

SUMMARY
Reef-building corals and many other cnidarians are symbiotic with dinoflagellates of the genus Symbiodinium. It has long been known that the endosymbiotic algae transfer much of their photosynthetically fixed carbon to the host and that this can provide much of the host's total energy. However, it has remained unclear which metabolite(s) are directly translocated from the algae into the host tissue. We reexamined this question in the small sea anemone Aiptasia using labeling of intact animals in the light with 13C-bicarbonate, rapid homogenization and separation of animal and algal fractions, and analysis of metabolite labeling by gas chromatography-mass spectrometry. We found labeled glucose in the animal fraction within 2 min of exposure to 13C-bicarbonate, whereas no significant labeling of other compounds was observed within the first 10 min. Although considerable previous evidence has suggested that glycerol might be a major translocated metabolite, we saw no significant labeling of glycerol within the first hour, and incubation of intact animals with 13C-labeled glycerol did not result in a rapid production of 13C-glucose. In contrast, when Symbiodinium cells freshly isolated from host tissue were exposed to light and 13C-bicarbonate in the presence of host homogenate, labeled glycerol, but not glucose, was detected in the medium. We also observed early production of labeled glucose, but not glycerol, in three coral species. Taken together, the results suggest that glucose is the major translocated metabolite in dinoflagellate–cnidarian symbiosis and that the release of glycerol from isolated algae may be part of a stress response.
KEY WORDS: Aiptasi, coral biology, GC-MS, mutualism, photosynthate transfer, sea anemone
INTRODUCTION
Coral reefs provide enormous environmental and economic benefits: they protect coasts, harbor vast biodiversity, sequester carbon, support fisheries and create tourist destinations. The principal architects of these reefs are the coral animals (Phylum Cnidaria), whose ability to grow in nutrient-poor waters and deposit the calcareous skeletons that form the backbone of the reefs depends on their receipt of photosynthetically fixed carbon from their intracellular dinoflagellate symbionts (genus Symbiodinium). The algae can provide >90% of their photosynthate to the host, thus supplying >90% of the host's total energy requirements for maintenance, growth and calcium carbonate deposition (Falkowski et al., 1984); in turn, the host provides protection and passes a variety of other nutrients to the algae (Rahav et al., 1989).
Unfortunately, the dinoflagellate–cnidarian symbiosis is sensitive to environmental stress and thus is threatened by global warming, ocean acidification, sedimentation, pollution and disease. When the symbiosis is disrupted (known as coral bleaching because of the loss of photosynthesis-related pigments or the entire pigmented dinoflagellates), the host often dies, and the reef begins to erode. Major bleaching events have occurred multiple times in various parts of the world over the past 20 years, and there has been a loss of ~317,000 ha of live-coral coverage per year in the Indo-Pacific alone (Bruno and Selig, 2007). To improve the prospects for arresting or reversing the decline of coral reefs, a much more detailed understanding of the molecular and cellular biology of the dinoflagellate–cnidarian symbiosis is needed, including a better understanding of the metabolites transferred between alga and host and the mechanisms by which such transport is achieved and regulated.
Using 14CO2 and autoradiography, Muscatine and Hand demonstrated that carbon fixed by dinoflagellates is incorporated into cnidarian tissue (Muscatine and Hand, 1958). Since that pioneering study, numerous investigations have attempted to identify the transferred metabolite(s) using a wide variety of biological systems and experimental approaches. These studies have obtained a variety of results, as recently reviewed by others (Venn et al., 2008; Yellowlees et al., 2008; Davy et al., 2012). Briefly, the methods used have included: (1) detecting metabolites in the animal tissue that the animal itself was incapable of synthesizing; (2) isolating Symbiodinium cells from host tissue, exposing them to labeled substrate along with host homogenate or potential ‘host release factors’, and determining what compounds are released from the algae; and (3) exposing the intact holobionts to labeled compound(s), fractionating to separate host and algal components, and identifying the labeled compounds in each fraction. In addition, in the related Symbiodinium–giant-clam symbiosis, fluids have been withdrawn from intact organisms after labeling and then analyzed. In most studies, thin-layer chromatography and autoradiography have been used to identify the labeled compounds.
Many of these studies, including those examining the compounds released by isolated algae, have concluded that glycerol is the principal metabolite transferred from alga to host. However, glucose, succinate, fumarate, a variety of amino acids, and various lipids have also been detected in some studies and suggested to be among the major transferred compounds. In addition, several recent studies using intact systems have not found labeled glycerol. For example, in the giant clam, where the symbiotic algae are extracellular, significant concentrations of glucose, but not of glycerol, were detected in the hemolymph (Rees et al., 1993). However, Ishikura and co-workers did detect labeled glycerol when dinoflagellates freshly isolated from the clams were exposed to host homogenate or when the host mantle was damaged, even though exogenously supplied labeled glycerol was not converted rapidly to glucose in the amounts seen in the intact system (Ishikura et al., 1999). Similarly, when the anemone Anemonia viridis was fractionated after labeling the intact holobiont, the host fraction was found to contain 14C-labeled amino acids, glucose, malate, succinate and fumarate, but no detectable glycerol (Whitehead and Douglas, 2003). Taken together, these studies have raised the possibility that glycerol production and/or release is linked to damage to the symbiotic systems rather than being integral to the intact symbiosis.
It is possible that the diversity of results obtained in earlier studies reflects real differences in the compounds transferred in different organisms or under different conditions of testing. However, it also seems possible that technical difficulties have led to misleading results in many studies. Indeed, to a greater or lesser extent, all of the earlier studies have suffered from one or more of the following potential problems. (1) Algae isolated from host tissue may no longer behave normally, regardless of whether they are treated with host homogenate or artificial mixtures designed to mimic it. (2) The centrifugation steps used to separate host and algal fractions after labeling require many minutes of preparation time before analysis, often at room temperature or above, during which metabolism of the initially labeled and transferred compounds might continue. (3) Thin-layer chromatography requires standards to determine the identities of the compounds detected (thus causing a problem if any unexpected compounds are present in the mix) and also suffers from poor resolution of different compounds. (4) Autoradiograms can take weeks or months to develop (thus hindering any kind of iterative experimentation) and do not provide information on the proportion of the pool of each compound that is labeled, but rather just on the size of the labeled sub-pool.
To overcome such limitations, we sought an approach that would allow observations to be made on intact holobiont animals with minimal disturbance prior to or during the experimental period. In addition, we wanted to be able to both sample and separate host from algal fractions sufficiently rapidly that the chances of confusion by secondary metabolic conversion would be minimized. Finally, we wanted a method of analysis that allows rapid, quantitative detection of both the labeled and unlabeled pools of many metabolites (thus allowing the fractions labeled to be determined) and does so with sufficient sensitivity that labeling can be detected even after very short exposures to the label.
One such analytical method employs gas chromatography with mass spectrometry (GC-MS), which can detect >150 polar metabolites in a single run of ~1 h (Roessner et al., 2000; Lisec et al., 2006). Before analysis, samples are derivatized using trimethylsilyl groups, which decreases the boiling points of many organic compounds, including amino acids, organic acids, sugars, sugar alcohols and aromatic amines. This boiling-point depression allows the derivatized compounds to traverse a GC capillary column at relatively low temperatures. In addition, many hydrophobic compounds can be detected by saponification followed by derivatization (Blumenberg et al., 2007). Metabolic profiles of organisms under different conditions can be determined and can illuminate metabolic pathways (Christensen et al., 2000), and analysis of a sample can be completed in hours instead of weeks, allowing a much more rapid progression of experiments.
Although sensitive on its own, GC-MS becomes even more powerful when combined with 13C-isotope labeling. 13C increases the mass-to-charge (m/z) ratio of molecules and their ionized fragments, which can be detected by the MS. However, 13C incorporation does not change the retention time of a metabolite on the GC capillary column, meaning that the fraction of each metabolite that is labeled and the number of labeled carbons can be determined by examining the m/z ratios of the material emerging from the GC at a given elution time (Jiao et al., 2003). From the MS fragmentation patterns, it is possible to determine which positions of a given compound are labeled, allowing testing of hypotheses about the pathways responsible for the production of that compound (Sauer, 2006).
Using 13C labeling and the GC-MS analytical platform, we developed a protocol for analyzing metabolite transfer that met the criteria indicated above. This protocol yielded data suggesting strongly that, at least in the organisms and conditions examined, glucose is by far the predominant photosynthetic product passed from the alga into the host tissues.
MATERIALS AND METHODS
Reagents
Artificial seawater (ASW) was made by mixing Coral Pro Salt (Red Sea, Houston, TX) with deionized water at 3.35%. IMK medium for algal culture was obtained from Nihon Pharmaceutical (Tokyo, Japan). Most GC-MS extractions used ultrapure water from a Milli-Q system (Millipore, Billerica, MA, USA); others used ASW. Ribitol, LC-MS-grade methanol, HPLC-grade CHROMASOLV chloroform, [U-13C]glucose, [U-13C]glycerol and [13C]NaHCO3 were obtained from Sigma-Aldrich (St Louis, MO, USA). The strong photosynthesis poison DCMU [3-(3,4-dichlorophenyl)-1,1-dimethylurea] (Vandermeulen et al., 1972) and the inhibitor of gluconeogenesis 1-thioglycerol (Seltzer et al., 1986) were also obtained from Sigma-Aldrich. [12C]NaHCO3, 2% methoxyamine (MOX)-HCl in pyridine, and N-methyl-N-trimethylsilyl tri-fluoroacetamide (MSTFA) in pyridine were obtained from Fisher Scientific (Waltham, MA, USA). Other compounds used as GC-MS standards were obtained from Fisher Scientific or Sigma-Aldrich.
Animals and culture conditions
Most experiments used clonal stock CC7 of Aiptasia pallida (Sunagawa et al., 2009) containing its endogenous clade A Symbiodinium. One experiment used a clonal population (H1) derived from a single anemone of Hawaiian origin (and thus presumably of species A. pulchella; S. R. Santos, personal communication) containing its endogenous clade B Symbiodinium. Some experiments used aposymbiotic (dinoflagellate-free) animals derived from CC7. To produce these animals, CC7 anemones were first incubated in ASW at 4°C for 4 h (resulting in ejection of a mass of material containing many dinoflagellates) and then in ASW containing 50 μm l−1 DCMU at 24°C under fluorescent lights (20–40 μE m−2 s−1) for several days. [All light levels indicated are given as photosynthetically active radiation, measured with a GMSW-SS quantum meter (Apogee, Logan, UT).] These steps were repeated until all animals were conspicuously pale (two to three cycles), after which animals were kept in ASW containing 2–5 μmol l−1 DCMU for several weeks under fluorescent lights with a 12 h:12 h light:dark cycle (20–40 μE m−2 s−1). Animals in several separate tanks were then washed to remove DCMU, placed under normal culture conditions (see below) for several weeks, and examined periodically by fluorescence microscopy to detect the chlorophyll fluorescence of any residual dinoflagellates. Tanks in which all animals appeared dinoflagellate-free were used as sources of aposymbiotic animals.
For routine culture, anemones were maintained in ASW at approximately 24°C with light provided by a window and/or Cool White fluorescent bulbs at ~20–40 μE m−2 s−1 (light:dark ≈ 12 h:12 h) and feeding several times weekly with freshly hatched Artemia nauplii. For 7–14 days before use in an experiment, anemones were starved. During the 2 days immediately before an experiment, animals were rinsed repeatedly in ASW to remove any algae attached to their outer surfaces and inspected microscopically to ensure that all food had been digested.
Samples of two soft corals and one hard coral were obtained from commercial sources [Discosoma sp. (Garf, Boise, ID, USA), Cladiella sp. (Dolphin Pet Village, Campbell, CA, USA) and Acropora millepora (Coalition of Reef Lovers, Pago Pago, American Samoa)] and maintained in saltwater aquaria (light:dark ≈ 12 h:12 h) until just before use.
Symbiodinium culture and isolation from host tissue
Derivation of the clonal, axenic Symbiodinium strain SSB1 from a Hawaiian anemone (our clonal line H2; presumed A. pulchella) will be described in detail elsewhere. SSB1 algae were grown in IMK medium (Ishikura et al., 2004) at ~24°C under Cool White fluorescent lights at 20–40 μE m−2 s−1 (light:dark ≈ 12 h:12 h). To isolate algae for experiments, the culture was centrifuged at 24°C for 10 min at 1000 g. The brown fraction of the pellet was re-suspended in ASW and re-centrifuged twice. The final pellet was re-suspended in ASW to a final concentration of ~6×106 cells ml−1.
To isolate algae from host tissue, clone CC7 anemones grown as described above were homogenized with a PowerGen 125 Homogenizer (Fisher Scientific) and then centrifuged at 24°C for 10 min at 1000 g. The brown fraction of the pellet was rinsed, re-suspended in ASW and re-centrifuged twice. The final pellet was re-suspended in ASW to a final concentration of ~6×106 cells ml−1 and used immediately.
Tracing photosynthetic 13C incorporation
In our standard protocol, starved and washed anemones (see above) were pre-incubated in the dark for 24 h. They were then either used directly in experiments or pre-incubated further in the light (190 μE m−2 s−1) for 2.0–4.5 h. Experiments were initiated by exposing the animals to either [12C]NaHCO3 or [13C]NaHCO3 at 10 mg ml−1 in ASW either in the dark or in the light at 190 μE m−2 s−1. Experiments were performed in a fume hood to minimize possible contamination of other animal and dinoflagellate stocks by gaseous 13CO2. At each time point, sampling was performed in triplicate with three to five animals used for each sample (necessary to get sufficient biomass for adequate analysis). For each sample, the several animals were rinsed quickly with Milli-Q water, then disrupted in 1 ml Milli-Q water using a PowerGen 125 Homogenizer for ~3 s (which was ~2 s beyond the time at which intact pieces of the animal were visible). The homogenizer was washed thoroughly between uses. (Some homogenizations were performed in ASW rather than Milli-Q water. The results were similar, but the use of Milli-Q water eliminated the need to desalt, improved GC peak shape and increased the life of the capillary GC column.) Four hundred microliters of each homogenate were mixed with 400 μl Milli-Q water, 1.5 ml methanol and 5 μl of a 2 mg ml−1 ribitol stock solution (to provide an internal standard), and the sample was flash-frozen in liquid nitrogen. All methanol-containing solutions were handled in glassware to minimize possible contamination by material leached from plastics. The samples of total homogenate were frozen within ~1 min of initial sampling.
A second 400-μl aliquot of each homogenate was applied to a 25 mm diameter, 2.7 μm pore-size glass-fiber filter (Millipore) that had been thoroughly rinsed in methanol and Milli-Q water. Vacuum was applied, and the filtrate was collected in a 4 ml glass vial attached to the bottom of a custom-made filter holder (supplementary material Fig. S1). The filter was rinsed twice with 200 μl Milli-Q water, collecting the additional filtrates also in the same vial. Methanol (1.5 ml) was added to the vial along with 5 μl of the ribitol stock. The filter was placed in a separate vial containing 800 μl Milli-Q water, 1.5 ml methanol and 5 μl of the ribitol stock, and both vials were flash-frozen in liquid nitrogen. These fractionated samples were frozen within ~2.5 min of initial sampling.
Prior to analysis, the sample vials were thawed and incubated at 70°C for 45 min to extract metabolites, with a gentle shaking every 5 min. (More vigorous shaking resulted in disassociation of the glass-fiber filters and a loss of yield because the dissociated filters retained most of the liquid.) Vials were centrifuged at 2500 g for 10 min at 4°C to pellet insoluble materials, and each supernatant was decanted into a fresh vial containing 700 μl Milli-Q water and 750 μl HPLC-grade chloroform for extraction. Each vial was vortexed for 10 s and then centrifuged at 2500 g for 10 min at 4°C to separate the fractions. A 275 μl aliquot of the water/methanol layer (the more polar fraction) was separated from the chloroform layer (the less polar fraction), transferred to glass GC vial inserts and dried in vacuo in a Speedvac (Thermo Fisher Scientific, Waltham, MA, USA) at 35°C for up to 2 h.
Samples were derivatized as described by Roessner et al. (Roessner et al., 2000). A volume of 40 μl MOX in pyridine was added to each sample vial, followed by incubation in a shaker for 2 h at 37°C. We then added 70 μl MSTFA in pyridine, followed by incubation for 30 min at 37°C with shaking. For analysis, 1 μl of each derivatized sample was injected into a Varian Saturn 3900 GC (Varian, Palo Alto, CA, USA) using a Varian 8400 auto-sampler with a split ratio of 10:1 for 1.5 min, 100:1 for 1.5 min and 2:1 for 51 min; an injector temp of 220°C; and a constant flow of helium (purity 4.5 or better; Air Products, Allentown, PA, USA) at 1.7 ml min−1. The 30-m long, 0.25-mm internal-diameter Thermo TG-5MS column (Thermo Fisher Scientific) was held at 70°C for 5 min, then ramped to 310°C at 5°C min−1, and then held at 310°C for 1 min. Between sample injections, the system was returned to 70°C and equilibrated for 6 min, and an extensive needle-washing routine was used. The spectra were collected on a Varian Saturn 2000 MS, scanning between m/z 40 and 650. The MS was mass-tuned before running each batch of samples with manufacturer-supplied perfluorotributylamine (FC-43). Retention times and mass spectra were compared against the NIST02 library (National Institute of Standards and Technology, Gaithersburg, MD, USA) and our own custom library to identify compounds. Saturn GC/MS Workstation v5.5 software (Varian) was used to visualize spectra, integrate areas under peaks and search libraries.
On some occasions, the chloroform fraction (less polar compounds) was also analyzed. First, it was rinsed with Milli-Q water (retaining the aqueous phase for control purposes) and evaporated. Next, 500 μl of 1 mol l−1 KOH in LC-MS-grade methanol were added to the dry sample, and the mixture was saponified at 70°C for 24 h. The reaction was terminated by adding 1 mol l−1 HCl in water, and the samples were then extracted with 500 μl of chloroform. A 275 μl aliquot of the aqueous phase for each sample was added to a glass GC vial insert, the aqueous phase from the earlier extraction was added to another vial insert, and all samples were evaporated in a Speedvac at 35°C for 2 h or until dry. All inserts were sealed in vials and stored at −80°C until analyzed via GC-MS in the same manner as the polar fractions (Blumenberg et al., 2007; Arning et al., 2008).
To apply the method to soft corals, fleshy portions of the colonies were removed from the maintenance aquaria and placed in separate tanks of ASW just before labeling in the light (190 μE m−2 s−1) with 10 mg ml−1 13C-bicarbonate. Homogenization and filtration were as described above for anemones. For A. millepora, ~5-cm-long nubbins were removed from the maintenance aquaria and exposed to 13C-bicarbonate as described above. Tissue was then removed by scrubbing with a wire brush, but recovery was far from complete, and it took several nubbins to obtain enough material for homogenization, extraction and detection. We did not have sufficient material for a successful separation of animal and algal fractions by filtration.
Analysis of material released by isolated dinoflagellates
Freshly isolated dinoflagellates in ASW (see above) were mixed 1:3 either with ASW or with anemone homogenate (i.e. the supernatant from the initial centrifugation performed to isolate the dinoflagellates, but centrifuged twice more at 1000 g to remove any residual dinoflagellates). Another sample of the homogenate was mixed 3:1 with ASW to constitute a homogenate-only control. In all cases, 13C-bicarbonate was added to achieve a final concentration of 10 mg ml−1. The samples were then incubated in the light (190 μE m−2 s−1) for 1 h. Each sample was then centrifuged at 1000 g for 10 min at 24°C, the supernatants were collected, and the algal pellets (where present) were resuspended in ASW back to the volume of suspension before centrifugation. The supernatants and samples of resuspended algae were then mixed separately with Milli-Q water, methanol and ribitol-standard solution to achieve the concentrations used in our standard protocol (see above). Extraction and subsequent analysis then proceeded as described above. The GC chromatograms were generated by integration across nearly all of the MS spectrum (m/z 90 to 650) and peak areas (15.0 min peak for glycerol; 29.95 min peak for glucose) were determined by integration across each peak using Workstation v5.5 (Varian). The fractions of the glycerol and glucose that were labeled were then determined by the peak-ratio method as described below.
Determination of relative labeling by a peak-ratio method
To determine and compare relative labeling levels for a given compound at particular times and conditions of labeling, a distinctive mass fragment was chosen, giving preference to fragments with higher abundances and/or higher m/z ratios; the rightward tails caused by naturally occurring 13C and heavier isotopes of silicon (see Fig. 2 and supplementary material Fig. S2) were ignored. The magnitude of each m/z reading was recorded at the point in the GC chromatogram at which the sum of the readings for the unlabeled and labeled fragments was largest. When both fully labeled and partially labeled fragments were observed, the former was used in the analysis. The magnitude of the m/z reading for the labeled compound was divided by the sum of itself and the magnitude of the m/z reading for the unlabeled compound to derive the peak ratios used in the analyses. An exact binomial test on the actual counts was used to determine 95% confidence intervals for the peak-ratio data points. When two data points were to be compared (e.g. different labeling conditions or times of labeling) and a P-value was needed, Fisher's exact tests were performed to compare the actual counts. For example, to determine whether the glucose labeling observed at early time points in Fig. 4 was significant, the values obtained for exposure to 13C-bicarbonate in the light were compared in this way to the values obtained for exposure to 13C-bicarbonate in the dark and those for exposure to 12C-bicarbonate in the light. A difference in labeling was considered significant if the counts observed for one data point differed from those for the other at P≤0.05. Statistical tests were performed using R (R Development Core Team, 2009).
Fig. 2.

GC-MS identification of photosynthetically produced glucose and of a glucose fragment useful in quantitating the incorporation of 13C into newly synthesized glucose. CC7 anemones were incubated with 10 mg ml−1 12C-bicarbonate in the light for 10 min (A,D), with 10 mg ml−1 13C-bicarbonate in the dark for 10 min (B,E) or with 10 mg ml−1 13C-bicarbonate in the light for 10 min (C,F), and homogenates were prepared and analyzed as described in the Materials and methods. (A–C) Chromatograms produced by summing the MS peak intensities from 310 to 330 m/z for each elution time shown, showing a peak identified as glucose by comparing its elution time and MS spectrum (shown in part in D–F) to those observed in previous work and in our own glucose-standard runs (supplementary material Fig. S2). (D–F) Portions of the MS spectrum (m/z 310–330) of the material eluting from the GC at 29.95 min. Note that the m/z values are those for the derivatized glucose fragment; the rightward tail on each peak is due to naturally occurring 13C (~1.1%) and the isotopic distribution of the silicon in the silate groups used for derivatization (see supplementary material Fig. S2 legend for more details).
Fig. 4.
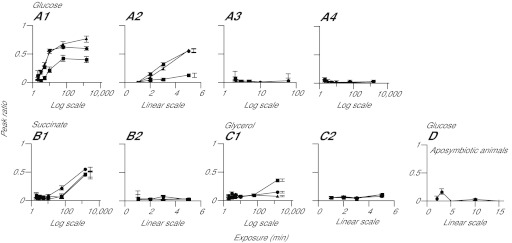
Detection of labeled glucose, but not of other compounds, in host tissue at early times after the exposure of symbiotic anemones to light and 13C-bicarbonate. (A–C) CC7 anemones were incubated for various times in the light or dark with 13C-bicarbonate or 12C-bicarbonate, as indicated, and were then homogenized and analyzed as described in the Materials and methods. The peak-ratio method was used to determine the distributions of 13C-labeled glucose (A), succinate (B) and glycerol (C) in the total homogenate (circles), the filtrate (triangles), and the material retained on the filter (squares). (1) Dark pre-incubation followed by incubation in the light with 13C-bicarbonate; (2) light pre-incubation for ~2 h followed by incubation in the light with 13C-bicarbonate; (3) dark pre-incubation followed by incubation in the dark with 13C-bicarbonate; (4) dark pre-incubation followed by incubation in the light with 12C-bicarbonate. (D) As in panel A1 except using aposymbiotic anemones. Time axes are in log or linear scales, as indicated. Error bars (all panels) represent 95% confidence intervals based on the three biological replicates in each case; some error bars in A2, B1 and C1 are offset from the corresponding data points to allow easier discrimination between the symbols.
RESULTS
Establishment of the experimental system
We performed a variety of tests to establish that the experimental system would function as intended. First, we confirmed that standards for various metabolites likely to be of interest eluted from our GC at discrete times that were consistent with expectations from prior work (supplementary material Fig. S2A–C, Table 1, and data not shown). Moreover, the standards yielded m/z peaks in the MS that could readily be used for quantitation (e.g. full-length glycerol and succinate and a four-carbon fragment of glucose; supplementary material Fig. S2D,F,G; note that the m/z values are those for the derivatized compounds and see legend for more details). As expected, 13C-labeled standards eluted from the GC at the same times as the 12C compounds and yielded m/z peaks that were shifted by the expected amounts (e.g. +3 for glycerol and +4 for the four-carbon glucose fragment; supplementary material Fig. S2E,H); this shift allows determination of the fractions labeled using a peak-ratio method, as described below and in the Materials and methods.
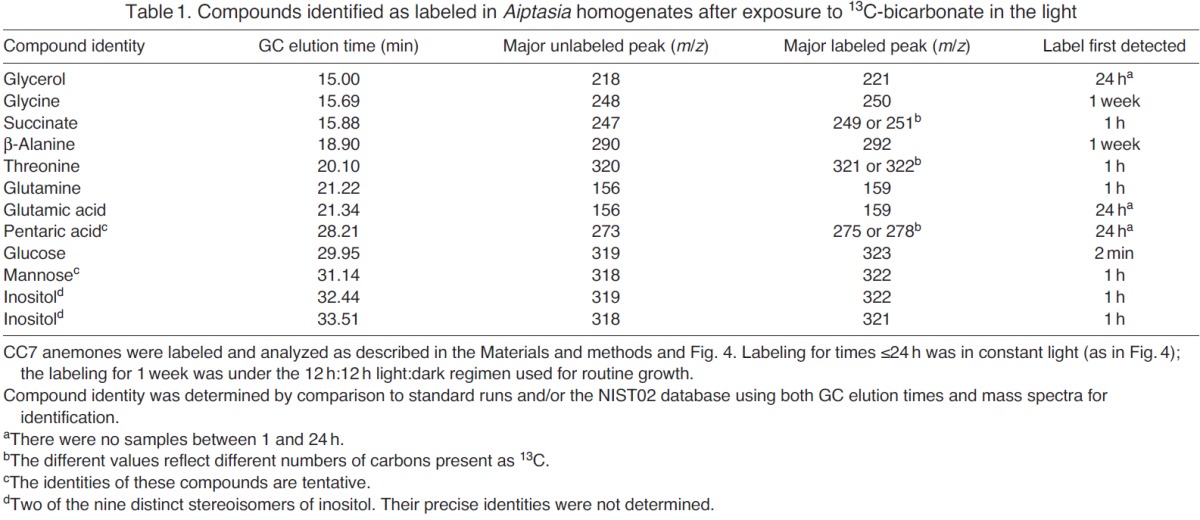
Table 1.
Compounds identified as labeled in Aiptasia homogenates after exposure to 13C-bicarbonate in the light
Next, we verified that, as expected, GC-MS analysis of an anemone homogenate could detect ~100 metabolites with discrete elution times (Fig. 1); a typical compound had a peak width of 3–10 s (of the 54 min GC-MS run). We examined more closely the GC peak presumed (from its elution time and m/z spectrum) to represent glucose. This peak increased significantly when symbiotic animals were incubated for 10 min in the light (compare Fig. 2A,C with 2B), suggesting that there were only low levels of free glucose in the starved animals in the dark, so that photosynthetically produced glucose could be readily detected. In the absence of 13C label or in the dark, the m/z peak for the four-carbon fragment was at 319 (Fig. 2D,E), but after labeling with 13C-bicarbonate for 10 min in the light, this peak was partially replaced by one with m/z 323 (Fig. 2F), as expected. This shift allows determination of the fractions of glucose labeled in time-course experiments by a peak-ratio method, dividing the sum of intensities in the 323 m/z peak by the sums of intensities in both the 319 and 323 m/z peaks [323/(319+323) ratio]; the same method can be applied to glycerol [221/(218+221) ratio; supplementary material Fig. S2D,E], succinate [251/(247+251) ratio; supplementary material Fig. S2F] and other metabolites.
Fig. 1.

GC-MS chromatograms showing detection of multiple metabolites with a wide range of GC elution times in an anemone homogenate. CC7 anemones were incubated with 13C-bicarbonate in the light for 10 min, and a homogenate was prepared and analyzed as described in the Materials and methods. (A) For each GC elution time, the height shown is the sum of all of the MS intensities recorded for m/z 90–650. Inset: magnified image of the results for elution times of 29.5–30.5 min; a glucose standard eluted at 29.95±0.05 min on the column used (supplementary material Fig. S2C). (B) The same chromatogram after log transformation to deemphasize the background (intensities <6000 are not displayed), compress the peaks, and thus better show the multitude of peaks detected. Intensitydisplayed=log10(intensity recorded/1000–5) or simply 0 when intensity recorded was ≤6000.
We also carefully tested our methods for sample collection. Using the protocol described in detail in the Materials and methods, we found that we could collect animals that previously had not been perturbed (except by exposure to light and/or the 13C-bicarbonate label), homogenize them, and flash-freeze samples of the homogenate within ~1 min. In addition, we could filter samples of the homogenate, rinse, and flash-freeze samples of both the filtrate and the filter (with retained material) within an additional ~1.5 min (thus, ~2.5 min from the time of initial collection of the animals). As expected, examination by fluorescence microscopy revealed many seemingly intact and fluorescent (by their intrinsic chlorophyll fluorescence) dinoflagellate cells on the top of the filter but not on its bottom or in the filtrate. Some host cells appeared to remain intact and were retained on the filter, but the amount of this material was clearly small in relation to both the dinoflagellate material retained on the filter and the amount of host material (cytoplasm plus organelles) that had passed through into the filtrate. To address the possibility that the homogenization and filtration protocol might cause disruption of the dinoflagellate cells and/or leaching of metabolites from them, we performed tests using both Symbiodinium freshly isolated from host tissue and Symbiodinium cells from culture. In both cases, it appeared that the dinoflagellate cells remained largely or entirely intact and released little of their newly synthesized glucose after labeling in isolation from host tissue (Fig. 3 and data not shown); note that the freshly isolated dinoflagellates had in fact passed through the PowerGen 125 Homogenizer twice (during homogenization of the host and again during the mock homogenization) without losing their integrity.
Fig. 3.

Retention of labeled glucose by isolated Symbiodinium cells. Algal cells freshly isolated from CC7 anemones (see Materials and methods) were incubated in artificial seawater (ASW) with 10 mg ml−1 13C-bicarbonate for 60 min in the light (190 μE m−2 s−1) and then rinsed and resuspended in Milli-Q water. The samples were then treated with the homogenizer, filtered, and analyzed as performed with samples of host material (see Materials and methods). The peak-ratio method (see Materials and methods and Fig. 2) was then used to determine the distribution of newly synthesized, labeled glucose. Error bars represent 95% confidence intervals based on the three biological replicates.
Rapid transfer of newly synthesized glucose, but not other metabolites, from dinoflagellate to host tissue
To begin analyzing potentially transferred metabolites, we first examined samples of homogenate of symbiotic CC7 anemones that had been incubated for 1 week (light:dark ≈ 12 h:12 h) in the presence of 13C-bicarbonate. We detected 97 GC peaks containing >300 major MS fragments and examined them for possible peak shifts. This essentially unbiased screen of small polar metabolites detected 21 candidate compounds (i.e. GC peaks with 13C incorporation), of which 12 could be identified with confidence using standard runs and the NIST02 library (Table 1 and data not shown). The other nine compounds were present in low abundance and/or had low labeling levels and so were not readily identified.
We then used shorter labeling times to determine when these various compounds first became detectably labeled; the entire unbiased screen (as above) was re-run at the earlier times to ensure that we did not miss a compound that was labeled at early but not at later times. In experiments in which 13C-bicarbonate was added just as the animals were shifted from dark to light, significant labeling of glucose (followed as its four-carbon fragment), but not of other compounds, could be detected in the homogenate and the animal fraction within 3 min (Fig. 4A1,B1,C1, Table 1, and data not shown). When light exposure began ~2 h before addition of 13C-bicarbonate, labeling of the glucose fragment could be detected within 2 min (Fig. 4A2). As expected, no significant labeling of glucose was observed in the dark (Fig. 4A3), in the absence of 13C-bicarbonate (Fig. 4A4), or when aposymbiotic animals were incubated in the light with 13C-bicarbonate (Fig. 4D). Surprisingly, the proportions of glucose labeled were higher in the homogenates and animal fractions than in the dinoflagellate fractions, particularly at the early time points (Fig. 4A1,A2); possible explanations for this result are considered in the Discussion.
No significant labeling of other compounds in the homogenate or animal fraction was detected in the 10 min or earlier samples (Fig. 4B1,C1, Table 1), and exposure to light prior to labeling did not change this result (Fig. 4B2,C2 and data not shown). Significant labeling of a variety of compounds (but not all) was detected in the 1 h sample (Fig. 4B1, Table 1). Particularly striking in light of earlier work (see Introduction) was that no significant labeling of glycerol was detected in the 1 h or earlier samples (Fig. 4C), and that even in the 24 h sample, labeling of glycerol was confined almost entirely to the dinoflagellate fraction (Fig. 4C1).
We tested several other dinoflagellate–cnidarian combinations to ask whether the essentially exclusive early labeling and transfer of glucose were an idiosyncratic feature of clone CC7 (A. pallida) with its endogenous clade A Symbiodinium. Results with our Aiptasia line H1 (putative A. pulchella with an endogenous clade B Symbiodinium; see Materials and methods) were very similar to those with clone CC7: after 5 min exposure to light and 13C-bicarbonate, glucose was extensively labeled, and the labeled material was largely in the animal fraction (Fig. 5A). After pre-incubation with light, label could be detected in glucose in the homogenate even after 2 min of labeling (data not shown). Significant labeling of glycerol or other compounds was not detected at these times. The results with three coral species also appeared to be similar: substantial labeling of glucose, but not of glycerol or the other compounds examined, was observed after 5 min of exposure to light and 13C-bicarbonate (Fig. 5B and data not shown), and, in the one case in which we had enough material to do the test, the labeled glucose appeared to be accumulating preferentially in the animal fraction (Fig. 5C). Thus, the preferential transfer of glucose from alga to host appears to be a common feature of dinoflagellate–cnidarian symbioses, at least under the particular conditions used in our experiments.
Fig. 5.

Rapid transfer of newly synthesized glucose into host tissue in other Symbiodinium–cnidarian symbioses. (A) Aiptasia strains CC7 (gray bars) and H1 (white bars) were pre-incubated in the dark, exposed to 13C-bicarbonate in the light for 5 min, and homogenized and analyzed for glucose and other potentially labeled compounds as described in the Materials and methods and Fig. 2. (B,C) Coral samples (see Materials and methods) were exposed to 13C-bicarbonate in the light for 5 min, homogenized and analyzed as in panel A. (B) Glucose peak ratios [323/(319+323)] in total homogenates of (1) Discosoma sp., (2) Cladiella sp. and (3) Acropora millepora. (C) The Discosoma sample was fractionated by filtration (as with anemone samples) and the peak-ratio method was used to determine the distribution of newly synthesized glucose. Error bars (all panels) represent 95% confidence intervals based on the three biological replicates in each case.
Blockage of glucose labeling by an inhibitor of photosynthesis but not by an inhibitor of gluconeogenesis
As expected, treatment of anemones with the potent photosynthesis inhibitor DCMU before and during labeling with 13C-bicarbonate effectively eliminated incorporation of label into glucose or other compounds (Fig. 6A and data not shown). In contrast, incubation with 1-thioglycerol (an inhibitor of gluconeogenesis from glycerol) before and during labeling with 13C-bicarbonate had little or no effect on the appearance of 13C-labeled glucose or its transfer into the animal tissue (Fig. 6B). Although we cannot be certain that 1-thioglycerol effectively blocked gluconeogenesis in the anemones, this result is consistent with the failure to detect labeled glycerol at the early time points and suggests that the labeled glucose observed in the animal fraction cannot be derived by gluconeogenesis after a transfer of glycerol from the dinoflagellate to the host. Further support for this conclusion was provided by the failure to detect labeled glucose after incubating CC7 anemones for 15 min in ASW containing 50 mmol l−1 [U-13C]glycerol (data not shown).
Fig. 6.

Blockage of glucose labeling by an inhibitor of photosynthesis but not by an inhibitor of gluconeogenesis. (A) CC7 anemones were treated with 50 μmol l−1 DCMU (white bars) or not (gray bars) for 15 min before and during 5 min of exposure to 13C-bicarbonate in the light. The animals were then homogenized, fractionated and analyzed by GC-MS for the distribution of newly synthesized glucose as described in the Materials and methods and Fig. 2. (B) CC7 anemones were treated with 25 mmol l−1 1-thioglycerol (TG) (white bars) or not (gray bars) for 15 min before and during 15 min of exposure to 13C-bicarbonate in the light. The animals were then analyzed as in panel A. Error bars (both panels) represent 95% confidence intervals based on the three biological replicates in each case.
Release of glycerol but not glucose by freshly isolated dinoflagellates and its stimulation by host homogenate
Our failure to detect early labeling and translocation of glycerol in studies with intact organisms prompted us to ask whether we would detect glycerol release in experiments using dinoflagellates freshly isolated from host tissue (as in many previous studies; see Introduction). When such algae were incubated with 13C-bicarbonate in ASW in the light for 1 h, both the algal cells and the medium contained significant amounts of glycerol (Fig. 7A), and much of this glycerol was labeled (Fig. 7B), indicating that it had been synthesized during the labeling period. When the incubation was performed in host homogenate instead of ASW, the total amount of glycerol in each fraction was elevated (Fig. 7A), and the labeling indices were again high (Fig. 7B). As the total amount of labeled glycerol is approximately the product of the bar height in Fig. 7A and the bar height in Fig. 7B, the data taken together indicate that both the synthesis and release of glycerol were stimulated ~2.5-fold by host homogenate, similar to what was observed in the earlier studies (which could not measure either the total amount of glycerol or the fraction labeled but only the total amount of labeled glycerol).
Fig. 7.
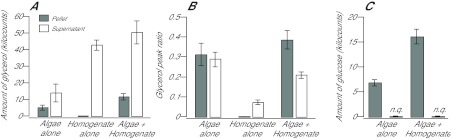
Release of glycerol but not glucose by freshly isolated dinoflagellates and its stimulation by host homogenate. Freshly isolated algae in ASW, host homogenate without algae, and freshly isolated algae mixed with host homogenate were prepared and incubated with 13C-bicarbonate in the light as described in the Materials and methods. Each sample was then centrifuged, and the algal pellets (where present) and supernatants were analyzed by GC-MS. (A,C) Total amounts of glycerol (A) and glucose (C) in pellets (gray bars) and supernatants (white bars) as determined by integration under the GC peaks (see Materials and methods). n.q., the amounts of glucose found in the supernatants were not large enough to be detected above background by the automated peak-integration software. (B) Glycerol labeling in samples from A as determined by the 221/(221+218) peak ratio. Error bars in A and C are 95% confidence intervals calculated from the standard errors of the means of the three biological replicates; error bars in B are 95% confidence intervals determined using an exact binomial test on the data from the three biological replicates (see Materials and methods).
Surprisingly, host homogenate that had been incubated without algae showed nearly as much total glycerol as when algae had been present during the incubation (Fig. 7A), although, as expected, the fraction labeled was very small (Fig. 7B). [Note that most or all of the apparent labeling of the glycerol in the homogenate-only sample in Fig. 7B is actually a quirk of the peak-ratio method as applied to molecules, such as this fragment of derivatized glycerol, that have a high ratio of trimethylsilyl groups to carbon. As shown in supplementary material Fig. S2, naturally occurring 13C, 29Si and 30Si result in enough material at m/z 221 that the 221/(218+221) ratio is ~0.08 even for a sample of unlabeled glycerol.] When we monitored glucose instead of glycerol in these experiments, we observed an apparent stimulation of glucose production (total cellular amounts of glucose were higher), but no detectable glucose release, by the algal cells (Fig. 7C, see also Fig. 3). Taken together, these data suggest that the glycerol observed in host homogenate and that released by the algae during the incubations do not result from metabolic conversion of released glucose to glycerol and thus presumably represent a substantial pool of glycerol in host cytoplasm in the intact organisms (see the presumed glycerol peak at 15.0 min in Fig. 1), glycerol released from the algae during homogenization and centrifugation (perhaps as part of a stress response), or both. In any case, the observation that inclusion of algae in the host homogenate during the incubation with 13C-bicarbonate has little effect on the total amount of glycerol, while substantially increasing its fraction labeled (Fig. 7A,B), suggests that newly synthesized (and thus labeled) and previously present glycerol exchange across the dinoflagellate surface during the course of the incubation.
We performed an experiment similar to that shown in Fig. 7A,B but using dinoflagellates that had been grown in culture rather than isolated from host tissue. Little or no stimulation of glycerol synthesis or release by host homogenate was observed (data not shown), suggesting that this behavior depends on the physiological state of dinoflagellates that had been living in hospite.
DISCUSSION
Improved methods for analyzing metabolite translocation in dinoflagellate–cnidarian symbiosis
The methods used here were adapted from those recently developed for other systems (Roessner et al., 2000; Lisec et al., 2006; Sauer, 2006) and have several advantages over those used in earlier cnidarian studies. First, labeling was performed on intact, minimally perturbed organisms and used the stable isotope 13C, thus avoiding the complications of working with radioactive 14C. Second, using a commercially available motorized homogenizer and a custom-built filtration apparatus, we were able to achieve very rapid sampling, homogenization, separation of algal and host-tissue fractions, and freezing of samples, thus making it unlikely that identification of the primary translocated compound(s) could be obscured by metabolic transformations occurring after translocation. Third, the rapidity of GC-MS analysis allowed us to go from sample to data in <8 h (as opposed to waiting weeks for an autoradiogram to develop), allowing rapid iteration of experiments when desired and opening the door to the use of ‘reef metabolomics’ as a diagnostic procedure.
Finally, the GC-MS analytical system is very powerful. Elution times can be reproduced with precision on the order of 1 s, and ≥100 polar metabolites can be resolved as discrete GC peaks. Indeed, even compounds co-eluting from the GC can be deconvolved on the basis of their differing mass spectra. These properties allow many compounds to be identified at least tentatively using freely available standard libraries of elution times and mass spectra. As a result, although we began this study with several likely candidate metabolites based on earlier work, we were also able to conduct an essentially unbiased screen that should have detected any previously unsuspected small-molecule metabolite that was synthesized and translocated from dinoflagellate to host in significant quantities. Also valuable is the ability to detect and quantitate both the unlabeled and labeled molecules of a compound of interest, which allows discrimination between a small fraction of labeling in a large pool and a large fraction of labeling in a small pool.
The analytical methods used here could be further improved, although it seems unlikely that such improvements would affect the biological conclusions of this study. For example, use of a more powerful mass spectrometer (with more scans per second over a greater m/z range) should allow more compounds to be detected and more easily identified. Greater mass precision should also allow discrimination between the m/z increase of a fragment due to incorporated 13C and the background due to the isotopes of silicon, which could allow detection of incipient labeling of the fragment even very early in the labeling period. A tandem MS method that re-fragments molecules could improve identification of compounds and allow tracing of the positions in a compound that first incorporate a label. Finally, the use of liquid rather than gas chromatography in conjunction with the MS would obviate the need to use trimethylsilyl derivatization and thus also reduce the background, although this method can also fail to detect some compounds because of ion suppression (Annesley, 2003) or incompatibilities between compounds and ionization modes.
Identification of glucose as the major translocated metabolite in the organisms examined
When clone CC7 anemones were exposed to 13C-bicarbonate in the light, we observed a rapid synthesis and transfer to the host of labeled glucose, with >50% of the free glucose within the host tissue becoming labeled within 10 min. The absence of such labeling in the dark, in aposymbiotic anemones, and in anemones treated with DCMU establishes that this glucose was produced photosynthetically and not by one of the several carbon-fixation pathways found in animals (Stern, 1948). We observed no similar rapid labeling and translocation of glycerol, succinate or other compounds. Thus, we infer that, at least in this organism, and under the growth and labeling conditions used, glucose is by far the predominant product of dinoflagellate photosynthesis that is translocated to the host. However, our examination of the nonpolar fractions was less complete and does preclude the possibility that one or more lipids are also translocated.
Although we have investigated other organisms in less detail, we did obtain very similar results with a different line of Aiptasia (putatively a different species, and containing Symbiodinium from a different major clade) and with three species of coral. These results suggest that glucose may in general be the primary translocated compound in dinoflagellate–cnidarian symbioses. Some earlier studies also detected glucose as an apparently transferred metabolite in dinoflagellate–cnidarian symbiosis. Notably, in a careful study in which intact Anemonia viridis anemones were labeled with 14C-bicarbonate, Whitehead and Douglas found glucose and several other small molecules as prominently labeled metabolites in the host tissue (Whitehead and Douglas, 2003). Glucose also appears to be the major compound released by Symbiodinium to the host in the undisturbed dinoflagellate–giant-clam symbiosis (Ishikura et al., 1999). However, further investigations of this issue will clearly be necessary. For example, it will be of great interest to determine whether methods similar to those used here detect other translocated compounds in different Symbiodinium–cnidarian combinations or when the organisms are subjected to different conditions such as different temperatures, light regimens or bicarbonate concentrations.
We were initially surprised to observe that the percentage of labeled glucose in the animal fraction actually increased more quickly than did that in the algal fraction (Fig. 4A1,A2). This difference may reflect in part the presence at the start of labeling of a small pool of free glucose in the cytoplasm of the previously starved and dark-incubated host (Fig. 2A,B), so that the newly imported (and labeled) glucose makes up a higher percentage of the total, and of a somewhat larger pool in the algae (as evident in the size of the GC peak; data not shown). However, on this model, it is then also necessary to hypothesize that the newly synthesized glucose is in some way segregated from the larger pool of unlabeled glucose in the dinoflagellate in order to explain the high percentage labeling of the glucose that has been translocated to the host. Thus, it appears that there must be highly efficient mechanisms for translocation of newly synthesized glucose across the several membranes separating the chloroplast interior from the host cytoplasm, a conclusion that is not surprising given previous evidence that >90% of new photosynthetic product is transferred from dinoflagellate to host (Falkowski et al., 1984). The percentages labeled (of glucose and other compounds) do eventually become similar in the algal and host fractions (e.g. after a week of labeling in our experiments; data not shown), indicating that whatever mechanism is responsible for the rapid export of glucose does also allow some newly produced photosynthate to remain within the algal cell.
Release of glycerol, but not glucose, by dinoflagellates isolated from host tissue
Although numerous previous studies have suggested that glycerol plays a major role in the translocation of dinoflagellate photosynthate to the host (Muscatine, 1967; Muscatine and Cernichiari, 1969; Trench, 1971; Venn et al., 2008; Yellowlees et al., 2008; Davy et al., 2012), our studies of the intact system do not support this model. Instead, we found that the percentage labeling of glycerol lagged far behind the percentage labeling of glucose in the animal tissue, seemingly precluding the possibility that transfer of newly synthesized glycerol from alga to host is followed by metabolic conversion of glycerol to glucose within the host.
In contrast, when we performed experiments designed to mimic earlier studies using algae freshly isolated from host tissue and incubated in the absence or presence of host homogenate, we obtained results consistent with the earlier studies. In particular, we observed: (1) substantial pools of total glycerol in both the host-cytoplasm fraction and the algal cells; (2) a rapid synthesis of labeled glycerol upon exposure to13C-bicarbonate in the light; (3) a rapid release of newly synthesized glycerol to the external environment; (4) a stimulation of both glycerol synthesis and release by host homogenate; and (5) an apparently abundant movement of glycerol also from homogenate back into the algal cells [as also observed previously (Grant et al., 1997)]. In contrast, although glucose synthesis by isolated algae was also stimulated by host homogenate, no glucose release was detected.
Although we have not definitively ruled out the possibility that the host cytoplasm contains a large pool of free glycerol prior to perturbation of the system, this seems unlikely given the rapidity with which the isolated algae (as opposed to algae in hospite) synthesize new glycerol and release it to the external environment (Fig. 7B). Thus, we think it more likely that most of the glycerol observed in our own and earlier experiments is produced by the algae as part of a stress response, a phenomenon also seen in other organisms (Westfall et al., 2004). This conclusion appears consistent both with other evidence that isolated algae are indeed under stress (Goiran et al., 1997) and with the observations in giant clams, where glycerol was detected in the hemolymph only when the system had been disturbed (Rees et al., 1993; Ishikura et al., 1999). It also appears consistent with our failure to observe glycerol synthesis or release when cultured dinoflagellates were tested; presumably these algae were already adapted to the conditions of culture, so that no stress response was elicited in our experiments.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Erik Lehnert, Pavel Aronov and Allis Chien for their feedback on this project; Jan DeNofrio, Liz Hambleton, Veena Singla and Sabrina Fullhart for providing anemones and cultured Symbiodinium and for help in identifying the corals used; and Annika Guse, Jan DeNofrio and the two anonymous reviewers for helpful comments on the manuscript. We also thank Wolf Frommer and the Carnegie Institution for Science, Department of Plant Biology, for generous access to the GC-MS and for high-purity compressed gases. Finally, we thank the late Chris Farrar (Varian) for generous training and help with equipment repair.
LIST OF ABBREVIATIONS
- ASW
artificial seawater
- DCMU
3-(3,4-dichlorophenyl)-1,1-dimethylurea
- GC
gas chromatography
- MOX
methoxyamine
- MS
mass spectrometry
- MSTFA
N-methyl-N-trimethylsilyl tri-fluoroacetamide
FOOTNOTES
Supplementary material available online at http://jeb.biologists.org/cgi/content/full/215/19/3467/DC1
FUNDING
This study was supported by the National Science Foundation [EAGER grant IOS-1138275], the Gordon and Betty Moore Foundation [grant 2629] and the National Institutes of Health [training grant 5 T32 HG000044]. Deposited in PMC for release after 12 months.
REFERENCES
- Annesley T. M. (2003). Ion suppression in mass spectrometry. Clin. Chem. 49, 1041-1044 [DOI] [PubMed] [Google Scholar]
- Arning E. T., Birgel D., Schulz-Vogt H. N., Holmkvist L., JØrgensen B. B., Larson A., Peckmann J. (2008). Lipid biomarker patterns of phosphogenic sediments from upwelling regions. Geomicrobiol. J. 25, 69-82 [Google Scholar]
- Blumenberg M., Seifert R., Petersen S., Michaelis W. (2007). Biosignatures present in a hydrothermal massive sulfide from the Mid-Atlantic Ridge. Geobiology 5, 435-450 [Google Scholar]
- Bruno J. F., Selig E. R. (2007). Regional decline of coral cover in the Indo-Pacific: timing, extent, and subregional comparisons. PLoS ONE 2, e711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen B., Thykaer J., Nielsen J. (2000). Metabolic characterization of high- and low-yielding strains of Penicillium chrysogenum. Appl. Microbiol. Biotechnol. 54, 212-217 [DOI] [PubMed] [Google Scholar]
- Davy S. K., Allemand D., Weis V. M. (2012). Cell biology of cnidarian–dinoflagellate symbiosis. Microbiol. Mol. Biol. Rev. 76, 229-261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas F., Aussel C., Pernet P., Martin C., Giboudeau J. (1994). Gas chromatography applied to the lactulose-mannitol intestinal permeability test. J. Chromatogr. B Biomed. Appl. 654, 276-281 [DOI] [PubMed] [Google Scholar]
- Falkowski P. G., Dubinsky Z., Muscatine L., Porter J. W. (1984). Light and the bioenergetics of a symbiotic coral. Bioscience 34, 705-709 [Google Scholar]
- Goiran C., Allemand D., Galgani I. (1997). Transient Na+ stress in symbiotic dinoflagellates after isolation from coral-host cells and subsequent immersion in seawater. Mar. Biol. 129, 581-589 [Google Scholar]
- Grant A. J., Rémond M., People J., Hinde R. (1997). Effects of host-tissue homogenate of the scleractinian coral Plesiastrea versipora on glycerol metabolism in isolated symbiotic dinoflagellates. Mar. Biol. 128, 665-670 [Google Scholar]
- Ishikura M., Adachi K., Maruyama T. (1999). Zooxanthellae release glucose in the tissue of a giant clam, Tridacna crocea. Mar. Biol. 133, 665-673 [Google Scholar]
- Ishikura M., Hagiwara K., Takishita K., Haga M., Iwai K., Maruyama T. (2004). Isolation of new Symbiodinium strains from tridacnid giant clam (Tridacna crocea) and sea slug (Pteraeolidia ianthina) using culture medium containing giant clam tissue homogenate. Mar. Biotechnol. (NY) 6, 378-385 [DOI] [PubMed] [Google Scholar]
- Jiao Z., Baba T., Mori H., Shimizu K. (2003). Analysis of metabolic and physiological responses to gnd knockout in Escherichia coli by using C-13 tracer experiment and enzyme activity measurement. FEMS Microbiol. Lett. 220, 295-301 [DOI] [PubMed] [Google Scholar]
- Lisec J., Schauer N., Kopka J., Willmitzer L., Fernie A. R. (2006). Gas chromatography mass spectrometry-based metabolite profiling in plants. Nat. Protoc. 1, 387-396 [DOI] [PubMed] [Google Scholar]
- Muscatine L. (1967). Glycerol excretion by symbiotic algae from corals and Tridacna and its control by the host. Science 156, 516-519 [DOI] [PubMed] [Google Scholar]
- Muscatine L., Cernichiari E. (1969). Assimilation of photosynthetic products of zooxanthellae by a reef coral. Biol. Bull. 137, 506-523 [DOI] [PubMed] [Google Scholar]
- Muscatine L., Hand C. (1958). Direct evidence for the transfer of materials from symbiotic algae to the tissues of a coelenterate. Proc. Natl. Acad. Sci. USA 44, 1259-1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2009). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; [Google Scholar]
- Rahav O., Dubinsky Y., Achituv Y., Falkowski P. G. (1989). Ammonium metabolism in the zooxanthellate coral, Stylophora pistillata. Proc. R. Soc. Lond. B 236, 325-337 [Google Scholar]
- Rees T. A. V., Fitt W. K., Baillie B., Yellowlees D. (1993). A method for temporal measurement of hemolymph composition in the giant clam symbiosis and its application to glucose and glycerol levels during a diel cycle. Limnol. Oceanogr. 38, 213-217 [Google Scholar]
- Roessner U., Wagner C., Kopka J., Trethewey R. N., Willmitzer L. (2000). Technical advance: simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J. 23, 131-142 [DOI] [PubMed] [Google Scholar]
- Sauer U. (2006). Metabolic networks in motion: 13C-based flux analysis. Mol. Syst. Biol. 2, 62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer W. K., Dhariwal G., Mckelvey H. A., McCabe E. R. B. (1986). 1-Thioglycerol: inhibitor of glycerol kinase activity in vitro and in situ. Life Sci. 39, 1417-1424 [DOI] [PubMed] [Google Scholar]
- Stern J. R. (1948). Carbon dioxide fixation in animal tissues. Biochem. J. 43, 616-624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunagawa S., Wilson E. C., Thaler M., Smith M. L., Caruso C., Pringle J. R., Weis V. M., Medina M., Schwarz J. A. (2009). Generation and analysis of transcriptomic resources for a model system on the rise: the sea anemone Aiptasia pallida and its dinoflagellate endosymbiont. BMC Genomics 10, 258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trench R. K. (1971). The physiology and biochemistry of zooxanthellae symbiotic with marine coelenterates. II. Liberation of fixed 14C by zooxanthellae in vitro. Proc. R. Soc. Lond. B 177, 237-250 [Google Scholar]
- Vandermeulen J. H., Davis N. D., Muscatine L. (1972). The effect of inhibitors of photosynthesis on zooxanthellae in corals and other marine invertebrates. Mar. Biol. 16, 185-191 [Google Scholar]
- Venn A. A., Loram J. E., Douglas A. E. (2008). Photosynthetic symbioses in animals. J. Exp. Bot. 59, 1069-1080 [DOI] [PubMed] [Google Scholar]
- Westfall P. J., Ballon D. R., Thorner J. (2004). When the stress of your environment makes you go HOG wild. Science 306, 1511-1512 [DOI] [PubMed] [Google Scholar]
- Whitehead L. F., Douglas A. E. (2003). Metabolite comparisons and the identity of nutrients translocated from symbiotic algae to an animal host. J. Exp. Biol. 206, 3149-3157 [DOI] [PubMed] [Google Scholar]
- Yellowlees D., Rees T. A. V., Leggat W. (2008). Metabolic interactions between algal symbionts and invertebrate hosts. Plant Cell Environ. 31, 679-694 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.