

Abstract
Many areas of biochemistry and molecular biology, both fundamental and applications-orientated, require an accurate construction, representation and understanding of the protein molecular surface and its interaction with other, usually small, molecules. There are however many situations when the protein molecular surface gets in physical contact with larger objects, either biological, such as membranes, or artificial, such as nanoparticles. The contribution presents a methodology for describing and quantifying the molecular properties of proteins, by geometrical and physico-chemical mapping of the molecular surfaces, with several analytical relationships being proposed for molecular surface properties. The relevance of the molecular surface-derived properties has been demonstrated through the calculation of the statistical strength of the prediction of protein adsorption. It is expected that the extension of this methodology to other phenomena involving proteins near solid surfaces, in particular the protein interaction with nanoparticles, will result in important benefits in the understanding and design of protein-specific solid surfaces.
Introduction
Many areas of biochemistry and molecular biology, both fundamental, e.g., protein folding [1], protein conformational stability [2], inter- and intra- protein interactions [3], molecular recognition [4] and docking [5]; as well as applications-orientated, e.g., drug design [6], [7], protein and peptide solubility [8], crystal packing [9], and enzyme catalysis [10]; require an accurate construction, representation and understanding of the protein molecular surface and its interaction with other, usually small, molecules.
The applications enumerated above, almost exclusively focused on biomolecular interactions, necessitate the construction of the molecular surface at a resolution scale similar to the size of the molecule that interacts with the protein, e.g., up to 5Å, which is approximately the dimension of a large solvent molecule. There are however many situations when the protein molecular surface is in physical contact with larger objects, either biological or artificial. For instance, many biomolecular interactions occur on cell membranes, e.g., involving lipid rafts with sizes much larger than that of the water molecule [11]. Also, the long range self-assembly of proteins, e.g., cytoskeleton formation [12], formation of amyloid plaques and tangles [13], occurs through biomolecular recognition of larger areas on the molecular surface. Biomolecules also interact with solid surfaces on which they are immobilized, either by design, or unintentionally [14], [15], for applications as diverse as biomaterials [14], [16], chromatography [17] membrane research [18], [19], biomedical micro- and nano-devices [20], [21], such as biosensors [22], microarrays [23], [24] and lab-on-a-chip devices [25], where the preservation of the bioactivity of the immobilized proteins is paramount. More recently, nanoparticle research has become interested in the study of the interaction between proteins and artificial objects similar with their size, or larger [26]. Indeed, the nanoparticle:protein interaction can either amplify the beneficial effects of nanoparticles, e.g., protein aggregation around a nanoparticle can create a ‘protein corona’ [27], [28], which could be essential for the nanoparticle uptake in the cell, where its therapeutic action can unfold [29]; or conversely, it can induce the change of conformation and consequently the bioactivity of the proteins attached to the nanoparticle [30], [31], thus cascading in nanoparticle-induced nanotoxicity [32], [33].
The probing of the protein molecular surface with probes with larger radii has also fundamental motivations. The general consensus regarding the protein structure is based on the concept of the “hydrophobic core,” which states that the hydrophobic amino acids aggregate, via hydrophobic-hydrophobic attraction, towards the core of the protein, leaving the outbound protein sheath more hydrophilic. This central concept needs constant and thorough qualification, as proteins have extremely diverse and complex geometries. Recently, several reports [34]–[36], which tested the “hydrophobic core” concept using fractal analysis, found that all the major structural classes of proteins have an amount of ‘unused’ hydrophobicity, thus showing that they are not as optimally packed as they are supposed to be. The representation of biomolecular surfaces, especially for proteins, encounters serious difficulties, even for the simplest globular proteins, due to the complexity, lack of symmetry and irregularity of the distribution of atoms. The construction of the protein molecular surfaces uses its biomolecular structure [37], revealed through X-ray crystallography or NMR studies and archived in databases, such as Protein Database, PDB [38]. The first algorithms employed for the construction of molecular surfaces [39]–[41] used virtual probes to determine the position of the points of contact with protein atoms, thus generating an ‘envelope’ of the protein, which is a representation of its molecular surface. While other algorithms [4], [42]–[62] for the construction of molecular surfaces are reported to be more computer resources-efficient, the use of virtual probes has the conceptual advantage of being more intuitive, as the in silico probing mimics specific or non-specific biomolecular events.
The ubiquity and importance of these interactions, for reasons that are both fundamental and industrial, e.g., pharmaceutical, biomaterials, biomedical devices, suggest that the accurate representation of protein molecular surfaces as probed by probes with large dimensions is fully warranted. To this end, the aim of this contribution is to assess the impact of the probing resolution on the construction of protein molecular surfaces; demonstrate the benefits of this approach for the understanding of the interaction between proteins and large nano-objects; and propose new research avenues that can capitalize on this methodology.
Methods
Proteins
The structures of 35 proteins (Table 1) have been selected from the Protein Data Bank [38]. The set spans several representative sets of proteins, namely lactalbumin (dataset 1, in Table 1), lactoglobulin (dataset 2), lysozyme (dataset 3), ribonuclease (dataset 4), hemoglobin (dataset 5), albumin (dataset 6) and antibody (dataset 7). The protein datasets also cover a large range of molecular weights (14 to 148 kDa), residues (123 to 1344), isoelectric points (4.5 to 11) and shapes (globular, ellipsoidal, Y-shaped). Five representative proteins, i.e., lysozyme, ribonuclease, hemoglobin, albumin and IgG (in bold in Table 1) have been selected for graphical presentations. Complete results, with conclusions in accord with those for the selected proteins, are presented in File S1.
Table 1. Proteins used for the analysis of molecular surfaces.
| Dataset No. RMSD(residues) | No. | Protein name | PDB code | Atoms | Residues | Chains |
| 1 | 1 | α lactalbumin | 1A4V | 1092 | 123 | 1 |
| 2 | 2 | porcine β-lactoglobulin | 1EXS | 1248 | 160 | 1 |
| 3 | bovine β-lactoglobulin | 1BEB | 2473 | 324 | 2 | |
| 3, Subset 3.1 RMSD(129) = 0.6481 | 4 | hen egg-white lysozyme | 1LYZ | 1001 | 129 | 1 |
| 5 | turkey egg-white lysozyme | 135L | 994 | 129 | 1 | |
| 6 | hen egg-white lysozyme | 2LYM | 1001 | 129 | 1 | |
| 7 | triciclic lysozyme | 2LZT | 1001 | 129 | 1 | |
| 3, Subset 3.2 RMSD(164) = 0.24 | 8 | mutant of phage T4 lysozyme | 1L35 | 1305 | 164 | 1 |
| 9 | T4 lysozyme | 1LYD | 1309 | 164 | 1 | |
| 4 RMSD(124) = 0.1655 | 10 | ribonuclease-A | 8RAT | 951 | 124 | 1 |
| 11 | ribonuclease-A | 1RBX | 956 | 124 | 1 | |
| 12 | bovine ribonuclease-A | 3RN3 | 957 | 124 | 1 | |
| 13 | ribonuclease-A | 1AFU | 1894 | 248 | 2 | |
| 5, Subset 5.1 RMSD(287) = 0.8877 | 14 | human oxyhemoglobin | 1HHO | 2192 | 287 | 2 |
| 15 | human carbonmonoxy hemoglobin | 2HCO | 2192 | 287 | 2 | |
| 16 | horse deoxyhemoglobin | 2DHB | 2201 | 287 | 2 | |
| 5, Subset 5.2 RMSD(574) = 1.501 | 17 | human hemoglobin A | 1BUW | 4342 | 574 | 4 |
| 18 | human hemoglobin (W37A) | 1Y4F | 4368 | 574 | 4 | |
| 19 | hemoglobin mutant (W37A) | 1A01 | 4368 | 574 | 4 | |
| 20 | human hemoglobin (W37E) | 1Y4P | 4376 | 574 | 4 | |
| 21 | hemoglobin mutant (W37Y) | 1A00 | 4382 | 574 | 4 | |
| 22 | human hemoglobin (W37Y) | 1Y46 | 4382 | 574 | 4 | |
| 23 | human deoxyhemoglobin | 2HHB | 4384 | 574 | 4 | |
| 24 | human hemoglobin (W37G) | 1Y4G | 4366 | 574 | 4 | |
| 25 | hemoglobin mutant (V1M) | 1A0U | 4386 | 574 | 4 | |
| 26 | hemoglobin mutant (V1M) | 1A0Z | 4386 | 574 | 4 | |
| 27 | recombinant hemoglobin | 1C7D | 4396 | 576 | 3 | |
| 6 RMSD(585) = 2.3740 | 28 | human serum albumin complexed with octadecanoic acid | 1E7I | 4496 | 585 | 1 |
| 29 | recombinant human serum albumin | 1UOR | 4617 | 585 | 1 | |
| 30 | human serum albumin | 1E78 | 4302 | 585 | 1 | |
| 31 | human serum albumin | 1AO6 | 4600 | 585 | 1 | |
| 32 | human serum albumin | 1BM0 | 4600 | 585 | 1 | |
| 7 | 33 | immunoglobulin | 1IGY | 10002 | 1294 | 4 |
| 34 | immunoglobulin | 1IGT | 10196 | 1316 | 4 | |
| 35 | intact human IgG B12 | 1HZH | 10355 | 1344 | 4 |
To quantify the similarity between the members of a class, for all sets, or subsets, i.e., for lysozyme and hemoglobins, the Root-Mean-Square Deviation (RMSD) has been calculated using the protein structure comparison service Fold at the European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm).[63]
A subset of the hemoglobin class,[64] comprising eight mutant structures of the deoxy forms of the protein, with the same number of residues (574), but with (i) the Trp37 residue, i.e., 1A0U and 1A0Z, for the crystal form 1 and 2, respectively; and with residues replacing the Trp37 residue by (ii) Tyr37, i.e., structures 1Y46 and 1A00, for crystal 1 and 2, respectively; (iii) Ala37, i.e., 1Y4F and 1A01, for crystal form 1 and 3, respectively; (iv) Glu37, i.e., 1Y4P, for crystal form 1; and (v) Gly37, i.e., 1Y4G for crystal form 1; was used to test the fine definition of the molecular surface for very similar proteins. This sub-set is indicated in bold in Table 1. The full results regarding the quantification of properties on the molecular surfaces are presented in File S2 and the results regarding the quantitative measure of the sequence (sequence identity) and structural (RMSD after structural alignment where possible) are also presented in File S3.
Treatment of charges
The charges of the atoms in amino acids have been calculated applying a semi empirical method (PM3, as implemented in HyperChem, from HyperCube Inc.) on model tripeptides Gly-X-Gly (where X is the respective amino acid), following a geometry optimization step that used a molecular mechanics force field (Amber, as implemented in HyperChem). The charges have been calculated for the whole range of pH in 0.1 pH increments, assuming the ionized, or non-ionized structures at the respective pKa (calculated as implemented in Discovery Studio software, from Accelerys Inc.) of the side chains and interpolating the charges along the pH scale according to acid-base equilibrium relationships. The charge-related molecular surface properties have been calculated for each tested protein at its isoelectric point. The values of the calculated charges for each amino acid versus pH, as well as a detailed explanation of their calculation, are presented in File S4 and File S5.
Hydrophobicity
Among the many hydrophobicity scales proposed in the literature, the present analysis used the hydrophobicity as defined by Wimley and White [65]. Briefly, the hydrophobic character, or lack of, of an amino acid, is estimated by the enthalpy of the transfer of a peptide through a lipid membrane (ΔGwm), calculated from the thermodynamic measurements of the actual transfer of model penta-peptides that have embedded the amino acid of interest. Both hydrophobicity and hydrophilicity have been calculated, and the protein amphiphilicity is the algebraic sum of hydrophobicity, expressed in negative numbers, and hydrophilicity, expressed in positive numbers.
Molecular surfaces
Because of its conceptual benefits, i.e., the virtual probing of the molecular surfaces mimics the actual contact between the protein and a real object, the original Connolly’s algorithm [39]-[41] has been used to construct protein molecular surfaces. The algorithm has been upgraded to record the geometry of the molecular surface, protein amphiphilicity, hydrophobicity, hydrophilicity and charges, both positive and negative. Briefly, the algorithm records the position of the points of contact between a virtual rolling probing ball with a set radius and the atoms on the molecular surface of the protein, or alternatively the points placed at a distance equivalent to the van der Waals radius of the respective atoms.
The spatial distribution of the amphilicity, hydrophobicity and hydrophilicity on the protein molecular surface was determined through the allocation, at the point of contact, of the value for the respective amino acid, weighted with the ratio of the probed surface per the total area of the amino acid. A similar procedure was used for mapping the spatial distribution of the charges. The procedure involves the allocation of the specific charge weighted with the ratio between the probed atomic area and the total atomic area. The procedure is similar to the one used by Scarsi et al. [60], with the difference that only the actual property is recorded, instead of the interaction with the probe and that the charges are also accounted for.
The probing of the protein molecular surface was performed with probe with increasing radii, from 1.4 Å to 20 Å, because beyond a certain value of the radii the variation of the properties is negligible; and because, for flat solid surfaces, the actual real solid radius of an engineering grade-flat solid surface is close to this value [66]. The increase of the probe radii results in a large ratio of the area created due to the re-entry points of the probe and the overall molecular surface. Because our analysis uses the quantification of physico-chemical properties on the molecular surface at the points of contact, we used only the contact area in our calculations and graphical representation.
The calculations were run using Connolly’s original software code [39] upgraded for the quantification of physico-chemical properties and with a Windows Graphical User Interface. The 4D points (the x,y,z coordinates and the molecular property) were visualized using DS Viewer Pro (from Accelerys Inc.).
Protein properties on the molecular surface
The characterization of the protein molecular surface requires the quantification of several properties on the molecular surface: (i) global properties, namely, total surface, total charges and total amphiphilicity, hydrophobicity and hydrophilicity, as well as the area-per-volume ratio; (ii) property surface densities, namely, charge, amphiphilic, hydrophobic and hydrophilic density, calculated by dividing the respective total property to the total biomolecular area; and (iii) property specific surface densities, calculated as in (ii), but dividing the respective property, e.g. positive charge, to the area that property turns up, e.g., positive charged area. A synthetic view of all parameters is presented in Table 2.
Table 2. Definition of the properties measured on the protein molecular surface.
| No. | Property | Symbol | Method of calculation | Units | |
| 1. Global properties | |||||
| 1.1 | Molecular weight | Mw | PDB | Da | |
| 1.2 | Total number of atoms | Na | PDB | - | |
| 1.3 | Total number of residues | Nr | PDB | - | |
| 1.4 | Total probed area | A | Connolly | Å2 | |
| 1.5 | Volume | V | Eq. 2 | Å3 | |
| 1.6 | Shape factor | V/A | Eq. 2 | Å | |
| 1.7 | Total positive charge | PC_t | Connolly upgrade | e | |
| 1.8 | Total negative charge | NC_t | Connolly upgrade | e | |
| 1.9 | Total hydrophilicity | Phi_t | Connolly upgrade | kcal mol-1 | |
| 1.10 | Total hydrophobicity | Pho_t | Connolly upgrade | kcal mol-1 | |
| 1.11 | Total positively charged area | A_pc | Connolly upgrade | Å2 | |
| 1.12 | Total negatively charged area | A_nc | Connolly upgrade | Å2 | |
| 1.13 | Total hydrophilic area | A_phi | Connolly upgrade | Å2 | |
| 1.14 | Total hydrophobic area | A_pho | Connolly upgrade | Å2 | |
| 2. Property surface densities | |||||
| 2.1 | Positive charge density | PC_d | PC_t/A | e Å-2 | |
| 2.2 | Negative charge density | NC_d | NC_t/A | e Å-2 | |
| 2.3 | Hydrophilic density | Phi_d | Phi_t/A | kcal mol-1Å-2 | |
| 2.4 | Hydrophobic density | Pho_d | Pho_t/A | kcal mol-1Å-2 | |
| 3. Property specific densities | |||||
| 3.1 | Positive charge specific density | PC_sd | PC_t/A_pc | e Å-2 | |
| 3.2 | Negative charge specific density | NC_sd | NC_t/A_nc | e Å-2 | |
| 3.3 | Hydrophilic specific density | Phi_sd | Phi_t/A_phi | kcal mol-1Å-2 | |
| 3.4 | Hydrophobic specific density | Pho_sd | Pho_t/A_pho | kcal mol-1Å-2 | |
Statistical correlation between molecular surface properties and protein interfacial processes
The statistical strength of the correlation between the protein surface concentration on various solid surfaces and the respective protein physico-chemical parameters was firstly estimated by the Pearson Product-Moment Correlation Coefficient (PPMCC), as implemented in the Statistica software, from StatSoft Inc. The protein parameters taken into consideration, calculated on the protein molecular surface, as well as comprising the totality of the residues, were amphiphilicity, hydrophobicity and hydrophilicity, and their derived surface densities.
The PPMCC calculation was applied to a reduced set of proteins out of the initial set of 35, i.e., the five model proteins mentioned above plus α lactalbumin and β lactoglobulin (in italics in Table 1) for which comprehensive data regarding protein adsorption could be found in the Biomolecular Adsorption Database (BAD)[25], totaling 279 valid data points. PPMCC was calculated for all data points, and separately for hydrophilic and hydrophobic solid surfaces. The amphiphilicity of solid surfaces is usually quantified by the contact angle of a small (1µl) water droplet, which is the angle made by the intersection of the contour of the gas/liquid interface with the solid surface. While in general solid surfaces are considered hydrophobic if exhibit contact angles above 90°, in the particular case of protein adsorption the adsorbing solid surfaces are hydrophobic for contact angles above 45°,[25] with those below considered hydrophilic. With this threshold, the protein adsorption data for hydrophilic solid surfaces comprises 172 data points and for hydrophobic solid surfaces comprise 107 data points.
A piecewise, multilinear regression procedure with breakpoint, reported before [25], was applied to all data points for both hydrophobic and hydrophilic solid surfaces, as well as separately for the two subsets, i.e., hydrophobic and hydrophilic solid surfaces, respectively. The regression provided a measure of the correlation between the output variable, i.e., protein concentration on adsorbing solid surfaces; and sets of input variables comprising (i) protein concentration in solution; (ii) solid surface amphiphilicity measured by the respective contact angle; (iii) buffer parameters, i.e., pH, ionic strength; (iv) global bulk parameters of the protein, i.e., isoelectric point, molecular weight; (v) global molecular surface of the protein, i.e., molecular area, surface-to-volume ratio; (vi) hydrophobicity parameters derived from the probing of the molecular surface, i.e., hydrophobicity density, hydrophobicity specific density, and ratio between hydrophobicity and hydrophilicity, all derived for different probing radii; and (vii) charge parameters derived from the probing the molecular surface, i.e., positive charge density, positive charge specific density, and ratio between positive and negative charge, all derived for different probing radii.
Results and Discussion
Areas and surface-to-volume ratio of the protein
Many biomolecules, in particular proteins, are similar in size with the nanostructures present on artificial or natural surfaces, or with nano-objects, e.g., nanoparticles. Figure 1 represents a brief comparison between the molecular surface of five proteins with different sizes, with several examples of artificial nano-objects, either ‘flat’ solid surfaces or particles. The following discussion will focus on five representative proteins, i.e., lysozyme, ribonuclease, hemoglobin, albumin and IgG, which have very different molecular weights, i.e., from 129 to 1344 residues (Table 1, in bold); and shapes, i.e., globular, ellipsoidal and Y-shaped.
Figure 1. Molecular surface of several proteins (middle panel) and several artificial nano-surfaces and objects.

The amphiphilic (blue – hydrophilic; red - hydrophobic) molecular surface is mapped at 1.4 Å (water molecule dimensions) geometrical resolution, for five proteins, from left to right and top to bottom: lysozyme, ribonuclease; hemoglobin; IgG and albumin. The artificial surfaces are as follows: Top left: TEM image (side view) of a defect propagating from layer to layer in otherwise perfectly flat Pt/Rh multi-layered surface; feature: 50×100 Å. Top right: TEM image of gold nanoparticles; features: 10–25 Å. Bottom left: SEM image of a set of SiO2 pillars with gold caps; features: 150 – 300 Å. Bottom right: SEM image of SiO2 nano-wires grown from vapor phase; minimum feature: sub-500 Å.
As it can be easily inferred from Figure 1, and as in a classical fractality problem, the shape and extent of the molecular surface area depend, on one hand, on the characteristics of the molecule, e.g., structure, number of atoms; and, on the other, on the radius of the probing ball, be that virtual or real. Figure 2 presents a schematic view of the evolution of the constructed molecular surface as a function of the probe radius.
Figure 2. Schematic of different molecular surfaces obtained when probing a protein with different probe radii.
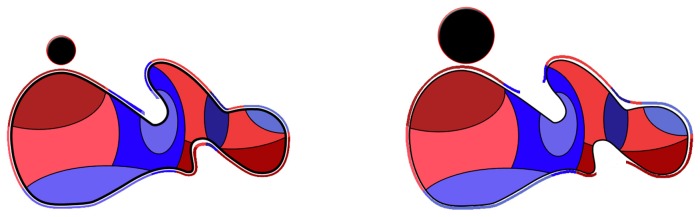
Larger probes (right) cannot visit some inner areas of the protein, as well as some parts of the residues.
The probing of the molecular surface creates non-contiguous surfaces, especially at large radii, as explained in Figure 2. Alternatively, holes can be the result of the actual structure of the protein, independently of the size of the probe radius, as presented in Figure 1.
Figure 3 presents the quantification of the area of the molecular surface and the surface-to-volume ratio for different probe radii for the five chosen representative proteins, i.e., lysozyme (1LYZ), ribonuclease (1AFU), hemoglobin (1Y4F), albumin (1AO6) and IgG (1HZH). These proteins have vastly different molecular weights, i.e., from 129 to 1344 residues (Table 1, in bold).
Figure 3. Variation of the molecular surface area and surface-to-volume ratio with the radius of the probe.

Molecular surface area (top) and surface-to-volume (bottom) for five model proteins: 1LZY = lysozyme; 1AFU = ribonuclease-A; 1Y4F = human hemoglobin; 1AO6 = human serum albumin; and 1HZH = intact human IgG.
The probed area decreases monotonically with the increase of the probe radius, but after a certain radius value, which depends on the size of the protein, it reaches a plateau. The relative decrease of the probed area is more pronounced for large proteins. For instance, at the plateau, the total area decreases to about 40% and 25%, for IgG (1HZH) and for albumin (1AO6), respectively, compared to their area obtained through the contact with a probe with a 1.4Å radius. In the first approximation, the probe radius after which the protein probed area does not vary substantially is slightly larger than the largest distance between two atoms of the respective protein, i.e., approximately 60 Å and 150 Å, for 40 Å-large lysozyme (1LYZ) and 100-120 Å-large IgG (1HZH) or albumin (1AO6), respectively. Most proteins larger than 50kDa usually form two or more domains independently folded [67]. The evolution from single to several globular domains with the increase of the molecular weight of the protein leads to an increase in the roughness of the molecular surface, rather than the change of its overall shape.[68] There are however proteins (not considered here) which exhibit highly elongated shapes, e.g., fibrinogen, but other very large proteins present specialized structures such as coiled coils, e.g., myosin with its very convoluted (and dynamic) shape[69] or the collagen triple helix.[67] Therefore, the difference in the evolution of the decrease of the probed area versus probe radius for proteins with different sizes appears to be the result of either the increased roughness of the molecular surface, or the departure from the globular shape (e.g., for IgG).
For the set of proteins studied here, the molecular surface area can be estimated with very good statistical quality (R2 = 0.98) as a function of its molecular weight (or number of atoms) and the probe radius, as follows:
| (1) |
where A is the probed area on the protein molecular surface (Å2); N is the number of atoms in the protein; R is the probe radius (Å); and a, b and c are fitting constants, which have values, for the set of 35 proteins considered, of a = 4.36; b = 0.95; c = 0.165. This relationship (Eq. 1) predicts that for a very large probe radius, i.e., R→∞, which is equivalent to a flat surface, the protein probed area is nearly proportional (i.e., c/R→0; 0.95<b<1) with the number of atoms in the protein, or by extension, to its molecular weight. In reality, engineering-grade-flat solid surfaces exhibit nanometer-range roughness, e.g., with features of around 20 Å[66].
Surface-to-volume ratio
Because the probing of the molecular surface, when performed with probes with large radii, generates non-contiguous molecular surfaces, the actual volume of the protein has to be calculated as a sum of the volumes of the constituent atoms, rather than the volume inside the molecular surface. Consequently, the molecular surface-area-to-volume ratio, or simply the surface-to-volume ratio is given by
| (2) |
where V is the volume of the protein, proportional with the number of atoms, N; v is the average atomic volume of the protein atoms[67]; and a, b and c are the constants in Eq. 1. A consequence of Eq. 2 is that the surface-to-volume ratio of larger proteins will decrease more with the increase of the probe radius than that of smaller proteins. Figure 3 (bottom) presents the evolution of the surface-to-volume versus the probe radius for five representative model proteins.
The graphical representation of the molecular surface (Figure 1 and an example for ribonuclease in Figure 4) allows for some qualitative considerations. For simple, globular, small-to-medium proteins, e.g., lysozyme (1LYZ), ribonuclease (1AFU), the use of small radii, e.g., 1.4 Å, results in contiguous molecular surfaces with distinct negative and positive patches (red and blue, respectively, in Figure 1, middle top). Conversely, for large proteins with more complex shapes, e.g., IgG (1HZH), (Figure 1, middle left) the probing with small probes will result in non-contiguous molecular surfaces. Eventually, the use of probes with larger radii results in non-contiguous molecular surfaces even for smaller proteins, e.g., ribonuclease (Figure 4), and the subsequent decrease of the exposed area.
Figure 4. Physico-chemical properties of ribonuclease represented on its molecular surface.

The probing is performed with increasingly large probe radius (from left to right), as charges (top row, red = negative, blue = positive), and amphiphilicity (bottom row; blue = hydrophilic; red = hydrophilic).
Significance
The above results and discussion lead to the following partial conclusions.
Above a certain roughness of the solid surface that interacts with a protein, i.e., in the range of 10–20 Å, the contact area of the protein with the solid surface, which is proportional with the molecular weight of the protein, remains constant versus the probe radius. Consequently, for applications seeking the amplification of the protein-solid surface interactions, assumed to be proportional with the contact area between the protein and the solid surface, e.g., liquid chromatography, and notwithstanding the flexibility of the protein (to be briefly discussed later), negligible effects are expected for a solid surface roughness above 10 Å for small globular proteins and 20 Å for large proteins.
The contact area represents only a fraction of the total protein molecular surface and this fraction is much smaller for large probe radii. As hydrophobic amino acids have the propensity to aggregate towards the center of the protein[70] , it follows that the contact area of the probe, be that virtual or real, with the “hydrophobic core” will decrease with the increase of the probe radii. Consequently, very flat hydrophobic solid surfaces are expected to be inefficient for hydrophobicity-controlled protein immobilization; or, alternatively, they will induce important conformational changes of the proteins if the hydrophobic solid surface reaches the contact with the hydrophobic amino acids localized inside the protein core.
Amphiphilicity and charge on the molecular surface
Amphiphilicity and charges on the molecular surfaces
A full portrayal of the protein molecular surface entails both the geometrical position of the points of contact and the description of the physico-chemical parameters at those positions. Qualitative, yet insightful observations can be gathered from the inspection of the representation of the charges and amphiphilicity on the protein molecular surfaces, as a function of the probe radius, such as presented for ribonuclease in Figure 4. The charged molecular surface comprises largely, but not exclusively, negative charges, due to the more exposed oxygen atoms in carboxy groups. This propensity decreases, relatively, with the increase of the radius of the probe (Figure 4, top row), as the contiguous negative charged molecular surface is ruptured due to the impossibility of larger probes to reach the negatively charged regions towards the core, e.g., negative oxygen atoms in amide groups; while the positively charged areas of the few amino groups placed away from the protein core remain largely unchanged. Conversely, the molecular surfaces (Figure 4, bottom row) remain largely, and evenly, hydrophilic with the increase of the probe radius, as the probe will touch atoms with amphilicities assigned according to their parent amino acid, which those that are hydrophilic predominantly placed away from the protein core.
Hydrophobic and negatively charged areas
This qualitative description is supported by quantitative data, presented in Figure 5 for the five model proteins discussed before. The full account of the calculations is presented in File S1.
Figure 5. Hydrophobicity-related areas; and negatively charged-related areas modulated by the probe radius.

Left: hydrophobic area (a1, top); ratio of hydrophobic per total area (a2, middle); and relative decrease of the hydrophobic area, reported to its maximum extent at minimum probe radius, (a3, bottom) for five model proteins: 1LZY = lysozyme; 1AFU = ribonuclease-A; 1Y4F = human hemoglobin; 1AO6 = human serum albumin; and 1HZH = intact human IgG. Right: negatively charged area (b1, top); ratio of negatively charged per total area (b2, middle); and relative decrease of the negatively charged area, reported to its maximum extent at minimum probe radius (b3, bottom) for the same model proteins.
The overall hydrophobic and the negatively charged areas (Figure 5a1 and 5b1, respectively; logarithmic scales) decrease, as expected, with the increase of the probe radius, similarly with the decrease of the overall area (Figure 3 top). Interestingly, the ratio of hydrophobic-to-total area (Figure 5a2; logarithmic scale) remains nearly constant for IgG, and, to a lesser extent, for ribonuclease and hemoglobin, with albumin and lysozyme decreasing constantly. Conversely, the ratio of negatively charged-to-total area (Figure 5b2) presents a monotonic decrease, and in a much tighter range than hydrophobicity. Finally, both the ratio of the hydrophobic and negatively charged areas divided to their maximum value (at 1.4Å) with the increase of the probe radius, as presented in Figures 5a3 and 5b3, respectively, present a monotonic decrease, with the evolution of charges more protein-specific than that of hydrophobicity.
Amphiphilic, hydrophobic, hydrophilic and charge densities
The variation of the densities of the physico-chemical properties, i.e., amphiphilicity, hydrophobicity, hydrophilicity; and total, negative and positive charges, with the probe radius (Figure 6) provides a finer protein-specific analysis.
Figure 6. Amphiphilic, hydrophobic and hydrophilic densities; and total, positive and negative densities modulated by the probe radius.

Left: Impact of the probe radius on the amphiphilic (a1, top), hydrophobic (a2, second from the top) and hydrophilic (a3, third from the top) densities, i.e., reported to the total area of the protein; and of the hydrophobic (a4, forth from the top) and hydrophilic (a5, fifth from the top) specific densities, i.e., reported to their respective areas for five model proteins: 1LZY = lysozyme; 1AFU = ribonuclease-A; 1Y4F = human hemoglobin; 1AO6 = human serum albumin; and 1HZH = intact human IgG. Right: Impact of the probe radius on the total (b1, top), positive (b2, second from top), and negative (b3, third from top) densities, reported to the total area of the protein; and of positive (b4, forth from top), and negative (b5, fifth from top) specific density, reported to their respective areas for the same five model proteins.
In general, the amphiphilic density (Figure 6a1) is increasing, mildly, with the increase of the probe radius, after a threshold around 3Å, which is equivalent to more hydrophilic areas being exposed by larger probes compared to smaller ones. While following this general trend, albumin (1AO6) presents however a large increase of the amphiphilic density with the probe radius, which could explain the very good blocking, protein-repelling properties of this protein. The evolution of the hydrophobic density with the probe radius (Figure 6a2) is more protein-specific. Indeed, the hydrophobic density of IgG (1HZH) and ribonuclease (1AFU) remain constant, the hydrophobic density of albumin (1AO6) and hemoglobin (1Y4F) decrease steeply until approximately 3Å after which remain constant, and finally the hydrophobic density of lysozyme presents a monotonic decrease with the probe radius. Interestingly, the hydrophobic density of IgG (1HZH) remains four times higher than that of all other proteins, thus explaining its propensity for adsorption on solid surfaces. Because it represents a large proportion of the overall amphiphilicity, the evolution of the hydrophilic density (Figure 6a3) presents similarities with that of amphiphilic densities. These findings indicate that the ‘hydrophobic core’ concept is in general valid, as inferred from the increase of the amphiphilic (and hydrophilic) density with the increase of the probe radius. The level of protection of the hydrophobic core from the exposure to probes as a function of their size varies largely from protein to protein: (i) small, globular proteins are gradually exposing less hydrophobic regions to gradually larger probes; (ii) large proteins with pseudo-globular shapes stop becoming less hydrophobic for probe diameters similar to the protein size; and (iii) large protein with non-globular shapes present low level of protection of the hydrophobic core.
In contrast with the evolution of amphiphilic density (Figure 6a1), the total charge density (Figure 6b1) is decreasing monotonically towards zero with the increase of the probe radius, , which is equivalent to less overall charged areas being visited by large probes than by small ones. While it would be expected that an increase of the amphiphilic density would by linked to an increase in the charging of the respective areas, this apparent contradiction can be understood observing separately the evolution of the constitutive elements, i.e., the positive and negative charge density.
Indeed, the positive charge densities (Figure 6b2), presents a similar evolution with the hydrophilic density, i.e., a gradual increase of positive charge density with the size of the probe radius. On the other hand, with one exception (albumin) the evolution of the negative charge density is rather uneventful, with only very small decreases of the density. As it was the case before, albumin presents an exceptional increase of both positive and negative charge density, which can explain its exceptional evolution with regard to hydrophilic density. The evolution of the property specific density, i.e., the hydrophobic, hydrophilic, positive and negative charge values quantified on the molecular surfaces, then divided by their specific areas, offer a different perspective into the variation of physico-chemical properties on the protein molecular surfaces. With few notable exceptions, the hydrophobic and hydrophilic specific densities (Figure 6a4 and Figure 6a5) do not vary substantially. However, mirroring the evolution of the respective densities, IgG (1HZH) presents an increase, albeit moderate, of its hydrophobic specific density; and albumin (1AO6) presents a considerable increase of its hydrophilic specific density. The positive and negative charge specific density (Figure 6b4 and 6b5) replicate the evolution of their overall counterparts, including the exceptional behavior of albumin.
Molecular surfaces of single-point mutants
The high-resolution X-ray structures of the deoxy forms of four recombinant hemoglobins in which a Trp residue has been replaced with Tyr, Ala, Glu, or Gly, have been reported[64] and recently the structures have further refined.[71] As it was found that no significant mutation-induced changes in tertiary structure were detected, we used this restricted sub-dataset to test the fine definition of the molecular surface for very similar proteins. The evolution of the hydrophobicity and charges on the respective molecular surfaces modulated by the variation of the probing radius is represented in Figure 7 as ratios between various properties. Interestingly, the evolution of the ratios of hydrophobic/hydrophilic areas and hydrophobicity/hydrophilicity are essentially indistinguishable for the hemoglobin structures considered. In contrast, the ratios of positive/negative areas and that positive/negative charges are quite different from one hemoglobin structure to another, especially for the latter. This difference between the hydrophobicity- and charge-based ratios suggests that atom-based properties have the potential of describing more specifically the molecular surface than amino acid-based properties.
Figure 7. Ratio of the molecular surface properties for single-point mutants (haemoglobin structures) as a function of the probe radius.

The top panels represent the ration between the hydrophobic and positive areas, respectively, reported to the hydrophilic and negative areas, respectively. The bottom panels represent the ratios of the respective properties. The members of the sub-set are as follows: 1Y4F (W37A); 1A01 (W37A); 1Y4P (W37E); 1A00 (W37Y); 1Y46 (W37Y); 1Y4G (W37G); 1A0U (V1M); and 1A0Z (V1M).
Significance
The partial conclusions flowing from the analysis of the impact of the probe radius on the molecular surface properties are as follows:
While the present analysis shows that the “hydrophobic core” model stands valid for the proteins studied, in many cases this concept requires serious qualifications, as proteins appear to be specific regarding the propensity for protecting their hydrophobic core. In this context, a recent contribution[72] dealing with the quantification of the shape and distribution of the hydrophobicity of disordered proteins, which play a significant role in many biological processes, showed the lack of a well-formed hydrophobic core unlike that of the globular proteins.
Similarly with the “hydrophobic core” concept, it appears that proteins have a “positive charge core”. This positive charge core is however less evident, and it is arguably of a lesser importance than the hydrophobic core, due to the long range of electrostatic interactions, compared with the short range hydrophobic ones.
Although it is expected that the amphiphilicity and the charges of the protein on their molecular surface are correlated, e.g., a higher charged area will be more hydrophilic, these two sets of parameters are specific enough to deny a univocal relationship between them.
An atom-level description of the amphiphilicity, proposed recently[73] would allow arguably a more precise treatment of molecular surface, and even open the possibility of deriving “hydrophobic potentials”, as proposed before, e.g.,[74], similarly with electrostatic potential (but with very different mathematical formalism).
The probing of the protein with a large ball in silico is conceptually the closest to the interaction between a protein and a real nanoparticle. This conceptual commonality could open ways of designing nanoparticles that are tailored to elicit a desired response from the protein:nanoparticle complex, as it was proposed recently [26], thus turning the phenomenon of protein corona from a deleterious effect into a powerful nanoengineering tool. Indeed, while the concept of hydrophobic core and the existence of hydrophobic patches is well established, much less attention has been paid to the distribution of hydrophobic-complementary (or charge-complementary) “patches” on nano-surfaces. Because the proteins are actually not as flexible[67] as usually thought, the design of hydrophobic- and/or charge complementary nanosurfaces is conceivable, and possibly achievable.
Correlation between molecular surface properties and protein adsorption
Protein adsorption
The quantification of the physico-chemical properties, in particular amphiphilicity, on the protein molecular surface raises the expectation that these parameters could be correlated with measures of the interaction between a protein and a solid surface, e.g., protein adsorption on solid surfaces, protein interaction with lipid membranes, protein aggregation in large fibrilar structures. Among these, protein adsorption is arguably the best documented. Recently, data regarding the protein adsorption published in the open literature in the last half a century have been archived in a Biomolecular Adsorption Database [25], which register the amount of protein adsorbed on a particular solid surface, the structural properties of that protein (most relevant for this study, the structure deposited in the PDB database and component residues), as well as the solid surface contact angles, properties of the fluid media, method of measurement, etc.
Statistical strength of the correlation between molecular surface parameters and protein adsorption
The Pearson Product-Moment Correlation Coefficient (PPMCC) represents the strength of a statistical linear correlation between two variables, with 1, or -1, the former for both variables increasing, representing perfectly linear correlations. In the context of this study, the closer the PPMCC value is to 1 (or -1), the higher the predictive power is for the protein physico-chemical parameter assumed as predictor of protein adsorbed mass on a solid surface. The evolution of PPMCC with the probe radius (Figure 8) demonstrates that the molecular surface-based properties, such as amphiphilicity, hydrophobicity and hydrophilicity, are vastly better predictors than the same properties calculated from all residues, exposed or not to the molecular surface. More specifically, the results reveal specific features of different physico-chemical parameters, as follows.
Figure 8. Pearson Product-Moment Correlation Coefficient (PPMCC) of the relationship between protein parameters the protein adsorbed mass.

Protein properties are bulk amphiphilicity (H_bulk), and amphiphilicity (AA_amph). The adsorbed mass of the respective proteins (as reported in Biomolecular Adsorption Database [25]). PPMCC calculations are presented for all surfaces (top); hydrophobic (i.e., contact angle > 45°middle); and hydrophilic (i.e., contact angle < 45°bottom).
For all solid surfaces (Figure 8, top) the molecular surface-based amphiphilicity presents a PPMCC around 0.7–0.8, compared with the PPMCC of the ‘bulk’ amphiphilicity, which has a small (0.16) constant value irrespective of the probe radius. This high statistical strength is remarkable, having in mind the extreme spread of the protein and solid surface properties, and experimental data (buffers, methods of measurement, etc.), as well as the fact that PPMCC assumes a linear relationship between the tested parameters, while protein adsorption is clearly a non-linear phenomenon.
For hydrophobic solid surfaces (Figure 8, middle) the statistical relevance of molecular surface amphiphilicity is high, in the region of ±0.7, and decreasing only slightly with the probe radius. The ‘bulk’ counterpart, with PPMCC values close to 0, has no statistical relevance. The slight decrease of the amphiphilicity-related PPMCC can be understood in the context of the tug-of-war between (i) protein molecular surface hydrophobicity, which increases the propensity for its adsorption on hydrophobic solid surfaces; and (ii) protein molecular surface hydrophilicity which increases the protein solubility, and consequently allows for higher concentrations of protein in solution, which in turn increases protein adsorption.
For hydrophilic solid surfaces, (Figure 8, bottom) the statistical relevance of molecular surface-derived amphiphilicity is lower than for hydrophobic solid surfaces, albeit still substantial (0.5–0.6). This decrease of the PPMCC is the result of the protein adsorption on hydrophilic solid surfaces being governed to a lesser extent by hydrophobic interactions.
Multilinear regressions of the correlation between molecular surface parameters and protein adsorption
The results of the piecewise linear regression that connects the molecular surface properties, calculated for the maximum probing radius (20 Å) and adsorbing solid surface properties (Figure 9) demonstrates more compellingly the statistical relevance of this relationship, in particular regarding solid surface hydrophobicity.
Figure 9. Statistical strength of the piecewise linear regression between molecular surface properties and concentration of protein on adsorbing surfaces measured as the fit between observed vs. predicted data.
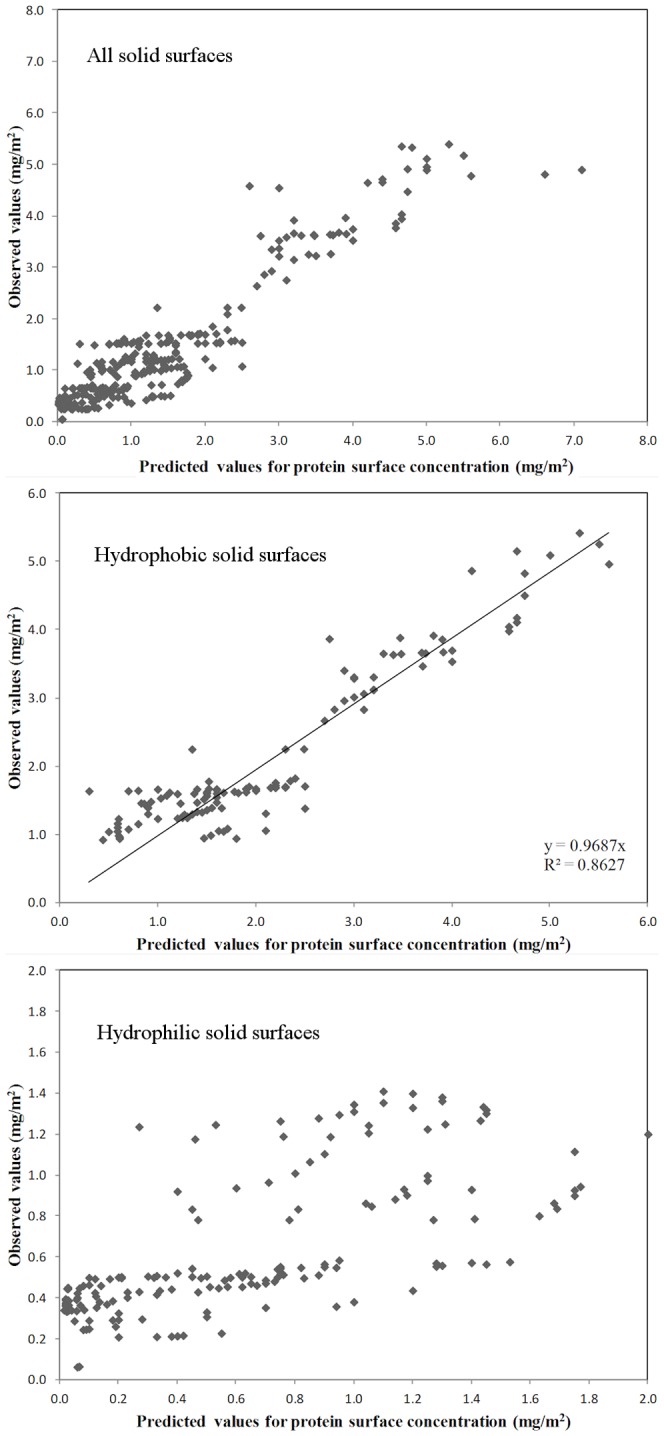
Comparison presented for all surfaces, i.e., hydrophobic and hydrophilic surfaces (top); hydrophobic surfaces only (middle) and hydrophilic surfaces (bottom). The fitted line in the middle panel represents the linear regression between predicted and observed data, forced to pass through origin, and not the actual multilinear regression with breakpoint best fit
The quasi-nonlinear feature of the piecewise linear regression, i.e., one multilinear relationship until a set breakpoint, followed by a different linear relationship after, succeeds in fitting well all available data, (Figure 9 top) for both hydrophobic and hydrophilic solid surfaces, and for a large span of protein concentrations on the adsorbing solid surfaces, i.e., close to 8 mg/m2. However, while the overall regression coefficient is reasonably high, i.e., R2 = 0.8758, a closer inspection of the comparison of predicted vs. observed data reveals that the fit starts to lose its quality for higher concentrations on the solid surface, e.g., higher than 4 mg/m2. This loss of quality can be due to the fact that, at high surface concentrations, i.e., higher than those required for a complete coverage of the surface, the correlation between protein and adsorbing solid surface properties loses its physical meaning, because the protein does not interact with the solid surface, but with other proteins immobilized on the solid surface. It is also interesting to note that including in the analysis the charge-related properties does not bring any improvement in the statistical quality of the fit.
The piecewise linear regression performed for hydrophobic solid surfaces results in much better overall fit, as suggested by Figure 9 middle panel, despite a slight decrease of the correlation factor, possibly due to the smaller pool of data. As before, the addition, or deletion of charge-related variables is rather inconsequential for the quality of the statistical fit.
Finally, the linear regression performed for the sub-set of hydrophilic solid surfaces (Figure 9 bottom) has the poorest quality, due to the lower relevance of hydrophobicity-related properties for protein adsorption on hydrophilic solid surfaces.
Significance
Following the establishing of the statistical relevance of amphiphilicity quantified on the protein molecular surface, this could be used further for finding relationships between the protein parameters, and those of the solid surfaces the proteins interact with, on one side; and the result of the interaction, on the other side. For instance, if other relevant parameters, e.g., pH and ionic strength of the liquid; topography, zeta potential, and surface tension of the solid surfaces; are included in the statistical correlation, protein adsorption could be better predicted, and protein-specialized materials could be designed.
The flexibility of the protein could impact on the validity of the analysis based on protein structures that are assumed to be rigid in contact with probing objects that are rigid. Indeed, it was elegantly demonstrated [30], [31] that proteins with very different shapes, i.e., albumin and fibrinogen, present opposite denaturation behavior when presented to nanoparticles with different radii. Also, it has been demonstrated [28] that the size of nanoparticles play an important role in determining the nanoparticle coronas on different particles of identical materials. However, it has to be noted that the cases mentioned above are extreme ones; and that, despite the general perception, proteins are rather rigid, plexiglass-like, at their core. [67]
Conclusion
The mapping and the quantification of the physico-chemical properties on the molecular surfaces of proteins using probes with increasing sizes offers insights into the interaction of proteins with nano-sized objects, or more generally with artificial solid surfaces. The geometrical and physico-chemical mapping of the molecular surfaces for a set of model proteins comprising various classes offered examples of this analysis, such as the protein-specific propensity for protecting the hydrophobic core. The relevance of the molecular surface-derived properties has been demonstrated via the calculation of the statistical strength of the prediction of protein adsorption. It is expected that the extension of this methodology to other protein:solid surface phenomena, in particular the interaction of nanoparticles, will result in important benefits in the understanding and design of protein-tailored solid surfaces.
Supporting Information
(XLSX)
(XLSX)
(DOC)
(DOC)
(XLSX)
Funding Statement
This work has been supported by the Defense Advanced Research Projects Agency (DARPA), under SymBioSys Program, Grant Contract No. F30602-00-2-0614); and by the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement no. 214538, project Bio-Inspired Self-assembled Nano-Enabled Surfaces (BISNES). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Brockwell DJ, Smith DA, Radford SE (2000) Protein folding mechanisms: new methods and emerging ideas. Curr Opin Struct Biol 10: 16–25. [DOI] [PubMed] [Google Scholar]
- 2. Takano K, Yamagata Y, Yutani K (2001) Contribution of polar groups in the interior of a protein to the conformational stability. Biochemistry (Mosc) 40: 4853–4858. [DOI] [PubMed] [Google Scholar]
- 3. Jones S, Thornton JM (1996) Principles of protein-protein interactions. Proc Natl Acad Sci USA 93: 13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Janin J, Chothia C (1990) The structure of protein-protein recognition sites. J Biol Chem 265: 16027–16030. [PubMed] [Google Scholar]
- 5. Bonvin AMJJ (2006) Flexible protein-protein docking. Curr Opin Struct Biol 16: 194–200. [DOI] [PubMed] [Google Scholar]
- 6. Gordon EM, Barrett RW, Dower WJ, Fodor SPA, Gallop MA (1994) Applications of combinatorial technologies to drug discovery. II: Combinatorial organic synthesis, libray screening strategies, and future directions. Journal of Medicinal Chemistry 37: 1385–1401. [DOI] [PubMed] [Google Scholar]
- 7. Eyrisch S, Helms V (2009) What induces pocket openins on protein surface patches involved in protein-protein interactions? J Comput Aided Mol Des 23: 73–86. [DOI] [PubMed] [Google Scholar]
- 8. Sharp K, Nicholls A, Fine R, Honig B (1990) Reconciling the magnitude of the microscopic and macroscopic hydrophobic effects. Science 252: 106–109. [DOI] [PubMed] [Google Scholar]
- 9. Richards FM (1977) Areas, Volumes, Packing, and Protein Structure. Annual Review of Biophysics and Bioengineering 6: 151–176. [DOI] [PubMed] [Google Scholar]
- 10.Fersht A (1985) Enzyme Structure and Mechanism.: W.H. Freeman and Company.
- 11. Nicolau DV Jr, Burrage K, Parton RG, Hancock JF (2006) Identifying Optimal Lipid Raft Characteristics Required To Promote Nanoscale Protein-Protein Interactions on the Plasma Membrane. Mol Cell Biol 26: 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bretschneider T, Anderson K, Ecke M, Müller-Taubenberger A, Schroth-Diez B, et al. (2009) The Three-Dimensional Dynamics of Actin Waves, a Model of Cytoskeletal Self-Organization. Biophys J 96: 2888–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kawabata S, Higgins GA, Gordon JW (1991) Amyloid plaques, neurofibrillary tangles and neuronal loss in brains of transgenic mice overexpressing a C-terminal fragment of human amyloid precursor protein. Nature 354: 476–478. [DOI] [PubMed] [Google Scholar]
- 14. Kasemo B (2002) Biological surface science. Surf Sci 500: 656–677. [Google Scholar]
- 15. Folch A, Toner M (2000) Microengineering of cellular interactions. Annu Rev Biomed Eng 2: 227–256. [DOI] [PubMed] [Google Scholar]
- 16. Langer R, Peppas NA (2003) Advances in biomaterials, drug delivery, and bionanotechnology. AICHE J 49: 2990–3006. [Google Scholar]
- 17. Nagase K, Kobayashi J, Okano T (2009) Temperature-responsive intelligent interfaces for biomolecular separation and cell sheet engineering. J R Soc Interface 6: 293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang B, Zhang L, Bae SC, Granick S (2008) Nanoparticle-induced surface reconstruction of phospholipid membranes. Proc Natl Acad Sci USA 105: 18171–18175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dawson KA, Salvati A, Lynch I (2009) Nanoparticles reconstruct lipids. Nat Nanotechnol 4: 84–85. [DOI] [PubMed] [Google Scholar]
- 20. Mukhopadhyay R (2006) Devices to drool for. Anal Chem 78: 7379–7382. [DOI] [PubMed] [Google Scholar]
- 21. Hawkins KR, Steedman MR, Baldwin RR, Fu E, Ghosal S, et al. (2007) A method for characterizing adsorption of flowing solutes to microfluidic device surfaces. Lab Chip 7: 281–285. [DOI] [PubMed] [Google Scholar]
- 22. Wilson R, Nicolau DV (2011) Separation-Free Detection of Biological Molecules Based On Plasmon-Enhanced Fluorescence. Angew Chem Int Ed 50: 2151–2154. [DOI] [PubMed] [Google Scholar]
- 23. Ayeyard J, Hedegaard T, Bilenberg B, Nicolau DV (2010) Microfabricated magnetic bead polydimethylsiloxane microarrays. Microelectron Eng 87: 760–764. [Google Scholar]
- 24. Filipponi L, Sawant PD, Fulga F, Nicolau DV (2009) Microbeads on microposts: An inverted architecture for bead microarrays. Biosens Bioelectron 24: 1850–1857. [DOI] [PubMed] [Google Scholar]
- 25. Vasina EN, Paszek E, Nicolau DV Jr, Nicolau DV (2009) The BAD project: data mining, database and prediction of protein adsorption on surfaces. Lab Chip 9: 891–900. [DOI] [PubMed] [Google Scholar]
- 26. Lynch I, Dawson KA (2008) Protein-nanoparticle interactions. Nano Today 3: 40–47. [Google Scholar]
- 27. Cedervall T, Lynch I, Lindman S, Berggard T, Thulin E, et al. (2007) Understanding the nanoparticle-protein corona using methods to quantify exchange rates and affinities of proteins for nanoparticles. Proc Natl Acad Sci USA 104: 2050–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lundqvist M, Stigler J, Elia G, Lynch I, Cedervall T, et al. (2008) Nanoparticle size and surface properties determine the protein corona with possible implications for biological impacts. Proc Natl Acad Sci USA 105: 14265–14270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cabaleiro-Lago C, Lynch I, Dawson KA, Linse S (2010) Inhibition of IAPP and IAPP((20–29)) Fibrillation by Polymeric Nanoparticles. Langmuir 26: 3453–3461. [DOI] [PubMed] [Google Scholar]
- 30. Roach P, Farrar D, Perry CC (2005) Interpretation of protein adsorption: Surface-induced conformational changes. J Am Chem Soc 127: 8168–8173. [DOI] [PubMed] [Google Scholar]
- 31. Roach P, Farrar D, Perry CC (2006) Surface tailoring for controlled protein adsorption: Effect of topography at the nanometer scale and chemistry. J Am Chem Soc 128: 3939–3945. [DOI] [PubMed] [Google Scholar]
- 32. Lynch I, Salvati A, Dawson KA (2009) Protein-nanoparticle interactions. What does the cell see? Nat Nanotechnol 4: 546–547. [DOI] [PubMed] [Google Scholar]
- 33. Walczyk D, Bombelli FB, Monopoli MP, Lynch I, Dawson KA (2010) What the Cell "Sees" in Bionanoscience. J Am Chem Soc 132: 5761–5768. [DOI] [PubMed] [Google Scholar]
- 34. Banerji A, Ghosh I (2009) A new computational model to study mass inhomogeneity and hydrophobicity inhomogeneity in proteins. Eur Biophys J Biophy 38: 577–587. [DOI] [PubMed] [Google Scholar]
- 35.Banerji A, Ghosh I (2009) Revisiting the Myths of Protein Interior: Studying Proteins with Mass-Fractal Hydrophobicity-Fractal and Polarizability-Fractal Dimensions. Plos One 4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Banerji A, Ghosh I (2011) Fractal symmetry of protein interior: what have we learned? Cell Mol Life Sci 68: 2711–2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Connolly ML (1983) Solvent-accesible surfaces of proteins and nucleic acids. Ann Rev BiophysBioeng 221: 709–713. [DOI] [PubMed] [Google Scholar]
- 38. Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, et al. (2000) The Protein Data Bank. Nucleic Acids Res 28: 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Connolly ML (1983) Analytical molecular surface calculation. J Appl Crystallogr 16: 548–558. [Google Scholar]
- 40. Connolly ML (1983) Solvent-accessible surfaces of proteins and nucleic-acids. Science 221: 709–713. [DOI] [PubMed] [Google Scholar]
- 41. Connolly ML (1985) Molecular Surface Triangulation. J Appl Crystallogr 18: 499–505. [Google Scholar]
- 42. Can T, Chen C-I, Wang Y-F (2006) Efficient molecular surface generation using level-set methods. J Mol Graphics Model 25: 442–454. [DOI] [PubMed] [Google Scholar]
- 43.Dodd LR, Theodorou DN (1991) Analytical treatment of the volume and surface area of molecules formed by an arbitrary collection of unequal spheres intersected by planes. Mol Phys 72 : :1313 –1345. [Google Scholar]
- 44. Edelsbrunner H, Mücke EP (1994) Three-dimensional alpha shapes. TOG 13: 43–72. [Google Scholar]
- 45. Eisenhaber F, Argos P (1993) Improved strategy in analytic surface calculation for molecular systems : handling of singularities and computational efficiency. J Comput Chem 14: 1272–1280. [Google Scholar]
- 46. Eisenhaber F, Lijnzaad P, Argos P, Sander C, Scharf M (1995) The double cubic lattice method: Efficient approaches to numerical integration of surface area and volume and to dot surface contouring of molecular assemblies. J Comput Chem 16: 273–284. [Google Scholar]
- 47. Gibson KD, Scheraga HA (1988) Surface area of the intersection of three spheres with unequal radii A simplified analytical formula. Mol Phys 64: 641–644. [Google Scholar]
- 48. Kim D, Cho C-H, Cho Y, Ryu J, Bhak J, et al. (2008) Pocket extraction on proteins via the Voronoi diagram of spheres. J Mol Graphics Model 26: 1104–1112. [DOI] [PubMed] [Google Scholar]
- 49. Kim D-S, Cho C-H, Kim D, Cho Y (2006) Recognition of docking sites on a protein using β-shape based on Voronoi diagram of atoms. Comput Aid Des 38: 431–443. [Google Scholar]
- 50. Kinjo AR, Horimoto K, Nishikawa K (2005) Predicting absolute contact numbers of native protein structure from amino acid sequence. Proteins: Struct Funct Bioinform 58: 158–165. [DOI] [PubMed] [Google Scholar]
- 51. Kinjo AR, Nishikawa K (2005) Recoverable one-dimensional encoding of three-dimensional protein structures. Bioinformatics 21: 2167–2170. [DOI] [PubMed] [Google Scholar]
- 52. Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157: 105–132. [DOI] [PubMed] [Google Scholar]
- 53. Le Grand SM, Merz KM Jr (1993) Rapid approximation to molecular surface area via the use of Boolean logic and look-up tables. J Comput Chem 14: 349–352. [Google Scholar]
- 54. Lin SL, Nussinov R (1996) Molecular recognition via face center representation of a molecular surface. J Mol Graphics 14: 78–90. [DOI] [PubMed] [Google Scholar]
- 55. Putta S, Beroza P (2007) Shapes of Things: Computer Modeling of Molecular Shape in Drug Discovery. Curr Top Med Chem 7: 1514–1524. [DOI] [PubMed] [Google Scholar]
- 56. Richards FM (1974) The interpretation of protein structures: Total volume, group volume distributions and packing density. J Mol Biol 82: 1–14. [DOI] [PubMed] [Google Scholar]
- 57. Rosen M, Lin SL, Wolfson H, Nussinov R (1998) Molecular shape comparisons in searches for active sites and functional similarity. Protein Eng 11: 263–277. [DOI] [PubMed] [Google Scholar]
- 58. Ryu J, Parka R, Kim D-S (2007) Molecular surfaces on proteins via beta shapes. Comput Aid Des 39: 1042–1057. [Google Scholar]
- 59. Sanner MF, Olson AJ, Spehner J-C (1996) Reduced Surface: An Efficient Way to Compute Molecular Surfaces. Biopolymers 38: 305–320. [DOI] [PubMed] [Google Scholar]
- 60. Scarsi M, Majeux N, Caflisch A (1999) Hydrophobicity at the surface of proteins. Proteins: Struct Funct Gen 37: 565–575. [DOI] [PubMed] [Google Scholar]
- 61. Wang H, Levinthal C (1991) A vectorized algorithm for calculating the accessible surface area of macromolecules. J Comput Chem 12: 868–871. [Google Scholar]
- 62. Zauhar RJ, Morgan RS (1990) Computing the electric potential of biomolecules: Application of a new method of molecular surface triangulation. J Comput Chem 11: 603–622. [Google Scholar]
- 63. Krissinel E, Henrick K (2004) Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr Sect D Biol Crystallogr 60: 2256–2268. [DOI] [PubMed] [Google Scholar]
- 64. Kavanaugh JS, Weydert JA, Rogers PH, Arnone A (1998) High-resolution crystal structures of human hemoglobin with mutations at tryptophan 37β: Structural basis for a high-affinity T-state. Biochemistry (Mosc) 37: 4358–4373. [DOI] [PubMed] [Google Scholar]
- 65. Wimley WC, White SH (1996) Experimentally determined hydrophobicity scale for proteins at membrane interfaces. Nat Struct Biol 3: 842–848. [DOI] [PubMed] [Google Scholar]
- 66.Sawant PD, Nicolau DV (2005) Line and two-dimensional fractal analysis of micrographs obtained by atomic force microscopy of surface-immobilized oligonucleotide nano-aggregates. Appl Phys Lett 87.
- 67. Erickson HP (2009) Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol Proc Online 11: 32–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Serdyuk IN, Galzitskaya OV, Timchenko AA (1997) Roughness of the globular protein surface. Biofizika 42: 1206–1207. [PubMed] [Google Scholar]
- 69. Nicolau DV, Solana G, Kekic M, Fulga F, Mah-Anivona C, et al. (2008) Surface hydrophobicity modulates the operation of actomyosin-based dynamic nanodevices (vol 23, pg 10846, 2007). Langmuir 24: 4420–4420. [DOI] [PubMed] [Google Scholar]
- 70. Tsai CJ, Lin SL, Wolfson HJ, Nussinov R (1997) Studies of protein-protein interfaces: A statistical analysis of the hydrophobic effect. Protein Sci 6: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kavanaugh JS, Rogers PH, Arnone A (2005) Crystallographic evidence for a new ensemble of ligand-induced allosteric transitions in hemoglobin: The T-to-THigh quaternary transitions. Biochemistry (Mosc) 44: 6101–6121. [DOI] [PubMed] [Google Scholar]
- 72. Rawat N, Biswas P (2012) Hydrophobic moments, shape, and packing in disordered proteins. J Phys Chem B 116: 6326–6335. [DOI] [PubMed] [Google Scholar]
- 73. Cristea PD, Arsene O, Tuduce R, Nicolau D (2012) Protein Surface Functional Imaging. Mater Sci Forum 721: 319–324. [Google Scholar]
- 74. Kellogg GE, Semus SF, Abraham DJ (1991) HINT - A new method of empirical hydrophobic field calculation for ComFA. J Comput-Aided Mol Des 5: 545–552. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(XLSX)
(XLSX)
(DOC)
(DOC)
(XLSX)