

Abstract
A highly efficient and stereoselective arylation of in situ generated azavinyl carbenes affording 2,2-diaryl enamines at ambient temperatures has been developed. These transition metal carbenes are directly produced from readily available and stable 1-sulfonyl-1,2,3-triazoles in the presence of a rhodium carboxylate catalyst. In several cases, the enamines generated in this reaction can be cyclized into substituted indoles employing copper catalysts.
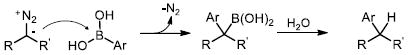
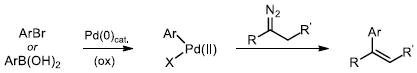
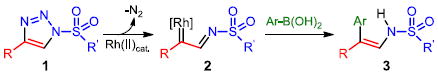
Utilization of diazo compounds as versatile reactive species has become a cornerstone in modern organic synthesis.1 In particular, the diverse reactivity of rhodium carbene complexes derived from diazo compounds has gained considerable attention.2 Despite the multitude of reactions developed for rhodium carbenes, there are only a few examples of their direct arylation, which are limited to intramolecular processes.3 The Wang4a and Barluenga4b groups recently reported that diazo compounds (either directly, or generated in situ from tosylhydrazones)5 react with arylboron nucleophiles in the absence of a transition metal catalyst (eq. 1). Furthermore, related arylation reactions of diazo compounds via migratory carbene insertion6 into Pd-aryl intermediates have been described (eq. 2).7 Herein we report a stereoselective Rh(II)-catalyzed reaction of arylboronic acids with azavinyl carbenes, derived from 1-sulfonyl-1,2,3-triazoles 1, to access a variety of functional enamine products 3 (eq. 3).
Previous work (Barluenga and Wang):
 |
(1) |
 |
(2) |
This work:
 |
(3) |
It is known that certain 1,2,3-triazoles can undergo ring-chain tautomerization in solution to form the corresponding diazo imines.8,9f Initially we attempted to capture the diazo tautomer of 1 by reaction with phenylboronic acid, effectively mimicking the Barluenga and Wang reaction (eq. 1). However, no reaction was observed, even under forcing conditions. We attribute the failure of this transformation to a low concentration of the putative diazo tautomer in solution.
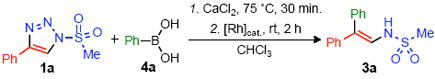
We have recently demonstrated that readily available 1-sulfonyl-1,2,3-triazoles 1 can serve as direct precursors to rhodium(II) azavinyl carbenes 2.9 These structurally unique carbenes share many similarities to the well-studied diazoester derivatives; however, the ready availability and stability of the triazole allows for simple experimental setup and handling. Following our recent success with the Rh(II)-catalyzed transformations of 1-sulfonyl-1,2,3-triazoles 1, we attempted a rhodium-catalyzed arylation utilizing organoboronic acids. We hypothesized that the pendant nitrogen lone pair of the sulfonyl imine of azavinyl carbene 2 could participate in the formation of a boronate complex; a subsequent transfer of the nucleophilic organic group could then afford the arylated product. To this end, we examined the reaction of phenylboronic acid 4a and triazole 1a in the presence of 0.5 mol% of various Rh(II)-catalysts and CaCl2 as an additive (Table 1). We were pleased to find that the arylated enamine 3a was formed at room temperature as the single product.
Table 1.
Catalyst Optimization for the Arylation of 1a.a
| ||
|---|---|---|
| entry | catalyst | Yield (%)b |
| 1 | Rh2(oct)4 | <5 |
| 2 | Rh2(piv)4 | 69 |
| 3 | Rh2(esp)2 | 50 |
| 4 | Rh2(S-PTAD)4 | 85 |
| 5 | Rh2(S-NTTL)4 | 75 |
Performed on a 0.30 mmol scale using 0.5 mol% Rh-cat. and 1.0 equiv of 4a in 1.0 mL CHCl3.
Yield determined by LC/MS using acetanilide as an internal standard.
Chloroform was identified as the optimum solvent for the generation of Rh(II)-azavinyl carbenes during previous studies, hence, no solvent optimization was performed.
Although most of the Rh(II)-carboxylate complexes were effective for the arylation of 1a, Rh2(S-PTAD)4 10 was found to be an optimal catalyst (85% yield of 3a, entry 4, Table 1). During optimization of the reaction conditions we discovered that CaCl2 was an essential additive for the success of this transformation.11 A short desiccation period (30 min) is necessary for the formation of an aryl boroxine (ArBO)3 which is likely the active arylating species (vide infra). Moreover, in the absence of CaCl2, we observed the significant formation of side-products12 and catalyst deactivation.
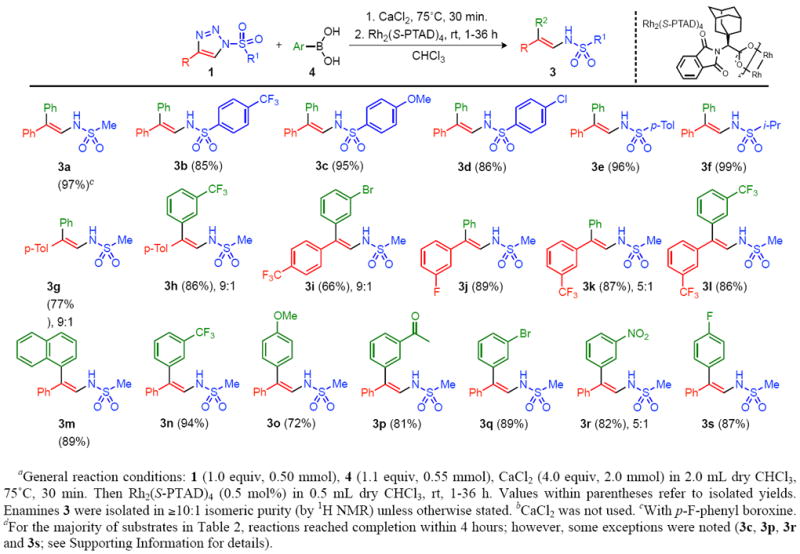
With the optimized conditions in hand, we examined the scope of this new reaction with respect to variously substituted 1-sulfonyl-1,2,3-triazoles 1 and arylboronic acids 4, leading to enamine products 3 (Table 2).
Table 2.
Substrate Scope of the Rhodium(II)-Catalyzed Formation of Substituted Enamines.
|
Variation of the 1-sulfonyl group at N-1 of triazole 1 did not affect the efficiency of the reaction. Accordingly, both aliphatic (3a and 3f) and aromatic (3b-3e) sulfonyl groups were tolerated in the reaction with phenylboronic acid (85-97% yield).
The reaction of 1-mesyl-4-phenyl-1,2,3-triazole 1a with substituted arylboronic acids 4 to produce unsymmetrical 2,2-diaryl enamines 3 raised the question of stereoselectivity of the process. To our delight, we found that the enamine products 3g-3m were obtained in high isomeric purity, indicating a highly stereoselective arylation reaction. The geometry of the double bond was assigned by performing single crystal X-ray analysis of enamine 3g (Figure 1).13
Figure 1.
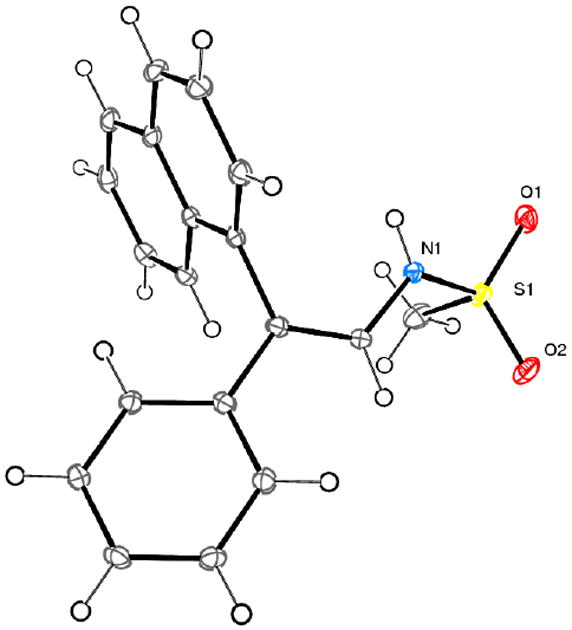
Crystal structure of enamine 3g.
As shown in Table 2, sterically encumbered (3g), electron deficient (3h, 3j, and 3l), electron rich (3i), and halogen containing (3k and 3m) boronic acids worked effectively (72-94% yield) for the construction of diversely functionalized 2,2-diaryl enamines. Although ortho-substituted arylboronic acids undergo this arylation process, the reactions were sluggish leading to low conversions. Furthermore, employing triazoles 1 bearing different aryl groups at the C-4 position furnished the corresponding enamine products 3n-3s in high yields. Both electron-donating (3n, 3o) and electron-withdrawing (3p-3s) substituents were tolerated in the arylation reaction with a series of substituted arylboronic acids (Table 2).
Although this arylation reaction proceeded with high stereoselectivity, we noted that in several cases (cf. Table 2) mixtures of isomers were obtained upon isolation (by NMR: 9:1 - 5:1). The apparent reduction of stereoselectivity likely arises from a facile tautomerization of an initial syn product. Rapid isolation of products was required in order to obtain enamines of high isomeric purity (see Supporting Information).
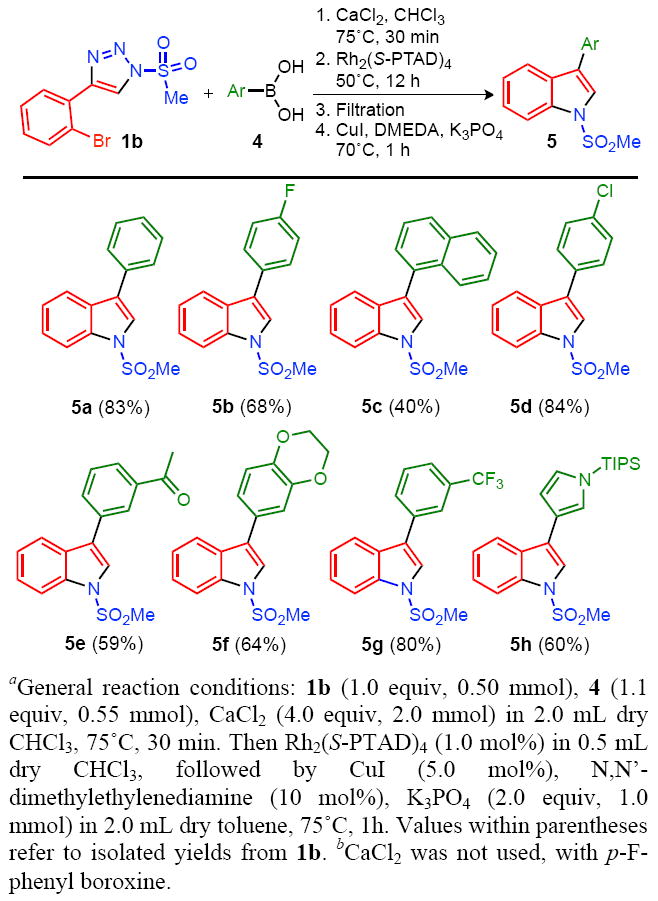
Enamines are endowed with a wealth of reactivity and can serve as versatile precursors for the construction of heterocyclic frameworks.14 Accordingly, we envisioned that the 2,2-diaryl enamine products 3 could be employed in the synthesis of aryl-substituted indoles via a metal-catalyzed cyclization. To this end, we subjected triazole 1b to the Rh-catalyzed arylation with a series of arylboronic acids, followed by a copper(I)-catalyzed intramolecular N-arylation of the enamine intermediate (Table 3).
Table 3.
Synthesis of 3-aryl Indoles via an Arylation-Cyclization Sequence.
|
Gratifyingly, triazole 1b underwent the Rh(II)-catalyzed arylation reaction efficiently, although increased temperatures (50°C), extended reaction times (12 h) and higher catalyst loadings (1.0 mol%) were required. After filtration, the derived enamine products agreeably cyclized into the anticipated N-sulfonyl-3-aryl-indoles 5 employing the copper(I)-catalyzed conditions developed by Buchwald and co-workers15 (Table 3). This cyclization requires syn-orientation of the o-Br-phenyl substituent and the N-sulfonyl amino group, which is readily achieved by a rapid isomerization of the enamine double bond (vide supra). As demonstrated in Table 3, several arylboronic acids 4 were tolerated in this two-pot synthetic sequence. Thus, halogen-containing indoles 5b and 5d, acetyl derivative 5e and heteroaryl-substituted indole 5h were obtained in good yields.
A plausible mechanism of this novel Rh(II)-catalyzed arylation reaction is shown in Figure 2. The ring-chain tautomerization of triazole 1 delivers diazoimine 1’, which then reacts with the Rh-carboxylate catalyst to form azavinyl carbene complex 2. We propose that the lone-pair of the sulfonylimine nitrogen in 2 will reversibly coordinate to the empty pz-orbital of the boron atom,16 forming complex 6. The active arylation species is believed to be a trimeric arylboroxine complex since compounds 3a, 3m and 5b were obtained without the use of CaCl2 from commercially available aryl boroxines. The stereochemical outcome of this reaction is likely dictated by the irreversible facial-selective delivery of the aryl group from intermediate 6 to obtain 7. Finally, the dissociation of rhodium and subsequent protonolysis would afford the enamine product 3. The alternative, a concerted arylation mechanism which also accounts for the observed stereochemical outcome, can not currently be ruled out.
Figure 2.
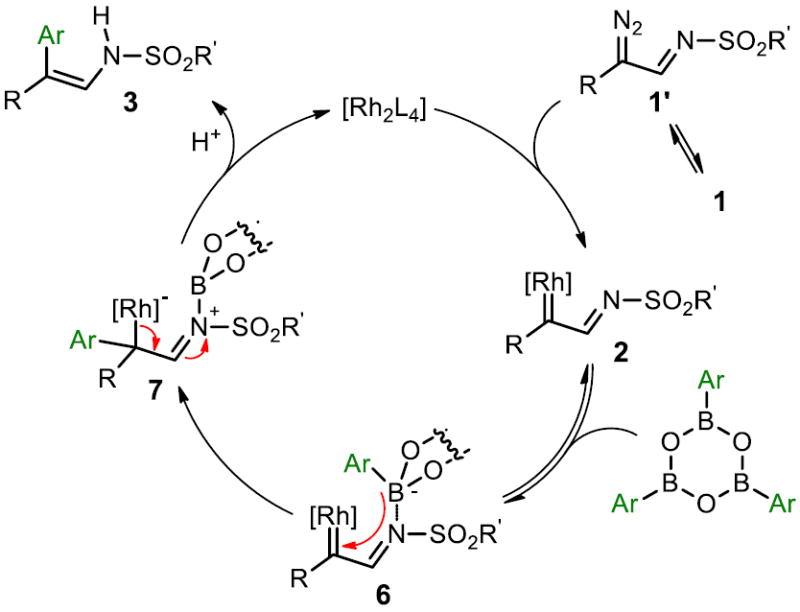
Proposed Mechanism for the Arylation of 1.
The mild, efficient and stereoselective Rh(II)-catalyzed arylation of 1-sulfonyl-1,2,3-triazoles described here provides convenient access to 2,2-diaryl enamines. This sequence of two reliable steps (synthesis of 1-sulfonyl-1,2,3-triazoles followed by their arylation with boronic acids) in effect carboaminates alkynes, regio- and stereoselectively introducing two functional groups into the acetylenic backbone. The synthetic utility of the enamine products was demonstrated by a copper-catalyzed cyclization to form a range of 3-aryl indoles. Further studies of the scope and mechanism of this reaction are underway in our laboratory and will be reported in due course.
Supplementary Material
Acknowledgments
This work was supported by the National Science Foundation (CHE-0848982) and the National Institute of General Medical Sciences, National Institutes of Health (GM-087620). N.S. also acknowledges a postdoctoral fellowship from the Swedish Research Council (VR). We also acknowledge Prof. A. Rheingold and Dr. C. Moore for X-ray crystallographic assistance.
Footnotes
Supporting Information. Experimental procedures, characterization data, NMR spectra and crystallographic data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
References
- 1.(a) Ye T, McKervey MA. Chem Rev. 1994;94:1091. [Google Scholar]; (b) Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds. Wiley; New York, NY: 1998. [Google Scholar]; (c) Zhang Z, Wang J. Tetrahedron. 2008;64:6577. [Google Scholar]
- 2.For reviews see: Padwa A, Austin DJ. Angew Chem Int Ed Engl. 1994;33:1797.. Padwa A, Weingarten MD. Chem Rev. 1996;96:223. doi: 10.1021/cr950022h.. Doyle MP, Forbes DC. Chem Rev. 1998;98:911. doi: 10.1021/cr940066a.. Hodgson DM, Pierard FYTM, Stupple PA. Chem Soc Rev. 2001;30:50.. Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861. doi: 10.1021/cr0200217.. Lebel H, Marcoux J-F, Molinaro C, Charette AB. Chem Rev. 2003;103:977. doi: 10.1021/cr010007e.. Davies HML, Manning JR. Nature. 2008;451:417. doi: 10.1038/nature06485.. Doyle MP, Duffy R, Ratnikov M, Zhou L. Chem Rev. 2010;110:704. doi: 10.1021/cr900239n.. Davies HML, Morton D. Chem Soc Rev. 2011;40:1857. doi: 10.1039/c0cs00217h.
- 3.(a) Taber DF, Ruckle RE., Jr J Am Chem Soc. 1986;108:7686. doi: 10.1021/ja00284a037. [DOI] [PubMed] [Google Scholar]; (b) Hrytsak M, Etkin N, Durst T. Tetrahedron Lett. 1986;27:5679. [Google Scholar]; (c) Hrytsak M, Durst T. J Chem Soc Chem Commun. 1987:1150. [Google Scholar]; (d) Doyle MP, Shanklin MS, Pho HQ, Mahapatro SN. J Org Chem. 1988;53:1017. [Google Scholar]; (e) Padwa A, Austin DJ, Hornbuckle SF, Semones MA. J Am Chem Soc. 1992;114:1874. [Google Scholar]
- 4.(a) Peng C, Zhang W, Yan G, Wang J. Org Lett. 2009;11:1667. doi: 10.1021/ol900362d. [DOI] [PubMed] [Google Scholar]; (b) Barluenga J, Tomás-Gamasa M, Aznar F, Valdés C. Nat Chem. 2009;1:494. doi: 10.1038/nchem.328. [DOI] [PubMed] [Google Scholar]
- 5.(a) Barluenga J, Valdés C. Angew Chem Int Ed. 2011;50:7486. doi: 10.1002/anie.201007961. [DOI] [PubMed] [Google Scholar]; (b) Shao Z, Zhang H. Chem Soc Rev. 2012;41:560. doi: 10.1039/c1cs15127d. [DOI] [PubMed] [Google Scholar]
- 6.For pioneering work on the Pd-catalyzed arylation of diazo species, see: Greenman KL, Carter DS, Van Vranken DL. Tetrahedron. 2001;57:5219.
- 7.(a) Barluenga J, Moriel P, Valdés C, Aznar F. Angew Chem Int Ed. 2007;46:5587. doi: 10.1002/anie.200701815. [DOI] [PubMed] [Google Scholar]; (b) Barluenga J, Tomás-Gamasa M, Moriel P, Aznar F, Valdés C. Chem Eur J. 2008;14:4792. doi: 10.1002/chem.200800390. [DOI] [PubMed] [Google Scholar]; (c) Peng C, Wang Y, Wang J. J Am Chem Soc. 2008;130:1566. doi: 10.1021/ja0782293. [DOI] [PubMed] [Google Scholar]; (d) Peng C, Zhang W, Yan G, Wang J. Org Lett. 2009;11:1667. doi: 10.1021/ol900362d. [DOI] [PubMed] [Google Scholar]; (e) Barluenga J, Escribano M, Aznar F, Valdés C. Angew Chem Int Ed. 2010;49:6856. doi: 10.1002/anie.201003450. [DOI] [PubMed] [Google Scholar]; (f) Zhao X, Jing J, Lu K, Zhang Y, Wang J. Chem Commun. 2010;46:1724. doi: 10.1039/b925590g. [DOI] [PubMed] [Google Scholar]; (g) Tsoi Y-T, Zhou Z, Chan ASC, Yu W-Y. Org Lett. 2010;12:4506. doi: 10.1021/ol101796t. [DOI] [PubMed] [Google Scholar]; (h) Peng C, Yan G, Wang Y, Jiang Y, Zhang Y, Wang J. Synthesis. 2010:4154. [Google Scholar]; (i) Zhang Y, Wang J. Eur J Org Chem. 2011:1015. [Google Scholar]
- 8.(a) Dimroth O. Ann. 1909;364:183. [Google Scholar]; (b) Gilchrist TL, Gymer TE. Adv Heterocycl Chem. 1974;16:33. [Google Scholar]; (c) Harmon RE, Stanley F, Jr, Gupta SK, Johnson J. J Org Chem. 1970;35:3444. [Google Scholar]; (d) Regitz M, Himbert G. Tetrahedron Lett. 1970:2823. [Google Scholar]; (e) Chuprakov S, Hwang FW, Gevorgan V. Angew Chem Int Ed. 2007;46:4757. doi: 10.1002/anie.200700804. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Chuprakov S, Gevorgyan V. Org Lett. 2007;9:4463. doi: 10.1021/ol702084f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Horneff T, Chuprakov S, Chernyak N, Gevorgyan V, Fokin VV. J Am Chem Soc. 2008;130:14972. doi: 10.1021/ja805079v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chuprakov S, Kwok SW, Zhang L, Lercher L, Fokin VV. J Am Chem Soc. 2009;131:18034. doi: 10.1021/ja908075u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Grimster NP, Zhang L, Fokin VV. J Am Chem Soc. 2010;132:2510. doi: 10.1021/ja910187s. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zibinsky M, Fokin VV. Org Lett. 2011;13:4870. doi: 10.1021/ol201949h. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Chuprakov S, Malik JA, Zibinsky M, Fokin VV. J Am Chem Soc. 2011;133:10352. doi: 10.1021/ja202969z. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Selander N, Fokin VV. J Am Chem Soc. 2012;134:2477. doi: 10.1021/ja210180q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reddy RP, Lee GH, Davies HML. Org Lett. 2006;8:3437. doi: 10.1021/ol060893l. [DOI] [PubMed] [Google Scholar]
- 11.It should be noted that the use of phenylboronic acid yields 3a without the addition of a desiccant (commercial samples contain approximately 75% of the corresponding boroxine by NMR).
- 12.The major side-products observed were α-amino ketones that were likely formed by an OH-insertion of the Rh-carbenoids. See: Miura T, Biyajima T, Fujii T, Murakami M. J Am Chem Soc. 2012;134:194. doi: 10.1021/ja2104203.
- 13.The crystal structure of 3g has been deposited at the Cambridge Crystallographic Data Centre (CCDC 883835). The geometry of all other enamine products 3 was assigned by analogy.
- 14.For a recent review of reactivity of enamides, see Carbery DR. Org & Biomol Chem. 2008;6:3455. doi: 10.1039/b809319a.; For general syntheses of indoles: Barluenga J, Valdés C. In: Modern Heterocyclic Chemistry. Alvarez-Builla J, Vaquero JJ, Barluenga J, editors. Vol. 1. Wiley-VCH; Weinheim: 2011. pp. 377–531.; Krüger K, Tillack A, Beller M. Adv Synth Catal. 2008;350:2153.. Humphrey GR, Kuethe JT. Chem Rev. 2006;106:2875. doi: 10.1021/cr0505270.. Alsabeh PG, Lundgren RJ, Longobardi LE, Stradiotto M. Chem Comm. 2011;47:6936. doi: 10.1039/c1cc11874a.. Larock RC, Yum EK, Refvik MD. J Org Chem. 1998;63:7652.
- 15.(a) Surry DS, Buchwald SL. Chem Sci. 2010;1:13. doi: 10.1039/C0SC00107D. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Klapars A, Huang XH, Buchwald SL. J Am Chem Soc. 2002;124:7421. doi: 10.1021/ja0260465. [DOI] [PubMed] [Google Scholar]
- 16.Attempts to arylate rhodium(II) carbenoids derived from phenyl diazoacetate with phenyl boronic acid failed, indicating the importance of a pendant coordinating nitrogen atom.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.