

Abstract
Traumatic brain injury (TBI) is a progressive disease process underlain by dynamic and interactive biochemical mechanisms; thus, large-scale and unbiased assessments are needed to fully understand its highly complex pathobiology. Here, we report on a new high-capacity label-free proteomic platform to evaluate the post-TBI neuroproteome. Six orthogonal separation stages and data-independent MS were employed, affording reproducible quantitative assessment on 18 651 peptides across biological replicates. From these data 3587 peptides were statistically responsive to TBI of which 18% were post-translationally modified. Results revealed as many as 484 proteins in the post-TBI neuroproteome, which was fully nine times the number determined from our prior study of focal cortical injury. Yet, these data were generated using 25 times less brain tissue per animal relative to former methodology, permitting greater anatomical specificity and proper biological replication for increased statistical power. Exemplified by these data, we discuss benefits of peptide-centric differential analysis to more accurately infer novel biological findings testable in future hypothesis-driven research. The high-capacity label-free proteomic platform is designed for multi-factor studies aimed at expanding our knowledge on the molecular underpinnings of TBI and to develop better diagnostics and therapeutics.
Keywords: Data-independent, Label free, Neuroproteomics, Neurotrauma, TBI
1 Introduction
Traumatic brain injury (TBI) is the number one cause of injury-related death (53 000 per annum) and morbidity in the United States across all ages, races and incomes [1]. Though initiated by a traumatic event, TBI is actually a long-lasting disease process underlain by evolving degenerative and reparative mechanisms as are reviewed elsewhere [2–4]. Further, insults and outcomes vary greatly across individuals, encumbering TBI patient care and treatment. Thus, novel diagnostics and therapeutics are required, which selectively inform on and treat biochemical processes relevant to the individual. To this end, an unbiased assessment of interactive molecular dynamics is needed across anatomy and time following various modelled brain injuries. Such data will clarify the pathobiological relevance of prospective biofluid markers and advise on the type and effective administration of TBI therapeutics.
Recent proteomic studies reveal numerous TBI marker candidates; e.g., Hanrieder et al. found 70 TBI-responsive proteins in human cerebral spinal fluid (CSF) using an isobaric-labelling approach and Cadosch et al. reported 59 serum and 23 CSF TBI-responsive proteins in humans using 2D-PAGE analysis [5, 6]. However, the biological relevance of many prospective markers remains speculative lacking knowledge of their relationships with interactive pathobiological mechanisms evolving across space and time after TBI. While targeted studies are warranted, results lack global data that inform on molecular interactions associated with relevant post-injury mechanisms.
Ideally, neuroproteomics offers an unbiased approach to characterize the wide-scope biochemical response to TBI; however, conventional techniques have been limited in their capacity to reveal more than the most prominent of protein change. For example, our study from only 6 years ago showed just 55 proteins as altered at 2 days following focal TBI [7]. Indeed, the TBI-responsive neuroproteome remains at under 150 proteins following several recent studies using 2D-PAGE or more modern iTRAQ (isobaric tags for relative and absolute quantitation) LC-MS methods [5, 6, 8–11]. These data, while relevant for biomarker discovery, poorly inform on underlying mechanisms. Thus, greater analytical capacity is warranted to expand our knowledge of the TBI-responsive neuroproteome. Here, we report on a new high-capacity peptide-centric platform for differential analysis following TBI. We incorporated six orthogonal separation steps with data-independent MS. Using label-free quantification, the platform is intended to easily scale to more complex experimental designs and provide increase informative power to further our knowledge of post-TBI molecular dynamics.
2 Materials and methods
2.1 Controlled cortical impact brain injury
Male Sprague–Dawley rats (~450 g) were purchased from Harlan Laboratories. Animals were anesthetized using 4% isoflurane and maintained at 2%. A 5 mm craniotomy was made 3.5 mm from bregma and 3 mm lateral. Using an Impact One Stereotaxic Impactor (Leica), we administered a contusional brain injury with a 3 mm flat-tip impactor translating perpendicular to the brain surface at 4.0 m/s to a depth of 2 mm for 0.5 s. After suturing the skin, bacitracin and 2% lidocaine were applied. Two days post injury, animals were re-anesthetized and decapitated. Brains from injured and uninjured control animals (n = 6/group) were removed from the skull, rinsed with chilled PBS and snap-frozen in liquid nitrogen. All procedures followed the Guide for the Care and Use of Laboratory Animals (U.S. Department of Health and Human Services) in accordance with the Virginia Commonwealth University Institutional Animal Care and Use Committee protocol approval.
2.2 Sample processing
Neocortical tissue from outside the glial scar, proximal to the lesion was dissected at −20°C, referred to hereafter as the perilesion (PL). Each dissection (2 × 2 × 1 mm3) was homogenized for 30 s within 300 μL of a non-denaturing buffer: 150 mM ammonium bicarbonate with 10% Halt Protease and Phosphatase Inhibitor Cocktail (Thermo). The lysate was incubated for 90 min at 4°C with vortexing every 30 min. After centrifugation at 10 000 × g and 4°C for 10 min, the supernatant was collected (matrix fraction). The pellet was washed twice in 150 μL of non-denaturing buffer, vortexing for 5 min, and then centrifuged. The pellet was then re-suspended in 300 μL of a denaturing buffer: 1% Triton X-100, 1 M NaCl, 5 mM EDTA, 5 mM EGTA and 10 mM DTT brought up in non-denaturing buffer. The lysate was incubated for 90 min at 4°C with vortexing every 30 min., centrifuged and then collected (membrane fraction).
A Pierce 660 assay was used to determine protein concentration (Thermo). A 50-μg aliquot of each lysate was concentrated using an Orgosol Detergent Out kit (G Biosciences). Samples were reduced for 30 min at 90°C by adding fresh DTT to 10 mM, and followed by alkylation for 30 min by adding fresh iodoacetamide to 18 mM. After adjusting the pH to 8 with ammonium hydroxide, LC-MS grade trypsin (Promega) was added at a 1:100 enzyme-to-protein ratio digestion at 37°C for 16 h. Digests were transferred to autosampler vials and dried by speed vacuum. Peptides were reconstituted in 50 mM ammonium formate (pH 10) for 2DLC/IMS/MS analysis.
2.3 2DLC/IMS/MS analysis
Biological replicates (n = 6/group) were injected (8 μg on column, two technical replicates) in a group interspersed order onto a 2D NanoAcquity system (Waters). Separation was performed with an On-Line RP/RP 2D Separations kit (Waters). Five fractions (10.0, 14.8, 17.3, 21.0 and 45.0% ACN balanced with 20 mM ammonium formate, pH 10) were sequentially trapped and gradient eluted with 0.1% formic acid modified ACN and water: 8 to 10% ACN in 3 min; to 17% in 25 min; to 28% in 20 min; to 45% in 12 min; to 85% in 3 min; held for 3 min; to 8% in 1 min; equilibrated for 15 min. Eluting peptides were electrosprayed via a 10 μm id PicoTip emitter (New Objective) into a Synapt G2 hybrid mass spectrometer (Waters) with ion mobility enabled: 600 m/s wave velocity; 40 V wave height; 25 000 nominal MS resolving power. An external mass correction standard of 250 pM [Glu1]-fibrinopeptide B was sampled every 30 s. Data were acquired using a data-independent analysis (DIA) mode [12, 13]. The instrument switched between precursor (m/z 350 to 1300) and product (m/z 50 to 2000) ion scan functions at 2 Hz. Fragmentation was performed with a collision energy ramp from 27 to 50 V. Mass calibration and quality control digest analysis were performed daily to affirm system performance.
2.4 Informatics
Data were processed using Waters PLGS software (v.2.5) as detailed elsewhere [14]. Briefly, ion data were merged from technical replicates and tabulated by monoisotopic reduced mass and peak apex retention time. Ion tables were searched against a Uniprot KB Rattus database (rel. 2011_06) using these parameters: trypsin specificity; one missed cleavage; a minimum of two peptides per protein ID each with a minimum of three fragment ions; fixed carbamidomethylation; variable methionine oxidation, phosphorylation, acetylation and methylation; neutral loss of ammonia or water; 5 ppm precursor ion tolerance, 15 ppm product ion tolerance. Peptide annotations were filtered to a 1% false detection rate using a reversed-decoy database method.
Annotated ion tables were imported into IsoQuant software for peptide alignment across samples: 10 ppm mass tolerance; 1 min time tolerance (Tenzer S., to be published). Peptide intensity data were merged when split by methionine oxidation, missed cleavage, neutral loss of water or ammonia during experimentation. Data were filtered for replicating ions measured in ≥4 of 6 biological replicates per group. We chose this moderate level of replication to balance the desires for sufficient statistical power and comprehensive assessment of the TBI-responsive neuroproteome. Intensity values were log2 transformed and normalized (Loess) using DanteR software (v.1.0.1) [15]. Data were imputed for non-random missing values (reproducibly absent based on treatment) using a simulated Gaussian distribution about the lowest measure in a group using the median variance for that group [16]. ANOVA testing was performed at peptide and protein levels (α = 0.05) and visualized within DanteR. Protein data were generated using a peptide-to-protein roll-up function in DanteR [17]. Cell localization ontology was mined using DAVID Bioinformatics Resources (v.6.7) [18]. Pearson correlation and principal component analysis were performed and visualized using DanteR.
2.5 Immunoblot analysis
Protein-balanced samples were resolved using the NuPAGE 4–12% Bis-Tris gel system with MOPS buffer (Invitrogen). Proteins were then transferred to PVDF membrane (Millipore) via a semi-dry method using NuPAGE transfer buffer. Membranes were probed with the following primary antibodies: cytochrome oxidase-1 (Mitosciences); dynamin-related protein-1 (Origene); glycogen phosphorylase-BB (Abcam); mitofilin (Mitosciences); pan neurofascin (NeuroMab); syntaxin-binding protein-1 (Stxbp1) (Abcam); small ubiquitin-related modifier-1 (Cell Signaling); synaptotagmin-1 (BD Biosciences); vinculin (Millipore). We used horse-radish peroxidase-conjugated secondary antibodies and the SuperSignal West Pico chemiluminescence kit (Thermo) for detection. We acquired 16-bit images on an Image Station 4000MM Pro CCD imager and measured net band intensity (Carestream). Blots used for differential comparison (n = 5/group) were re-probed with anti-mouse beta-actin (Abcam) for controlling load error. These data were load normalized in ratio to corresponding beta-actin data. NF data with unequal group variance were assessed using a Mann–Whitney rank sum test (α = 0.05); all other data were assessed using a Student’s t-test (α = 0.05) within SigmaPlot (v.12.2).
3 Results and discussion
3.1 Proteomic localization
Given the complexity of biological change following TBI, we have devised a new high-capacity separations platform to afford more comprehensive quantification of the post-TBI neuroproteome (Fig. 1). Multidimensional separations and peptide-level quantification affords assessment of important post-translational dynamics such as protein translocation and modification. Location is a key consideration in neuroproteomics studies, both at the anatomical and sub-cellular level. In this study, we focused on change within the PL, an area of surviving cortical grey matter outside the glial scar surrounding the injury-induced lesion (Fig. 1A). While beyond the scope of the discussion here, the PL is of interest for the study of synaptic perturbation and remodelling following TBI. In such studies, protein translocation is often more functionally relevant than the total magnitude of change in a protein. We developed a simple but efficient extraction protocol to resolve proteins based on matrix or membrane localization. In addition to providing an additional mode of separation, measures between these sub-proteomes can reveal functionally relevant shifts in protein localization. We recently reported examples of this in regard to altered mitochondrial energetics in the developing brain after environmental exposure [19]. Proteomic data in that study revealed dose-dependent translocation between cytosolic and mitochondrial membrane compartments. For example, the total abundance of hexokinase 1 did not change with exposure; however, its localization was significantly altered, which reflected an increase in aerobic metabolism. In the present study, we assessed the performance of sub-proteome enrichment. Using six representative proteins, we attained 96 ± 2% enrichment for matrix-associated (Fig. 2A) and 94 ± 4% for membrane-associated (Fig. 2B) proteins. Across both sub-proteomes, over three-fourths of all proteins were found to have a relevant localization (Fig. 2C and D). Yet, fully 22% of proteins are known to translocate between these two compartments, representing a functionally important post-translational dynamic that can only be monitored when measured independently.
Figure 1.
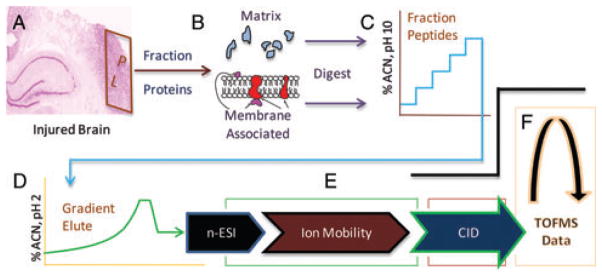
High-capacity label-free platform for differential neuroproteomic analysis. (A) Brain complexity requires selective anatomical sampling, here from tissue adjacent to the lesion; i.e., the perilesion (PL). (B) Subcellular fractionation is a biologically informative means to resolve the neuroproteome, here between matrix- and membrane-associated components. Multidimensional peptide separations provides a multiplicative increase in capacity to resolve sample components, here by 2D reversed-phase chromatography at pH 10 (C) and pH 2 (D) and by ion mobility (E) and mass-to-charge separation (F) within a hybrid mass spectrometer.
Figure 2.

Subcellular fractionation further resolves the proteome and informs on protein translocation. (A) Immunoblot data to assess selective enrichment of matrix proteins: glycogen phosphorylase-BB (Gpbb); small ubiquitin-related modifier-1 (Sumo-1); dynamin-related protein-1 (Drp1). (B) Immunoblot data to assess selective enrichment of membrane proteins: cytochrome oxidase-1 (Cox1); mitofilin; Synaptotagmin-1 (Syt1). Results are reported as mean ± SE percent of total net intensity across matrix and membrane fractions; n = 3/group. Charts of GO ontology annotated localization for the matrix (C) and membrane (D) enriched proteomes.
3.2 Orthogonal peptide separations
MS-based proteomics employs ‘bottom-up’ analysis as a necessity, increasing molecular complexity through restrictive protease digestion (e.g., trypsin). To address the analytical challenge this presents, we integrated a platform with 2D liquid-phase and 2D gas-phase peptide separations for a multiplicative increase in analytical capacity. Once optimized, tandem reversed-phase separations at pH 10 and pH 2 (Fig. 1C and D) were used to distribute peptide mass across five sequential gradient elution profiles, reducing the flux of co-eluting peptides for improved quantitative precision [20]. Data were acquired using DIA methodology [12, 13], which increased duty-cycle compared to conventional data-dependent analysis (DDA). Each precursor scan was followed by one product ion scan of all co-eluting peptides, allowing a rapid 2 Hz precursor acquisition rate. Thus, we were able to integrate chromatographic peak area for lower abundant peptides eluting in <10 s, which requires careful processing when using DDA [21, 22]. Ion mobility performed ahead of the fragmentation cell provided additional separation of co-eluting peptides, which simplified product to precursor ion alignment (Fig. 1E). Ultimately, each peptide was characterized by localization, retention time, drift time and reduced mass properties (Fig. 1B–F) affording the molecular selectivity needed to effectively align measures across biological replicates. Applying a replication filter across the 26 860 unique peptide measures, a total of 18 651 from 1806 protein products (Supporting Information Table 1) were quantified across a minimum of four of six biological replicates.
3.3 Differential analysis
Label-free quantification using DIA acquired data is more precise and complete relative to DDA spectral counting methods [17, 23]. Of the 18 651 peptides, only 16% had missed more than one measure out of six biological replicates, providing ample power to perform statistical testing. However, 15% of data were left-censored, a method-independent problem in proteomic studies where measures are reproducibly absent from one experimental group [17]. These non-random, biologically relevant events occur when a peptide falls below the detection limit. Standard imputation methods (e.g., k-nearest neighbours) are inappropriate for censored events that are neither random nor independent of intensity. Fixed value substitution or leaving values blank biases or prohibits variance testing across groups. Here, we imputed a set of Gaussian values distributed based on the median variance for that experimental group and a mean equal to the lowest detected measure for that experimental group [16]. With this approach our analysis accounted for peptide measures falling below the detection limit as a function of treatment while using a conservative, group-relevant estimate of variance. Results from statistical testing revealed 3567 peptides with a significant group mean difference between TBI and control (Fig. 3A). Of these, 634 were differentially modified (154 phosphorylated, 155 acetylated and 325 methylated peptides), which potentially have unique biological relevance and may diverge from other peptides to the same protein (see Section 3.5).
Figure 3.

Evaluation of the TBI-responsive neuroproteome. Variance analysis results are reported at peptide (A) and protein (B) levels, plotted by p-value against fold-change from control (x-axis). An α = 0.05 significance cutoff is drawn. Results of Pearson correlation analysis are reported with a heatmap display (C) across replicate control and TBI samples, with close correlation represented as similarity of colour. The first two principal components from principal component analysis testing are plotted (D) across treatment (larger markers in centre) and peptide factors (smaller marks left and right).
Applying a peptide-to-protein roll-up function, these data revealed perturbation among 379 unique protein products and 105 protein families indicating at least 484 responsive proteins in the TBI neuroproteome (Fig. 3B, Supporting Information Table 2). Homology among protein families often confounds proteomic results; however, performing differential analysis at the peptide level permitted separate assessment of isoform specific and homologous sequences [22]. Interestingly, of the 3567 differential peptides, 78% were isoform specific, which was increased from 66% across all 18 651 peptides suggesting an enrichment of isoform selective data. All together, study results showed a large difference between TBI and control neuroproteomes visualized here following Pearson correlation analysis (Fig. 3C). Replicates for each group were clearly delineated following non-supervised principal component analysis (Fig. 3D). Indeed, the first principal component accounted for an impressive 75% of total variance, reflecting a strong effect of treatment on the neuroproteome. The next two principal components correlated to within-group variability that accounted for only 8.7% of total variance.
3.4 Expanded TBI-responsive neuroproteome
Ultimately, our intention was to develop a more powerful differential proteomic platform able to expand our knowledge of the TBI neuroproteome. Thus, in this study we likewise assessed the TBI neuroproteome at 2 days following focal cortical impact injury as published previously with use of a lower capacity gel-based method [7]. Results here represented a ninefold increase in the number of revealed TBI-responsive proteins (484 versus 55), while the ratio between increased to decreased measures remained the same at just under 2:1 (Fig. 4). Of the previously reported differential proteins, 95% were confirmed having the same direction of response to TBI in the present study. Peptide-centric analysis performed here further resolved isoform specific responses for phosphoglycerate kinase 1 (not 2) and aldolase C (not A) that were previously confounded. Together these results affirmed more extensive characterization of a common proteome with the present platform. The improved TBI neuroproteome coverage was achieved first through greater analytical capacity and sensitivity afforded by the present multidimensional platform and second by the use of a statistical significance threshold rather than a fold-change cutoff in defining the differential TBI neuroproteome. The first improvement is appreciated applying the previously used twofold differential cutoff to the present data revealing 169 TBI-responsive proteins, or three times the number found previously (55). These findings are more impressive given that 25 times less tissue and 14 times less protein mass per animal were analyzed in this study, with no need to pool samples as before. Greater efficiency here further allowed for the ready use of appropriate biological replication and concomitantly a statistical differential threshold (p <0.05), which revealed the remainder of the 484 TBI-responsive proteins found with a significant group-mean difference (79% with >1.5-fold change). Improved analytical efficiency also affords greater anatomical specificity in the design of neuroproteomic studies.
Figure 4.
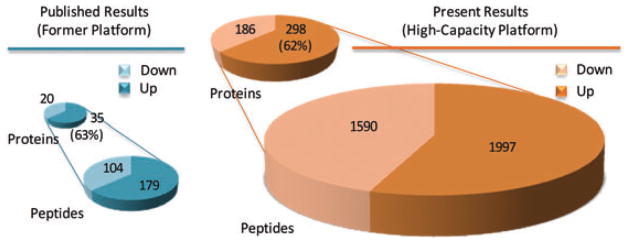
Expanding the TBI-responsive neuroproteome. Differential analysis results are compared from former lower capacity gel-based methods [7], and the new high-capacity platform described in this study. Data represent the TBI-responsive neuroproteome assessed at 2 days following focal brain injury to somatosensory cortex.
The label-free approach used here is also practical for more complex multi-factor experimental designs. Neurotrauma research requires the ability to manipulate multiple factors of insult, time post injury, brain region and therapeutic treatment. The trade-off here is in the time needed to analyze samples sequentially rather than multiplexed analysis used with labelled quantitative proteomics. To put this into perspective, instrumental analysis time using this platform was equivalent to that spent on generating samples and in processing and analyzing the data. Another important consideration with this platform is that careful attention must be paid to minimize experimental variance. For example, upgraded climate control systems were installed to maintain a constant laboratory temperature. Mobile phase pH, instrument calibration and platform performance were checked routinely, and room traffic was minimized. We were able to control the median technical variance to an acceptable 12% across 1 wk of acquisition, though further refinement of technical precision is an ongoing interest.
3.5 Increased information for biological inference
Peptide-centric statistical assessment is a crucial feature of our new neuroproteomics approach, which enhances interpretation of protein dynamics following TBI and their biological relevance. Here, we provide several illustrative examples. First, five TBI-responsive peptides were reported in the matrix fraction for the protein vinculin. Four peptides suggested increased vinculin following TBI while one suggests a decreased response. Closer inspection revealed that the decreased peptide has a known phosphoserine modification site (721), though its biological role remains unknown [24]. Yet, the existence of this motif provided rationale for separating this peptide for inferential purposes even though we did not detect the modified form. The remaining data suggested an increase in cytosolic vinculin, an integrin complexing protein involved in actin coupling with the plasma membrane, which is particularly relevant to growth cone elongation [25]. Increased vinculin at the membrane would suggest greater growth cone motility. However, increased cytosolic vinculin found here and affirmed by immunoblot results (Fig. 5A) suggests destabilization and retraction of neuronal processes, as more of the protein is autoinhibited in the matrix [26].
Figure 5.
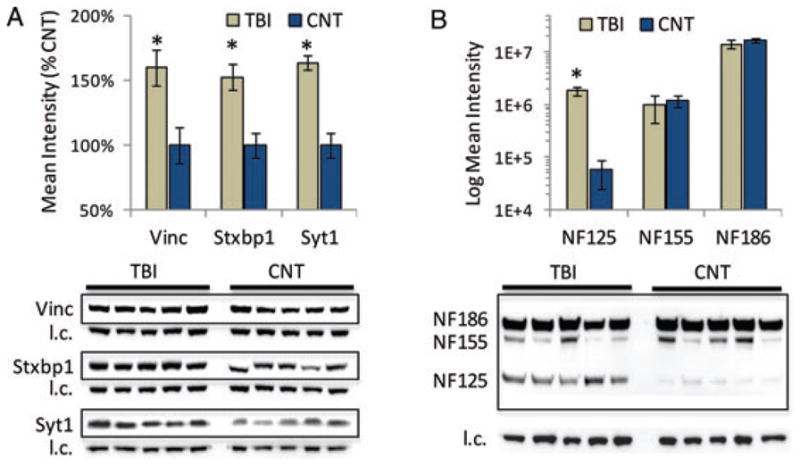
Validating response to TBI for exemplar proteins. (A) Immunoblot results for vinculin (Vinc), syntaxin-binding protein 1 (Stxbp1) and synaptotagmin 1 (Syt1), three responsive proteins relevant to synaptic dynamics following TBI. (B) Immunoblot results for three neurofascin (NF) isoforms in response to TBI (log(10) intensity scale). Blot images of target proteins are shown boxed with their corresponding beta-actin load control (l.c.) images provided below. Values reported as mean ± SE; n = 5/group. *p <0.05 compared to control.
The relative magnitude of vinculin response is confounded here by inter-peptide variance (44%), a common limitation of ‘bottom-up’ proteomics [17]. Roll-up functions as used here with DanteR software address this issue by averaging peptide intensities, with cross-replicate scaling, to achieve a unified protein measure [15]. While a valuable tool, automated roll-up functions do not consider potential biological confounds in selecting which peptide measures to use. For example, vinculin is reported to increase by 27% over control based on results applying the roll-up function to all data. Removing the divergent peptide measure discussed above, however, increased the result to 55% over control, which approximated the 52% increase measured by immunoblot analysis (Fig. 5A). Data for Stxbp1 offered a more striking example of the difference between protein- and peptide-centric analyses. Protein measures generated with the roll-up function did not reach significance for Stxbp1. Yet, seven increased and three decreased Stxbp1 peptides were significantly responsive to TBI. The three decreased peptides were all post-translationally modified (one phosphorylated and two methylated), which justified their consideration as independent Stxbp1 responses to injury. Immunoblot results then affirmed the TBI-induced increase in Stxbp1 abundance inferred from the remaining seven peptides (Fig. 5A). Importantly, the relative change measured for vinculin and Stxbp1 was less than twofold, a commonly used but arbitrary threshold. Indeed, to the best of our knowledge, we are the first to report vinculin and Stxbp1 perturbation following TBI. Yet identifying more of the TBI neuroproteome will ultimately expand our knowledge of the pathobiology.
Peptide-level data can also be used to differentiate between isoforms, which often is important when inferring biological relevance. For example, in our previous study, we found a differential response for the synaptotagmin (Syt) protein family; however, we were unable to resolve a response for specific isoforms. Syt1 and Syt2 are 75% homologous and perform similar functions; however, they are differently associated with excitatory and inhibitory synapses, respectively [27]. Results here show five responsive peptides increased in the cytosol – three specific to Syt1, one to Syt2 and a fifth lacking specificity. These data suggest that both isoforms increased as confirmed for Syt1 by immunoblot results (Fig. 5A). This broadened our interpretation to both excitatory and inhibitory synaptic change. Increased synapotagmin would generally suggest greater synaptic activity given its role in regulating synaptic vesicle cycling during neurotransmission. However, our data specifically showed increased matrix synaptotagmin, an atypical localization for this membrane protein. Synaptotagmins are known to be proteolyzed following TBI by calpain, which releases the large cytoplasmic domain [28]. Our data also revealed decreased phosphorylation at T-202, suggesting less WNK1 activation and reduce vesicle cycling [29]. Together, localization and peptide-level information suggests decreased synaptic activity, which fits with reduced neurotransmission in the PL expected 2 days following focal TBI [30].
Inference from peptide-centric results can also reveal dynamics of lesser abundant isoforms among more dominant but non-responsive homologs. For example, of 28 neurofascin peptides measured from the membrane fraction only one was responsive to injury (VYSDTVQGQLR, p = 0.0004). Further, just this one peptide was specific for a smaller, uncharacterized neurofascin variant (accession: D3ZW56). Using a pan-neurofascin antibody, the known NF186 and NF155 forms of neurofascin were found unresponsive; however, we measure a smaller variant that was significantly differential at 2 days following TBI (p = 0.0009, Fig. 5B). The smaller, less well studied but commonly observed variant (NF125 here forward) is reported to migrate between 120 and 140 kDa on-gel, depending on post-translational modification [31–34]. Retained at the plasma membrane during oligodendrocyte process elongation, NF125 is shed from the membrane upon contact with an axon [33]. Given prominent demyelination at 2 days following injury [35], the increased membrane-bound level of NF125 found here suggests an upregulated response to re-establish oligodendrocyte contact with axons. Reeves et al. similarly reported an increase in NF125 within white matter following diffuse brain injury, affirming relevance to fibre demyelination after TBI [34]. However, Maier et al. suggest that NF125 may be a metaloprotease cleaved product of oligodendrocyte specific NF155 [33]. The isoform selective peptide from our data challenges their interpretation, given a distinct 10.0208 Da difference between corresponding peptides for NF125 and NF155. Results here suggest for future research a role for this distinct NF125 isoform in demyelinating pathobiology, and perhaps more broadly in the transient GalC stage of oligodendrocyte differentiation.
4 Concluding remarks
Results from this study demonstrate improved efficacy for large-scale, peptide-centric analysis of the neuroproteomic response to brain injury. We describe a high-capacity label-free quantitative platform practical for use in multi-factor experimental designs. Greater efficiency permitted analysis in a small neuroanatomical region, while providing up to nine times more differential information than available only 6 years ago. With appropriate biological replication, the present study had sufficient power to deduce as small as 25% peptide change with treatment. We affirmed a sub-twofold TBI response for three representative proteins, signifying TBI responses that would have been missed using an arbitrary twofold threshold. Such precision is particularly advantageous in studying neuroproteomics response to more subtle mild or diffuse TBI. Results here also illustrate the value of localization and peptide-centric assessment when inferring biological relevance. This powerful proteomic platform facilitates unbiased study of the complex, interactive biochemical response to brain injury needed to further the development and evaluation of new diagnostics and therapeutics.
Supplementary Material
Acknowledgments
We thank M. Fox and L. Phillips for gifted antibodies; P. Ottens for programming expertise; S. Tenzer for providing and assisting with Isoquant Software. This work was funded in part by the National Institutes of Health grant NS055012.
Abbreviations
- CSF
cerebral spinal fluid
- DDA
data-dependent analysis
- DIA
data-independent analysis
- PL
peri-lesion
- Stxbp1
syntaxin-binding protein-1
- Syt
Synaptotagmin
- TBI
traumatic brain injury
Footnotes
The authors have declared no conflict of interest.
Additional supporting information may be found in the online version of this article at the publisher’s web-site
References
- 1.Coronado VG, Xu L, Basavaraju SV, McGuire LC, Wald MM, Faul MD, Guzman BR, Hemphill JD. MMWR Surveill Summ. 2011;60:1–32. [PubMed] [Google Scholar]
- 2.Masel BE, DeWitt DS. J Neurotrauma. 2010;27:1529–1540. doi: 10.1089/neu.2010.1358. [DOI] [PubMed] [Google Scholar]
- 3.Garcia AN, Shah MA, Dixon CE, Wagner AK, Kline AE. PM R. 2011;3:S18–S27. doi: 10.1016/j.pmrj.2011.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McAllister TW. Dialogues Clin Neurosci. 2011;13:287–300. doi: 10.31887/DCNS.2011.13.2/tmcallister. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hanrieder J, Wetterhall M, Enblad P, Hillered L, Bergquist J. J Neurosci Methods. 2009;177:469–478. doi: 10.1016/j.jneumeth.2008.10.038. [DOI] [PubMed] [Google Scholar]
- 6.Cadosch D, Thyer M, Gautschi OP, Lochnit G, Frey SP, Zellweger R, Filgueira L, Skirving AP. ANZ J Surg. 2010;80:542–547. doi: 10.1111/j.1445-2197.2010.05268.x. [DOI] [PubMed] [Google Scholar]
- 7.Kobeissy FH, Ottens AK, Zhang Z, Liu MC, Denslow ND, Dave JR, Tortella FC, Hayes RL, Wang KK. Mol Cell Proteomics. 2006;5:1887–1898. doi: 10.1074/mcp.M600157-MCP200. [DOI] [PubMed] [Google Scholar]
- 8.Yang X, Yang S, Wang J, Zhang X, Wang C, Hong G. Brain Inj. 2009;23:830–840. doi: 10.1080/02699050903196670. [DOI] [PubMed] [Google Scholar]
- 9.Ding Q, Vaynman S, Souda P, Whitelegge JP, Gomez-Pinilla F. Eur J Neurosci. 2006;24:1265–1276. doi: 10.1111/j.1460-9568.2006.05026.x. [DOI] [PubMed] [Google Scholar]
- 10.Crawford F, Crynen G, Reed J, Mouzon B, Bishop A, Katz B, Ferguson S, Phillips J, Ganapathi V, Mathura V, Roses A, Mullan M. J Neurotrauma. 2012;29:246–260. doi: 10.1089/neu.2011.1789. [DOI] [PubMed] [Google Scholar]
- 11.Opii WO, Nukala VN, Sultana R, Pandya JD, Day KM, Merchant ML, Klein JB, Sullivan PG, Butterfield DA. J Neurotrauma. 2007;24:772–789. doi: 10.1089/neu.2006.0229. [DOI] [PubMed] [Google Scholar]
- 12.Blackburn K, Mbeunkui F, Mitra SK, Mentzel T, Goshe MB. J Proteome Res. 2010;9:3621–3637. doi: 10.1021/pr100144z. [DOI] [PubMed] [Google Scholar]
- 13.Geromanos SJ, Vissers JP, Silva JC, Dorschel CA, Li GZ, Gorenstein MV, Bateman RH, Langridge JI. Proteomics. 2009;9:1683–1695. doi: 10.1002/pmic.200800562. [DOI] [PubMed] [Google Scholar]
- 14.Danton GH, Dietrich WD. J Neuropathol Exp Neurol. 2003;62:127–136. doi: 10.1093/jnen/62.2.127. [DOI] [PubMed] [Google Scholar]
- 15.Flouris A, Vardavas C, Metsios G, Tsatsakis A, Koutedakis Y. Am J Physiol Lung Cell Mol Physiol. 2010;298:L3–L12. doi: 10.1152/ajplung.00215.2009. [DOI] [PubMed] [Google Scholar]
- 16.Chich JF, David O, Villers F, Schaeffer B, Lutomski D, Huet S. J Chromatogr B Analyt Technol Biomed Life Sci. 2007;849:261–272. doi: 10.1016/j.jchromb.2006.09.033. [DOI] [PubMed] [Google Scholar]
- 17.Karpievitch Y, Stanley J, Taverner T, Huang J, Adkins JN, Ansong C, Heffron F, Metz TO, Qian WJ, Yoon H, Smith RD, Dabney AR. Bioinformatics. 2009;25:2028–2034. doi: 10.1093/bioinformatics/btp362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang da W, Sherman BT, Lempicki RA. Nat Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 19.Fuller BF, Cortes DF, Landis MK, Yohannes H, Griffin HE, Stafflinger JE, Bowers MS, Lewis MH, Fox MA, Ottens AK. Environ Health Perspect. 2012;120 doi: 10.1289/ehp.1104857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Doneanu CE, Xenopoulos A, Fadgen K, Murphy J, Skilton SJ, Prentice H, Staples M, Chen W. MAbs. 2012;4:24–44. doi: 10.4161/mabs.4.1.18748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chelius D, Bondarenko PV. J Proteome Res. 2002;1:317–323. doi: 10.1021/pr025517j. [DOI] [PubMed] [Google Scholar]
- 22.Liao Z, Wan Y, Thomas SN, Yang AJ. Anal Chem. 2012;84:4535–4543. doi: 10.1021/ac300510t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Neilson KA, Ali NA, Muralidharan S, Mirzaei M, Mariani M, Assadourian G, Lee A, van Sluyter SC, Haynes PA. Proteomics. 2011;11:535–553. doi: 10.1002/pmic.201000553. [DOI] [PubMed] [Google Scholar]
- 24.Olsen JV, Blagoev B, Gnad F, Macek B, Kumar C, Mortensen P, Mann M. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 25.Carlstrom LP, Hines JH, Henle SJ, Henley JR. BMC Biol. 2011;9:82. doi: 10.1186/1741-7007-9-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bakolitsa C, Cohen DM, Bankston LA, Bobkov AA, Cadwell GW, Jennings L, Critchley DR, Craig SW, Liddington RC. Nature. 2004;430:583–586. doi: 10.1038/nature02610. [DOI] [PubMed] [Google Scholar]
- 27.Fox MA, Sanes JR. J Comp Neurol. 2007;503:280–296. doi: 10.1002/cne.21381. [DOI] [PubMed] [Google Scholar]
- 28.Liu MC, Akle V, Zheng W, Dave JR, Tortella FC, Hayes RL, Wang KK. Biochem J. 2006;394:715–725. doi: 10.1042/BJ20050905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee BH, Min X, Heise CJ, Xu BE, Chen S, Shu H, Luby-Phelps K, Goldsmith EJ, Cobb MH. Mol Cell. 2004;15:741–751. doi: 10.1016/j.molcel.2004.07.018. [DOI] [PubMed] [Google Scholar]
- 30.Kroppenstedt SN, Sakowitz OW, Thomale UW, Unterberg AW, Stover JF. J Neurotrauma. 2002;19:1421–1432. doi: 10.1089/089771502320914651. [DOI] [PubMed] [Google Scholar]
- 31.Schafer DP, Bansal R, Hedstrom KL, Pfeiffer SE, Rasband MN. J Neurosci. 2004;24:3176–3185. doi: 10.1523/JNEUROSCI.5427-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pomicter AD, Shroff SM, Fuss B, Sato-Bigbee C, Brophy PJ, Rasband MN, Bhat MA, Dupree JL. Brain. 2010;133:389–405. doi: 10.1093/brain/awp341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Maier O, van der Heide T, Johnson R, de Vries H, Baron W, Hoekstra D. Exp Cell Res. 2006;312:500–511. doi: 10.1016/j.yexcr.2005.11.014. [DOI] [PubMed] [Google Scholar]
- 34.Reeves TM, Greer JE, Vanderveer AS, Phillips LL. Brain Pathol. 2010;20:1055–1068. doi: 10.1111/j.1750-3639.2010.00412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ottens AK, Golden EC, Bustamante L, Hayes RL, Denslow ND, Wang KK. J Neurochem. 2008;104:1404–1414. doi: 10.1111/j.1471-4159.2007.05086.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.