

Abstract
Interleukin 17 (IL-17) promotes expression of chemokines and cytokines via induction of gene transcription and post-transcriptional stabilization of mRNA. We show that IL-17 enhanced the stability of CXCL1 and other mRNAs through a pathway that involves Act1, TRAF2 or TRAF5 and the splicing factor SF2/ASF. TRAF2/TRAF5 were necessary for IL-17 to signal CXCL1 mRNA stabilization. Furthermore, IL-17 promoted formation of complexes between TRAF5/TRAF2, Act1 and SF2/ASF. Overexpression of SF2/ASF shortened while depletion of SF2/ASF prolonged CXCL1 mRNA half-life. SF2/ASF bound chemokine mRNA in unstimulated cells while the SF2/ASF-mRNA interaction was markedly diminished following stimulation with IL-17. These findings define an IL-17-induced signaling pathway that links to the stabilization of selected mRNAs through Act1, TRAF2/5 and the RNA binding protein SF2/ASF.
Introduction
Interleukin 17A (IL-17A) is a proinflammatory cytokine produced primarily by CD4+ T helper cells (TH17 cells) that are distinct from the classical T helper type 1 (TH1) and T helper type 2 (TH2) cells1,2. While IL-17 is required for host defense against bacterial and fungal infection, it is also known for its role in the pathogenesis of certain human and animal autoimmune inflammatory diseases including rheumatoid arthritis, inflammatory bowel disease and experimental autoimmune encephalomyelitis1-3. IL-17 coordinates tissue inflammation through the elevated expression proinflammatory cytokines and chemokines, which collectively determine the magnitude and character of the response.
Amongst the earliest responses to injury is the expression of chemokine genes in cell populations resident within the injured tissue whose products specifically regulate the trafficking of neutrophils4,5. These endogenously derived peptides contribute to many features of chemotactic recruitment and hence their regulation is of paramount importance6. Neutrophil-specific chemokines are members of the CXC family, contain the ELR motif and act through CXCR1 and CXCR2 (CXCL1 – 8 in humans and CXCL1, CXCL2 and CXCL5 in the mouse). IL-17 is well documented to promote strong expression of CXCL1 and related chemokines in fibroblasts and epithelial cell populations7-9 and is known to signal, at least in part through activation of NF-κB via the adaptor proteins Act1 and TRAF6 (refs. 10-13). Though increases in transcription driven by NF-κB are a requisite feature of the induced expression of CXCL1, the regulation of the mRNA half-life is also a critical determinant of the magnitude of expression14,15. mRNAs that are highly unstable prevent the inappropriate expression of the encoded protein in resting cells but often require stimulus-induced prolongation of half-life during inflammatory stimulation. Interestingly, the mechanisms through which IL-17 promotes enhanced mRNA stability in non-myeloid cell populations appear to be distinct from those operating downstream of Toll Like Receptors (TLRs) in myeloid cells16,17.
The major paradigm for instability in cytokine, chemokine and growth factor mRNAs involves adenine uridine rich regions (AREs) within the 3′ UTR of the message that are recognized by proteins functioning to orchestrate the sequential deadenylation, decapping and ultimately exonucleolytic degradation of the RNA18,19. Among the best studied of these sequence motifs is the core pentamer AUUUA often flanked by additional A or U residues. A number of proteins exhibiting recognition specificity for this motif have been identified and shown to have capacity for either promoting or diminishing the rate of mRNA degradation. These include the elav family member HuR, AUF1 (hnRNP D), Tristetraprolin (TTP), and KH domain containing splicing regulatory factor KSRP19. Though the role of such RNA binding proteins in regulating mRNA half-life is well-recognized, the diversity of factors and the signal transduction pathways downstream of extracellular stimuli that modulate their activities remains poorly defined.
In the present report, we identify an mRNA stability signaling pathway that couples to the IL-17R via the downstream signaling intermediate Act1. Upon IL-17 stimulation, Act1 formed a complex with TRAF5 (or TRAF2) and both TRAFs were required for IL-17-mediated stabilization of CXCL1 mRNA. IL-17 also induced association of the RNA splicing regulatory factor known as SF2/ASF with the complex of Act1 and TRAF5/TRAF2. SF2/ASF promoted chemokine mRNA instability in sequence specific fashion and its depletion resulted in prolonged mRNA half-life. SF2/ASF bound chemokine mRNA in unstimulated cells but the SF2/ASF-mRNA interaction was markedly diminished following stimulation with IL-17. These findings define a separate IL-17-initiated signaling pathway linked to the stabilization of a subset of mRNAs in non-myeloid cell populations.
Results
TRAF2 and TRAF5 promote enhanced stability of CXCL1 mRNA
TRAF6 has been shown to be essential for the ability of IL-17 to activate target gene transcription via NF-κB11,12, yet it is not required for the ability to promote prolongation of CXCL1 mRNA half-life16. To test the role of other members of the TRAF family, we employed an experimental system that utilizes reporter plasmids in which transgene transcription is controlled by a tetracycline responsive element through the action of the tet transactivator protein (tTA). HeLa cells stably expressing the tTA protein were transfected with a reporter construct (KCΔ4) containing the CXCL1 coding region and a truncated 3′UTR conferring instability that can be attenuated following stimulation with IL-17 (ref. 17). When this construct was transfected in HeLa tet-off cells, the transcript was readily detected but upon transcriptional blockade by addition of doxycyline (dox), the message decayed rapidly with a half-life of 25-50 min. IL-17 provided at the same time as dox extended the half-life to 2-3 h (Fig. 1a). In cells co-transfected with the reporter and expression plasmids encoding TRAF2 or TRAF5, the message was also stabilized, suggesting that TRAF2 and TRAF5 each have the ability to mediate CXCL1 mRNA stability (Fig. 1b). As reported previously, similar results were not seen in cells transfected to express TRAF6 (ref. 16). The ability of TRAF5 to stabilize mRNA was relatively selective for the CXCL1 3′UTR sequence motif since the half-life of another unstable reporter containing the 3′UTR from GM-CSF mRNA was not changed in cells over-expressing TRAF5 (Fig. 1c). These findings suggest that TRAF2 or TRAF5 serve to couple IL-17R signals with the control of chemokine mRNA half-life.
Figure 1. TRAF2 or TRAF5 expression selectively prolongs CXCL1 mRNA half life.

a. HeLa Tet-off cells were transfected with the reporter plasmid KCΔ4 and treated with dox alone or in the presence of IL-17 (25 ng/ml) for the indicated times prior to quantification of residual CXCL1 and GAPDH mRNA by RNA hybridization. The autoradiograph was scanned and quantified using ImageJ software and the abundance of CXCL1 mRNA was normalized to GAPDH mRNA in each sample. The results of 3 independent experiments were used to determine the half-lives under resting and stimulated conditions and are presented as the mean +/- 1 S.D. b. HeLa tet-off cells were co-transfected with KCΔ4 and vector alone (pcDNA3) or plasmids encoding TRAF2, TRAF5, or TRAF6. Cultures were treated with dox (1 μg/ml) for the indicated times prior to quantification of individual TRAFs by immunoblot and CXCL1 mRNA as described in a. Half-lives were calculated from 3 independent experiments. c. HeLa tet-off cells were co-transfected with reporter plasmids KCΔ4 or KC-GM-CSF along with pcDNA3 or the TRAF5 expression plasmid. Dox was added and the remaining CXCL1 and GAPDH (not shown) mRNA was quantified and half-lives determined from 3 independent experiments as described in a.
IL-17-induced CXCL1 mRNA stability requires TRAF2/TRAF5
To further test the hypothesis that TRAF2 or TRAF5 may be important components of IL-17 signaling leading to selective mRNA stabilization, mouse embryo fibroblasts (MEFs) deficient for both TRAF2 and TRAF5 were evaluated for the ability of IL-17 to promote enhanced stability of CXCL1 mRNA. In many non-myeloid cells, including MEFs, tumor necrosis factor (TNF) is a potent transcriptional stimulus for the CXCL1 gene but is unable to promote message stability9,20. This enables a comparison of mRNA half-life in cells with or without the addition of a stabilizing stimulus such as IL-17. In wild-type MEFs, TNF treatment for 2 h resulted in endogenous CXCL1 mRNA transcripts that decay rapidly following addition of the transcriptional inhibitor actinomycin D (ActD) (Fig. 2a). When the cells are stimulated with TNF and IL-17 in combination (IL-17 alone is a poor transcriptional stimulus for CXCL1), the half-life of the induced CXCL1 mRNA was markedly prolonged (Fig. 2a). IL-17 was unable to stabilize CXCL1 mRNA in MEFs from mice that were deficient in both TRAF2 and TRAF5. Sensitivity for IL-17-driven mRNA stabilization was restored by reconstitution of the double-knockout cells with both TRAF2 and TRAF5 (Fig. 2b). Other IL-17 sensitive mRNAs including those encoding CSF3 and IκBζ also exhibit comparable behavior in TRAF2/TRAF5 double knockout cells (Supplementary Fig. 1a). When MEFs from mice singly deficient in either TRAF2 or TRAF5 were tested in this experiment, there was no change in IL-17-induced mRNA stabilization (not shown).
Figure 2. TRAF2 and TRAF5 are required for IL-17-induced CXCL1 mRNA stabilization.

a. Wild type and TRAF2-/-,TRAF5-/- MEFs were treated with TNF (10 ng/ml) alone or in the presence of IL-17 for 2 h prior to the addition of ActD (5 μg/ml). Remaining CXCL1 and GAPDH (not shown) mRNA was determined by RNA hybridization and the half-lives calculated as described in the legend to Figure 1a from 4 independent experiments. b. TRAF2/TRAF5 deficient MEFs and TRAF-reconstituted MEFs were treated with TNF+IL-17 (10 ng/ml each) for 2 hrs prior to addition of ActD (5 μg/ml) and remaining CXCL1 and GAPDH (not shown) mRNA was quantified and half-lives calculated as in figure 1 from 3 independent experiments. c. HeLa tet-off cells were co-transfected with control siRNA or siRNA specific for TRAF2 or TRAF5 along with the KCΔ4 reporter. Separate cultures were used to determine residual TRAF expression by immunoblot. The cells were treated with dox alone or along with IL-17 and CXCL1 and GAPDH (not shown) mRNA was quantified and half-lives calculated as described in a. Similar results were obtained in 2 independent experiments.
We also tested the requirement for TRAF2 and/or TRAF5 in cytokine-induced mRNA stabilization using the HeLa tet-off system by depleting the expression of either or both factors using specific siRNAs. Transfection of siRNAs targeting TRAF2 or TRAF5 resulted in reduction of the proteins as compared with cultures treated with non-specific control siRNA (Fig. 2c). KCΔ4 mRNA half-life was similar in both untreated and control siRNA-treated cells and IL-17 treatment resulted in message stabilization as seen in prior experiments. As in single knockout MEFs, the reporter mRNA half-life was unchanged in cultures treated with siRNAs targeting either TRAF2 or TRAF5 and sensitivity to IL-17 was retained. However, IL-17-induced stabilization of KCΔ4 mRNA was markedly reduced in cells treated with siRNAs targeting both TRAF2 and TRAF5 (Fig. 2c). These results provide strong evidence that either TRAF2 or TRAF5 or both are required for IL-17 to prolong chemokine mRNA half-life.
Dominant interfering TRAF5 inhibited IL-17-induced mRNA stability
The observation that deficiency in both TRAF2 and TRAF5 was necessary to abolish the capacity of IL-17 to stabilize CXCL1 mRNA indicates the functional redundancy of TRAF2 and TRAF5 in this pathway. We then determined whether mutants of TRAF2, TRAF5 or TRAF6 that can block cytokine induced NF-κB activation via dominant-negative (dn) activity could also inhibit IL-17 signaling to mRNA stabilization. In HeLa tet-off cells co-transfected with KCΔ4 reporter transgene and the empty expression vector pcDNA3, IL-17 promotes enhanced stability of the message. Overexpression of dn versions of TRAF2 or TRAF6 have little impact on the cytokine induced prolongation of half-life despite effectively blocking IL-1-stimulated NF-κB activity as measured by a κB-dependent luciferase reporter (Supplementary Fig. 2 and data not shown). Overexpression of the dnTRAF5 construct, however, was able to block mRNA stabilization induced by IL-17. These findings suggest that TRAF5 may have a selective role in this signaling pathway.
IL-17 induces Act1 and TRAF5 interaction
TRAF2 and/or TRAF5 are best known to participate in signaling by members of the TNFR superfamily21. The requirement for TRAF2 or TRAF5 in IL-17 induced mRNA stability suggested that one or both might interact with Act1, the adaptor protein that is essential for most IL-17 responses9,11. HeLa tet-off cells were transfected with hemagglutinin (HA)-tagged versions of Act1 and FLAG-tagged versions of TRAF2, TRAF5, or TRAF6 and detergent extracts were used for immunoprecipitation using either non-specific mouse IgG or a FLAG-specific antibody. The immunoprecipitate (IP) was subsequently separated by SDS-PAGE and analyzed by immunoblot using HA antibody (Fig. 3a). While no signal was detected in the IP using nonspecific IgG, HA-Act1 was readily detectable in the anti-FLAG IPs in cells expressing any of the three FLAG-TRAFs. Because the evidence favors a predominant role for TRAF5 the remaining experiments are focused upon this TRAF family member. In cells expressing Act1 and TRAF5 as transgenes, we were unable to see IL-17-dependency for the Act1-TRAF5 interaction (data not shown). Therefore, we assessed the stimulus dependency of the interaction between endogenous Act1 and TRAF5 in non-transfected HeLa cells either untreated or treated with IL-17 by immunoprecipitation using TRAF5 antibody (Fig. 3b). IPs were separated by denaturing SDS-PAGE and subjected to immunoblot analysis using Act1 antibody. While Act1 was not detected in the IPs using non-specific IgG or in untreated cells, it is readily observed in anti-TRAF5 IPs from cells treated with IL-17 for 5, 15 or 30 min. We observed multiple anti-TRAF5 interactive bands, one of which reproducibly showed dependence on IL-17 stimulation. While TRAF5 that migrated at the expected molecular weight was present in all lanes, the IL-17-induced form migrated at a substantially higher position (approximately 85 kDa). Finally, we examined anti-Act1 IPs from MEFs treated with IL-17 by SDS-PAGE and immunoblotted TRAF5. Several Act1-interacting, anti-TRAF5-reactive bands were dependent on IL-17 stimulation (Fig. 3c). These bands migrated more slowly than unmodified TRAF5 but at positions comparable to those seen in the IPs using anti-TRAF5, suggesting that interaction of TRAF5 with Act1 may lead to some form of molecular modification. Act1 has been shown to have ubiquitin E3 ligase activity22, raising the possibility that the higher molecular forms observed might represent an ubiquitinated form of TRAF5.
Figure 3. IL-17 promotes interaction of Act1 and TRAF5.
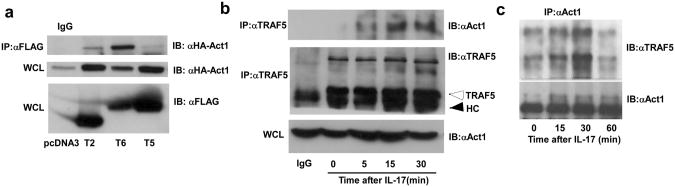
a. HeLa tet-off cells were co-transfected with FLAG-TRAF2, FLAG-TRAF5, or FLAG-TRAF6 along with HA-Act1 used for immunoprecipitation with FLAG antibody. The IPs were analyzed by SDS-PAGE and immunoblot with anti-HA. 40 μg protein from whole cell lysates (WCL) were analyzed by immunoblot using anti-FLAG to illustrate TRAF expression. Results are representative of 3 independent experiments. b. HeLa tet-off cells were treated with IL-17 for the indicated times and used for immunoprecipitation with TRAF5 antibody. The IPs were analyzed by SDS-PAGE and immunoblot with anti-Act1 or anti-TRAF5. The control IgG lane used WCL from cells treated with IL-17 for 5 minutes. 40 μg of each WCL were subjected to immunoblot for Act1. The unfilled arrow indicates the position of unmodified TRAF5 and the solid arrow indicates the position of antibody heavy chain (HC). Results are representative of 2 independent experiments. c. Wild type MEFs were treated with IL-17 for the indicated times and used for immunoprecipitation with anti-Act1. The IPs were analyzed by SDS-PAGE and immunoblot with anti-TRAF5 or anti-Act1. The results are representative of 3 independent experiments.
IL-17 induces interaction of Act1, TRAF5 and SF2/ASF
To extend our understanding of signals downstream of IL-17R, Act1, and TRAF5, we searched for TRAF5-interacting molecules that could provide a link between TRAF5 and the mRNA decay pathways. Using detergent extracts from TRAF2/TRAF5 double knockout MEFs reconstituted with FLAG-TRAF2 and FLAG-TRAF5, we performed SDS-PAGE on anti-FLAG IPs followed by silver staining and identified a band migrating at approximately 35 kDa that appeared to associate with the TRAF molecules only in cells treated with IL-17 (Fig. 4a). This band was identified by mass spectrometry as the arginine serine rich splicing factor SRSF1 also known as SF2/ASF. The ability of TRAF5 to interact with SF2/ASF was confirmed using anti-TRAF5 immunoprecipitation of extracts from HeLa cells either untreated or treated with IL-17 for 5, 15, or 30 min. Blots of SDS-PAGE separations developed with antibody to SF2/ASF revealed specific interaction that was dependent on stimulation with IL-17 and could be observed within 5 min (Fig. 4b). The specificity of the interaction of SF2/ASF was demonstrated using transiently expressed FLAG-tagged versions of TRAF2, TRAF4, TRAF5 and TRAF6. SF2/ASF was observed only in IPs for either TRAF2 or TRAF5 and was absent from anti-FLAG IPs using cells expressing tagged TRAF4 or TRAF6 (Fig. 4c). In contrast, TAK1, a kinase involved in NF-κB activation that is known to interact strongly with TRAF6 (ref. 23) did not interact with TRAF5 (Fig. 4d). Thus different TRAFs appear to function as segregation points for coupling with distinct downstream endpoints. To determine if the TRAF5-SF2/ASF complex might also contain Act1, HeLa cells were stimulated with IL-17 for up to 60 min and the extract subjected to IP with anti-Act1. The IPs from this experiment contain both TRAF5 and SF2/ASF in addition to Act1, suggesting that all three proteins are assembled in this initial signaling complex (Fig. 4e).
Figure 4. IL-17 induces a complex with TRAF2 or TRAF5 and SF2/ASF.

a. MEFs from TRAF2/TRAF5-deficient mice that were reconstituted with FLAG-TRAF2 and FLAG-TRAF5 were untreated or stimulated with IL-17 (10 ng/ml) for 1 hr. Cell lysates were immunoprecipitated with anti-FLAG and the IPs were subjected to SDS-PAGE followed by silver staining. The band identified by the filled arrow was subjected to in-gel trypsin digestion and LC-MS and the data analyzed using MASCOT software to search the NCBI non-redundant data base. b. HeLa tet-off cells were untreated or treated with IL-17 (25 ng/ml) for the indicated times prior to immunoprecipitation with anti-TRAF5. The IPs or 40 μg of WCL protein were analyzed by SDS-PAGE and immunoblot using anti-SF2/ASF. Results are representative of 2 independent experiments. c. HeLa tet-off cells were transfected to express FLAG-tagged versions of TRAF2, TRAF4, TRAF5 or TRAF6 and used for immunoprecipitation with anti-FLAG. The IPs or 40 μg WCL protein were analyzed by SDS-PAGE and immunoblot using anti-SF2/ASF or anti-FLAG to detect TRAFs. d. HeLa tet-off cells were co-transfected to express FLAG-TRAF5 or FLAG-TRAF6 along with HA-TAK1 and used for immunoprecipitation with anti-FLAG. IPs or 40 μg WCL protein were analyzed by SDS-PAGE and immunoblot using anti-HA for TAK1 and anti-FLAG for TRAFs. Results are representative of 2 independent experiments. e. Extracts from HeLa tet-off cells treated with IL-17 (50 ng/ml) for the indicated times immunoprecipitated with anti-Act1. The IPs and wcl extracts were analyzed by SDS-PAGE and immunoblot using antibodies to SF2/ASF, TRAF5, and Act1. Results are representative of 3 independent experiments.
SF2 modulates specific mRNA half life
The finding that SF2/ASF interaction with TRAF5 is IL-17-dependent and the coincidence of this event with stimulus-induced chemokine mRNA stabilization suggests that SF2/ASF plays some role in controlling the half-life of the mRNA and this is consistent with several prior studies24,25. To test the function of SF2/ASF in regulating CXCL1 mRNA half-life we first examined the consequences of modulating the abundance of SF2/ASF by overexpression or by siRNA-mediated knockdown. When an expression plasmid encoding SF2/ASF was co-transfected along with the KCΔ4 reporter into HeLa tet-off cells, the steady-state abundance of CXCL1 mRNA was diminished and the half-life was reduced by a factor of 2 (Fig. 5a). This effect was sequence specific since there was no change in the half-life of a second unstable reporter containing the 3′UTR from the GM-CSF mRNA. This reporter was not sensitive to IL-17-mediated stabilization in HeLa cells (data not shown). IL-17 is known to enhance expression of CSF3 and IκBζ mRNAs1,17. The half-lives of reporter mRNAs containing the 3′UTRs from the IκBζ and CSF3 mRNAs were also prolonged in response to IL-17 in HeLa tet-off cells and reduced in cells co-transfected with the SF2/ASF expression plasmid (Supplementary Fig. 1b,c). SF2/ASF was depleted in HeLa tet-off cells by transfection with either control or SF2/ASF-specific siRNA (Fig. 5b). While the KCΔ4 reporter mRNA half-life was prolonged in cells depleted of SF2/ASF, the decay of a second reporter containing an insensitive region from the CXCL1 mRNA (KC-Clu-P2)17 was unaltered. Finally, we utilized MEFs in which the expression of SF2/ASF can be depleted by treatment with Dox26. When these cells were treated for 48 h with Dox, SF2/ASF expression was eliminated and the half-life of TNF-induced endogenous CXCL1 mRNA was prolonged (Fig. 5c). Collectively, these results support the hypothesis that SF2/ASF promotes the decay of specific mRNAs depending upon sequences located in the 3′UTR.
Figure 5. SF2/ASF promotes enhanced decay of CXCL1 mRNA.

a. Hela tet-off cells were co-transfected with KCΔ4 or KC-GM-CSF and either vector alone (pcDNA3) or full length SF2/ASF. Dox (1 μg/ml) was added and residual CXCL1 and GAPDH (not shown) mRNA was quantified by RNA hybridization. Half-lives were calculated as described in the legend to figure 1 from 3 independent experiments. b. Hela tet-off cells were co-transfected with reporter plasmids KCΔ4 or KC-Clu-P2 along with control or SF2/ASF-specific siRNA. After 48 hrs, cells were treated with Dox (1 μg/ml) and CXCL1 and GAPDH (not shown) mRNA was quantified by RNA hybridization and half lives were calculated as in the legend to figure 1 from 2 independent experiments. 40 μg of WCL was used to measure SF2/ASF expression by immunoblot analysis. c. SF2/ASF knockout MEFs reconstituted with HA-tagged SF2/ASF under tet-off control were untreated or treated with Dox (1 μg/ml) for 2 days prior to stimulation with TNF (10 ng/ml) for 2 hrs followed by ActD (5 μg/ml) for the indicated times. CXCL1 and GAPDH (not shown) mRNA was measured by RNA hybridization and half-lives were calculated as in the legend to figure 1 from 2 independent experiments. HA-SF2/ASF was quantified in untreated and Dox-treated cells by immunblot using 40 μg WCL protein.
IL-17 modulates SF2/ASF binding to target mRNAs
SF2/ASF is known to be largely localized within the nucleus and this subcellular distribution pattern is controlled in part via phosphorylation of multiple serine residues in the arginine-serine rich carboxyl-terminal domain of the protein27,28. We determined if the N-terminal region missing the RS rich domain, which will localize more predominantly in the cytoplasm, could also mediate the effects on CXCL1 mRNA decay. While transient expression of a C-terminal deletion of SF2/ASF could modulate reporter (KCΔ4) half-life as well as the full-length version, an N-terminal deletion construct had no such activity (Fig. 6a). We reasoned that the critical RNA recognition motif in the N-terminal domain of SF2/ASF would be required for the RNA decay promoting activity. The modulation of KCΔ4-derived mRNA half-life was lost in HeLa tet-off cells transfected with a mutant version of the C-terminal deletion construct in which two phenylalanine residues critical for RNA binding are mutated (FF56/58DD)29 (Fig. 6b).
Figure 6. SF2/ASF binds CXCL1 mRNA.

a. HeLa tet-off cells were co-transfected with KCΔ4 and expression plasmids encoding empty vector (pcDNA3), C-terminal deletion (SF2/ASF-N), or N-terminal deletion (SF2/ASF-C) versions of SF2/ASF. Dox (1 μg/ml) was added and remaining CXCL1 and GAPDH (not shown) mRNA was quantified and half-lives were calculated as described in the legend to figure 1 using results from 3 independent experiments. b. HeLa tet-off cells were co-transfected with KCΔ4 reporter plasmid and vector alone (pcDNA3) or pcDNA3 encoding either the wild-type (SF2/ASF-N) or mutant (SF2/ASF-N-FF-DD) C terminal deletion variant of SF2/ASF. Dox (1 μg/ml) was added and remaining CXCL1 or GAPDH (not shown) mRNA was quantified and half-lives were calculated as in the legend to figure 1 using results from 2 independent experiments. c. HeLa tet-off cells were transfected with KCΔ4 or KC-Clu-P2 plasmids with or without either the wild-type or RNA binding mutant (FF-DD) version of SF2/ASF C-terminal deletion as indicated. In some experiments the cells were either untreated or treated with IL-17 for 30 min. Extracts were prepared and used for RNA immunoprecipitation with anti-SF2/ASF or anti-HA (for cells expressing the transgenic versions of SF2/ASF). RNA was prepared from IPs and used to quantify CXCL1 and GAPDH mRNA by real time RT-PCR. The data are presented as mRNA enrichment comparing ΔΔCt value for non-specific IgG with that for anti-SF2/ASF. Data are presented as the mean fold enrichment +/- ½ the range of two independent experiments.
As a further test of the functional linkage of SF2/ASF with chemokine mRNA half-life we determined if SF2/ASF can bind with CXCL1 mRNA and if so, whether the interaction exhibits sequence specificity and/or stimulus dependency. For this purpose extracts were prepared from KCΔ4-transfected HeLa tet-off cells as described previously and subjected to immunoprecipitation with antibody against SF2/ASF or with non-specific mouse IgG (RNA-IP). The IP pellets were used to prepare RNA and analyzed for relative abundance of CXCL1 reporter mRNA as compared to GAPDH mRNA using real time RT-PCR. In cells transfected with KCΔ4 reporter, transgene mRNA abundance in the IP with specific antibody was enriched 4-fold as compared with the non-specific IgG as determined by ΔΔCt. In contrast, essentially no enrichment of reporter mRNA was obtained in cells transfected with the SF2/ASF-insensitive reporter KC-Clu-P2 (Fig. 6c). In a second experiment we compared the IP-based enrichment for KCΔ4 sequence between untreated and IL-17-treated cells. Consistent with the hypothesis, the interaction of SF2/ASF with the mRNA was markedly reduced in cells treated with IL-17. Finally, the RNA binding mutant of SF2/ASF (FF56/58DD) exhibited substantial loss of KCΔ4 binding activity as compared with the non-mutant N-terminal fragment. Collectively, these findings support the hypothesis that SF2/ASF is a mediator of the decay of selected mRNA species via specific RNA interaction and this function is disrupted in response to IL-17 treatment, apparently as a consequence of interaction with TRAF5.
IL-17 promotes TRAF5 and SF2/ASF function in primary cells
Preceding experiments identified the role of TRAF2/5 and SF2/ASF in coupling IL-17 stimulation with mRNA stabilization in long term cultured cell lines. To evaluate the pathway in primary cells, we prepared cultures of kidney epithelial cells as described30. While TNF can induce modest expression of CXCL1 mRNA in these cells, the combination of TNF plus IL-17 provides a very robust amplification that is due at least in part to the prolongation of mRNA half-life (Fig. 7a). Several additional pro-inflammatory chemokines and cytokines also exhibit enhanced mRNA half-life under the same treatment conditions including MIP-2, MCP-1, and CSF3 (Fig. 7a). IL-17 promotes SF2/ASF interaction with TRAF5 in primary kidney epithelial cells as demonstrated by co-IP of SF2/ASF using TRAF5 antibody (Fig. 7b). Finally we evaluated the interaction of SF2/ASF with several IL-17-sensitive mRNAs by co-IP. Endogenous mRNAs encoding CXCL1, CSF3, and MIP2 were each enriched in anti-SF2/ASF IPs relative to nonspecific Ig (Fig. 7c). Furthermore, in each case the interaction was disrupted by treatment with IL-17. Hence IL-17 appears to utilize this pathway in primary mouse epithelial cells.
Figure 7. IL-17 promotes TRAF5 and SF2/ASF function in primary cells.

a. Primary kidney epithelial cells were treated with TNF (10 ng/ml) alone or in combination with IL-17 (25 ng/ml) for 2 hrs. ActD (5 μg/ml) was added to each dish for the indicated times and CXCL1, MIP-2, MCP-1, and GAPDH mRNA was quantified by RNA hybridization. CSF3 and GAPDH mRNA was quantified in the same total RNA samples using RT-PCR (27 cycles) for greater sensitivity. Results are representative of 2 independent experiments. b. Primary kidney epithelial cells were treated with nothing or with IL-17 (25 ng/ml) for the indicated times and used for immunoprecipitation with anti-TRAF5. The IPs or 40 μg of WCL protein were analyzed by SDS-PAGE immunoblot using anti-SF2/ASF. Results are representative of 2 separate experiments. c. Primary kidney epithelial cells were treated with TNF for 2 hrs. Separate cultures were then left untreated or treated with IL-17 (25 ng/ml) for 30 minutes. Extracts were used for RNA immunoprecipitation with anti-SF2/ASF. RNA was recovered from IPs and used to quantify CXCL1, CSF3, MIP-2 and GAPDH mRNAs by real time RT-PCR. The data are presented as mRNA enrichment comparing ΔΔCt value for non-specific IgG with that for anti-SF2/ASF. Results are representative of 3 independent experiments.
Discussion
IL-17 cooperates with TNF for induction of CXCL1 gene expression by stabilizing mRNA through a mechanism distinct from that employed in myeloid cells via TLR stimulation1,9,17. Signaling downstream of the IL-17R involves the adaptor protein Act1, which couples to NF-κB activation via binding with TRAF611,13,31. However, TRAF6 is not required for IL-17-mediated stabilization of CXCL1 mRNA16. Collectively these observations indicate that IL-17-mediated stabilization of CXCL1 mRNA involves a distinct signal transduction pathway32. IL-17 promotes interaction between Act1, TRAF2 or TRAF5, and the multifunctional RNA splicing regulatory factor SF2/ASF. SF2/ASF selectively enhances target mRNA decay via binding to sites within the 3′UTRs and this interaction is disrupted by IL-17, perhaps via TRAF5-dependent sequestration of limiting amounts of cytoplasmic SF2/ASF.
The identification of TRAF5 and/or TRAF2 in signal transduction downstream of IL-17R is noteworthy. Known primarily for their roles in TNFR family responses, the participation of these TRAFs in IL-17-mediated modulation of chemokine mRNA half life provides the first evidence for their participation in this signaling pathway. Interestingly, as in TNFR family signaling, these two proteins appear to provide redundant function in the IL-17 pathway since mRNA stabilization is compromised only in doubly deficient cells21,33. In this regard, a dn version of TRAF5 was the most effective in blocking IL-17-mediated mRNA stabilization suggesting that this is the more physiologic regulator. The hypothesis that these TRAFs are involved in IL-17 response is strongly supported by the ligand-dependency of TRAF2 and TRAF5 interactions with both Act1 and SF2/ASF. Indeed, all three proteins appear to assemble in the initial signaling complex following receptor stimulation.
The selectivity of TRAF2 and TRAF5 for interaction with SF2/ASF, in contrast to that of TRAF6 for TAK1, suggests the intriguing hypothesis that selective TRAF interaction provides an important segregation point in IL-17 signaling where the upstream events can be independently coupled to distinct functional endpoints (i.e., NF-κB versus mRNA stabilization). It is also noteworthy that the form of TRAF5 interacting with Act1 in IL-17-stimulated cells migrates at a significantly higher mobility in SDS-PAGE suggesting that it has been subject to post-translational modification. In this regard Act1 has been shown to possess E3 ubiquitin ligase activity encoded within the U box domain22 and it is thus tempting to speculate that TRAF5 may be subject to modification through such activity. TRAF6 is also an E3 ubiquitin ligase and this activity is critical for its function in promoting the activation of NFkB34,35 but neither TRAF2 or TRAF5 appears to have this activity36.
The initial identification of SF2/ASF as a candidate RNA binding protein involved in chemokine mRNA stability was based upon its ligand-dependent interaction with TRAF2 and TRAF5. Nevertheless, the finding that SF2/ASF has a significant role in the control of specific mRNA decay is not surprising given the dramatic range of functions that have been attributed to this molecule37,38. These include not only regulation of standard and alternative splicing but also nuclear-cytoplasmic transport28, message translational efficiency24, mRNA quality surveillance (nonsense mediated decay)39, and sequence specific mRNA decay24,25. In this regard, the manipulation of TRAF2/TRAF5 and SF2/ASF expression appear to selectively affect the behavior of mRNAs containing the IL-17-sensitive sequence motif in their 3′UTR17,40. Interestingly, the ability of SF2/ASF to modulate CXCL1 mRNA half life is dependent upon sequence selective RNA binding that can be modulated by IL-17 treatment. Since the interaction of SF2/ASF with TRAF2 or TRAF5 is dependent upon the stimulus, the simplest hypothesis is that IL-17 treatment results in sequestration of SF2/ASF. Though SF2/ASF is a highly abundant protein, the majority is localized in the nucleus so sequestration of cytoplasmic forms may be effective in modulation of function. Whether the TRAF-SF2/ASF interaction has other consequences beyond the effects on chemokine and cytokine mRNA half-life is unknown.
Our studies of CXCL1 mRNA instability have now identified two mechanisms that are distinguished by the responsible 3′UTR sequence motifs and the proteins that bind such sequences and modulate the rate of decay17,41. While the paradigm of ARE-dependent control of cytokine and chemokine mRNA half life via TTP is well established in myeloid cells19,42-44, the pathway is not restricted to the myeloid lineage and indeed both the ARE/TTP and the TRAF5-SF2/ASF mechanism may coexist within the same cell population17. It seems likely, however, that the SF2/ASF pathway for mRNA stabilization pathway is not operative in myeloid cells since CXCL1 mRNA is completely stable in TTP-deficient macrophages41. What is the rationale for the existence of multiple mechanisms operating to control stability of the same mRNA? The potential to encode multiple regulatory sequence determinants, even within a single mRNA, will enable cell type- and stimulus-specific response patterns comparable to the regulatory sequences governing inducible gene transcription. That this TRAF5-SF2/ASF mechanism is observed primarily in non-myeloid cell populations is noteworthy since these cells are the more common target for the action of IL-17-driven inflammatory neutrophil recruitment. While the physiologic significance of the IL-17-sensitive mechanism remains to be explored experimentally, it is likely to be important in IL-17-dependent pathogenesis since mRNA stabilization appears to be necessary for CXC chemokine expression and the associated recruitment of inflammatory neutrophils.
Methods
Reagents
G418, formamide, MOPS, salmon sperm DNA, and diethyl-pyrocarbonate were purchased from Sigma-Aldrich. DMEM, Dulbecco's PBS, penicillin and streptomycin were obtained from Central Cell Services of the Lerner Research Institute. FBS was purchased from BioWhittaker. PolyFect Transfection Reagent was obtained from Qiagen and Tri-Reagent was purchased from Molecular Research Center. Human and mouse rIL-17 and rTNF were purchased from R&D Systems. Anti-FLAG (clone M2) Ab was purchased from Sigma-Aldrich. Antibodies specific for TRAF2 (C20), TRAF5 (C-19), Act1 (H-300), SF2/ASF (96) and β-actin (C-11) were from Santa Cruz Biotechnology.
Cell Culture
HeLa tet-off cells were transiently transfected using Polyfect as described previously16,17. siRNA oligonucleotides targeting TRAF2 (5′-CGACAUGAACAUCGCAAGC-3′), TRAF5 (5′-CCAAGAACGCCUACAUUAA-3′) and SF2 (5′-ACGAUUGCCGCAUCUACGU-3′) were transfected using Lipofectamine 2000. TRAF2/TRAF5 DKO MEFs and reconstituted MEFs (a generous gift from H. Nakano, Juntendo University School of Medicine, Tokyo, Japan)45, and inducible tet-off SF2/ASF MEFs (from X. Fu, University of California, San Diego)26 were cultured in DMEM containing 10% FBS, penicillin, and streptomycin. Primary kidney epithelial cells were prepared as described previously30.
Plasmids
Plasmids encoding KCA4, KC-Clu-P2, TRAF2, TRAF4, TRAF5, TRAF6, dnTRAF2, dnTRAF5, dnTRAF6, and Act1 have been described40,46,47. Additional reporter plasmids containing the 5′UTR and coding region of mouse CXCL1 and the 3′UTRs from GM-CSF, CSF3, and IκBζ were generated by PCR and cloned in the pTRE2 plasmid. Flag-tagged TRAF2, TRAF4, and TRAF5, HA-tagged full length, N-terminus (1-196aa), and C-terminus (196-248aa) versions of SF2/ASF were generated by PCR and cloned into pcDNA3.1. The SF2/ASF mRNA binding motif mutant (F56D F58D)29 plasmid was generated using the QuikChange site-directed mutagenesis kit from Stratagene.
RNA hybridization, immunoblot and Immunoprecipitation
RNA hybridization, immunoblot and immunoprecipitation were performed as previously described48. Probes for RNA hybridization and antibodies for immunoblot and immunoprecipitation were described in Reagents.
RNA Immunoprecipitation
Immunoprecipitation to detect protein bound mRNA was carried out as described previously17,41. Transiently transfected HeLa cells or primary mouse kidney epithelial cells were washed with PBS and cross-linked with 0.1% formaldehyde for 15 min at 20°C prior to preparation of cell extracts and immunoprecipitation with the indicated antibodies as described in Reagents. Reversal of crosslinks was carried by adding 150 μl elution buffer (50 mM Tris-HCl, pH 7.0, 1% SDS, 5 mM EDTA, 10 mM DTT, 1 μl RNase inhibitor) and heating at 70°C for 1 h. RNA was extracted with TRIzol reagent and genomic DNA was digested using Turbo DNA-free kit (Ambion). First strand cDNA was synthesized with M-MLV reverse transcriptase (Promega). Real-time PCR was done with the ABI PRISM 7300 (Applied Biosystems).
Supplementary Material
Acknowledgments
This work was supported by USPHS awards R01CA039621 (TH) and R01HL098935 (XL), a Senior Investigator Award from the American Asthma Foundation (XL), and the David and Lindsay Morgenthaler Endowed Fellowship (DS).
Footnotes
Authorship Statement: MN, KB and CL performed experiments. DS designed, performed and interpreted experiments and participated in writing the manuscript. TH and XL designed and interpreted experiments and participated in writing the manuscript. All authors reviewed the final version of the manuscript.
References
- 1.Ouyang W, Kolls JK, Zheng Y. The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity. 2008;28:454–467. doi: 10.1016/j.immuni.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi: 10.1146/annurev.immunol.021908.132710. [DOI] [PubMed] [Google Scholar]
- 3.Kang Z, et al. Astrocyte-restricted ablation of interleukin-17-induced Act1-mediated signaling ameliorates autoimmune encephalomyelitis. Immunity. 2010;32:414–425. doi: 10.1016/j.immuni.2010.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Endlich B, Armstrong D, Brodsky J, Novotny M, Hamilton TA. Distinct temporal patterns of macrophage-inflammatory protein-2 and KC chemokine gene expression in surgical injury. J Immunol. 2002;168:3586–3594. doi: 10.4049/jimmunol.168.7.3586. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi Y. Neutrophil infiltration and chemokines. Crit Rev Immunol. 2006;26:307–16. doi: 10.1615/critrevimmunol.v26.i4.20. [DOI] [PubMed] [Google Scholar]
- 6.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 7.McAllister F, et al. Role of IL-17A, IL-17F, and the IL-17 receptor in regulating growth-related oncogene-alpha and granulocyte colony-stimulating factor in bronchial epithelium: implications for airway inflammation in cystic fibrosis. J Immunol. 2005;175:404–412. doi: 10.4049/jimmunol.175.1.404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Witowski J, et al. IL-17 stimulates intraperitoneal neutrophil infiltration through the release of GRO alpha chemokine from mesothelial cells. J Immunol. 2000;165:5814–5821. doi: 10.4049/jimmunol.165.10.5814. [DOI] [PubMed] [Google Scholar]
- 9.Hartupee J, Lu C, Novotny M, Li X, Hamilton TA. IL-17 enhances chemokine gene expression through mRNA stabilization. J Immunol. 2007;179:4135–4141. doi: 10.4049/jimmunol.179.6.4135. [DOI] [PubMed] [Google Scholar]
- 10.Kao CY, et al. Up-regulation of CC chemokine ligand 20 expression in human airway epithelium by IL-17 through a JAK-independent but MEK/NF-kappaB-dependent signaling pathway. J Immunol. 2005;175:6676–6685. doi: 10.4049/jimmunol.175.10.6676. [DOI] [PubMed] [Google Scholar]
- 11.Qian Y, et al. The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol. 2007;8:247–256. doi: 10.1038/ni1439. [DOI] [PubMed] [Google Scholar]
- 12.Schwandner R, Yamaguchi K, Cao Z. Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med. 2000;191:1233–1240. doi: 10.1084/jem.191.7.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaffen SL. Structure and signalling in the IL-17 receptor family. Nat Rev Immunol. 2009;9:556–567. doi: 10.1038/nri2586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohmori Y, Fukumoto S, Hamilton TA. Two structurally distinct kappaB sequence motifs cooperatively control LPS-induced KC gene transcription in mouse macrophages. J Immunol. 1995;155:3593–3600. [PubMed] [Google Scholar]
- 15.Biswas R, et al. Regulation of chemokine mRNA stability by lipopolysaccharide and IL-10. J Immunol. 2003;170:6202–6208. doi: 10.4049/jimmunol.170.12.6202. [DOI] [PubMed] [Google Scholar]
- 16.Hartupee J, et al. IL-17 signaling for mRNA stabilization does not require TNF receptor-associated factor 6. J Immunol. 2009;182:1660–1666. doi: 10.4049/jimmunol.182.3.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Datta S, et al. IL-17 Regulates CXCL1 mRNA Stability via an AUUUA/Tristetraprolin-Independent Sequence. J Immunol. 2010;184:1484–1491. doi: 10.4049/jimmunol.0902423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Garneau NL, Wilusz J, Wilusz CJ. The highways and byways of mRNA decay. Nat Rev Mol Cell Biol. 2007;8:113–126. doi: 10.1038/nrm2104. [DOI] [PubMed] [Google Scholar]
- 19.Anderson P. Post-transcriptional control of cytokine production. Nat Immunol. 2008;9:353–359. doi: 10.1038/ni1584. [DOI] [PubMed] [Google Scholar]
- 20.Tebo J, et al. IL-1-mediated stabilization of mouse KC mRNA depends on sequences in both 5′ and 3′ untranslated regions. J Biol Chem. 2000;275:12987–12993. doi: 10.1074/jbc.275.17.12987. [DOI] [PubMed] [Google Scholar]
- 21.Au PY, Yeh WC. Physiological roles and mechanisms of signaling by TRAF2 and TRAF5. Adv Exp Med Biol. 2007;597:32–47. doi: 10.1007/978-0-387-70630-6_3. [DOI] [PubMed] [Google Scholar]
- 22.Liu C, et al. Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Sci Signal. 2009;2:ra63. doi: 10.1126/scisignal.2000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang Z, Ninomiya-Tsuji J, Qian Y, Matsumoto K, Li X. Interleukin-1 (IL-1) receptor-associated kinase-dependent IL-1-induced signaling complexes phosphorylate TAK1 and TAB2 at the plasma membrane and activate TAK1 in the cytosol. Mol Cell Biol. 2002;22:7158–7167. doi: 10.1128/MCB.22.20.7158-7167.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delestienne N, et al. The splicing factor ASF/SF2 is associated with TIA-1-related/TIA-1-containing ribonucleoproteic complexes and contributes to post-transcriptional repression of gene expression. Febs J. 2010;277:2496–2514. doi: 10.1111/j.1742-4658.2010.07664.x. [DOI] [PubMed] [Google Scholar]
- 25.Lemaire R, et al. Stability of a PKCI-1-related mRNA is controlled by the splicing factor ASF/SF2: a novel function for SR proteins. Genes Dev. 2002;16:594–607. doi: 10.1101/gad.939502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin S, Xiao R, Sun P, Xu X, Fu XD. Dephosphorylation-dependent sorting of SR splicing factors during mRNP maturation. Mol Cell. 2005;20:413–425. doi: 10.1016/j.molcel.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 27.Cazalla D, et al. Nuclear export and retention signals in the RS domain of SR proteins. Mol Cell Biol. 2002;22:6871–6882. doi: 10.1128/MCB.22.19.6871-6882.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Huang Y, Yario TA, Steitz JA. A molecular link between SR protein dephosphorylation and mRNA export. Proc Natl Acad Sci USA. 2004;101:9666–9670. doi: 10.1073/pnas.0403533101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Caceres JF, Krainer AR. Functional analysis of pre-mRNA splicing factor SF2/ASF structural domains. Embo J. 1993;12:4715–4726. doi: 10.1002/j.1460-2075.1993.tb06160.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin J, et al. TLR8-mediated NF-kappaB and JNK activation are TAK1-independent and MEKK3-dependent. J Biol Chem. 2006;281:21013–21021. doi: 10.1074/jbc.M512908200. [DOI] [PubMed] [Google Scholar]
- 31.Chang SH, Park H, Dong C. Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem. 2006;281:35603–35607. doi: 10.1074/jbc.C600256200. [DOI] [PubMed] [Google Scholar]
- 32.Hamilton T, et al. Diversity in post-transcriptional control of neutrophil chemoattractant cytokine gene expression. Cytokine. 52:116–122. doi: 10.1016/j.cyto.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tada K, et al. Critical roles of TRAF2 and TRAF5 in tumor necrosis factor-induced NF-kappa B activation and protection from cell death. J Biol Chem. 2001;276:36530–36534. doi: 10.1074/jbc.M104837200. [DOI] [PubMed] [Google Scholar]
- 34.Chen ZJ. Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol. 2005;7:758–765. doi: 10.1038/ncb0805-758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chung JY, Lu M, Yin Q, Lin SC, Wu H. Molecular basis for the unique specificity of TRAF6. Adv Exp Med Biol. 2007;597:122–130. doi: 10.1007/978-0-387-70630-6_10. [DOI] [PubMed] [Google Scholar]
- 36.Yin Q, Lamothe B, Darnay BG, Wu H. Structural basis for the lack of E2 interaction in the RING domain of TRAF2. Biochemistry. 2009;48:10558–10567. doi: 10.1021/bi901462e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang Y, Steitz JA. SRprises along a messenger's journey. Mol Cell. 2005;17:613–615. doi: 10.1016/j.molcel.2005.02.020. [DOI] [PubMed] [Google Scholar]
- 38.Long JC, Caceres JF. The SR protein family of splicing factors: master regulators of gene expression. Biochem J. 2009;417:15–27. doi: 10.1042/BJ20081501. [DOI] [PubMed] [Google Scholar]
- 39.Sato H, Hosoda N, Maquat LE. Efficiency of the pioneer round of translation affects the cellular site of nonsense-mediated mRNA decay. Mol Cell. 2008;29:255–262. doi: 10.1016/j.molcel.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 40.Novotny M, Datta S, Biswas R, Hamilton T. Functionally independent AU-rich sequence motifs regulate KC (CXCL1) mRNA. J Biol Chem. 2005;280:30166–30174. doi: 10.1074/jbc.M502280200. [DOI] [PubMed] [Google Scholar]
- 41.Datta S, Biswas R, Novotny M, Pavicic P, Herjan T, Mandal P, Hamilton TA. Tristetraprolin regulates CXCL1 (KC) mRNA stability. J Immunol. 2008;180:2545–2552. doi: 10.4049/jimmunol.180.4.2545. [DOI] [PubMed] [Google Scholar]
- 42.Hitti E, et al. Mitogen-activated protein kinase-activated protein kinase 2 regulates tumor necrosis factor mRNA stability and translation mainly by altering tristetraprolin expression, stability, and binding to adenine/uridine-rich element. Mol Cell Biol. 2006;26:2399–2407. doi: 10.1128/MCB.26.6.2399-2407.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Winzen R, et al. Distinct domains of AU-rich elements exert different functions in mRNA destabilization and stabilization by p38 mitogen-activated protein kinase or HuR. Mol Cell Biol. 2004;24:4835–4847. doi: 10.1128/MCB.24.11.4835-4847.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winzen R, et al. Functional analysis of KSRP interaction with the AU-rich element of interleukin-8 and identification of inflammatory mRNA targets. Mol Cell Biol. 2007;27:8388–8400. doi: 10.1128/MCB.01493-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sakon S, et al. NF-kappaB inhibits TNF-induced accumulation of ROS that mediate prolonged MAPK activation and necrotic cell death. Embo J. 2003;22:3898–3909. doi: 10.1093/emboj/cdg379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 47.Qian Y, Zhao Z, Jiang Z, Li X. Role of NF kappa B activator Act1 in CD40-mediated signaling in epithelial cells. Proc Natl Acad Sci USA. 2002;99:9386–9391. doi: 10.1073/pnas.142294499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun D, Ding A. MyD88-mediated stabilization of interferon-gamma-induced cytokine and chemokine mRNA. Nat Immunol. 2006;7:375–381. doi: 10.1038/ni1308. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.