phosphorylated and thereby activated in this context have remained unresolved. Many studies have implicated ROCK and Rho GTPases in the activation of Limk1 (Scott and Olson, 2007); however, these are broadly acting factors making it unclear where the specificity of their action resides. Could BmprIb, which is putatively recruited into a complex with BmprII upon BMP binding, have a role in this process? We concentrated on examining how BmprIb regulates cofilin, because cofilin is a direct effector of actin dynamics (Bamburg and Bernstein, 2008).
Our results show that there is an inverse correlation between the activity of BmprIb and both the activity of cofilin and the rate of axon outgrowth. When signaling through BmprIb was increased, the activity of cofilin decreased and the growth rate of commissural axons slowed. Our studies have also implied that the ratio of active to inactive cofilin is a crucial part of the mechanism that controls the speed of axon growth. When BmprIb signaling was increased and the level of wt-cofilin concomitantly increased, this combination slowed the rate of commissural axon growth even further (Fig. 6I). This observation suggests that caBmprIb can inactivate both endogenous and ectopically expressed cofilin, thereby further antagonizing actin polymerization and decreasing the rate of axon growth. Finally, our studies have implicated Limk1 as the key intermediate by which BmprIb regulates cofilin. Only cofilinS3A, which functions independently of Limk1 regulation, can rescue the axon outgrowth defect caused by caBmprIb. Thus, if Limk1 activity is bypassed in the presence of caBmprIb, cofilin remains active, strongly implying that BmprIb acts through Limk1 to control the speed of commissural axon outgrowth (Fig. 6J).
BmprIb regulates the Limk1-cofilin pathway to regulate the rate of commissural axon outgrowth
How are the distinct activities of the BMPs translated by commissural neurons into different cellular processes? Our studies (Phan et al., 2010; Hazen et al., 2011; Hazen et al., 2012) have lent support to a model in which these activities are differentially translated at both the receptor and second messenger levels. In this model, a shared activity of type I Bmprs is to mediate the specification of the dorsal-most cell fates by activating the BMP receptor-regulated Smads (Hazen et al., 2012), the canonical second messenger intermediates (Heldin et al., 1997). BmprIb alone mediates the guidance activities of BMPs. BmprIb modulates the activity of the Limk1-cofilin pathway to control the rate at which the actin cytoskeleton polymerizes or treadmills (Fig. 6J) (Bernard, 2007). The ability of BMPs to re-orient commissural axons appears to be controlled through activation of the phosphoinositide-3-kinase pathway (Perron and Dodd, 2011).
These results imply that there is a mechanistic difference in the ability of BMPs to signal through type I Bmprs to specify cellular identity versus axon growth rate. The basis of this mechanistic difference is unresolved; the different BMP ligands present in the RP might differentially activate the type I Bmprs (Liem et al., 1997; Butler and Dodd, 2003). Alternatively, the intrinsic context in which the type I Bmprs are activated might change as commissural neural development proceeds. The correct ‘context’ for a specific activity could simply be determined by the composition of the Bmpr complex or the complement of second messengers present in commissural neurons. Thus, the presence of both type I Bmprs might activate the Smad transcription factors, whereas the presence of only BmprIb results in signaling through Limk1. The presence or absence of Limk1 may also be crucial in determining ‘context’. Limk1 is only present in post-mitotic spinal neurons (Phan et al., 2010). The direct interaction of Limk1 with BmprII (Foletta et al., 2003; Lee-Hoeflich et al., 2004) followed by BMP dimerization of the Bmpr complex might result in a downstream response that either bypasses or works in conjunction with the canonical Smad signaling pathway (Hazen et al., 2011; Hazen et al., 2012).
In summary, BmprIb is required with BmprII to regulate commissural axon outgrowth through the Limk1-cofilin pathway. These studies have shed light on the specific identity of the receptor complex that permits the diverse activities of the BMPs to be translated within commissural neurons and have also further described the intrinsic signaling pathway by which axon outgrowth can be accelerated in vivo. This mechanism is likely to have clinical relevance; the intrinsic mechanisms by which axon growth is inhibited in vivo must be overcome for the regeneration of neural circuitry to be successful. Moreover, the modulation of these mechanisms may provide a means of accelerating the regeneration of neural circuits, thereby speeding up the lengthy process of re-growing neural circuits in a human patient.
Acknowledgments
We thank Pico Caroni for the cofilin/Limk1 constructs; Kohei Miyazono for the constitutively active BmprIb construct; Bennett Novitch for Olig2 antibodies; Jane Johnson for the Math1::tauGfp mouse line; David Rowitch for the Math1:Cre mouse line; and Karen Lyons for the Bmpr1aflox/flox and Bmpr1b–/– mice. We would also like to thank Bennett Novitch, Stephen Tymanskyj and members of the Butler and Novitch laboratories for helpful discussions; and Bennett Novitch and Michele Frendo for comments on the manuscript.
Footnotes
Funding
This work was supported by fellowships from the Rose Hills Foundation and University of Southern California (USC) Undergraduate Research Fund [to J.E.L.]; a Ruth L. Kirschstein National Research Service Award Individual Predoctoral Fellowship [NS 058097 to K.Y.]; and grants from the March of Dimes Foundation [1-FY07-458 to S.J.B.] and the National Institutes of Health [NS 063999 to S.J.B.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.089524/-/DC1
References
- Arber S., Barbayannis F. A., Hanser H., Schneider C., Stanyon C. A., Bernard O., Caroni P. (1998). Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature 393, 805–809 [DOI] [PubMed] [Google Scholar]
- Augsburger A., Schuchardt A., Hoskins S., Dodd J., Butler S. (1999). BMPs as mediators of roof plate repulsion of commissural neurons. Neuron 24, 127–141 [DOI] [PubMed] [Google Scholar]
- Bamburg J. R., Bernstein B. W. (2008). ADF/cofilin. Curr. Biol. 18, R273–R275 [DOI] [PubMed] [Google Scholar]
- Bernard O. (2007). Lim kinases, regulators of actin dynamics. Int. J. Biochem. CellBiol. 39, 1071–1076 [DOI] [PubMed] [Google Scholar]
- Butler S. J., Dodd J. (2003). A role for BMP heterodimers in roof plate-mediated repulsion of commissural axons. Neuron 38, 389–401 [DOI] [PubMed] [Google Scholar]
- Butler S. J., Tear G. (2007). Getting axons onto the right path: the role of transcription factors in axon guidance. Development 134, 439–448 [DOI] [PubMed] [Google Scholar]
- Dickson B. J. (2002). Molecular mechanisms of axon guidance. Science 298, 1959–1964 [DOI] [PubMed] [Google Scholar]
- Dodd J., Morton S. B., Karagogeos D., Yamamoto M., Jessell T. M. (1988). Spatial regulation of axonal glycoprotein expression on subsets of embryonic spinal neurons. Neuron 1, 105–116 [DOI] [PubMed] [Google Scholar]
- Endo M., Ohashi K., Mizuno K. (2007). LIM kinase and slingshot are critical for neurite extension. J. Biol. Chem. 282, 13692–13702 [DOI] [PubMed] [Google Scholar]
- Evan G. I., Lewis G. K., Ramsay G., Bishop J. M. (1985). Isolation of monoclonal antibodies specific for human c-myc proto-oncogene product. Mol. Cell. Biol. 5, 3610–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foletta V. C., Lim M. A., Soosairajah J., Kelly A. P., Stanley E. G., Shannon M., He W., Das S., Massague J., Bernard O. (2003). Direct signaling by the BMP type II receptor via the cytoskeletal regulator LIMK1. J. Cell Biol. 162, 1089–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisert E. E., Jr, Frankfurter A. (1989). The neuronal response to injury as visualized by immunostaining of class III beta-tubulin in the rat. Neurosci. Lett. 102, 137–141 [DOI] [PubMed] [Google Scholar]
- Hamburger V., Hamilton H. L. (1992). A series of normal stages in the development of the chick embryo. 1951. Dev. Dyn. 195, 231–272 [DOI] [PubMed] [Google Scholar]
- Hazen V. M., Phan K. D., Hudiburgh S., Butler S. J. (2011). Inhibitory Smads differentially regulate cell fate specification and axon dynamics in the dorsal spinal cord. Dev. Biol. 356, 566–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hazen V. M., Andrews M. G., Umans L., Crenshaw E. B., 3rd, Zwijsen A., Butler S. J. (2012). BMP receptor-activated Smads confer diverse functions during the development of the dorsal spinal cord. Dev. Biol. 367, 216–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldin C. H., Miyazono K., ten Dijke P. (1997). TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature 390, 465–471 [DOI] [PubMed] [Google Scholar]
- Helms A. W., Abney A. L., Ben-Arie N., Zoghbi H. Y., Johnson J. E. (2000). Autoregulation and multiple enhancers control Math1 expression in the developing nervous system. Development 127, 1185–1196 [DOI] [PubMed] [Google Scholar]
- Imondi R., Jevince A. R., Helms A. W., Johnson J. E., Kaprielian Z. (2007). Mis-expression of L1 on pre-crossing spinal commissural axons disrupts pathfinding at the ventral midline. Mol. Cell. Neurosci. 36, 462–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K. J., Jessell T. M. (1999). The specification of dorsal cell fates in the vertebrate central nervous system. Annu. Rev. Neurosci. 22, 261–294 [DOI] [PubMed] [Google Scholar]
- Lee-Hoeflich S. T., Causing C. G., Podkowa M., Zhao X., Wrana J. L., Attisano L. (2004). Activation of LIMK1 by binding to the BMP receptor, BMPRII, regulates BMP-dependent dendritogenesis. EMBO J. 23, 4792–4801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liem K. F., Jr, Tremml G., Jessell T. M. (1997). A role for the roof plate and its resident TGFbeta-related proteins in neuronal patterning in the dorsal spinal cord. Cell 91, 127–138 [DOI] [PubMed] [Google Scholar]
- Liem K. F., Jr, Jessell T. M., Briscoe J. (2000). Regulation of the neural patterning activity of sonic hedgehog by secreted BMP inhibitors expressed by notochord and somites. Development 127, 4855–4866 [DOI] [PubMed] [Google Scholar]
- Matei V., Pauley S., Kaing S., Rowitch D., Beisel K. W., Morris K., Feng F., Jones K., Lee J., Fritzsch B. (2005). Smaller inner ear sensory epithelia in Neurog 1 null mice are related to earlier hair cell cycle exit. Dev. Dyn. 234, 633–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meberg P. J., Bamburg J. R. (2000). Increase in neurite outgrowth mediated by overexpression of actin depolymerizing factor. J. Neurosci. 20, 2459–2469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishina Y., Suzuki A., Ueno N., Behringer R. R. (1995). Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes Dev. 9, 3027–3037 [DOI] [PubMed] [Google Scholar]
- Mishina Y., Hanks M. C., Miura S., Tallquist M. D., Behringer R. R. (2002). Generation of Bmpr/Alk3 conditional knockout mice. Genesis 32, 69–72 [DOI] [PubMed] [Google Scholar]
- Moore S. W., Kennedy T. E. (2008). Dissection and culture of embryonic spinal commissural neurons. Curr. Protoc. Neurosci. 3, 3.20 [DOI] [PubMed] [Google Scholar]
- Novitch B. G., Chen A. I., Jessell T. M. (2001). Coordinate regulation of motor neuron subtype identity and pan-neuronal properties by the bHLH repressor Olig2. Neuron 31, 773–789 [DOI] [PubMed] [Google Scholar]
- Perron J. C., Dodd J. (2011). Inductive specification and axonal orientation of spinal neurons mediated by divergent bone morphogenetic protein signaling pathways. Neural Dev. 6, 36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan K. D., Hazen V. M., Frendo M., Jia Z., Butler S. J. (2010). The bone morphogenetic protein roof plate chemorepellent regulates the rate of commissural axonal growth. J. Neurosci. 30, 15430–15440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placzek M., Tessier-Lavigne M., Jessell T., Dodd J. (1990). Orientation of commissural axons in vitro in response to a floor plate-derived chemoattractant. Development 110, 19–30 [DOI] [PubMed] [Google Scholar]
- Scott R. W., Olson M. F. (2007). LIM kinases: function, regulation and association with human disease. J. Mol. Med. 85, 555–568 [DOI] [PubMed] [Google Scholar]
- Skaggs K., Martin D. M., Novitch B. G. (2011). Regulation of spinal interneuron development by the Olig-related protein Bhlhb5 and Notch signaling. Development 138, 3199–3211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C. S., William C. M., Tanabe Y., Jessell T. M., Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessier-Lavigne M., Placzek M., Lumsden A. G., Dodd J., Jessell T. M. (1988). Chemotropic guidance of developing axons in the mammalian central nervous system. Nature 336, 775–778 [DOI] [PubMed] [Google Scholar]
- Timmer J. R., Wang C., Niswander L. (2002). BMP signaling patterns the dorsal and intermediate neural tube via regulation of homeobox and helix-loop-helix transcription factors. Development 129, 2459–2472 [DOI] [PubMed] [Google Scholar]
- Wen Z., Han L., Bamburg J. R., Shim S., Ming G. L., Zheng J. Q. (2007). BMP gradients steer nerve growth cones by a balancing act of LIM kinase and Slingshot phosphatase on ADF/cofilin. J. Cell Biol. 178, 107–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wine-Lee L., Ahn K. J., Richardson R. D., Mishina Y., Lyons K. M., Crenshaw E. B., 3rd (2004). Signaling through BMP type 1 receptors is required for development of interneuron cell types in the dorsal spinal cord. Development 131, 5393–5403 [DOI] [PubMed] [Google Scholar]
- Yamauchi K., Phan K. D., Butler S. J. (2008). BMP type I receptor complexes have distinct activities mediating cell fate and axon guidance decisions. Development 135, 1119–1128 [DOI] [PubMed] [Google Scholar]
- Yi S. E., Daluiski A., Pederson R., Rosen V., Lyons K. M. (2000). The type I BMP receptor BMPRIB is required for chondrogenesis in the mouse limb. Development 127, 621–630 [DOI] [PubMed] [Google Scholar]
- Yi S. E., LaPolt P. S., Yoon B. S., Chen J. Y., Lu J. K., Lyons K. M. (2001). The type I BMP receptor BmprIB is essential for female reproductive function. Proc. Natl. Acad. Sci. USA 98, 7994–7999 [DOI] [PMC free article] [PubMed] [Google Scholar]