

Abstract
Familial hematuria (FH) is explained by at least four different genes (see below). About 50% of patients develop late proteinuria and chronic kidney disease (CKD). We hypothesized that MYH9/APOL1, two closely linked genes associated with CKD, may be associated with adverse progression in FH. Our study included 102 thin basement membrane nephropathy (TBMN) patients with three known COL4A3/COL4A4 mutations (cohort A), 83 CFHR5/C3 glomerulopathy patients (cohort B) with a single CFHR5 mutation and 15 Alport syndrome patients (cohort C) with two known COL4A5 mild mutations, who were categorized as “Mild” (controls) or “Severe” (cases), based on renal manifestations. E1 and S1 MYH9 haplotypes and variant rs11089788 were analyzed for association with disease phenotype. Evidence for association with “Severe” progression in CFHR5 nephropathy was found with MYH9 variant rs11089788 and was confirmed in an independent FH cohort, D (cumulative p value = 0.001, odds ratio = 3.06, recessive model). No association was found with APOL1 gene. Quantitative Real time PCR did not reveal any functional significance for the rs11089788 risk allele. Our results derive additional evidence supporting previous reports according to which MYH9 is an important gene per se, predisposing to CKD, suggesting its usefulness as a prognostic marker for young hematuric patients.
Introduction
The differential diagnosis in 2012 for familial hematuria (FH) includes mostly the COL4A3/A4 heterozygous carriers that exhibit thin basement membrane nephropathy (TBMN) with lifelong microscopic hematuria [1]–[5] and a newly described disease, CFHR5 nephropathy, with isolated C3 mesangial deposits [6], [7]. Other still unknown causes may exist.
Heterozygous mutations in the COL4A3/COL4A4 genes are said to account for up to 40–50% of families with TBMN [1]–[5]. CFHR5 nephropathy is also an autosomal dominant disease with an up to now unknown prevalence, although it is highly prevalent in Cyprus [6], [7]. We and others demonstrated that more than 50% of these patients develop added proteinuria and chronic kidney disease (CKD) after the age of 30 and with a broad phenotypic spectrum [8]–[10]. Interestingly, X-linked Alport syndrome cases (male patients) with mild mutations in the COL4A5 gene, often present as phenocopies of TBMN, with a wide spectrum of phenotypes [11]–[17]. We hypothesize that this great phenotypic heterogeneity is largely due to modifier genes [10].
Studies in animal models for TBMN and Alport syndrome support the existence of genetic loci that influence disease progression [18], [19]. In humans, studies by Tonna et al (2008) and Voskarides et al (2012) provide evidence that NPHS2-R229Q predisposes to proteinuria in TBMN [20], [21]. Additionally, Papagregoriou et al (2012) investigated potential microRNAs' target sites in hematuric patients and found that a miR-1207-5p binding site variant abolishes regulation of HBEGF and is associated with disease severity in CFHR5 nephropathy [22].
In this study, we took advantage of the extended founder effects we have observed among the Greek-Cypriot population, to investigate whether genetic polymorphisms of the MYH9/APOL1 region can act as modifiers for the FH phenotype. Genome-wide association studies (GWAS) have identified MYH9 (Myosin Heavy Chain 9) and its closely linked gene APOL1, as major susceptibility genes predisposing towards end stage kidney disease (ESKD), in various types of renal diseases (idiopathic focal segmental glomerulosclerosis, HIV-associated nephropathy, hypertensive nephrosclerosis, diabetic nephropathy, IgA nephropathy progression), in African-Americans, European-Americans, Europeans, Hispanic-Americans and Chinese [23]–[27]. For the present work our cohorts included patients with a monogenic primary form of FH, TBMN, Alport Syndrome and CFHR5 nephropathy. Our results demonstrate a likely contribution of MYH9 variant rs11089788 in the progression of CKD when co-inherited with CFHR5 nephropathy, but cast doubt on the recently alleged association with variation in the APOL1 gene that is closely linked to MYH9.
Materials and Methods
Study Cohorts - Clinical Assessment and Study Outcomes
The study cohorts are presented in Table 1. Patients of all four cohorts were categorized as having “Mild” or “Severe” disease. Our main cohorts include 102 adult TBMN patients, 83 adult CFHR5 patients and 15 Alport syndrome patients. These cohorts are unique worldwide because they include a large number of patients with common mutations due to founder effects. Among 102 TBMN patients, 77 carry mutation COL4A3-p.G1334 and 19 carry mutation COL4A3-p.G871C. All 83 CFHR5 patients carry the same exon 2–3 duplication. This situation offers an advantage as regards reduced genomic background complexity based on common ethnicity, thereby facilitating the search for genetic modifiers in relatively smaller cohorts. Since young individuals with apparently mild disease could develop severe disease when older, we excluded young patients without evidence of severe disease (Table 1). Mild disease was characterized by the presence of only microscopic hematuria and up to low grade proteinuria, repeatedly under 300 mg/day and no CKD. “Severe” disease was characterized by hematuria plus proteinuria ≥500 mg/day, or hematuria plus proteinuria plus CKD, or ESKD (Table 1). CKD was defined as an elevated serum creatinine over 1.5 mg/dL. Patients with borderline proteinuria and another concomitant renal disease (e.g., over five years of diabetes, vesicoureteral reflux etc), or severe patients at the extreme of body weight (outside ±2 SD of the cohort mean) were excluded. For Alport syndrome patients, disease severity is defined by most researchers according to age at ESKD, so the severe group included patients that reached ESKD below the age of 40 years. Cohort D was genotyped for replication purposes, including FH patients not genetically studied yet. The study was approved by the Cyprus National Bioethics Committee and participants gave their signed informed consent, unless they were included anonymously after testing for purely diagnostic purposes.
Table 1. Description of cohorts and patients under study.
| Patient group (cohort) | Origin | N | Mild | Severe | ||||||
| N (%) | Age: mean (SD) | Females: N (%) | with ESKD: N (%) | N (%) | Age: mean (SD) | Females: N (%) | with ESKD: N (%) | |||
| A. Heterozygous mutation carriers of COL4A3-G1334E or COL4A3-G871C or COL4A4-3854delG1 | Cyprus | 102 | 44 (43%) | 60.3 (±10.3) | 26 (59%) | 0 | 58 (57%) | 62.6 (±12.9) | 26 (44%) | 20 (34%) |
| B. Heterozygous mutation carriers of CFHR5 Exons 2–3 duplication2 | Cyprus | 83 | 48 (58%) | 57.5 (±12.9) | 29 (60%) | 0 | 35 (42%) | 58.4 (±11.1) | 8 (23%) | 20 (57%) |
| C. XLAS male patients, mutation carriers of COL4A5-P628L or COL4A5-G624D3 | Cyprus, Greece | 15 | 11 (41%) | 50.8 (±5.3) | 0 | 5 (45%) | 4 (59%) | 50.8 (±5.3) | 0 | 4 (100%) |
| D. Familial cases of MH4 | Cyprus, Greece | 67 | 33 (49%) | 53.8 (±8.9) | 26 (79%) | 0 | 34 (51%) | 56.4 (±12.8) | 13 (38%) | 11 (32%) |
Please note that cohort B1 is the male only patients with CFHR5 nephropathy.
MH: Microscopic Hematuria, ESKD: End Stage Kidney Disease, XLAS: X-linked Alport syndrome.
“Mild” patients born before 01/1963. Gender difference (Mild vs Severe) is not significant (p = 0.141).
“Mild” patients born before 01/1975. Gender difference (Mild vs Severe) is significant (p = 0.001).
“Severe” patients: ESKD≤40 yo.
“Mild” patients born before 01/1979. Gender difference (Mild vs Severe) is significant (p = 0.001).
Single Nucleotide Polymorphisms (SNPs) genotyping – Haplotype analysis
We genotyped MYH9 SNPs that were previously shown to be strongly associated with renal failure in Caucasian populations (Table 2; it includes information on the PCR-RFLP genotyping assays): two lying on the E1 risk haplotype (rs4821481 and rs4821480), one lying on the S1 risk haplotype (rs2413396) and the rs11089788 variant that is found in the 5′-end of MYH9 (IVS3 -5801C/A) – identified by a meta-analysis study – associated with serum creatinine levels in three Caucasian populations (Table 2).
Table 2. Information about the MYH9 non-coding SNPs and the non-synonymous APOL1 SNPs, genotyped in this study.
| SNP | Risk haplotype | Forward primer | Reverse primer | Tm (°C) | Restriction enzyme | Cleavage products (bp) | Known associations in Caucasian populations |
| MYH9 rs4821481 (T/C) | E1 | GAAGAGTACCCCGTATCTCAACAC | AGCATGAGGGCTTCTGCTTAACT | 65 | BfaI | 132+115 (T allele) | 1. Non-diabetic ESKD in Hispanic Americans [26] 2. Diabetic nephropathy in European Americans [33] |
| MYH9 rs4821480 (T/G) | E1 | CTCATGCTTGTTCTTGAGCTTG | AGAACAGAAAGCAAGGAGAGCAG | 66 | DraI | 261+291 (T allele) | 1. Non-diabetic ESKD in Hispanic Americans [26] 2. Diabetic nephropathy in European Americans [33] 3. Non-diabetic CKD in European Americans [33] |
| MYH9 rs2413396 * (T/C) | S1 | CCAGCACCTCCCCGTGA | GTGGAGAAGGTGATGCAGGAG | 65 | HaeII | 109+21 (C allele) | 1. Non-diabetic ESKD in Hispanic Americans [33] |
| MYH9 rs11089788 (C/A) | No strong LD (<0.3) with any neighboring SNPs, according to HapMap data | GATGTCCCATCCAATTGTTTTC | GATTATGGCCTAAAAAGGCAACTG | 59 | BslI | 230+158 (C allele) | 1. Association with serum creatinine levels in three Caucasian populations [33] |
| APOL1 E149K | - | CTGGTTTCTGAAAGAGTTTCCTCGGTTGAAAATT ** | GCTGTGATTCCCAACTCCATCCCAGGTTCCAA | 60 | MseI | 207+33 (K allele) | - |
| APOL1 M227I | - | CGTATTAGTCAGGATGGTCTCAATC | AAAGTTGGATATGTTCTCACCCAAA | 65 | BtgI | 505+115 (M allele) | - |
| APOL1 R254K | - | CTGGTCATCAAAAGCCTTGACAAATTGAAGGAGGTTA ** | ATTAACCCTCTCCACCTGTTCACCGCTTTCAGCTG | 67 | MseI | 201+35+4 (K allele) | - |
Genotyping of rs2413396 was performed according to [27].
Modified nucleotides for creating restriction sites are in bold and underlined.
DNA samples from 15 patients (six from cohort A, five from cohort B and four from cohort C), representing all three rs11089788 genotypes, were selected for PCR amplification (primers available on request) and DNA re-sequencing (in ABI PRISM™ 3130) of the last exon of APOL1, where most of non-synonymous SNPs are located, according to electronic databases (www.ensemble.org). Three of the five non-synonymous SNPs detected, were genotyped in cohorts A, B and D. Haplotype analysis was performed by Haploview software (http://www.broad.mit.edu/mpg/haploview/).
Statistical analysis
Genotypic statistical analysis and odds ratios calculations were performed by SPSS v.13 (IBM, USA). Both dominant and recessive models of inheritance were tested for significance where this was possible. P values were calculated by Pearson Chi-Square test. Fisher's Exact Test (2-sided) was used where genotype values less than 10 existed. The significance level, alpha, was set to 0.05. We decided to genotype the entire cohort B (it includes males and females with CFHR5 nephropathy), only for SNPs that would give a p value close to 0.1 for cohort B1 (B1 are only the males with CFHR5 nephropathy; mostly males proceed to CKD in this type of FH).
Real Time PCR
mRNA was isolated from peripheral blood leukocytes using the QIAamp RNA Blood Mini Kit (Qiagen, Germany) from: three male healthy volunteers genotyped as CC for rs11089788, three male healthy volunteers genotyped as AA for rs11089788 and three male “severe” hematuric patients genotyped as CC for rs11089788. RNA integrity and concentration were assessed by gel electrophoresis and spectrophotometrically (Nanodrop technologies), respectively. 200 ng of total RNA from each sample was reverse transcribed (ProtoScript™ by New England Biolabs, UK). The quantitative Real-Time PCR (qRT-PCR) amplifications were performed in duplicate on the LightCycler® system (Roche Diagnostics and Applied Sciences) using SYBR Green (Applied Biosystems, California, USA) in a reaction volume of 20 µl and primers according to Liang et al (2011) [28]. Relative quantification analysis was carried out on the LightCycler® Software 4.1. The endogenous reference gene GAPDH was used for normalisation of the results. Statistical significance was checked by independent t-test, through the SPSS v.13 package (IBM, USA).
Results
CC genotype of MYH9 rs11089788 confers significant risk for proteinuria and CKD
None of the analyzed MYH9 SNPs was significantly associated with phenotypes in any of our initial cohorts. In accordance with previous publications, MYH9 rs4821480 was found to be in complete linkage disequilibrium (LD) with MYH9 rs4821481, after genotyping 85 samples of cohort A; thus it was not genotyped any further. MYH9 rs2413396 was found in partial LD with MYH9 rs4821481 (see frequencies in Table 3, LD value was not calculated). MYH9 rs11089788 gave a p value close to 0.1 in cohort B1 (Table 4). Hence, cohort B (B = B1 plus the female patients, see Methods) and D were analyzed only for MYH9 rs11089788. For cohort B, MYH9 rs11089788 gave a p-value of 0.015 (OR = 3.18) under a recessive model. Cohort D replicated the significance for MYH9 rs11089788 (p = 0.024) under a recessive model of inheritance (Table 5). The significance was highly increased for the sum of cohorts B+D for MYH9 rs11089788 (p = 0.001 and OR = 3.06, see Table 5).
Table 3. Genotype distribution of the studied MYH9 variants, by cohort and by severity.
| SNP | MYH9 rs4821481 | MYH9 rs2413396 | MYH9 rs11089788 | |||||||
| Mild | TT | TC | CC | TT | TC | CC | AA | AC | CC | n |
| Cohort A | 28 (64%) | 14 (32%) | 2 (4%) | 29 (66%) | 14 (32%) | 1 (2%) | 14 (32%) | 21 (48%) | 9 (20%) | 44 |
| Cohort B1 | 17 (89%) | 2 (11%) | 0 (0%) | 16 (84%) | 3 (16%) | 0 (0%) | 6 (32%) | 9 (47%) | 4 (21%) | 19 |
| Cohort B | - | - | - | - | - | - | 15 (31%) | 22 (46%) | 11 (23%) | 48 |
| Cohort C | 8 (73%) | 3 (27%) | 0 (0%) | 8 (73%) | 3 (27%) | 0 (0%) | 3 (27%) | 3 (27%) | 5 (46%) | 11 |
Table 4. Genotype associations for the three MYH9 variants genotyped in this study.
| Variant | p value | OR (95% CI) | p-value | OR (95% CI) |
| MYH9 rs4821481 | TT+TC vs CC | TT vs TC+CC | ||
| Patients cohort A | 0.184* | ** | 0.572 | 0.79 (0.34, 1.80) |
| Patients cohort B1 | - | - | 1.000* | 1.06 (0.16, 7.06) |
| Patients cohort C | - | - | 0.516* | ** |
| Sum (A+B1+C) | 0.205* | ** | 0.487 | 0.78 (0.39, 1.58) |
| MYH9 rs2413396 | TT+TC vs CC | TT vs TC+CC | ||
| Patients cohort A | 1.000* | 1.54 (0.14, 17.50) | 0.825 | 1.10 (0.48, 2.50) |
| Patients cohort B1 | - | - | 0.716* | 1.52 (0.33, 7.04) |
| Patients cohort C | - | - | 0.516* | ** |
| Sum (A+B1+C) | 1.000* | 1.68 (0.15, 18.88) | 0.785 | 1.10 (0.56, 2.17) |
| MYH9 rs11089788 | AA+AC vs CC | AA vs AC+CC | ||
| Patients cohort A | 0.639* | 1.36 (0.53, 3.47) | 0.509 | 1.34 (0.56, 3.18) |
| Patients cohort B1 | 0.126* | 3.00 (0.79, 11.45) | 0.133* | 3.69 (0.79, 17.25) |
| Patients cohort B | 0.015 | 3.18 (1.24, 8.17) | 0.016 * | 4.85 (1.28, 18.36) |
| Patients cohort C | 0.604* | 0.40 (0.01, 5.15) | 0.235* | 0.125 (0.01, 1.72) |
| Sum (A+B+C) | 0.129 | 1.61 (0.87, 2.98) | 0.131 | 1.63 (0.86, 3.09) |
P-values were calculated by Pearson Chi-Square test. The whole CFHR5 cohort (B) was genotyped only for MYH9 rs11089788, the only SNP that gave p-value close to 0.1 for the male CFHR5 patients (B1).
P-values calculated by Fisher's Exact Test (2-sided) due to existence of genotypes values less than 10.
Odds ratio (OR) cannot be estimated due to zero genotypic values in the “Severe” category.
Table 5. MYH9 rs11089788 statistical analysis for a replicate cohort (D) and for the sum of cohorts B and D, that gave statistical significance.
| MYH9 rs11089788 | P value, Odds ratio | P value, Odds ratio | ||||
| Cohort | n | AA | AC | CC | AA+AC vs CC | AA vs AC+CC |
| D – “Mild” | 33 | 7 (21%) | 18 (55%) | 8 (24%) | P = 0.024, OR = 3.52 (1.24, 9.97) | P = 0.765, OR = 1.26 (0.37, 4.23) |
| D – “Severe” | 34 | 6 (18%) | 10 (29%) | 18 (53%) | ||
| B+D – “Mild” | 83 | 23 (28%) | 40 (48%) | 20 (24%) | P = 0.001, OR = 3.06 (1.54, 6.10) | P = 0.049, OR = 2.26 (0.99, 5.16) |
| B+D – “Severe” | 69 | 10 (15%) | 25 (36%) | 34 (49%) | ||
Patients in cohort D belong to families that segregate microscopic hematuria but no known mutation has been found in any of the genes COL4A3, COL4A4, or CFHR5, so far.
Non-synonymous SNPs found on the last exon of APOL1
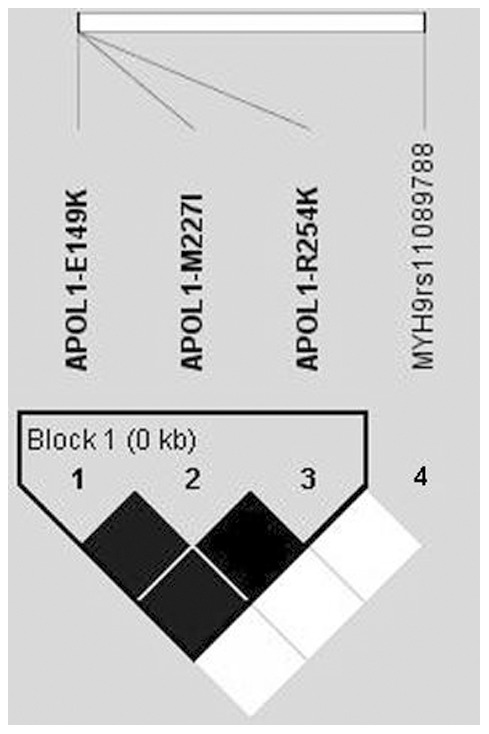
Re-sequencing of the last APOL1 exon revealed five non-synonymous SNPs: p.E149K (rs2239785), p.M227I (rs136175), p.R254K (rs136176), p.G270D (rs73403889) and p.D336N (rs16996616). Those SNPs are not among the published ones to be associated with CKD [29], [30]. APOL1 p.E149K, p.M227I and p.R254K [all reported with significant frequency only in Caucasian samples (http://hapmap.ncbi.nlm.nih.gov/)] were genotyped for the cohorts A, B and D. No significant association was observed (results not shown). All three APOL1 SNPs were in complete LD, constructing a haplotype block (results compatible with HapMap data), but they were not linked with rs11089788 (Figure 1).
Figure 1. Linkage disequilibrium plots show that E149K, M227I and R254K in APOL1 gene are in complete linkage disequilibrium (D′ = 1), but are not linked with MYH9 rs11089788.
Expression of MYH9 in rs11089788 “CC” and “AA” patients
No statistical significance was observed when comparing the means of the blood mRNA values among the genotypic groups, (see Methods, results not shown) despite that there was a trend for an increase of the CCs over the AAs.
Discussion
Genotype CC of rs11089788, located on the 5′-end of MYH9, was found to be significantly associated with bad progression in FH in two different cohorts. No significant association was observed for SNPs representing E1 και S1 haplotypes. This may be expected as these haplotypes were mainly associated with renal failure in populations out of Europe [23], [24]. On the other hand, MYH9 rs11089788 was previously associated with high serum creatinine levels in Europeans [25] and with progression of IgA nephropathy in Chinese [27]. CFHR5 nephropathy and IgA nephropathy possibly share common pathophysiology features due to their immune origin. Pending confirmation with other studies, our study suggests that rs11089788 is a negative prognostic factor in CFHR5 nephropathy and possibly in other glomerular hematuric diseases. This result was confirmed in a second cohort (D), including hematuric patients of various etiologies. Considering the two cohorts together, the results show that a hematuric patient with the CC genotype (recessive model) has a 3.06-fold increased likelihood (p = 0.001) for proteinuria and renal failure (Table 5). This enables us to speculate that certain FH types may share common risk factors as regards disease progression.
It is useful to underline that for certain types of FH, gender is an important cofounder in the disease progression in association with the modifier genes. In this study, difference in gender percentages is significant for cohort B (CFHR5 patients) and cohort D. In a previous publication we discuss in detail the high risk of male CFHR5 patients for progression to renal failure, comparing also the survival rates of renal function in the two genders [6], [17]. All in all however, in accordance with the previous literature, the significance we describe here is still valid when taking into consideration the gender.
Recent studies give evidence that three coding SNPs on the last exon of APOL1 gene, lying on extended haplotypes with the MYH9 SNPs (haplotypes E1, F1 and S1), may be the actual cause for predisposition to renal failure. On the other hand, O'Seaghdha et al (2011) found association of rs4821480 with CKD in European-Americans [31], but did not find an association with any of the APOL1 SNPs reported by Genovese et al (2010) and Tzur et al (2010), suggesting that variation in APOL1 may not be the complete explanation [29], [30]. We screened the last exon of APOL1, but the identified SNPs were not in LD with either of the two alleles of rs11089788, despite a strong LD existing between them (Fig. 1), this being compatible with results of O'Seaghdha et al (2011) [31]. Additionally, Freedman et al (2011) found that homozygotes for MYH9 E1 haplotype are protected from Diabetic Nephropathy when predisposed by FRMD3 SNPs; this is in contrast to the non-carriers of MYH9 E1 that are prone to Diabetic Nephropathy when carrying the FRMD3 SNPs. These data are indicative of more complex and unpredictable gene interactions and are supportive of our results, signifying the genetic modifying influence of MYH9 SNPs. Non-coding SNPs on MYH9 may have an unknown effect on MYH9 e.g. in its expression, even though we did not find such an effect by a quantitative real time PCR approach. Some of those SNPs may affect proper splicing of MYH9, as Nelson et al (2010) found recently for rs2413396 and rs4821480 [32].
Our finding for rs11089788 may prove useful in FH cases, with early and effective interventions at early life, before symptoms worsen. However, it remains unclear how certain non-coding MYH9 variants exert their epistatic role and are associated with renal failure. It does not escape our attention that despite the unique character of our cohorts as regards the reduced genetic complexity owing to the extensive founder mutations, the overall size of our patient cohorts is somewhat small, and unfortunately there is no easy way to remedy this inherent limitation of the population we serve. Future collaboration of multiple research centers will be necessary to increase the statistical power and replicate or annul our results.
Acknowledgments
The authors express their gratitude to all patients, relatives and volunteers who participated and made this study possible. We thank the many clinical colleagues that have recruited patients to this research project and we also thank the organizations that funded this project.
Funding Statement
The work was supported from the Cyprus Research Promotion Foundation grants: ΠEΝEK EΝΙΣX/0505/02, ΠEΝEK/EΝΙΣX/0308/08, NEW INFRASTRUCTURE/STRATEGIC/0308/24 (co-funding by the EU Structural Funds) and from the University of Cyprus articles 3/311 and 3/346, to CD. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Lemmink HH, Nillesen WN, Mochizuki T, Schroder CH, Brunner HG, et al. (1996) Benign familial hematuria due to mutation of the type IV collagen alpha4 gene. J Clin Invest 98: 1114–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Badenas C, Praga M, Tazon B, Heidet L, Arrondel C, et al. (2002) Mutations in theCOL4A4 and COL4A3 genes cause familial benign hematuria. J Am Soc Nephrol 13: 1248–1254. [DOI] [PubMed] [Google Scholar]
- 3. Buzza M, Wang YY, Dagher H, Babon JJ, Cotton RG, et al. (2001) COL4A4 mutation in thin basement membrane disease previously described in Alport syndrome. Kidney Int 60: 480–483. [DOI] [PubMed] [Google Scholar]
- 4. Kashtan CE (2005) Familial hematurias: what we know and what we don't. Pediatr Nephrol 20: 1027–1035. [DOI] [PubMed] [Google Scholar]
- 5. Slajpah M, Gorinsek B, Berginc G, Vizjak A, Ferluga D, et al. (2007) Sixteen novel mutations identified in COL4A3, COL4A4, and COL4A5 genes in Slovenian families with Alport syndrome and benign familial hematuria. Kidney Int 71: 1287–1295. [DOI] [PubMed] [Google Scholar]
- 6. Athanasiou Y, Voskarides K, Gale DP, Damianou L, Patsias C, et al. (2011) Familial C3 glomerulopathy associated with CFHR5 mutations: clinical characteristics of 91 patients in 16 pedigrees. Clin J Am Soc Nephrol 6: 1436–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gale DP, de Jorge EG, Cook HT, Martinez-Barricarte R, Hadjisavvas A, et al. (2010) Identification of a mutation in complement factor H-related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet 376: 794–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pierides A, Voskarides K, Athanasiou Y, Ioannou K, Damianou L, et al. (2009) Clinico-pathological correlations in 127 patients in 11 large pedigrees, segregating one of three heterozygous mutations in the COL4A3/COL4A4 genes associated with familial haematuria and significant late progression to proteinuria and chronic kidney disease from focal segmental glomerulosclerosis. Nephrol Dial Transplant 24: 2721–2729. [DOI] [PubMed] [Google Scholar]
- 9. Voskarides K, Damianou L, Neocleous V, Zouvani I, Christodoulidou S, et al. (2007) COL4A3/COL4A4 mutations producing focal segmental glomerulosclerosis and renal failure in thin basement membrane nephropathy. J Am Soc Nephrol 18: 3004–3016. [DOI] [PubMed] [Google Scholar]
- 10. Voskarides K, Pierides A, Deltas C (2008) COL4A3/COL4A4 mutations link familial hematuria and focal segmental glomerulosclerosis. glomerular epithelium destruction via basement membrane thinning? Connect Tissue Res 49: 283–288. [DOI] [PubMed] [Google Scholar]
- 11. Barker DF, Hostikka SL, Zhou J, Chow LT, Oliphant AR, et al. (1990) Identification of mutations in the COL4A5 collagen gene in Alport syndrome. Science 248: 1224–1227. [DOI] [PubMed] [Google Scholar]
- 12. Barker DF, Pruchno CJ, Jiang X, Atkin CL, Stone EM, et al. (1996) A mutation causing Alport syndrome with tardive hearing loss is common in the western United States. Am J Hum Genet 58: 1157–1165. [PMC free article] [PubMed] [Google Scholar]
- 13. Martin P, Heiskari N, Zhou J, Leinonen A, Tumelius T, et al. (1998) High mutation detection rate in the COL4A5 collagen gene in suspected Alport syndrome using PCR and direct DNA sequencing. J Am Soc Nephrol 9: 2291–2301. [DOI] [PubMed] [Google Scholar]
- 14. Chen Y, Zhu S, Zhang Y (2001) [Thin basement membrane nephropathy: a mutation in COL4A5 gene]. Zhonghua Nei Ke Za Zhi 40: 239–242. [PubMed] [Google Scholar]
- 15. Wilson JC, Yoon HS, Walker RJ, Eccles MR (2007) A novel Cys1638Tyr NC1 domain substitution in alpha5(IV) collagen causes Alport syndrome with late onset renal failure without hearing loss or eye abnormalities. Nephrol Dial Transplant 22: 1338–1346. [DOI] [PubMed] [Google Scholar]
- 16. Demosthenous P, Voskarides K, Stylianou K, Hadjigavriel M, Arsali M, et al. (2012) X-linked Alport syndrome in Hellenic families: phenotypic heterogeneity and mutations near interruptions of the collagen domain in COL4A5. Clin Genet 81: 240–248. [DOI] [PubMed] [Google Scholar]
- 17. Deltas C, Pierides A, Voskarides K (2012) The role of molecular genetics in diagnosing familial hematuria(s). Pediatr Nephrol 27: 1221–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Andrews KL, Mudd JL, Li C, Miner JH (2002) Quantitative trait loci influence renal disease progression in a mouse model of Alport syndrome. Am J Pathol 160: 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Beirowski B, Weber M, Gross O (2006) Chronic renal failure and shortened lifespan in COL4A3+/− mice: an animal model for thin basement membrane nephropathy. J Am Soc Nephrol 17: 1986–1994. [DOI] [PubMed] [Google Scholar]
- 20. Tonna S, Wang YY, Wilson D, Rigby L, Tabone T, et al. (2008) The R229Q mutation in NPHS2 may predispose to proteinuria in thin-basement-membrane nephropathy. Pediatr Nephrol 23: 2201–2207. [DOI] [PubMed] [Google Scholar]
- 21. Voskarides K, Arsali M, Athanasiou Y, Elia A, Pierides A, et al. (2012) Evidence that NPHS2-R229Q predisposes to proteinuria and renal failure in familial hematuria. Pediatr Nephrol 27: 675–679. [DOI] [PubMed] [Google Scholar]
- 22. Papagregoriou G, Erguler K, Dweep H, Voskarides K, Koupepidou P, et al. (2012) A miR-1207-5p binding site polymorphism abolishes regulation of HBEGF and is associated with disease severity in CFHR5 nephropathy. PLoS One 7: e31021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kao WH, Klag MJ, Meoni LA, Reich D, Berthier-Schaad Y, et al. (2008) MYH9 is associated with nondiabetic end-stage renal disease in African Americans. Nat Genet 40: 1185–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kopp JB, Smith MW, Nelson GW, Johnson RC, Freedman BI, et al. (2008) MYH9 is a major-effect risk gene for focal segmental glomerulosclerosis. Nat Genet 40: 1175–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pattaro C, Aulchenko YS, Isaacs A, Vitart V, Hayward C, et al. (2009) Genome-wide linkage analysis of serum creatinine in three isolated European populations. Kidney Int 76: 297–306. [DOI] [PubMed] [Google Scholar]
- 26. Behar DM, Rosset S, Tzur S, Selig S, Yudkovsky G, et al. (2010) African ancestry allelic variation at the MYH9 gene contributes to increased susceptibility to non-diabetic end-stage kidney disease in Hispanic Americans. Hum Mol Genet 19: 1816–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cheng W, Zhou X, Zhu L, Shi S, Lv J, et al. (2011) Polymorphisms in the nonmuscle myosin heavy chain 9 gene (MYH9) are associated with the progression of IgA nephropathy in Chinese. Nephrol Dial Transplant 26: 2544–2549. [DOI] [PubMed] [Google Scholar]
- 28. Liang S, He L, Zhao X, Miao Y, Gu Y, et al. (2011) MicroRNA let-7f inhibits tumor invasion and metastasis by targeting MYH9 in human gastric cancer. PLoS One 6: e18409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, et al. (2010) Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329: 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, et al. (2010) Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet 128: 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. O'Seaghdha CM, Parekh RS, Hwang SJ, Li M, Kottgen A, et al. (2011) The MYH9/APOL1 region and chronic kidney disease in European-Americans. Hum Mol Genet 20: 2450–2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nelson GW, Freedman BI, Bowden DW, Langefeld CD, An P, et al. (2010) Dense mapping of MYH9 localizes the strongest kidney disease associations to the region of introns 13 to 15. Hum Mol Genet 19: 1805–1815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cooke JN, Bostrom MA, Hicks PJ, Ng MC, Hellwege JN, et al. (2012) Polymorphisms in MYH9 are associated with diabetic nephropathy in European Americans. Nephrol Dial Transplant 27: 1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]