

Abstract
Transition metals are essential to many biological processes in almost all organisms from bacteria to humans. Their versatility, which arises from an ability to undergo reduction–oxidation chemistry, enables them to act as critical cofactors of enzymes throughout the cell. Accumulation of metals, however, can also lead to oxidative stress and cellular damage. The importance of metals to both enzymatic reactions and oxidative stress makes them key players in mitochondria. Mitochondria are the primary energy-generating organelles of the cell that produce ATP through a chain of enzymatic complexes that require transition metals, and are highly sensitive to oxidative damage. Moreover, the heart is one of the most mitochondrially-rich tissues in the body, making metals of particular importance to cardiac function. In this review, we focus on the current knowledge about the role of transition metals (specifically iron, copper, and manganese) in mitochondrial metabolism in the heart. This article is part of a Special Issue entitled ‘Focus on Cardiac Metabolism’.
Keywords: Transition metals, Heart, Mitochondria, Iron, Copper, Manganese
1. Introduction
Transition metals are necessary at trace levels for normal cellular function, and include iron, copper, manganese, zinc, selenium, chromium, nickel, and cobalt. These metals are absorbed from the diet by the duodenum and transported to sites of tissue utilization. There, they are used as cofactors in enzymatic reactions, as they can readily transition between oxidized and reduced states. The systemic and cellular levels of these metals must be actively monitored and regulated though, as their deficiency leads to loss of crucial enzyme activities, while their accumulation leads to inappropriate enzymatic reactions that generate reactive oxygen species (ROS), oxidative damage, and eventually cell death [1]. Due to the importance of transition metals to enzyme activity and their potential to generate ROS, they are particularly imperative in mitochondrial biology, which requires robust and undamaged enzymatic reactions to generate sufficient cellular ATP through electron transfer and oxidative phosphorylation. As the heart is a mitochondrially-rich tissue that requires abundant energy production to sustain continuous myocardial contractions, transition metals are thus also an important factor in cardiac metabolism. Although relatively few details are known about the role of transition metals in metabolism, the current available research suggests that transition metals are a key component of cardiac metabolism. In the following sections, we review the current knowledge about iron, copper, and manganese in cardiac mitochondrial metabolism.
2. Iron and mitochondrial metabolism in the heart
Iron is an essential transition metal that is required for normal cell physiology, and which plays a prominent role in cell metabolism, heme synthesis, cell proliferation, and cytochrome p450 enzyme activity. Its importance to so many catalytic processes stems from its redox reactivity, which enables it to transition between a reduced ferrous (Fe2+) and oxidative ferric (Fe3+) state [2]. However, this reactivity of iron also allows it to participate in the Fenton reaction as an electron donor to hydrogen peroxide [3], with subsequent production of a potent ROS species, the hydroxyl radical.
ROS are highly reactive molecules that contain oxygen and unpaired electrons, and which can react aberrantly with and damage macromolecules [4]. One type of ROS, the superoxide radical ( ), is generated during cellular respiration and protonated to make hydrogen peroxide, providing ample opportunity for iron to catalyze the generation of hydroxyl radicals. Cells contain antioxidant defense systems which are composed of enzymes that can convert ROS into inert compounds, but ROS accumulation can overload these systems and lead to cellular damage. Therefore, the levels of iron must be tightly regulated in order to provide a sufficient amount of iron for critical enzymatic processes without increasing ROS-induced damage.
2.1. Systemic and cellular iron regulation
The regulation of iron levels occurs at various sites and cellular levels within the body [5,6] (Fig. 1). Dietary iron is absorbed in the duodenum, where ferric iron is converted into ferrous iron by ferric reductases. Ferrous iron is then transported into enterocytes by divalent metal transporter 1 (DMT1). Dietary heme can also be imported into enterocytes, then converted to ferrous iron by heme oxygenase 1 (HO1). The export of ferrous iron by ferroportin is coupled to its conversion into ferric iron, which is catalyzed by hephaestin and ceruloplasmin. Export of iron into the bloodstream is inhibited by hepcidin, a circulating peptide produced by the liver which initiates internalization and lysosomal degradation of ferroportin [7]. Once in the bloodstream, ferric iron is stabilized from participating in redox reactions by binding to transferrin. Delivery of iron to tissues occurs through binding of transferrin to transferrin receptor 1 (TfR1) at the plasma membrane, which is internalized through clathrin-coated pits. In the cell, iron is either utilized, bound to ferritin and stored, or remains as a labile iron pool. Together, this system of iron absorption and transport is crucial to regulating systemic iron levels and avoiding iron deficiency or overload, as iron excretion is not regulated and instead occurs passively through cell sloughing or bleeding.
Fig. 1.
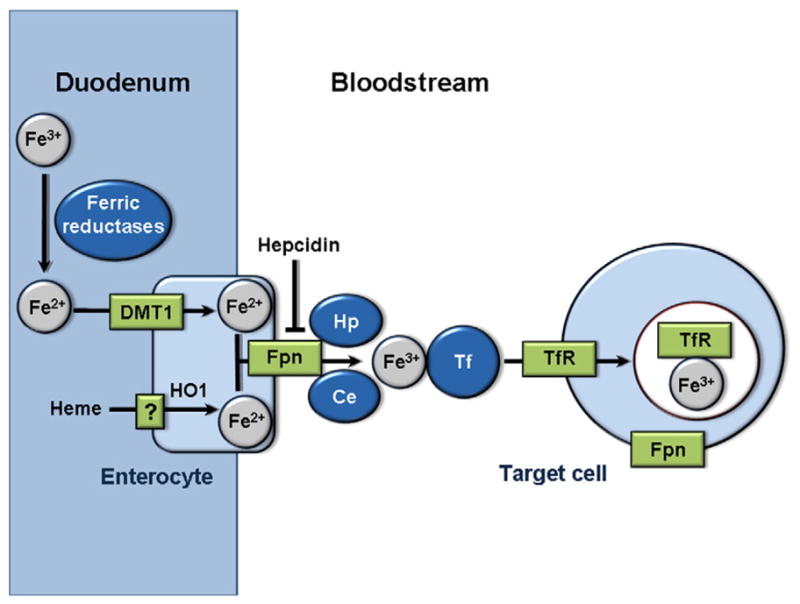
Systemic iron transport. Ferric iron (Fe3 +) from the diet is converted to ferrous iron (Fe2 +) in the duodenum by ferric reductases, then imported into enterocytes by DMT1. Heme from the diet is also imported into enterocytes and converted into ferrous iron by HO1. Ferrous iron is then exported into the bloodstream by Fpn. The export of ferrous iron is coupled to its conversion to ferric iron, which is catalyzed by hephaestin and ceruloplasmin. Iron export by Fpn can also be inhibited by the peptide hepcidin. In the bloodstream, ferric iron binds to transferrin, which enables import into target cells by binding to TfR. Ferric iron-bound TfR is then imported into cells by clathrin-coated pits. DMT1: Divalent metal transporter 1. Fpn: Ferroportin. Hp: Hephaestin. Ce: Ceruloplasmin. Tf: Transferrin. TfR: Transferrin receptor.
The dynamics of cellular iron metabolism are modified by iron-detecting transcriptional regulators called iron regulatory proteins (IRPs). IRP1 and 2 enable iron sensing in the cell, with appropriate modulation of iron import and export to control intracellular iron levels [8,9]. During iron depleted conditions, IRPs are active and bind to iron regulatory elements (IREs) to regulate mRNA levels of many proteins related to iron metabolism. IRPs bind to IREs in 3′ untranslated regions (UTRs) to increase mRNA levels through inhibition of transcript cleavage, while IRPs bind to IREs present in 5′ UTRs to inhibit translation. Specifically, IRP1 binds to 3′ IREs in TfR to stabilize its transcripts and increase iron import, and IRP2 binds to 5′ IREs in ferritin to inhibit its translation and decrease iron export. Overall, this regulation increases free iron available for utilization in the cell. When iron levels are restored, IRPs are inactivated, TfR mRNA is cleaved, and ferritin is no longer degraded, leading to decreased cellular free iron.
In addition to cellular iron, levels of mitochondrial iron are also specifically regulated. The mechanism of import of iron from the cytosol into the intermembrane space is unknown, but has been suggested to occur through transient interaction of TfR-containing endosomes with the outer mitochondrial membrane [10]. Iron is imported into the mitochondrial matrix by mitoferrin-1 and -2 (Mfrn1 and Mfrn2), which localize to the inner mitochondrial membrane [11] (Fig. 2). The transporter ATP-binding cassette (ABC) B10 has been shown to stabilize Mfrn1, and thus also seems to play a role in mitochondrial iron import [12]. An exporter of mitochondrial iron has not been identified, but ABCB8 has been found to facilitate export of iron from the mitochondria [13], and ABCB7 is involved in the maturation of cytosolic iron/sulfur cluster proteins [14]. Additionally, deletion of ABCB8 or ABCB7 results in mitochondrial iron accumulation [13,15]. Thus, although incompletely understood, maintenance of iron levels in the mitochondria is a regulated process.
Fig. 2.
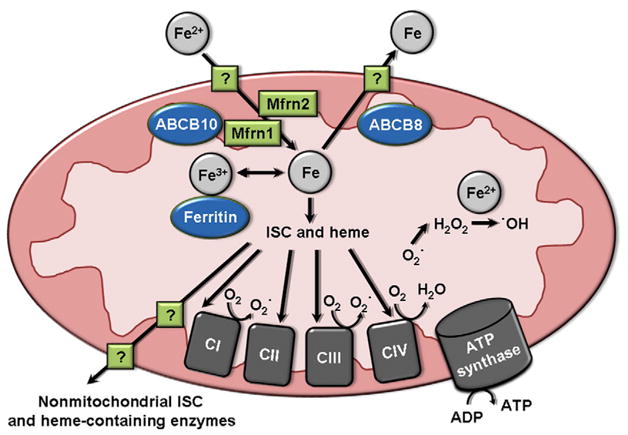
Iron signaling in the mitochondria. The mechanism of iron import into the outer mitochondrial membrane is not fully known, but iron is imported into the inner mitochondrial membrane by Mfrn1 (assisted by ABCB10) and Mfrn2. The mechanism of iron export from the mitochondria is also not known, but is facilitated by the inner mitochondrial membrane protein ABCB8. In the mitochondria, iron remains as a labile iron pool, is bound by ferritin and stored, or is incorporated into ISC- and heme-containing enzymes of mitochondrial complexes of the electron transport chain. ISC and heme are also exported from the mitochondria by an unknown mechanism, then incorporated into non-mitochondrial ISC- and heme-containing enzymes. Mitochondrial complexes I and III produce superoxide ROS by electron leakage to O2. This superoxide is protonated to form H2O2, which can then react with free iron to form hydroxyl radical ROS. CI–CIV: complexes I through IV. ISC: iron/sulfur cluster.
2.2. Cardiac iron overload and deficiency in mitochondrial metabolism
As a vigorously metabolically active tissue, the heart is a primary tissue target of iron delivery. The heart requires robust levels of ATP from oxidative metabolism to sustain continuous contractions [16], and thus cardiomyocytes are highly mitochondria-dense. Iron is required for iron/sulfur cluster (ISC) protein and heme-containing cytochrome components of the electron transport chain complexes I, II, III and IV that enable oxidative phosphorylation by ATP synthase within the mitochondria [17] (Fig. 2). Additionally, mitochondria are the cellular site of assembly of ISCs and heme, which are also required by several non-mitochondrial enzymes. Mitochondrial iron levels must be tightly regulated, since mitochondria are the site of produced from electron leakage to O2 by complexes I and III [18]. can lead to the generation of iron-catalyzed ROS, which can then damage proximal mitochondrial proteins and DNA, leading to mitochondrial injury and broader cellular dysfunction.
Several cardiovascular disorders are related to deregulated iron homeostasis. Iron deficiency and anemia [19], iron overload [20], and Friedreich’s ataxia (FRDA) [21], a disorder of altered iron homeostasis, have all been found to cause cardiomyopathies. Additionally, patient studies have identified an association among iron-related cardiovascular dysfunction and metabolic disruption. Conditions of aberrant iron deficiency and overload are correlated with exercise intolerance, an indication of mitochondrial and cardiac dysfunctions. Specifically, patients with iron deficiency and congenital heart disease or heart failure have reduced exercise tolerance [22,23]. Additionally, myocardial iron overload in thalassemic patients with heart failure is positively associated with exercise intolerance [24]. Iron status has also been linked to metabolic dysfunction, as iron levels and cardiovascular disease risk factors related to metabolism of glucose and lipids are correlated in women [25]. Together, these studies suggest that there is a clinical association between iron and metabolic function in the heart that warrants further investigation.
Studies utilizing rodent models have provided more direct evidence for a connection among aberrant iron levels and cardiac and metabolic dysfunctions. Several models of iron overload have uncovered an adverse effect of excess iron on cardiac mitochondria and metabolism. A mouse model of hemochromatosis, a condition of iron overload that leads to cardiomyopathy, can be recapitulated by the deletion of the human protein HFE, one of several genes that can cause clinical hemochromatosis when mutated [26]. HFE increases expression of hepcidin, an inhibitor of cellular iron release. HFE−/− mice exhibit skeletal muscle metabolic inflexibility with a preference for fatty acid oxidation, which are characteristics of diabetes [27]. This mouse model also has reduced glucose oxidation in cardiac muscle, as well as trends toward lower mitochondrial oxygen consumption and ATP levels, demonstrating that iron overload causes cardiac metabolic and mitochondrial dysfunctions similar to that found in diabetes. Acute and chronic iron overload in mice have also been found to increase iron in the heart, with accompanying ultrastructural damage to mitochondria and accumulation of electron-dense material, indicative of mitochondrial iron accumulation [28]. Iron overloaded-mice with cardiac iron accumulation and dysfunction also have a loss of mitochondrial DNA and transcription, reduced complex I and IV activity, and reduced mitochondrial respiration [29]. Thus, iron overload and its associated cardiac dysfunction are linked with damage to mitochondria and metabolic alterations in mice.
These findings have also been corroborated by in vitro studies conducted directly on isolated mitochondria or cells. Iron-loading of isolated mitochondria results in double-stranded mitochondrial DNA breaks and reduced mitochondrial transcription [30], as well as reduced mitochondrial respiration and ATP production [31]. In isolated rat cardiomyocytes, iron loading reduces complex I, II, and III activity and ATP levels [32,33]. Additionally, the iron chelators deferoxamine and deferiprone can reverse changes in cell contractility, mitochondrial complex activity and energy production in neonatal rat cardiomyocytes [34], supporting the finding that iron loading reduces mitochondrial function in cardiac cells.
Iron overload in mitochondria specifically has been found to be particularly damaging to cardiac and mitochondrial function. In a mouse model with cardiac-specific deletion of ABCB8, iron accumulates in the mitochondria [13]. These mice exhibit spontaneous cardiomyopathy, demonstrating that mitochondrial iron accumulation is detrimental to cardiac function. ABCB8 deletion also leads to increased mitochondrial damage and ROS, as well as reduced maturation of cytosolic iron–sulfur proteins. These studies have demonstrated that mitochondrial iron overload in particular can damage mitochondria and lead to wider cellular and cardiac dysfunctions.
In addition to iron overload, iron deficiency has also been found to be detrimental to cardiac and metabolic functions in rodent models. Rats fed a diet deficient in iron develop cardiac hypertrophy with disorganized myofilaments and sarcomeres [35,36]. These rats also have enlarged and damaged mitochondria with increased cytochrome c release, and exhibit reduced complex II activity. The changes seen in mitochondrial structure and function are similar to those seen with ischemia or hypoxia. Iron deficiency thus appears to mimic energy deficiency, likely due to reduced activity of respiratory complexes. In support of this, mitochondrial iron uptake has been found to be increased in the presence of ADP and inhibited with ATP [37], suggesting that mitochondrial iron is required in low energy conditions.
Given the apparent importance of regulated iron levels to proper cardiac function and metabolism, clinical iron detection in the heart may be useful in predicting cardiac metabolic aberrations. Iron levels can be detected noninvasively in the heart by cardiac magnetic resonance imaging (MRI), in which the presence of iron results in shortening of the relaxation parameter T2* [38]. MRI iron detection has been validated by measurement of iron concentration through inductively-coupled plasma atomic emission spectroscopy in ex vivo heart biopsies, and has been shown to be more accurate in predicting cardiac failure than measurement of serum ferritin levels [39]. MRI is also less invasive and more precise than tissue biopsies, which are necessarily small and do not accurately represent the iron status of the cardiac tissue as a whole. Correlation of iron detection by MRI with cardiac metabolic status will be an interesting focus of future research to determine whether noninvasive clinical iron measurements can reliably predict cardiac metabolic dysfunction.
2.3. Iron in the mitochondrial defects of Friedreich’s ataxia (FRDA) and doxorubicin-induced cardiotoxicity
The clearest clinical indication of a role for iron in cardiac metabolism arises from studies of FRDA, an inherited autosomal recessive disorder resulting in neurodegeneration and a heterogeneous manifestation of heart disease. The cause of the disorder in 98% of cases is the expansion of a GAA repeat in the first exon of the frataxin gene (FXN), which is a mitochondrial protein that is highly expressed in the heart and spinal cord [40]. The precise function of FXN is unknown, however there is pervasive evidence that it is involved in mitochondrial iron metabolism. Iron deposition has been found in patients with FRDA, who also exhibit deficiencies in mitochondrial ISC-containing complexes I, II, and III and the cytoplasmic ISC-containing enzyme aconitase [41,42]. FXN deficiency has also been found to lead to accumulation of iron and ROS in the mitochondria of fibroblasts from FRDA patients [43]. These findings suggest that FXN acts as a mitochondrial iron-binding protein that protects iron from mediating ROS production and delivers iron to ISCs.
A mouse model lacking FXN recapitulates the cardiac hypertrophy and neuronal defects found in FRDA cases, and also exhibits mitochondrial and cytosolic ISC enzyme deficiency followed by mitochondrial iron accumulation [44]. Moreover, a mouse model with conditional deletion of FXN has upregulation of TfR, downregulation of ferroportin and ferritin, and reduced ISC synthesis, amounting to a condition of iron overload with reduced iron utilization [45]. Together, the current research suggests that FXN deletion leads to decreases in the major iron utilization pathways of ISC and heme synthesis and iron storage, resulting in mitochondrial iron overload [46].
Studies on the yeast homolog of FXN, Yfh1p, also point to a role of FXN in the regulation of mitochondrial iron and ISC synthesis. Yeast cells with Yfh1p deletion (Δyfh1) have cellular and mitochondrial iron accumulation. Additionally, these cells have deficiencies in mitochondrial respiration and oxidative phosphorylation, as demonstrated by a lack of growth on fermentable carbon sources and a decrease in oxygen consumption [47]. Δyfh1 yeast cells also exhibit deficiencies in ISC enzymes and mitochondrial DNA [48]. These studies suggest that the FXN homolog Yfh1p is required for proper iron availability for ISC enzymes in yeast [49].
Additional enzymatic studies have demonstrated that FXN plays a role in providing iron for ISC and heme synthesis. In vitro studies have shown that FXN binds with ferrous iron and accessory proteins to enable sulfur incorporation into and assembly of ISCs [50], and this function of FXN is essential to activation of the cytoplasmic ISC enzyme aconitase [51]. Cellular studies have additionally revealed that delivery of iron to ISCs by FXN is essential for normal cell viability [52]. In addition to delivering iron to ISCs, FXN has also been found to provide iron to the heme synthesis enzyme ferrochelatase [53], and FXN deficiency results in reduced transcription of heme synthesis components [54]. Thus, FXN is necessary to provide iron for both ISC and heme synthesis in the mitochondria.
As FXN is necessary for ISC and heme biosynthesis and its deficiency leads to mitochondrial iron overload, it follows that many cases of Friedreich’s ataxia also present with metabolic defects. In vivo 31phosphorous magnetic resonance spectroscopy used in FRDA patients identified a decline in the maximum rate of muscle mitochondrial ATP production [55], suggesting that FXN deficiency decreases oxidative phosphorylation in these patients. Regardless of the presence of cardiac hypertrophy, FRDA patients were also found to have a reduced phosphocreatine/ATP ratio, indicating loss of high-energy phosphates [56]. Thus, the metabolic dysfunction in these patients appears to be the primary defect, which may play a role in the development of cardiac hypertrophy. Reversibly, increased FXN expression has been shown to enhance cellular metabolism. Overexpression of FXN in adipocytes leads to increased oxygen consumption and ATP [57], while increased expression of a FXN-TAT fusion protein in an FRDA mouse model leads to increased cardiac output, improved mitochondrial proliferation and structure, and increased energy production [58]. Moreover, a mouse model with transgenic overexpression of FXN has increased mitochondrial metabolism, reduced oxidative stress, and improved cardiac function following doxorubicin treatment [59]. Changes in FXN expression are therefore directly associated with altered oxidative metabolism.
These metabolic defects likely stem from ISC and heme assembly deficiency and mitochondrial iron loading that increase ROS and mitochondrial damage. Thus, several studies have attempted to treat FRDA aberrations with iron chelation or antioxidants. Treatment of a mouse model of FRDA with a mitochondria-permeable ligand conjugated to the iron chelator deferoxamine was successful in preventing iron overload and reducing cardiac hypertrophy [60], and fibroblasts from FRDA patients, which have iron overload and respiration deficiency, are rescued by deferoxamine [61]. Deferoxamine is the current iron chelator with the broadest clinical use, but is likely not beneficial for FRDA patients due to its limited mitochondrial access [62]. Thus, the challenge for clinical practice will be to find an approved iron chelator that can access the mitochondria.
In addition to iron chelators, antioxidants have also shown efficacy in treating characteristics of FRDA. Patients treated with antioxidants showed improved cardiac high-energy phosphate ratios of phosphocreatine to ATP and an overall increase in ATP production that was sustained for four years, with a slower progression of FRDA clinical features and improvement in cardiac function [63,64]. Moreover, in fibroblasts from FRDA patients, the mitochondrially-targeted antioxidants MitoQ and MitoVit E were found to be significantly more potent than non-targeted antioxidants in protecting against oxidative stress [65]. Thus, iron chelators and/or antioxidants may be promising treatments for FRDA if effective, clinically safe, and mitochondria-accessible compounds can be identified.
Another indication of the role of iron in cardiac mitochondrial metabolism arises from studies involving the antineoplastic chemotherapy drug doxorubicin, which is well-known to result in cardiotoxicity [66]. The precise nature of doxorubicin-induced cardiomyopathy is unknown. However, doxorubicin is known to cause damage to mitochondria and the mechanism may involve iron and/or oxidative stress. Specifically, doxorubicin is hypothesized to form a complex with iron that results in formation of hydroxyl radicals [67]. In support of a potential role for iron in doxorubicin-induced cardiotoxicity, overexpression of FXN, whose deficiency results in mitochondrial iron overload and iron-containing enzyme dysfunction, reduces oxidative stress and increases mitochondrial metabolism and cardiac function following doxorubicin treatment in mice [59]. Additionally, HFE−/− mice have increased iron deposition in response to doxorubicin, as well as increased mitochondrial damage compared to wild type mice [68], providing further evidence that iron likely contributes to doxorubicin-induced cardiac mitochondrial dysfunction.
The iron chelator dexrazoxane has been found to reduce doxorubicin-induced injury both in the clinic and in the laboratory. Dexrazoxane is the only clinically-approved drug that has been shown to reduce cardiac injury in patients taking doxorubicin [69]. In rats, dexrazoxane was also found to protect against doxorubicin-induced cardiomyopathy [70], and to prevent mitochondrial membrane depolarization in neonatal rat cardiomyocytes [67]. Moreover, dexrazoxane reverses the doxorubicin-induced increase in nuclear factor erythroid 2-related factor (Nrf2)-mediated stress response genes and reverses the decrease in expression of the mitochondrial genes adenylate kinase and creatine kinase, which are involved in ATP transfer [71]. Additionally, the protective effects of dexrazoxane have been suggested to be hypoxia- inducible factor (HIF)-dependent [72]. Iron is required for degradation of HIF, so chelation of iron may result in HIF upregulation and a following increase in antiapoptotic genes. Thus, iron chelation may be beneficial to increasing overall cell viability in doxorubicin-induced cardiomyopathy through multiple signaling pathways.
In conclusion, iron is a key player in mitochondrial metabolism, which is highly dependent on the proper function of several ISC-and heme-containing enzymes. Both iron deficiency and iron overload have been found to reduce mitochondrial and cardiac function through reductions in mitochondrial complex activity and increased oxidative stress damage. Additionally, FRDA has demonstrated a clinical significance of the effects of iron on mitochondrial metabolism, and iron may play a role in doxorubicin-induced cardiotoxicity. Iron chelators and antioxidants have shown some efficacy in reversing metabolic changes induced by iron overload, but more research is needed before these drugs are used in a wide clinical setting for this purpose. The prospect of using clinical MRI measurement of iron to help predict cardiac metabolic dysfunction also warrants further study. Moreover, additional investigation is needed into the precise mechanisms of the effects of iron on mitochondrial metabolism, the regulators of mitochondrial iron transport and utilization, and the role of iron in non-mitochondrial metabolic processes.
3. Copper and mitochondrial metabolism in the heart
Like iron, copper is a redox-active metal that can transition from a reduced Cu1 + form to an oxidized Cu2 + state. Copper is less abundant than iron, but is critical in many cellular processes including mitochondrial respiration, antioxidant capacity, and iron oxidation. Cellular copper levels must also be tightly controlled, as copper accumulation can lead to oxidative stress or inappropriate binding to macromolecules. However, limited information is known about the systemic regulation of copper (Fig. 3). Ctr1 imports dietary copper from the duodenum into enterocytes [73], and ATP7A exports copper from enterocytes into the bloodstream and also exports copper from other tissues, except the liver which contains ATP7B. Ctr1 also imports copper into the heart and is necessary for normal heart function, as specific deletion of Ctr1 in the mouse heart results in cardiac hypertrophy [74]. Additionally, the heart is capable of controlling systemic copper metabolism, as heart-specific deletion of Ctr1 results in increased serum copper levels and reduced hepatic copper stores. Most of the copper in serum is bound by ceruloplasmin, which helps to catalyze the oxidation of iron [75]. A mutation in ceruloplasmin has been linked to the systemic iron overload disorder hemosiderosis, suggesting that ceruloplasmin helps to regulate systemic iron homeostasis [76]. However, the implications of copper binding to ceruloplasmin on iron metabolism have not been studied in the heart.
Fig. 3.
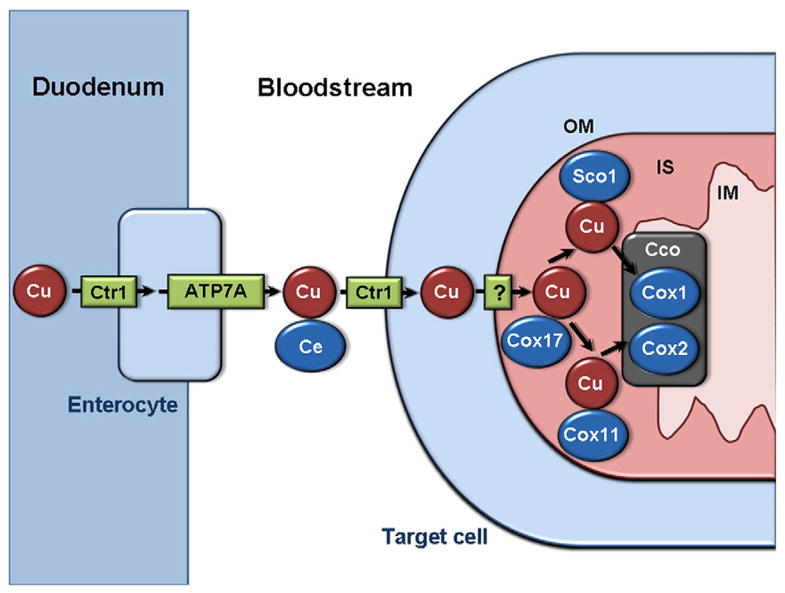
Systemic and mitochondrial copper transport. Copper from the diet is imported into enterocytes by Ctr1, then exported into the bloodstream by ATP7A. In the bloodstream, the majority of copper is bound to ceruloplasmin. Copper is transported into cells by Ctr1, and across the outer mitochondrial membrane by an unknown mechanism. In the inter-membrane space, copper is bound by Cox17, which delivers copper to Sco1 or Cox11. Sco1-bound copper is delivered to the Cox1 subunit of Cco. Cox11-bound copper is delivered to the Cox2 subunit of Cco. Cu: Copper. Ce: Ceruloplasmin. OM: outer mitochondrial membrane. IS: intermembrane space. IM: inner mitochondrial membrane. Cco: cytochrome oxidase.
In the mitochondria, copper is an essential component of complex IV, also known as cytochrome oxidase. Cu1+ is bound by Cox 17 in the intermembrane space, then transferred to Sco1 or Cox 11 for delivery to the Cox1 or Cox2 subunit of cytochrome oxidase, respectively [77] (Fig. 3). Although copper is crucial for cytochrome oxidase function, the transporters and regulators of mitochondrial copper are unknown. Additionally, the steady state levels of copper in the mitochondria are an order of magnitude more than the amount necessary for normal cytochrome oxidase function, suggesting that the mitochondria may also act as a copper storage organelle, although this has not been directly demonstrated.
Several studies have linked copper deficiency to respiration defects and cardiomyopathy. Mutations in Sco1 and Sco2 have been found to be associated with copper deficiency, cytochrome oxidase assembly defects, and hypertrophic cardiomyopathy [78]. Moreover, dietary copper deficiency in rats is linked to cardiac hypertrophy, decreased cardiac cytochrome oxidase activity, lowered oxygen consumption and mitochondrial membrane potential, increased mitochondrial biogenesis and ultrastructural damage, and ROS formation [79–88]. The cardiomyopathy induced by copper restriction can be reversed by copper repletion, which can also reverse the associated lipid accumulation and mitochondrial dysfunction [89]. There is also evidence that this improvement in function, at least in hypertrophy, requires cytochrome oxidase [90]. Alternatively, an oxidized copper chelator has been found to restore cardiac function in diabetic patients, and treatment of diabetic rats with this chelator is associated with a reversal in alterations in cardiac proteins involved in mitochondrial respiration and fatty acid oxidation [91]. These findings suggest that copper accumulation is deleterious in diabetic cardiomyopathy possibly through reductions in mitochondrial metabolism. Thus, carefully regulated levels of copper appear to be necessary for normal mitochondrial function.
Further research into copper regulation and its effects on mitochondria and metabolism is needed. Mitochondrial copper transporters have not been identified. Additionally, copper is a necessary cofactor for the cytosolic Cu/Zn-superoxide dismutase (SOD), but the role of this enzyme in cardiac mitochondrial metabolism has not been investigated. Moreover, although copper binds to the ferroreductase ceruloplasmin, no studies have yet identified an effect of ceruloplasmin disruption on cardiac iron or mitochondrial metabolism. Additional studies on copper in mitochondrial metabolism should yield many interesting findings about the role of this essential transitional metal in heart function.
4. Manganese and mitochondrial metabolism in the heart
Manganese is an essential trace metal and cofactor for the mitochondrial antioxidant enzyme MnSOD, as well as other enzymes such as pyruvate carboxylase and arginine synthase [92]. However, as with other transition metals, excessive exposure is deleterious and manganese accumulation is well-known to result in neurological damage. Manganese accumulation has also been suggested to reduce cardiac mitochondrial integrity and energy production through competitive binding to and inhibition of Mg2+- or Ca2+-dependent mitochondrial enzymes [93–95], although the implications of excess manganese on heart function have not been studied. Additionally, the regulation of manganese transport has not been extensively researched, but manganese is thought to be transported by proteins involved in Ca2+ and Fe2+ transport, including DMT1 and TfR [96].
Manganese is necessary to the function of MnSOD, which helps neutralize mitochondrial ROS by catalyzing the conversion of into O2 and H2O2. MnSOD deficiency results in increased oxidative stress in MnSOD+/− mice and rat cardiac fibroblasts [97,98]. Heterozygous deletion of MnSOD in mice also leads to enhanced cardiac mitochondrial oxidative damage, reduced complex I activity, and increased sensitivity to cell death [99]. Conversely, transgenic mice with MnSOD overexpression have preserved integrity of cardiac mitochondrial complex I and decreased doxorubicin-induced cardiomyopathy [100]. In a type 1 diabetic model, transgenic MnSOD mice have cardiac mitochondria with increased respiration and normalized mass, as well as improved cardiac contractility and function compared to nontransgenic mice [101]. Thus, MnSOD is suggested to be an important contributor to cardiac mitochondrial and metabolic functions through reductions in mitochondrial oxidative stress, but the mechanisms and clinical implications must be further investigated.
5. Conclusions
Overall, there is a growing body of research demonstrating the involvement of transition metals in mitochondrial metabolism of the heart through their roles in enzyme activity and regulation of oxidative stress. However, our knowledge about this topic is still limited and many questions remain. First, several transition metals, including zinc, have been virtually unstudied with regard to mitochondrial metabolism and their role in this process remains unknown. Second, crosstalk among pathways regulating metals would be expected given the role of transition metals in overlapping functions, but information about potential crosstalk is very limited, especially in the context of the heart. Third, the regulation of transition metal levels and transport by metabolic regulators would be expected, but has so far been largely unstudied. Further research into the role of transition metals in cardiac mitochondria is needed, and should reveal crucial information about cardiac metabolism and function, as well as potential new pharmacological targets for cardiovascular diseases.
Footnotes
Disclosures
None.
References
- 1.Nelson N. Metal ion transporters and homeostasis. EMBO J. 1999;18:4361–71. doi: 10.1093/emboj/18.16.4361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aisen P, Enns C, Wessling-Resnick M. Chemistry and biology of eukaryotic iron metabolism. Int J Biochem Cell Biol. 2001;33:940–59. doi: 10.1016/s1357-2725(01)00063-2. [DOI] [PubMed] [Google Scholar]
- 3.Wardman P, Candeias LP. Fenton chemistry: an introduction. Radiat Res. 1996;145:523–31. [PubMed] [Google Scholar]
- 4.Thannickal VJ, Fanburg BL. Reactive oxygen species in cell signaling. Am J Physiol Lung Cell Mol Physiol. 2000;279:L1005–28. doi: 10.1152/ajplung.2000.279.6.L1005. [DOI] [PubMed] [Google Scholar]
- 5.Wang J, Pantopoulos K. Regulation of cellular iron metabolism. Biochem J. 2011;434:365–81. doi: 10.1042/BJ20101825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hentze MW, Muckenthaler MU, Andrews NC. Balancing acts: molecular control of mammalian iron metabolism. Cell. 2004;117:285–97. doi: 10.1016/s0092-8674(04)00343-5. [DOI] [PubMed] [Google Scholar]
- 7.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, et al. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–3. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 8.Rouault TA. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat Chem Biol. 2006;2:406–14. doi: 10.1038/nchembio807. [DOI] [PubMed] [Google Scholar]
- 9.Pantopoulos K. Iron metabolism and the IRE/IRP regulatory system: an update. Ann N Y Acad Sci. 2004;1012:1–13. doi: 10.1196/annals.1306.001. [DOI] [PubMed] [Google Scholar]
- 10.Huang ML, Lane DJ, Richardson DR. Mitochondrial mayhem: the mitochondrion as a modulator of iron metabolism and its role in disease. Antioxid Redox Signal. 2011;15:3003–19. doi: 10.1089/ars.2011.3921. [DOI] [PubMed] [Google Scholar]
- 11.Richardson DR, Lane DJ, Becker EM, Huang ML, Whitnall M, Suryo Rahmanto Y, et al. Mitochondrial iron trafficking and the integration of iron metabolism between the mitochondrion and cytosol. Proc Natl Acad Sci U S A. 2010;107:10775–82. doi: 10.1073/pnas.0912925107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen W, Paradkar PN, Li L, Pierce EL, Langer NB, Takahashi-Makise N, et al. Abcb10 physically interacts with mitoferrin-1 (Slc25a37) to enhance its stability and function in the erythroid mitochondria. Proc Natl Acad Sci U S A. 2009;106:16263–8. doi: 10.1073/pnas.0904519106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ichikawa Y, Bayeva M, Ghanefar M, Potini V, Sun L, Mutharasan RK, et al. Disruption of ATP-binding cassette B8 in mice leads to cardiomyopathy through a decrease in mitochondrial iron export. Proc Natl Acad Sci U S A. 2012;109:4152–7. doi: 10.1073/pnas.1119338109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bekri S, Kispal G, Lange H, Fitzsimons E, Tolmie J, Lill R, et al. Human ABC7 transporter: gene structure and mutation causing X-linked sideroblastic anemia with ataxia with disruption of cytosolic iron–sulfur protein maturation. Blood. 2000;96:3256–64. [PubMed] [Google Scholar]
- 15.Cavadini P, Biasiotto G, Poli M, Levi S, Verardi R, Zanella I, et al. RNA silencing of the mitochondrial ABCB7 transporter in HeLa cells causes an iron-deficient phenotype with mitochondrial iron overload. Blood. 2007;109:3552–9. doi: 10.1182/blood-2006-08-041632. [DOI] [PubMed] [Google Scholar]
- 16.Huss JM, Kelly DP. Mitochondrial energy metabolism in heart failure: a question of balance. J Clin Invest. 2005;115:547–55. doi: 10.1172/JCI200524405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Atamna H, Walter PB, Ames BN. The role of heme and iron–sulfur clusters in mitochondrial biogenesis, maintenance, and decay with age. Arch Biochem Biophys. 2002;397:345–53. doi: 10.1006/abbi.2001.2671. [DOI] [PubMed] [Google Scholar]
- 18.Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027–31. doi: 10.1074/jbc.M304854200. [DOI] [PubMed] [Google Scholar]
- 19.van Veldhuisen DJ, Anker SD, Ponikowski P, Macdougall IC. Anemia and iron deficiency in heart failure: mechanisms and therapeutic approaches. Nat Rev Cardiol. 2011;8:485–93. doi: 10.1038/nrcardio.2011.77. [DOI] [PubMed] [Google Scholar]
- 20.Gujja P, Rosing DR, Tripodi DJ, Shizukuda Y. Iron overload cardiomyopathy: better understanding of an increasing disorder. J Am Coll Cardiol. 2010;56:1001–12. doi: 10.1016/j.jacc.2010.03.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Payne RM. The heart in Friedreich’s ataxia: basic findings and clinical implications. Prog Pediatr Cardiol. 2011;31:103–9. doi: 10.1016/j.ppedcard.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tay EL, Peset A, Papaphylactou M, Inuzuka R, Alonso-Gonzalez R, Giannakoulas G, et al. Replacement therapy for iron deficiency improves exercise capacity and quality of life in patients with cyanotic congenital heart disease and/or the Eisenmenger syndrome. Int J Cardiol. 2011;151:307–12. doi: 10.1016/j.ijcard.2010.05.066. [DOI] [PubMed] [Google Scholar]
- 23.Jankowska EA, Rozentryt P, Witkowska A, Nowak J, Hartmann O, Ponikowska B, et al. Iron deficiency predicts impaired exercise capacity in patients with systolic chronic heart failure. J Card Fail. 2011;17:899–906. doi: 10.1016/j.cardfail.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 24.Mavrogeni S, Gotsis E, Verganelakis D, Berdousis E, Dritsas A, Kolovou G, et al. Effect of iron overload on exercise capacity in thalassemic patients with heart failure. Int J Cardiovasc Imaging. 2009;25:777–83. doi: 10.1007/s10554-009-9491-9. [DOI] [PubMed] [Google Scholar]
- 25.Ramakrishnan U, Kuklina E, Stein AD. Iron stores and cardiovascular disease risk factors in women of reproductive age in the United States. Am J Clin Nutr. 2002;76:1256–60. doi: 10.1093/ajcn/76.6.1256. [DOI] [PubMed] [Google Scholar]
- 26.Fleming RE, Ponka P. Iron overload in human disease. N Engl J Med. 2012;366:348–59. doi: 10.1056/NEJMra1004967. [DOI] [PubMed] [Google Scholar]
- 27.Huang J, Jones D, Luo B, Sanderson M, Soto J, Abel ED, et al. Iron overload and diabetes risk: a shift from glucose to fatty acid oxidation and increased hepatic glucose production in a mouse model of hereditary hemochromatosis. Diabetes. 2011;60:80–7. doi: 10.2337/db10-0593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bartfay WJ, Butany J, Lehotay DC, Sole MJ, Hou D, Bartfay E, et al. A biochemical, histochemical, and electron microscopic study on the effects of iron-loading on the hearts of mice. Cardiovasc Pathol. 1999;8:305–14. doi: 10.1016/s1054-8807(99)00008-3. [DOI] [PubMed] [Google Scholar]
- 29.Gao X, Qian M, Campian JL, Marshall J, Zhou Z, Roberts AM, et al. Mitochondrial dysfunction may explain the cardiomyopathy of chronic iron overload. Free Radic Biol Med. 2010;49:401–7. doi: 10.1016/j.freeradbiomed.2010.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Asín J, Pérez-Martos A, Fernández-Silva P, Montoya J, Andreu AL. Iron(II) induces changes in the conformation of mammalian mitochondrial DNA resulting in a reduction of its transcriptional rate. FEBS Lett. 2000;480:161–4. doi: 10.1016/s0014-5793(00)01768-3. [DOI] [PubMed] [Google Scholar]
- 31.Kim M, Kim J, Cheon CI, Cho DH, Park JH, Kim KI, et al. Increased expression of the F(1)F(o) ATP synthase in response to iron in heart mitochondria. BMB Rep. 2008;41:153–7. doi: 10.5483/bmbrep.2008.41.2.153. [DOI] [PubMed] [Google Scholar]
- 32.Link G, Saada A, Pinson A, Konijn AM, Hershko C. Mitochondrial respiratory enzymes are a major target of iron toxicity in rat heart cells. J Lab Clin Med. 1998;131:466–74. doi: 10.1016/s0022-2143(98)90148-2. [DOI] [PubMed] [Google Scholar]
- 33.Link G, Konijn AM, Hershko C. Cardioprotective effect of alpha-tocopherol, ascorbate, deferoxamine, and deferiprone: mitochondrial function in cultured, iron-loaded heart cells. J Lab Clin Med. 1999;133:179–88. doi: 10.1016/s0022-2143(99)90011-2. [DOI] [PubMed] [Google Scholar]
- 34.Glickstein H, El RB, Link G, Breuer W, Konijn AM, Hershko C, et al. Action of chelators in iron-loaded cardiac cells: accessibility to intracellular labile iron and functional consequences. Blood. 2006;108:3195–203. doi: 10.1182/blood-2006-05-020867. [DOI] [PubMed] [Google Scholar]
- 35.Tanne Z, Coleman R, Nahir M, Shomrat D, Finberg JP, Youdim MB. Ultrastructural and cytochemical changes in the heart of iron-deficient rats. Biochem Pharmacol. 1994;47:1759–66. doi: 10.1016/0006-2952(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 36.Dong F, Zhang X, Culver B, Chew HG, Kelley RO, Ren J. Dietary iron deficiency induces ventricular dilation, mitochondrial ultrastructural aberrations and cytochrome c release: involvement of nitric oxide synthase and protein tyrosine nitration. Clin Sci (Lond) 2005;109:277–86. doi: 10.1042/CS20040278. [DOI] [PubMed] [Google Scholar]
- 37.Kim M, Song E. Effects of ATP and ADP on iron uptake in rat heart mitochondria. Anim Cells Syst. 2010;14:245–52. [Google Scholar]
- 38.Sanz J. Evolving diagnostic and prognostic imaging of the various cardiomyopathies. Ann N Y Acad Sci. 2012;1254:123–30. doi: 10.1111/j.1749-6632.2012.06490.x. [DOI] [PubMed] [Google Scholar]
- 39.Carpenter JP, He T, Kirk P, Roughton M, Anderson LJ, de Noronha SV, et al. On T2* magnetic resonance and cardiac iron. Circulation. 2011;123:1519–28. doi: 10.1161/CIRCULATIONAHA.110.007641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Campuzano V, Montermini L, Moltò MD, Pianese L, Cossée M, Cavalcanti F, et al. Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–7. doi: 10.1126/science.271.5254.1423. [DOI] [PubMed] [Google Scholar]
- 41.Rötig A, de Lonlay P, Chretien D, Foury F, Koenig M, Sidi D, et al. Aconitase and mitochondrial iron–sulphur protein deficiency in Friedreich ataxia. Nat Genet. 1997;17:215–7. doi: 10.1038/ng1097-215. [DOI] [PubMed] [Google Scholar]
- 42.Sanchez-Casis G, Cote M, Barbeau A. Pathology of the heart in Friedreich’s ataxia: review of the literature and report of one case. Can J Neurol Sci. 1976;3:349–54. doi: 10.1017/s0317167100025580. [DOI] [PubMed] [Google Scholar]
- 43.Pandolfo M. Frataxin deficiency and mitochondrial dysfunction. Mitochondrion. 2002;2:87–93. doi: 10.1016/s1567-7249(02)00039-9. [DOI] [PubMed] [Google Scholar]
- 44.Puccio H, Simon D, Cossée M, Criqui-Filipe P, Tiziano F, Melki J, et al. Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe–S enzyme deficiency followed by intramitochondrial iron deposits. Nat Genet. 2001;27:181–6. doi: 10.1038/84818. [DOI] [PubMed] [Google Scholar]
- 45.Richardson DR, Huang ML, Whitnall M, Becker EM, Ponka P, Suryo Rahmanto Y. The ins and outs of mitochondrial iron-loading: the metabolic defect in Friedreich’s ataxia. J Mol Med (Berl) 2010;88:323–9. doi: 10.1007/s00109-009-0565-x. [DOI] [PubMed] [Google Scholar]
- 46.Huang ML, Becker EM, Whitnall M, Suryo Rahmanto Y, Ponka P, Richardson DR. Elucidation of the mechanism of mitochondrial iron loading in Friedreich’s ataxia by analysis of a mouse mutant. Proc Natl Acad Sci U S A. 2009;106:16381–6. doi: 10.1073/pnas.0906784106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Babcock M, de Silva D, Oaks R, Davis-Kaplan S, Jiralerspong S, Montermini L, et al. Regulation of mitochondrial iron accumulation by Yfh1p, a putative homolog of frataxin. Science. 1997;276:1709–12. doi: 10.1126/science.276.5319.1709. [DOI] [PubMed] [Google Scholar]
- 48.Wilson RB, Roof DM. Respiratory deficiency due to loss of mitochondrial DNA in yeast lacking the frataxin homologue. Nat Genet. 1997;16:352–7. doi: 10.1038/ng0897-352. [DOI] [PubMed] [Google Scholar]
- 49.Zhang Y, Lyver ER, Knight SA, Pain D, Lesuisse E, Dancis A. Mrs3p, Mrs4p, and frataxin provide iron for Fe–S cluster synthesis in mitochondria. J Biol Chem. 2006;281:22493–502. doi: 10.1074/jbc.M604246200. [DOI] [PubMed] [Google Scholar]
- 50.Tsai CL, Barondeau DP. Human frataxin is an allosteric switch that activates the Fe–S cluster biosynthetic complex. Biochemistry. 2010;49:9132–9. doi: 10.1021/bi1013062. [DOI] [PubMed] [Google Scholar]
- 51.Bulteau AL, O’Neill HA, Kennedy MC, Ikeda-Saito M, Isaya G, Szweda LI. Frataxin acts as an iron chaperone protein to modulate mitochondrial aconitase activity. Science. 2004;305:242–5. doi: 10.1126/science.1098991. [DOI] [PubMed] [Google Scholar]
- 52.Schmucker S, Martelli A, Colin F, Page A, Wattenhofer-Donzé M, Reutenauer L, et al. Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron–sulfur assembly complex. PLoS One. 2011;6:e16199. doi: 10.1371/journal.pone.0016199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoon T, Cowan JA. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J Biol Chem. 2004;279:25943–6. doi: 10.1074/jbc.C400107200. [DOI] [PubMed] [Google Scholar]
- 54.Schoenfeld RA, Napoli E, Wong A, Zhan S, Reutenauer L, Morin D, et al. Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum Mol Genet. 2005;14:3787–99. doi: 10.1093/hmg/ddi393. [DOI] [PubMed] [Google Scholar]
- 55.Lodi R, Cooper JM, Bradley JL, Manners D, Styles P, Taylor DJ, et al. Deficit of in vivo mitochondrial ATP production in patients with Friedreich ataxia. Proc Natl Acad Sci U S A. 1999;96:11492–5. doi: 10.1073/pnas.96.20.11492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lodi R, Rajagopalan B, Blamire AM, Cooper JM, Davies CH, Bradley JL, et al. Cardiac energetics are abnormal in Friedreich ataxia patients in the absence of cardiac dysfunction and hypertrophy: an in vivo 31P magnetic resonance spectroscopy study. Cardiovasc Res. 2001;52:111–9. doi: 10.1016/s0008-6363(01)00357-1. [DOI] [PubMed] [Google Scholar]
- 57.Ristow M, Pfister MF, Yee AJ, Schubert M, Michael L, Zhang CY, et al. Frataxin activates mitochondrial energy conversion and oxidative phosphorylation. Proc Natl Acad Sci U S A. 2000;97:12239–43. doi: 10.1073/pnas.220403797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vyas PM, Tomamichel WJ, Pride PM, Babbey CM, Wang Q, Mercier J, et al. A TAT-frataxin fusion protein increases lifespan and cardiac function in a conditional Friedreich’s ataxia mouse model. Hum Mol Genet. 2012;21:1230–47. doi: 10.1093/hmg/ddr554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schulz TJ, Westermann D, Isken F, Voigt A, Laube B, Thierbach R, et al. Activation of mitochondrial energy metabolism protects against cardiac failure. Aging (Albany NY) 2010;2:843–53. doi: 10.18632/aging.100234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Whitnall M, Suryo Rahmanto Y, Sutak R, Xu X, Becker EM, Mikhael MR, et al. The MCK mouse heart model of Friedreich’s ataxia: alterations in iron-regulated proteins and cardiac hypertrophy are limited by iron chelation. Proc Natl Acad Sci U S A. 2008;105:9757–62. doi: 10.1073/pnas.0804261105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wong A, Yang J, Cavadini P, Gellera C, Lonnerdal B, Taroni F, et al. The Friedreich’s ataxia mutation confers cellular sensitivity to oxidant stress which is rescued by chelators of iron and calcium and inhibitors of apoptosis. Hum Mol Genet. 1999;8:425–30. doi: 10.1093/hmg/8.3.425. [DOI] [PubMed] [Google Scholar]
- 62.Richardson DR. Friedreich’s ataxia: iron chelators that target the mitochondrion as a therapeutic strategy? Expert Opin Investig Drugs. 2003;12:235–45. doi: 10.1517/13543784.12.2.235. [DOI] [PubMed] [Google Scholar]
- 63.Lodi R, Hart PE, Rajagopalan B, Taylor DJ, Crilley JG, Bradley JL, et al. Antioxidant treatment improves in vivo cardiac and skeletal muscle bioenergetics in patients with Friedreich’s ataxia. Ann Neurol. 2001;49:590–6. [PubMed] [Google Scholar]
- 64.Hart PE, Lodi R, Rajagopalan B, Bradley JL, Crilley JG, Turner C, et al. Antioxidant treatment of patients with Friedreich ataxia: four-year follow-up. Arch Neurol. 2005;62:621–6. doi: 10.1001/archneur.62.4.621. [DOI] [PubMed] [Google Scholar]
- 65.Jauslin ML, Meier T, Smith RA, Murphy MP. Mitochondria-targeted antioxidants protect Friedreich Ataxia fibroblasts from endogenous oxidative stress more effectively than untargeted antioxidants. FASEB J. 2003;17:1972–4. doi: 10.1096/fj.03-0240fje. [DOI] [PubMed] [Google Scholar]
- 66.Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–5. doi: 10.1056/NEJM199809243391307. [DOI] [PubMed] [Google Scholar]
- 67.Hasinoff BB, Schnabl KL, Marusak RA, Patel D, Huebner E. Dexrazoxane (ICRF-187) protects cardiac myocytes against doxorubicin by preventing damage to mitochondria. Cardiovasc Toxicol. 2003;3:89–99. doi: 10.1385/ct:3:2:89. [DOI] [PubMed] [Google Scholar]
- 68.Miranda CJ, Makui H, Soares RJ, Bilodeau M, Mui J, Vali H, et al. Hfe deficiency increases susceptibility to cardiotoxicity and exacerbates changes in iron metabolism induced by doxorubicin. Blood. 2003;102:2574–80. doi: 10.1182/blood-2003-03-0869. [DOI] [PubMed] [Google Scholar]
- 69.Hellmann K. Preventing the cardiotoxicity of anthracyclines by dexrazoxane. BMJ. 1999;319:1085–6. doi: 10.1136/bmj.319.7217.1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lebrecht D, Geist A, Ketelsen UP, Haberstroh J, Setzer B, Walker UA. Dexrazoxane prevents doxorubicin-induced long-term cardiotoxicity and protects myocardial mitochondria from genetic and functional lesions in rats. Br J Pharmacol. 2007;151:771–8. doi: 10.1038/sj.bjp.0707294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Thompson KL, Rosenzweig BA, Zhang J, Knapton AD, Honchel R, Lipshultz SE, et al. Early alterations in heart gene expression profiles associated with doxorubicin cardiotoxicity in rats. Cancer Chemother Pharmacol. 2010;66:303–14. doi: 10.1007/s00280-009-1164-9. [DOI] [PubMed] [Google Scholar]
- 72.Spagnuolo RD, Recalcati S, Tacchini L, Cairo G. Role of hypoxia-inducible factors in the dexrazoxane-mediated protection of cardiomyocytes from doxorubicin-induced toxicity. Br J Pharmacol. 2011;163:299–312. doi: 10.1111/j.1476-5381.2011.01208.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 73.Nose Y, Kim BE, Thiele DJ. Ctr1 drives intestinal copper absorption and is essential for growth, iron metabolism, and neonatal cardiac function. Cell Metab. 2006;4:235–44. doi: 10.1016/j.cmet.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 74.Kim BE, Turski ML, Nose Y, Casad M, Rockman HA, Thiele DJ. Cardiac copper deficiency activates a systemic signaling mechanism that communicates with the copper acquisition and storage organs. Cell Metab. 2010;11:353–63. doi: 10.1016/j.cmet.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cherukuri S, Potla R, Sarkar J, Nurko S, Harris ZL, Fox PL. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab. 2005;2:309–19. doi: 10.1016/j.cmet.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 76.Yoshida K, Furihata K, Takeda S, Nakamura A, Yamamoto K, Morita H, et al. A mutation in the ceruloplasmin gene is associated with systemic hemosiderosis in humans. Nat Genet. 1995;9:267–72. doi: 10.1038/ng0395-267. [DOI] [PubMed] [Google Scholar]
- 77.Kim BE, Nevitt T, Thiele DJ. Mechanisms for copper acquisition, distribution and regulation. Nat Chem Biol. 2008;4:176–85. doi: 10.1038/nchembio.72. [DOI] [PubMed] [Google Scholar]
- 78.Stiburek L, Vesela K, Hansikova H, Hulkova H, Zeman J. Loss of function of Sco1 and its interaction with cytochrome c oxidase. Am J Physiol Cell Physiol. 2009;296:C1218–26. doi: 10.1152/ajpcell.00564.2008. [DOI] [PubMed] [Google Scholar]
- 79.Liao Z, Medeiros DM, McCune SA, Prochaska LJ. Cardiac levels of fibronectin, laminin, isomyosins, and cytochrome c oxidase of weanling rats are more vulnerable to copper deficiency than those of postweanling rats. J Nutr Biochem. 1995;6:385–91. doi: 10.1016/0955-2863(95)80007-y. [DOI] [PubMed] [Google Scholar]
- 80.Nath R. Copper deficiency and heart disease: molecular basis, recent advances and current concepts. Int J Biochem Cell Biol. 1997;29:1245–54. doi: 10.1016/s1357-2725(97)00060-5. [DOI] [PubMed] [Google Scholar]
- 81.Medeiros DM, Jiang Y, Klaahsen D, Lin D. Mitochondrial and sarcoplasmic protein changes in hearts from copper-deficient rats: up-regulation of PGC-1alpha transcript and protein as a cause for mitochondrial biogenesis in copper deficiency. J Nutr Biochem. 2009;20:823–30. doi: 10.1016/j.jnutbio.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 82.Zeng H, Saari JT, Johnson WT. Copper deficiency decreases complex IV but not complex I, II, III, or V in the mitochondrial respiratory chain in rat heart. J Nutr. 2007;137:14–8. doi: 10.1093/jn/137.1.14. [DOI] [PubMed] [Google Scholar]
- 83.Johnson WT, Newman SM. Hearts in adult offspring of copper-deficient dams exhibit decreased cytochrome c oxidase activity, increased mitochondrial hydrogen peroxide generation and enhanced formation of intracellular residual bodies. J Nutr Biochem. 2007;18:97–104. doi: 10.1016/j.jnutbio.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 84.Li Y, Wang L, Schuschke DA, Zhou Z, Saari JT, Kang YJ. Marginal dietary copper restriction induces cardiomyopathy in rats. J Nutr. 2005;135:2130–6. doi: 10.1093/jn/135.9.2130. [DOI] [PubMed] [Google Scholar]
- 85.Chen X, Jennings DB, Medeiros DM. Impaired cardiac mitochondrial membrane potential and respiration in copper-deficient rats. J Bioenerg Biomembr. 2002;34:397–406. doi: 10.1023/a:1021258204921. [DOI] [PubMed] [Google Scholar]
- 86.Mao S, Medeiros DM, Wildman RE. Cardiac hypertrophy in copper-deficient rats is owing to increased mitochondria. Biol Trace Elem Res. 1998;64:175–84. doi: 10.1007/BF02783334. [DOI] [PubMed] [Google Scholar]
- 87.Johnson WT, Johnson LK. Copper deficiency inhibits Ca2+-induced swelling in rat cardiac mitochondria. J Nutr Biochem. 2009;20:248–53. doi: 10.1016/j.jnutbio.2008.02.009. [DOI] [PubMed] [Google Scholar]
- 88.Rossi L, Lippe G, Marchese E, De Martino A, Mavelli I, Rotilio G, et al. Decrease of cytochrome c oxidase protein in heart mitochondria of copper-deficient rats. Biometals. 1998;11:207–12. doi: 10.1023/a:1009274131473. [DOI] [PubMed] [Google Scholar]
- 89.Elsherif L, Wang L, Saari JT, Kang YJ. Regression of dietary copper restriction-induced cardiomyopathy by copper repletion in mice. J Nutr. 2004;134:855–60. doi: 10.1093/jn/134.4.855. [DOI] [PubMed] [Google Scholar]
- 90.Zuo X, Xie H, Dong D, Jiang N, Zhu H, Kang YJ. Cytochrome c oxidase is essential for copper-induced regression of cardiomyocyte hypertrophy. Cardiovasc Toxicol. 2010;10:208–15. doi: 10.1007/s12012-010-9080-0. [DOI] [PubMed] [Google Scholar]
- 91.Jüllig M, Chen X, Hickey AJ, Crossman DJ, Xu A, Wang Y, et al. Reversal of diabetes-evoked changes in mitochondrial protein expression of cardiac left ventricle by treatment with a copper(II)-selective chelator. Proteomics Clin Appl. 2007;1:387–99. doi: 10.1002/prca.200600770. [DOI] [PubMed] [Google Scholar]
- 92.Aschner M, Erikson KM, Herrero Hernández E, Hernández EH, Tjalkens R. Manganese and its role in Parkinson’s disease: from transport to neuropathology. Neuromolecular Med. 2009;11:252–66. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gunter TE, Gerstner B, Lester T, Wojtovich AP, Malecki J, Swarts SG, et al. An analysis of the effects of Mn2+ on oxidative phosphorylation in liver, brain, and heart mitochondria using state 3 oxidation rate assays. Toxicol Appl Pharmacol. 2010;249:65–75. doi: 10.1016/j.taap.2010.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Miller KB, Newman SM, Caton JS, Finley JW. Manganese alters mitochondrial integrity in the hearts of swine marginally deficient in magnesium. Biofactors. 2004;20:85–96. doi: 10.1002/biof.5520200203. [DOI] [PubMed] [Google Scholar]
- 95.Miller KB, Caton JS, Finley JW. Manganese depresses rat heart muscle respiration. Biofactors. 2006;28:33–46. doi: 10.1002/biof.5520280104. [DOI] [PubMed] [Google Scholar]
- 96.Au C, Benedetto A, Aschner M. Manganese transport in eukaryotes: the role of DMT1. Neurotoxicology. 2008;29:569–76. doi: 10.1016/j.neuro.2008.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wenzel P, Schuhmacher S, Kienhöfer J, Müller J, Hortmann M, Oelze M, et al. Manganese superoxide dismutase and aldehyde dehydrogenase deficiency increase mitochondrial oxidative stress and aggravate age-dependent vascular dysfunction. Cardiovasc Res. 2008;80:280–9. doi: 10.1093/cvr/cvn182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lijnen PJ, van Pelt JF, Fagard RH. Downregulation of manganese superoxide dismutase by angiotensin II in cardiac fibroblasts of rats: association with oxidative stress in myocardium. Am J Hypertens. 2010;23:1128–35. doi: 10.1038/ajh.2010.128. [DOI] [PubMed] [Google Scholar]
- 99.Van Remmen H, Williams MD, Guo Z, Estlack L, Yang H, Carlson EJ, et al. Knockout mice heterozygous for Sod2 show alterations in cardiac mitochondrial function and apoptosis. Am J Physiol Heart Circ Physiol. 2001;281:H1422–32. doi: 10.1152/ajpheart.2001.281.3.H1422. [DOI] [PubMed] [Google Scholar]
- 100.Yen HC, Oberley TD, Gairola CG, Szweda LI, St Clair DK. Manganese superoxide dismutase protects mitochondrial complex I against adriamycin-induced cardiomyopathy in transgenic mice. Arch Biochem Biophys. 1999;362:59–66. doi: 10.1006/abbi.1998.1011. [DOI] [PubMed] [Google Scholar]
- 101.Shen X, Zheng S, Metreveli NS, Epstein PN. Protection of cardiac mitochondria by overexpression of MnSOD reduces diabetic cardiomyopathy. Diabetes. 2006;55:798–805. doi: 10.2337/diabetes.55.03.06.db05-1039. [DOI] [PubMed] [Google Scholar]