

Summary
Many neuropsychiatric symptoms of fragile X syndrome (FXS) are believed to be a consequence of altered regulation of protein synthesis at synapses. We discovered that lovastatin, a drug that is widely prescribed for treatment of high cholesterol, can correct excess hippocampal protein synthesis in themouse model of FXS and can prevent one of the robust functional consequences of increased protein synthesis in FXS, epileptogenesis. These data suggest that lovastatin is potentially disease modifying, and could be a viable prophylactic treatment for epileptogenesis in FXS.
Seizures and EEG abnormalities were noted in some of the earliest studies of autism (Kanner, 1943; Tuchman et al., 2010), and it is now appreciated that approximately 30% of the ASD population have epilepsy. The etiologic heterogeneity of autism and epilepsy have made it difficult to understand the molecular mechanisms linking the two disorders. One approach is to focus on syndromic disorders that have a known genetic etiology and valid animal models. In the current study we have used a mouse model of FXS, a single-gene developmental disorder characterized by increased incidence of both autism and epilepsy (Berry-Kravis, 2002; Musumeci et al., 1999). The Fmr1−/y (KO) mouse exhibits robust epilepsy phenotypes, both in vitro as measured by ictal-like discharges in hippocampal slices and neocortical hyperexcitability, and in vivo as measured by increased susceptibility to audiogenic seizures (AGS) (Yan et al., 2004).
The FMR1 gene encodes the translational repressor FMRP (fragile X mental retardation protein). Pathological changes observed in FXS are believed to stem in part from an elevation of basal protein synthesis downstream of an extracellular signal regulated kinase (ERK1/2) signaling pathway (Bhakar et al., 2012). ERK1/2 is a key member of the larger MAP kinase (MAPK) signaling pathway which is involved in regulation of multiple biochemical processes including the initiation of cap-dependent translation. At the head of this intracellular cascade lies the small GTPase Ras. The Ras/MAPK pathway is a major regulator of cell growth, and thus a strong inhibitor of Ras-ERK1/2 would have deleterious consequences on the developing brain. However, a previous study reported that lovastatin, an HMG-CoA reductase inhibitor in widespread use for the treatment of hypercholesterolemia in both children and adults, could correct cognitive deficits caused by excess Ras activity in the mouse model of neurofibromatosis type 1 (Li et al., 2005). Lovastatin can achieve a mild reduction in Ras-ERK1/2 activation by interfering with the recruitment of Ras to the membrane, a process that is required to transition from the inactive GDP-bound form to the active GTP-bound form (Kloog et al., 1999; Schafer et al., 1989). The interaction of Ras with the membrane requires the posttranslational addition of a farnesyl group to the C-terminus and lovastatin inhibits Ras farnesylation by targeting the upstream mevalonate pathway (Fig. 1a) (Li et al., 2005; Mendola and Backer, 1990; Schafer et al., 1989). We therefore wondered if lovastatin could prevent pathological changes in FXS that lie downstream of excessive ERK-mediated protein synthesis and contribute to epileptogenesis. Unless otherwise stated, we tested lovastatin in male KO and WT mice in the C57BL/6 background.
Figure 1.
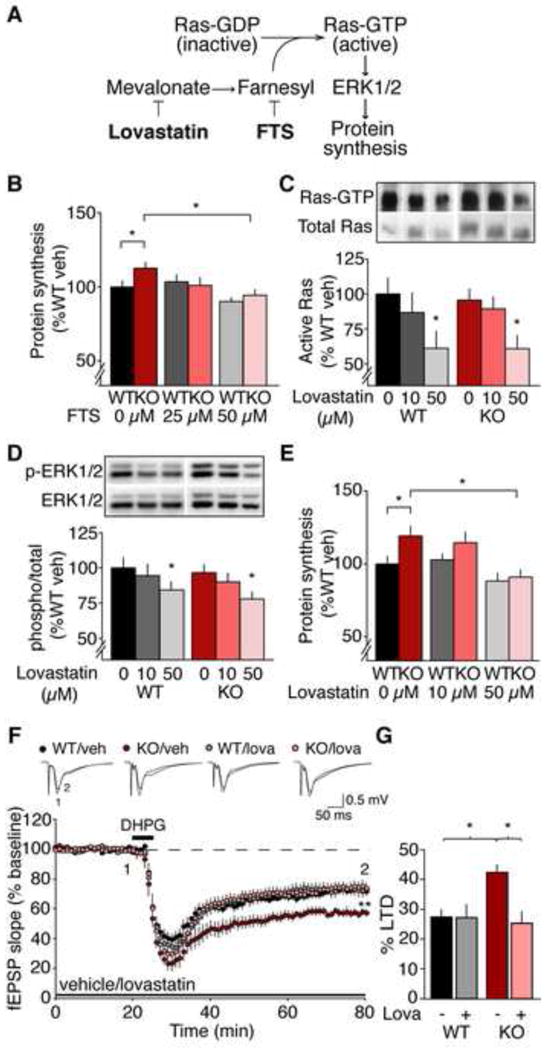
Lovastatin inhibits Ras-ERK1/2 signaling, normalizes excessive protein synthesis, and corrects exaggerated mGluR-LTD in the Fmr1 KO hippocampus. (A) Model of the mechanism by which lovastatin and FTS reduce Ras-ERK1/2 activation and normalize protein synthesis in the Fmr1 KO. (B) Application of FTS corrects excessive protein synthesis in the Fmr1 KO (WT: veh 100 ± 4%, 25 μM 104 ± 5%; 50 μM 90 ± 2%; KO: veh 113 ± 4%, 25 μM 101 ± 5%; 50 μM 94 ± 4%; ANOVA treatment *p = 0.0017; WT vs. KO veh *p = 0.041, KO veh vs. 50 μM *p = 0.00002; n = 15). A small but significant reduction of protein synthesis is also observed with 50 μM FTS in WT (*p = 0.0316). (C) Lovastatin inhibits Ras activation in hippocampal slices (WT: veh 100 ± 11%, 10 μM 87 ± 14%; 50 μM 61 ± 12%; KO: veh 96 ± 8%, 10 μM 89 ± 8%; 50 μM 61 ± 9%; ANOVA treatment *p = 0.0266; WT *p = 0.039, KO *p = 0.015; n = 6). (D) Lovastatin downregulates ERK1/2 (WT: veh 100 ± 7%, 10 μM 94 ± 8%; 50 μM 84 ± 5%; KO: veh 97 ± 5%, 10 μM 90 ± 6%; 50 μM 78 ± 5%; ANOVA treatment *p = 0.0037; WT *p = 0.035, KO *p = 0.008; n = 15). (E) Lovastatin normalizes protein synthesis in Fmr1 KO hippocampal slices (WT: veh 100 ± 5%, 10 μM 103 ± 4%; 50 μM 88 ± 5%; KO: veh 119 ± 6%, 10 μM 115 ± 7%; 50 μM 91 ± 4%; ANOVA genotype x treatment *p = 0.0332; WT vs. KO veh *p = 0.019, KO veh vs. 50 μM *p = 0.011; n = 11). (F) LTD was induced with 50 μM R,S-DHPG and extracellular recordings were performed in area CA1. In the presence of vehicle, greater LTD is observed in the Fmr1 KO versus WT (WT veh 72.5 ± 2.5%, KO veh 57.5 ± 2.5%, *p = 0.005, n = 9–10). 50 μM lovastatin significantly reduces LTD magnitude in the Fmr1 KO to WT levels (WT lova 72.7 ± 4.4%, KO lova 74.5 ± 3.4%; KO veh vs. lova *p < 0.001, n =11–13), but has no significant effect on LTD in the WT (WT veh vs. lova p = 0.869). Field potential traces are averages of all experiments, and were taken at times indicated by numerals; Scale bars = 0.5 mV, 5 ms. (G) Lovastatin significantly reduces LTD magnitude in the Fmr1 KO to WT levels (ANOVA genotype x treatment *p = 0.021). LTD magnitude was assessed by a comparison of the averaged last 5 minutes pre-DHPG and the last 5 min of recordings (minutes 55–60 post-DHPG). N = animals. Error bars = s.e.m.
Results
Lovastatin normalizes excessive protein synthesis in the Fmr1KO
We showed previously that partial inhibition of ERK1/2 was sufficient to restore normal levels of protein synthesis in the Fmr1 KO hippocampus (Osterweil et al., 2010). To determine if reduction of Ras signaling has the same effect, we used farnesyl thiosalicylic acid (FTS), which dislodges Ras from the membrane (Fig. 1A) (Weisz et al., 1999). Hippocampal slices were prepared from juvenile Fmr1 KO and wild type (WT) mice and pre-incubated in 25, 50 μM FTS or vehicle for 30 minutes. Protein synthesis was measured for another 30 min via incorporation of a 35S-labeled methionine/cysteine (35S-Met/Cys) mix. We found that 50 μM FTS reduces protein synthesis in the Fmr1KO down to WT levels (WT vs. KO veh *p = 0.041, KO veh vs. 50 μM FTS *p = 0.00002) (Fig. 1B).
We next asked if lovastatin could inhibit Ras in our slices as has been shown in other systems. Hippocampal slices were prepared from WT and Fmr1 KO, then incubated with 10–50 μM lovastatin or vehicle. GTP-bound Ras was isolated using a GST-pull down assay, and compared to total Ras. Our results show that 50 μM lovastatin significantly reduces the amount of active Ras in both WT and Fmr1 KO hippocampal slices (WT *p = 0.039, KO *p = 0.015) (Fig. 1C).
Western blot analysis revealed that 50 μM lovastatin also significantly reduces ERK1/2 activation in both WT and Fmr1 KO slices (WT *p = 0.035, KO *p = 0.008) (Fig. 1D). To examine the consequence of this modest reduction in ERK1/2 activity on hippocampal protein synthesis, we performed metabolic labeling on WT and Fmr1KO slices in the presence of 10–50 μM lovastatin. We found that inhibition of the Ras-ERK1/2 pathway by 50 μM lovastatin is sufficient to restore WT levels of protein synthesis in hippocampal slices from the Fmr1 KO (WT vs. KO veh *p = 0.019, KO veh vs. 50 μM *p = 0.011) (Fig. 1E). Consistent with previous findings, there was no difference in basal levels of Ras and ERK1/2 in the Fmr1 KO hippocampus, suggesting that the excessive protein synthesis in the Fmr1 KO is due to a hypersensitivity to, not hyperactivation of, the Ras-ERK1/2 signaling pathway (Osterweil et al., 2010). To see if the effects of FTS and lovastatin on protein synthesis might be due to inhibition of the homologous GTPase Rheb instead of Ras (Makovski et al., 2012), we measured the phosphorylation (activation) of p70S6 kinase (p70S6K) and S6 ribosomal protein (see Supplemental Results). We failed to detect changes in these downstream markers of Rheb activity (Figs S1, S2), suggesting that inhibition of Rheb is not likely to be the mechanism of action for FTS or lovastatin in our preparation.
An electrophysiological signature of altered hippocampal protein synthesis in the Fmr1 KO mouse is exaggerated long-term synaptic depression (LTD) induced by activation of metabotropic glutamate receptor 5 (mGluR5) (Huber et al., 2002). We therefore examined the effect of lovastatin on LTD induced by the mGluR5 agonist 3,5-dihydroxyphenylglycine (DHPG) in hippocampal area CA1 of WT and Fmr1 KO mice. Slices were incubated in vehicle or 50 μM lovastatin, extracellular field potentials were recorded in area CA1 in response to Schaffer collateral stimulation, and mGluR-LTD was induced with a 5 minute bath application of 50 μM DHPG with the experimenter blind to genotype and treatment. We found that lovastatin corrects the exaggerated mGluR-LTD observed in the Fmr1 KO to WT levels (WT veh vs. KO veh *p = 0.005, KO veh vs. lova *p < 0.001), but had no significant effect on mGluR-LTD in the WT (WT veh vs. lova p = 0.869)(Fig. 1F, G). These electrophysiological findings corroborate the biochemical results, and support the conclusion that lovastatin can correct excessive protein synthesis in the Fmr1KO.
Lovastatin prevents mGluR-induced epileptogenesis in hippocampal slices
Previous studies in slices of hippocampal area CA3 have shown that a lasting consequence of mGluR5- and ERK-mediated protein synthesis is the generation of epileptiform activity (Merlin et al., 1998; Zhao et al., 2004). We therefore investigated the effect of lovastatin in this model of epileptogenesis. Intracellular recordings were made from CA3 pyramidal cells in WT hippocampal slices ± 1 h pre-incubation with 50 μM lovastatin. Single cell behavior and network discharge behavior can be differentiated in these intracellular recordings in that the occurrence, rhythm, and duration of network discharges are not affected by depolarizing or hyperpolarizing the recorded cell (see (Taylor et al., 1995). In control slices, addition of DHPG first elicited short (~500 ms) synchronized discharges, which gradually extended to reach an average duration of 4.359 ± 0.14 sec at 60 min (Fig. 2A). Pre-incubation with 50 μM lovastatin significantly reduced the average duration of synchronized discharges to 0.618 ± 0.03 sec at 60 min (*p = 0.0000218; Fig. 2A,C). These results suggest that lovastatin is sufficient to block the induction of mGluR-mediated epileptiform activity in hippocampal slices.
Figure 2. Lovastatin blocks mGluR-mediated epileptiform bursting in the Fmr1 KO hippocampus.
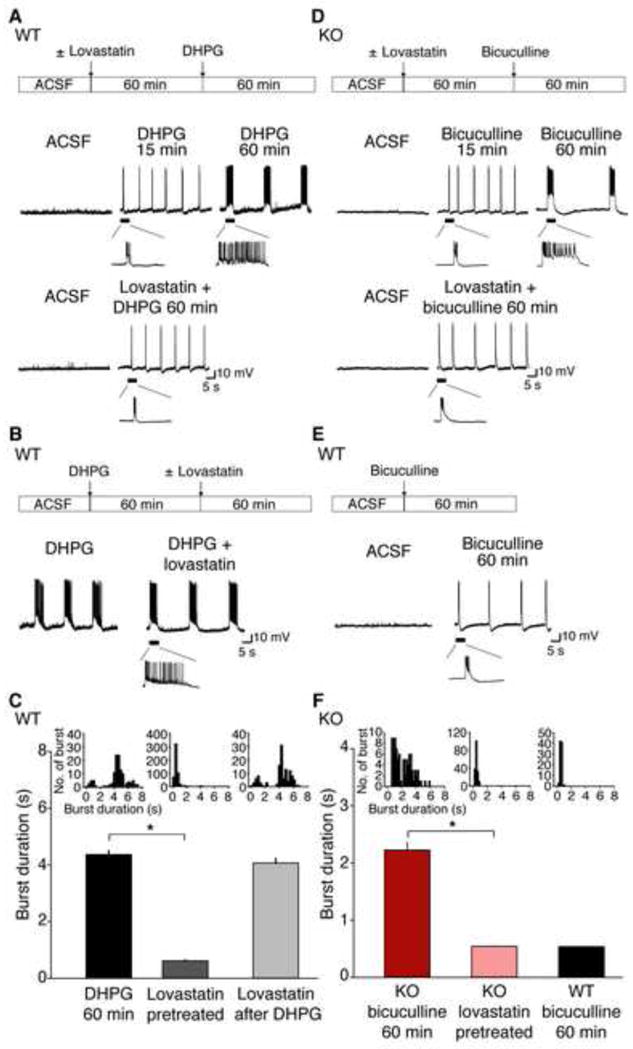
Intracellular recordings were performed on CA3 pyramidal neurons in WT and Fmr1 KO hippocampal slices. (A) In WT slices, 50 μM R,S-DHPG leads to bursting from CA3 neurons, which progresses to epileptiform discharges by 60 min. This epileptiform activity is prevented by a 60 min pre-treatment with 50 μM lovastatin. (B) Addition of lovastatin after 60 min post-DHPG did not affect the duration of epileptiform discharges. (C) Mean epileptiform burst durations from WT slices under the following conditions: at 60 min post-DHPG (4.359 ± 0.14 sec; n = 125 discharges, 6 slices from 6 animals), lovastatin-pretreated (0.618 ± 0.03 sec; n = 545 discharges, 12 slices from 7 animals), and lovastatin after DHPG (4.066 ± 0.15 sec; n = 124 discharges, 5 slices from 5 animals). Slices pretreated with lovastatin showed significant reduction in burst duration compared to vehicle-pretreated slices (*p = 0.0000218) while there was no significant difference in burst duration between DHPG 60 min and lovastatin after DHPG (p = 0.072). (D) In Fmr1 KO slices, synaptic mGluR activation by spontaneous activity in bicuculline induces prolonged epileptiform discharges, which are blocked with 60 min pre-treatment with 50 μM lovastatin. (E) In contrast to Fmr1 KO slices, bicuculline fails to induce prolonged epileptiform discharges in WT slices. (F) Plot of mean epileptiform burst durations from Fmr1 KO slices 60 min post-bicuculline ± 50 μM lovastatin, and WT slices 60 min post-bicuculline. Fmr1 KO slices pretreated with lovastatin showed significant reduction in burst duration (0.528 ± 0.01 sec; n = 182 discharges; 10 slices from 5 animals) compared to vehicle-pretreated slices (2.227 ± 0.13 sec; n = 90 discharges; 8 slices from 6 animals; *p = 0.000022) while there was no significant difference in burst duration between lovastatin pretreated Fmr1 KO slices and bicuculline treated WT slices (0.537 ± 0.01 sec; n = 88 discharges; 5 slices from 5 animals; p = 0.995). Insets indicate summary frequency histograms of synchronized discharges in each experimental condition. Error bars = s.e.m.
The epileptiform discharges observed after sustained DHPG application persist after the agonist is removed. This maintenance phase is not affected by inhibition of protein synthesis or ERK1/2, suggesting that translation plays a specific role in epileptogenesis (Zhao et al., 2004). Similarly exposure of slices to 50 μM lovastatin 60 min after DHPG had no effect on average burst duration (average duration 4.066 ± 0.15 sec; p = 0.072; Fig. 2B,C). These results are consistent with the action of lovastatin as an inhibitor of ERK-mediated protein synthesis (Merlin et al., 1998).
In the Fmr1 KO, spontaneous activity is sufficient to elicit epileptiform discharges in hippocampal CA3, suggesting a hypersensitive response to mGluR5-ERK1/2 activation (Chuang et al., 2005). Thus, unlike WT slices which require DHPG to induce ictal discharges, epileptiform activity can be induced in Fmr1 KO slices simply by applying the GABA-A receptor antagonist bicuculline to increase recurrent network activity (Bianchi et al., 2009). Intracellular recordings were therefore performed on bicuculline treated Fmr1 KO slices ± 1 h pre-incubation of 50 μM lovastatin. In untreated KO slices, bicuculline elicited short (< 1s) synchronized discharges that increased progressively in duration over the course of 60 min (average duration 2.227 ± 0.13 sec at 60 min). This progression was prevented by pre-incubation in lovastatin (Fig. 2D,F). The average burst duration of Fmr1 KO slices in lovastatin (0.528 ± 0.01 sec at 60 min) was not significantly different from discharges elicited by bicuculline in the WT (0.537 ± 0.01 sec; p = 0.995; Fig. 2E,F). These results show that acute application of lovastatin blocks epileptogenesis in the Fmr1 KO hippocampus.
Lovastatin dampens hyperexcitability in the Fmr1 KO visual cortex
Increased excitability has been observed in the neocortex of the Fmr1 KO. This phenotype is caused by specific changes in excitatory synaptic transmission and neuronal membrane excitability, and can be rescued by inhibition or reduced expression of mGluR5 (Hays et al., 2011). To investigate if lovastatin could similarly reduce cortical hyperexcitability, we prepared acute slices from visual cortex and measured persistent activity in layer 5 pyramidal neurons. Spiking activity was evoked with electrical stimulation of the underlying white matter every 30 sec for a total number of 60 trials in artificial cerebrospinal fluid (ACSF) plus vehicle, followed by 60 trials in ACSF plus 50 μM lovastatin (Fig. 3A). We found that the barrage of action potentials evoked by stimulation of white matter is significantly longer in the Fmr1 KO (Fig. 3B,C), but is reduced to WT levels by the application of 50 μM lovastatin (Fig. 3B,C). Analysis of the total number of action potentials evoked revealed a significant dampening effect of lovastatin in Fmr1 KO but not WT slices (WT vs. KO veh *p = 0.0258, KO veh vs. lova *p = 0.0001; Fig. 3D–E). These results suggest that lovastatin corrects the hyperexcitability in the Fmr1 KO visual cortex.
Figure 3. Lovastatin reduces excitability in the Fmr1 KO visual cortex.
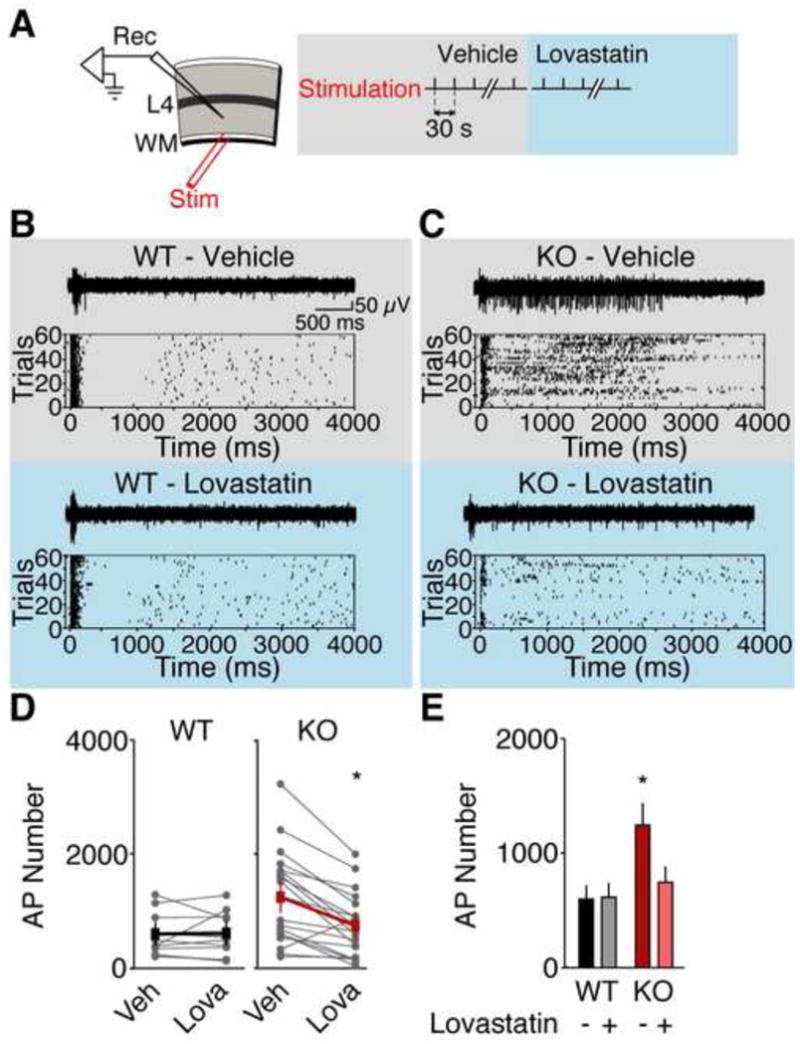
(A) Extracellular recordings were performed in layer 5 of visual cortical slices. Trains of action potentials (APs) were evoked with 60 trials of white matter stimulation. Responses were collected in ACSF plus vehicle, then ACSF plus 50 μM lovastatin. Representative traces and raster plots of recordings from WT (B) and Fmr1 KO (C) Prolonged firing in the Fmr1 KO is corrected with 50 μM lovastatin application. (D) Lovastatin significantly reduces firing in Fmr1 KO but not WT visual cortical slices. (E) The mean number of action potentials is significantly higher in Fmr1 KO slices versus WT slices, and 50 μM lovastatin restores normal firing to the Fmr1 KO slices (WT veh 100 ± 20%, WT lova 103 ± 20%, KO veh 203 ± 44%, KO lova 113 ±29%; ANOVA genotype x treatment *p = 0.0022; WT vs. KO veh *p = 0.0258, KO veh vs. lova *p = 0.0001, WT veh vs. lova p = 0.8412; WT n = 10 slices from 5 animals, KO n = 19 slices from 9 animals). Error bars represent s.e.m.
Lovastatin corrects AGS in the Fmr1KO
We next tested whether lovastatin could ameliorate AGS observed in the Fmr1 KO in vivo. Fmr1 KO and WT mice were injected intraperitoneally (i.p.) with 30 mg/kg lovastatin and, after one hour, exposed to a 130 dB stimulus for 2 minutes. Four stages of increasing AGS severity were scored: wild running, clonic seizure, tonic seizure, or death (Osterweil et al., 2010; Yan et al., 2004). Vehicle-treated Fmr1 KO mice exhibited a 74% incidence of AGS, and no seizures were observed in vehicle-treated WT mice (WT vs. KO *p = 0.0002). Acute injection of lovastatin reduced the incidence of AGS to 28% in the Fmr1 KO (KO veh vs. lov *p = 0.009) and significantly lessened the severity (Fig. 4A). A higher dose of 100 mg/kg lovastatin showed a similar reduction in AGS incidence (KO veh vs. lov 85% to 23%; *p = 0.005) and severity (KO veh vs. lov *p = 0.015).
Figure 4. Lovastatin significantly reduces AGS incidence and severity in the Fmr1 KO.
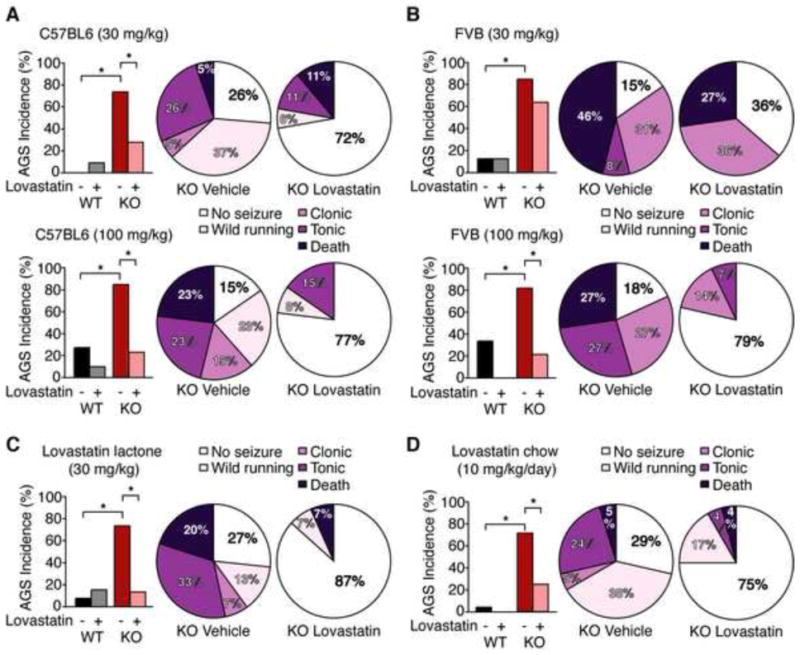
Fmr1 KO and WT mice on the C57BL/6 (A,C,D) or FVB (B) backgrounds were treated as indicated, tested for AGS, and scored for wild running, clonic seizure, tonic seizure, and death. (A) Injection of 30 mg/kg lovastatin acid significantly reduces AGS incidence in the Fmr1 KO on the C57BL/6 background (*p = 0.009; n = 18–19). Pie charts show a significant shift in the severity distribution of Fmr1 KO mice treated with vehicle vs. 30 mg/kg lovastatin (WT vs. KO veh *p = 0.002, WT vs. KO lov p = 0.999; KO veh vs. lov *p = 0.041). Injection of a higher100 mg/kg dose of lovastatin also significantly reduces AGS incidence (*p = 0.005; n = 13) and attenuates AGS severity (KO veh vs. lov *p = 0.015; WT vs. KO veh. *p = 0.04; WT vs. KO lov p = 0.999) in Fmr1 KO mice on the C57BL/6 background. (B) In Fmr1 KO mice bred on the FVB background, injection of 30 mg/kg lovastatin does not significantly reduce AGS incidence (KO veh 85%, KO lov 64%, p = 0.357) or severity (KO veh vs. lov p = 0.862), however a higher dose of 100 mg/kg lovastatin significantly reduces both AGS incidence (KO veh 82%, KO lov 21%, *p = 0.005; n = 11–14) and severity (KO veh vs. lov *p = 0.022; WT vs. KO veh *p = 0.016; WT vs. KO lov p = 0.999). (C) Injection of 30 mg/kg lovastatin in the lactone form significantly reduces AGS incidence (*p = 0.009; n = 15) and severity (KO veh vs. lov *p = 0.009; WT vs. KO veh *p = 0.005; WT vs. KO lov p = 0.999) in Fmr1 KO mice. (D) 48 h feeding of 0.01% lovastatin chow significantly reduces AGS incidence (KO con 71%, KO lov 25%, *p = 0.003; n = 21–24) and severity (KO veh vs. lov *p = 0.016; WT vs. KO veh *p = 0.0001; WT vs. KO lov p = 0.461) in Fmr1 KO mice. N = animals.
To determine whether the effect of lovastatin on AGS susceptibility is independent of the genetic background, we performed another set of experiments using Fmr1 KO mice in the seizure prone FVB strain. Consistent with previous studies, we observed an 85% incidence of AGS in vehicle treated Fmr1 KO FVB mice (Yan et al., 2004), which was significantly higher than the 11% incidence observed in vehicle treated WT mice (*p = 0.002). Interestingly, Fmr1 KO FVB mice injected with 30 mg/kg lovastatin still exhibit a 64% incidence of AGS incidence, which is not significantly different from the vehicle treated group (p = 0.357). The severity of AGS is similarly unaffected by this treatment (p = 0.862) (Fig. 4B).
Previous work has shown that higher doses of mGluR5 inhibitors are required to reduce AGS in Fmr1 KO mice bred on the FVB/NJ background versus the C57BL/6J x FVB/NJ hybrid background (Yan et al., 2005). In addition, a difference in lovastatin metabolism has been observed between different mouse strains (Gegg et al., 2005). Based on this literature, we repeated our experiments using a higher dose of lovastatin. The results show that an increased dose of 100 mg/kg lovastatin significantly lowers AGS incidence (KO veh 82%, KO lov 21%, *p = 0.005) and reduces AGS severity (KO veh vs. lov *p = 0.022) in FVB Fmr1 KO mice (Fig. 4B). Thus, although the effective dose range differs, lovastatin is effective in correcting AGS in Fmr1 KO mice bred on multiple background strains.
For human use, lovastatin is administered as a pro-drug in its lactone form, which is metabolized into an active conformation (lovastatin acid) (Lambert et al., 1996). We therefore wished to confirm that lovastatin lactone could also reduce AGS incidence and severity in the Fmr1 KO. Consistent with our previous results, we found that lovastatin lactone (30 mg/kg) significantly reduces AGS incidence from 73% to 20% (*p = 0.009) and reduces AGS severity (KO veh vs. lov *p = 0.009) in the Fmr1 KO (Fig. 4C).
We also wanted to verify that oral administration could similarly correct AGS in the Fmr1 KO. To address this question, WT and Fmr1 KO mice were fed standard rodent chow supplemented with 0.01% lovastatin, which corresponds to a dose of 10 mg/kg/day (Yamada et al., 2000). After a 48 hour exposure to either lovastatin chow or control chow with the same formulation, mice were tested for AGS. Our results show that this oral administration of lovastatin significantly reduces AGS incidence (KO con 71%, KO lov 25%, *p = 0.003) and severity (KO veh vs. lov *p = 0.016) in the Fmr1 KO mouse (Fig. 4D). These results suggest that oral administration of pharmaceutical grade lovastatin is effective in correcting AGS susceptibility in the Fmr1 KO.
Discussion
Based on the insights that an ERK1/2 signaling pathway lies upstream of the excessive hippocampal protein synthesis in the Fmr1KO (Osterweil et al., 2010) and that lovastatin can, among other actions, inhibit Ras-ERK1/2 signaling in hippocampal neurons (Li et al., 2005), we wondered if lovastatin could correct core biochemical and electrophysiological consequences of the loss of the translational repressor FMRP in FXS. We thought this was a particularly exciting prospect because lovastatin is widely prescribed, has a known safety profile, and is approved for use in children (to treat hypercholesterolemia). Our results show that lovastatin can indeed correct excessive protein synthesis and prevent the emergence of epileptiform activity in the Fmr1KO hippocampus in vitro, and can protect the Fmr1 KO mice from AGS in vivo.
It has been suggested that seizures in children with developmental disorders such as FXS could worsen the progression and severity of other symptoms including autism (Berry-Kravis, 2002; Tuchman et al., 2010). Once manifest, seizures can be controlled with conventional anticonvulsant medications, but these drugs themselves have side effects that can exacerbate other symptoms, such as cognitive impairment. Further, prodromal changes may be as deleterious for brain development as full-blown seizures. Therefore, preventing epileptogenesis is an important goal,.
Statins have been studied previously in rodent models of epilepsy in which seizures are initiated or kindled in vivo by administration of drugs or by direct electrical stimulation of the temporal lobes. Some studies have suggested that statins can lessen seizure activity (Lee et al., 2008; Ramirez et al., 2011; Xie et al., 2011), whereas others have reported no effect (van Vliet et al., 2011) or an exacerbation of seizures (Serbanescu et al., 2004). Beneficial effcts have typically been attributed to reduced brain inflammation associated with seizures. In contrast to previous work, we have used a genetically engineered mouse model of FXS in which seizure phenotypes are not caused by artificial induction of inflammatory responses and neurodegeneration. Our findings point to an entirely different mechanism of action, by which lovastatin corrects multiple Fmr1 KO phenotypes including, but not limited to in vivo seizure activity by downregulating Ras-ERK1/2 and protein synthesis.
It should be noted, however, that the connection between seizure phenotypes in the mouse model and childhood epilepsy in humans with FXS remains to be firmly established. For example, AGS may be largely a reflex event driven by activation of brainstem nuclei with limited relevance to cortical electroencephalographic seizures (Raisinghani and Faingold, 2003). Arguing against this point of view is the observation that audio stimulation rapidly elicits (within 2 min) generalized tonic-clonic seizures in most Fmr1 KO mice, showing that hyperexcitability extends beyond brainstem neurons. Nevertheless, epileptogenesis in fragile X patients is manifest as the appearance of spontaneous seizures, which are not observed in the mouse model. The issue of whether the pathogenesis of epilepsy is shared by mice and humans lacking FMRP could be addressed if the effects of a prophylactic treatment were compared in the two species. Our findings that lovastatin, already approved for use in humans, can prevent epileptogenesis and hyperexcitability in the Fmr1 KO suggest that carefully controled human clinical trials are warranted.
As a negative regulator of the mevalonate pathway, lovastatin can impact the production of multiple compounds involved in intracellular signaling, including ubiquinone, dolichol, and isoprenoids (Goldstein and Brown, 1990). Therefore, we cannot rule out the possibility that some beneficial effects of lovastatin could be due to actions other than the reduction in Ras-ERK1/2 signaling, including a reduction in the farnesylation of other target proteins. However, like lovastatin, inhibitors of Ras or ERK1/2 are sufficient to normalize protein synthesis, and downregulation of the ERK1/2 pathway also blocks hippocampal epileptogenesis and eliminates AGS in the Fmr1KO (Chuang et al., 2005; Osterweil et al., 2010). Thus, although the precise molecular mechanisms by which lovastatin confers benefit remain to be determined, the weight of the evidence suggests that the effects are due to a reduction in Ras-ERK1/2 signaling.
This study was focused on the seizure phenotype because it has both construct (genotypic) and face (phenotypic) validity with the human disorder (with the caveats raised above). However, previous work has suggested that multiple fragile X symptoms can arise from the same core pathophysiology (Bear et al., 2004; Bhakar et al., 2012). The finding that lovastatin normalizes mGluR-LTD in the Fmr1 KO hippocampus to WT levels (Fig. 1F) is consistent with this idea. This result is intriguing, however, as protein synthesis inhibitors cannot correct the exaggerated mGluR-LTD in the Fmr1KO (reviewed in (Bhakar et al., 2012). The implication is that lovastatin is exerting additional beneficial effects on the Fmr1 KO beyond the reduction of protein synthesis. What these effects are remains to be determined, however it should be noted that the selective reduction of mGluR-LTD in the Fmr1 KO has been observed in a previous study using lithium (Choi et al., 2011).
It will be of interest to assess in future studies the effect of lovastatin treatment on the full spectrum of fragile X phenotypes. Indeed, the findings that lovastatin can selectively quiet cortical hyperexcitability in the Fmr1 KO (Fig. 3) as well as exaggerated LTD suggest it could potentially improve sensory and cognitive functions. Moreover, there is growing appreciation that disruptions in Ras signaling (Krab et al., 2008) and synaptic protein synthesis (Kelleher and Bear, 2008) may lie at the core of many autisms of unknown etiology, suggesting that lovastatin could have therapeutic utility in other ASDs as well as in other symptom domains.
Experimental Procedures
Mice
Fmr1 KO (Jackson Labs) and WT littermates, bred on the C57BL/6 or FVB background strains, were group housed and maintained in a 12:12 h light:dark cycle. All animals were treated in accordance with NIH, MIT and SUNY guidelines.
Metabolic labeling
Experiments were performed on male WT and Fmr1 KO mice (P25-P32), blind to genotype, as described previously (Osterweil et al., 2010). Details appear in supplemental methods online.
Immunoblotting
Immunoblotting was performed blind to genotype and treatment, as described previously (Osterweil et al., 2010) using primary antibodies to Ras (Pierce), p-ERK1/2 (Thr202/Tyr204), ERK1/2, p-p70S6K (Thr389), p70S6K, p-S6 (Ser240/244), and S6 (Cell Signaling Technology), and HRP-conjugated secondary antibodies (GE Healthcare).
Ras activation assay
Slices (4–6 per animal) were prepared exactly as for metabolic labeling, and Ras-GTP isolated using the Active Ras Pull-Down and Detection kit (Pierce) according to manufacturer’s instructions.
Hippocampal LTD
Experiments were performed blind to genotype, as described previously (Huber et al., 2002). Lovastatin (50 μM) was dissolved in vehicle comprising 0.05% DMSO in artificial cerebrospinal fluid (ACSF). Slices were exposed to drug or vehicle from 30 minutes prior to application of DHPG until the end of the experiment. Details appear in supplemental methods online.
Hippocampal CA3 recordings
Epileptiform activity was elicited using DHPG or bicuculline and intracellular CA3 recordings were performed as previously described (Chuang et al., 2005). Details appear in supplemental methods online.
Cortical slice electrophysiology
Slices of visual cortex were prepared from P16–P21 WT or Fmr1 KO animals, blind to genotype, as described previously (Philpot et al., 2001). Submerged slices were maintained at 30°C in a modified ACSF that closely mimics physiological CSF (in mM: 124 NaCl, 3.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 10 dextrose, 0.8 MgCl2, 1 CaCl2, saturated with 95% O2 and 5% CO2) and promotes spiking in response to electrical stimulation. Stimulation electrodes (clustered bipolar tungsten, FHC) were positioned in white matter and extracellular recordings performed in layer 5 using glass recording electrodes (~1 MΩ). Baseline responses were collected every 30 s, using a stimulus intensity of 35–80 μA, 0.2 ms duration. Responses were collected in ACSF plus vehicle, then ACSF + 50 μM lovastatin. Extracellular recordings made using Axopatch 200B (Axon Instruments) were amplified 1000 times, filtered between 0.3 and 3 Hz, and digitized at 25 kHz. Further details are provided in Supplemental Information.
AGS
Experiments were performed using male WT and Fmr1 KO littermates (P17–25), blind to genotype, as described previously (Osterweil et al., 2010). For acute exposure experiments, mice were injected i.p. with drug or vehicle and returned to their home cages for 1 h. For oral administration experiments, mice were weaned onto standard rodent chow (Bio-serv) formulated with 100 mg/kg IC lovastatin (40 mg tablets, Mylan Inc) or identical chow containing no lovastatin, and allowed to feed ad libitum for 48 hours. Immediately prior to testing, mice were transferred to a quiet (< 60 dB ambient sound) room for 1 hour, then transferred to a transparent plastic test chamber. After 1 minute of habituation, mice were exposed to a 130 dB stimulus (recorded sampling of a modified personal alarm, Radioshack model 49–1010) for 2 minutes, and the incidence and severity of AGS was scored. Further details are provided in Supplemental Information.
Supplementary Material
Highlights.
Epilepsy is one consequence of excess protein synthesis in the Fmr1−/y mouse
Lovastatin inhibits Ras-ERK1/2 and normalizes protein synthesis
Lovastatin prevents epileptogenesis in the Fmr1−/y mouse
Lovastatin, approved for human use, is potentially disease modifying in FXS
Acknowledgments
For excellent technical and administrative support, and for valuble discussions: D. D. Krueger, K. Oram, S. Meagher, A. Heynen, E. Sklar, E. Greene-Colozzi, K. Reinhold, G. Conley, E. Gisin, S. Lacy, L. Khibnik, B. Dolan, B. D. Auerbach, R. W. Schecter, S. F. Cooke, and J. Gavornik. This work was supported in part by grants from FRAXA, NIMH, NICHD, and the Simons Foundation.
Footnotes
Competing Interests Statement: Mark Bear discloses a financial interest in Seaside Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bear MF, Huber KM, Warren ST. The mGluR theory of fragile X mental retardation. Trends Neurosci. 2004;27:370–377. doi: 10.1016/j.tins.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E. Epilepsy in fragile X syndrome. Dev Med Child Neurol. 2002;44:724–728. doi: 10.1017/s0012162201002833. [DOI] [PubMed] [Google Scholar]
- Bhakar AL, Dolen G, Bear MF. The Pathophysiology of Fragile X (and What It Teaches Us about Synapses) Annu Rev Neurosci. 2012;35:417–443. doi: 10.1146/annurev-neuro-060909-153138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi R, Chuang SC, Zhao W, Young SR, Wong RK. Cellular plasticity for group I mGluR-mediated epileptogenesis. J Neurosci. 2009;29:3497–3507. doi: 10.1523/JNEUROSCI.5447-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CH, Schoenfeld BP, Bell AJ, Hinchey P, Kollaros M, Gertner MJ, Woo NH, Tranfaglia MR, Bear MF, Zukin RS, et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 2011;1380:106–119. doi: 10.1016/j.brainres.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang SC, Zhao W, Bauchwitz R, Yan Q, Bianchi R, Wong RK. Prolonged epileptiform discharges induced by altered group I metabotropic glutamate receptor-mediated synaptic responses in hippocampal slices of a fragile X mouse model. J Neurosci. 2005;25:8048–8055. doi: 10.1523/JNEUROSCI.1777-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gegg ME, Harry R, Hankey D, Zambarakji H, Pryce G, Baker D, Adamson P, Calder V, Greenwood J. Suppression of autoimmune retinal disease by lovastatin does not require Th2 cytokine induction. J Immunol. 2005;174:2327–2335. doi: 10.4049/jimmunol.174.4.2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein JL, Brown MS. Regulation of the mevalonate pathway. Nature. 1990;343:425–430. doi: 10.1038/343425a0. [DOI] [PubMed] [Google Scholar]
- Hays SA, Huber KM, Gibson JR. Altered neocortical rhythmic activity states in Fmr1 KO mice are due to enhanced mGluR5 signaling and involve changes in excitatory circuitry. J Neurosci. 2011;31:14223–14234. doi: 10.1523/JNEUROSCI.3157-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Gallagher SM, Warren ST, Bear MF. Altered synaptic plasticity in a mouse model of fragile X mental retardation. Proc Natl Acad Sci U S A. 2002;99:7746–7750. doi: 10.1073/pnas.122205699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanner L. Autistic disturbances of affective contact. Nervous Child. 1943;2:217–250. [PubMed] [Google Scholar]
- Kelleher RJ, 3rd, Bear MF. The autistic neuron: troubled translation? Cell. 2008;135:401–406. doi: 10.1016/j.cell.2008.10.017. [DOI] [PubMed] [Google Scholar]
- Kloog Y, Cox AD, Sinensky M. Concepts in Ras-directed therapy. Expert Opin Investig Drugs. 1999;8:2121–2140. doi: 10.1517/13543784.8.12.2121. [DOI] [PubMed] [Google Scholar]
- Krab LC, Goorden SM, Elgersma Y. Oncogenes on my mind: ERK and MTOR signaling in cognitive diseases. Trends Genet. 2008;24:498–510. doi: 10.1016/j.tig.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Lambert M, Lupien PJ, Gagne C, Levy E, Blaichman S, Langlois S, Hayden M, Rose V, Clarke JT, Wolfe BM, et al. Treatment of familial hypercholesterolemia in children and adolescents: effect of lovastatin. Canadian Lovastatin in Children Study Group. Pediatrics. 1996;97:619–628. [PubMed] [Google Scholar]
- Lee JK, Won JS, Singh AK, Singh I. Statin inhibits kainic acid-induced seizure and associated inflammation and hippocampal cell death. Neurosci Lett. 2008;440:260–264. doi: 10.1016/j.neulet.2008.05.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, Cannon TD, Silva AJ. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol. 2005;15:1961–1967. doi: 10.1016/j.cub.2005.09.043. [DOI] [PubMed] [Google Scholar]
- Makovski V, Haklai R, Kloog Y. Farnesylthiosalicylic acid (salirasib) inhibits Rheb in TSC2-null ELT3 cells: a potential treatment for lymphangioleiomyomatosis. Int J Cancer. 2012;130:1420–1429. doi: 10.1002/ijc.26139. [DOI] [PubMed] [Google Scholar]
- Mendola CE, Backer JM. Lovastatin blocks N-ras oncogene-induced neuronal differentiation. Cell Growth Differ. 1990;1:499–502. [PubMed] [Google Scholar]
- Merlin LR, Bergold PJ, Wong RK. Requirement of protein synthesis for group I mGluR-mediated induction of epileptiform discharges. J Neurophysiol. 1998;80:989–993. doi: 10.1152/jn.1998.80.2.989. [DOI] [PubMed] [Google Scholar]
- Musumeci SA, Hagerman RJ, Ferri R, Bosco P, Dalla Bernardina B, Tassinari CA, De Sarro GB, Elia M. Epilepsy and EEG findings in males with fragile X syndrome. Epilepsia. 1999;40:1092–1099. doi: 10.1111/j.1528-1157.1999.tb00824.x. [DOI] [PubMed] [Google Scholar]
- Osterweil EK, Krueger DD, Reinhold K, Bear MF. Hypersensitivity to mGluR5 and ERK1/2 leads to excessive protein synthesis in the hippocampus of a mouse model of fragile X syndrome. J Neurosci. 2010;30:15616–15627. doi: 10.1523/JNEUROSCI.3888-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philpot BD, Sekhar AK, Shouval HZ, Bear MF. Visual experience and deprivation bidirectionally modify the composition and function of NMDA receptors in visual cortex. Neuron. 2001;29:157–169. doi: 10.1016/s0896-6273(01)00187-8. [DOI] [PubMed] [Google Scholar]
- Raisinghani M, Faingold CL. Identification of the requisite brain sites in the neuronal network subserving generalized clonic audiogenic seizures. Brain Res. 2003;967:113–122. doi: 10.1016/s0006-8993(02)04232-4. [DOI] [PubMed] [Google Scholar]
- Ramirez C, Tercero I, Pineda A, Burgos JS. Simvastatin is the statin that most efficiently protects against kainate-induced excitotoxicity and memory impairment. J Alzheimers Dis. 2011;24:161–174. doi: 10.3233/JAD-2010-101653. [DOI] [PubMed] [Google Scholar]
- Schafer WR, Kim R, Sterne R, Thorner J, Kim SH, Rine J. Genetic and pharmacological suppression of oncogenic mutations in ras genes of yeast and humans. Science. 1989;245:379–385. doi: 10.1126/science.2569235. [DOI] [PubMed] [Google Scholar]
- Serbanescu I, Ryan MA, Shukla R, Cortez MA, Snead OC, 3rd, Cunnane SC. Lovastatin exacerbates atypical absence seizures with only minimal effects on brain sterols. J Lipid Res. 2004;45:2038–2043. doi: 10.1194/jlr.M400097-JLR200. [DOI] [PubMed] [Google Scholar]
- Taylor GW, Merlin LR, Wong RK. Synchronized oscillations in hippocampal CA3 neurons induced by metabotropic glutamate receptor activation. J Neurosci. 1995;15:8039–8052. doi: 10.1523/JNEUROSCI.15-12-08039.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuchman R, Cuccaro M, Alessandri M. Autism and epilepsy: historical perspective. Brain Dev. 2010;32:709–718. doi: 10.1016/j.braindev.2010.04.008. [DOI] [PubMed] [Google Scholar]
- van Vliet EA, Holtman L, Aronica E, Schmitz LJ, Wadman WJ, Gorter JA. Atorvastatin treatment during epileptogenesis in a rat model for temporal lobe epilepsy. Epilepsia. 2011;52:1319–1330. doi: 10.1111/j.1528-1167.2011.03073.x. [DOI] [PubMed] [Google Scholar]
- Weisz B, Giehl K, Gana-Weisz M, Egozi Y, Ben-Baruch G, Marciano D, Gierschik P, Kloog Y. A new functional Ras antagonist inhibits human pancreatic tumor growth in nude mice. Oncogene. 1999;18:2579–2588. doi: 10.1038/sj.onc.1202602. [DOI] [PubMed] [Google Scholar]
- Xie C, Sun J, Qiao W, Lu D, Wei L, Na M, Song Y, Hou X, Lin Z. Administration of simvastatin after kainic acid-induced status epilepticus restrains chronic temporal lobe epilepsy. PLoS One. 2011;6:e24966. doi: 10.1371/journal.pone.0024966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Shinnoh N, Taniwaki T, Ohyagi Y, Asahara H, Horiuchi, Kira J. Lovastatin does not correct the accumulation of very long-chain fatty acids in tissues of adrenoleukodystrophy protein-deficient mice. J Inherit Metab Dis. 2000;23:607–614. doi: 10.1023/a:1005634130286. [DOI] [PubMed] [Google Scholar]
- Yan QJ, Asafo-Adjei PK, Arnold HM, Brown RE, Bauchwitz RP. A phenotypic and molecular characterization of the fmr1-tm1Cgr fragile X mouse. Genes Brain Behav. 2004;3:337–359. doi: 10.1111/j.1601-183X.2004.00087.x. [DOI] [PubMed] [Google Scholar]
- Yan QJ, Rammal M, Tranfaglia M, Bauchwitz RP. Suppression of two major Fragile X Syndrome mouse model phenotypes by the mGluR5 antagonist MPEP. Neuropharmacology. 2005;49:1053–1066. doi: 10.1016/j.neuropharm.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Zhao W, Bianchi R, Wang M, Wong RK. Extracellular signal-regulated kinase 1/2 is required for the induction of group I metabotropic glutamate receptor-mediated epileptiform discharges. J Neurosci. 2004;24:76–84. doi: 10.1523/JNEUROSCI.4515-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.