


Abstract
Genome-wide gene expression analyses of the human somatic cell cycle have indicated that the set of cycling genes differ between primary and cancer cells. By identifying genes that have cell cycle dependent expression in HaCaT human keratinocytes and comparing these with previously identified cell cycle genes, we have identified three distinct groups of cell cycle genes. First, housekeeping genes enriched for known cell cycle functions; second, cell type-specific genes enriched for HaCaT-specific functions; and third, Polycomb-regulated genes. These Polycomb-regulated genes are specifically upregulated during DNA replication, and consistent with being epigenetically silenced in other cell cycle phases, these genes have lower expression than other cell cycle genes. We also find similar patterns in foreskin fibroblasts, indicating that replication-dependent expression of Polycomb-silenced genes is a prevalent but unrecognized regulatory mechanism.
INTRODUCTION
Genome-wide studies of gene expression throughout the cell division cycle have revealed several genes that are differentially expressed (1–9), but have also indicated that the set of cycling genes differs between primary and cancer cells (3). Primary cells are, however, inherently difficult to synchronize, for example, only 40–50% of foreskin fibroblast cells in culture can be synchronized by serum starvation or double thymidine block (3). Although sophisticated statistics may partially overcome lack of synchronization (3), a large population of asynchronous or arrested cells results in high background gene expression noise. Consequently, more cycling genes can be detected in a highly synchronous culture than in a culture where at most 50% of the cells are synchronized. Moreover, as the only human cell line—in addition to primary fibroblasts (1,3,4)—profiled for cell cycle expression so far is the cervical cancer cell line HeLa (2,5), it is unclear to what extent cell type-specific factors affect reported differences in cycling genes.
We have used the human keratinocyte cell line HaCaT to address this question. Specifically, by measuring the gene expression profiles of double thymidine synchronized HaCaT cells, we identified three major groups of cycling genes. First, a set of genes with housekeeping characteristics, strong enrichment for known cell cycle functions and overlap with previously identified cell cycle genes. Second, a set of genes with cell type-specific characteristics, enrichment for HaCaT-specific functions and poor overlap with previously identified cell cycle genes. Third, a set of genes that has the mark for Polycomb silencing: histone H3 lysine 27 tri-methylation (H3K27me3). We show that this third set of genes is expressed in a replication-dependent manner, as the genes are upregulated during S phase in a pattern related to DNA replication timing. Consistent with being epigenetically silenced in other cell cycle phases, these genes are generally lower expressed than are other cell cycle expressed genes. We also find similar patterns in foreskin fibroblasts synchronized by serum starvation, indicating that replication-dependent expression of Polycomb-silenced genes is a prevalent but unrecognized regulatory mechanism.
MATERIALS AND METHODS
HaCaT cell culture and synchronization
HaCaT cells were plated at 10% confluence (1 × 106 cells) in 150-mm tissue culture dishes in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS). Cells were arrested in the interphase G1/S by double thymidine block; briefly, cells were treated with 2 mM of thymidine for 18 h, released from the arrest for 10 h and arrested a second time with 2 mM of thymidine for additional 18 h. After treatment, media was replaced, and cells were collected at 3-h intervals for up to 33 h, covering approximately two cell cycles. Synchrony was monitored by flow cytometry analysis of propidium iodide-stained cells and by cell counting. Quantification of cells in each phase was done with the MultiCycle DNA cell cycle analysis software (Phoenix Flow Systems Inc., San Diego, CA, USA) combined with the cell counting results.
HeLa cell culture and synchronization
Adherent HeLa cells were plated in 150-mm culture dishes in DMEM with 10% of FBS, 2 mM of glutamine, 0.1 mg/ml of gentamicin and 1.25 µg/ml of fungizone. Cells at 60–70% confluence were arrested in the G2/M transition with 100 ng/ml of nocodazole for 17 h. The mitotic cells were then collected by manual shake-off, washed twice and re-plated in fresh DMEM to progress through the cell cycle. Cells were harvested from culture dishes by trypsinization every 30 min for the first 2 h and then every 3 h from 3 to 24 h after release. Phosphate-buffered saline containing 3% of FBS was added to inactivate the trypsin.
HeLa cells were pelleted and resuspended in 100 µl of RNAlater (Applied Biosystems/Ambion, Austin, TX, USA). All pellets were kept at 4°C overnight and were stored at −80°C before use. Verification of the cell cycle stage was determined by analysing the DNA content of propidium iodide-stained cells by a BD FACSAria (BD Biosciences, San Jose, CA, USA) flow cytometer. Quantification of cells in each phase was done with FlowJo (Tree Star Inc., OR, USA).
cRNA synthesis and microarray hybridization—HaCaT
Total RNA was extracted using the High Pure RNA Isolation Kit (Roche Applied Science, Indianapolis, IN, USA) and the manufacturer’s protocol. RNA from synchronous cells was reverse transcribed into cDNA (cDNA synthesis Kit, Invitrogen, Carlsbad, CA, USA), which was used as a template for the RNA polymerase Enzo (Affymetrix, Santa Clara, CA, USA) to synthesize dUTP–dCTP biotinylated cRNA. The labelled cRNA was hybridized to Affymetrix oligonucleotide arrays (HG-U133 Set) under conditions specified by the manufacturer.
Microarray analyses—HeLa
Total RNA was prepared using the mirVana miRNA Isolation Kit (Ambion) according to the manufacturer’s protocol. The integrity and stability of RNA samples were assessed by Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). We used the Illumina TotalPrep RNA amplification Kit (Ambion) to amplify RNA for hybridization on Illumina BeadChips. Total RNA from each sample was used to synthesize first strand cDNA by reverse transcription. After the second strand cDNA synthesis and cDNA purification steps, the in vitro transcription to synthesize cRNA was prepared overnight for 12 h. The gene expression profiles were measured using Illumina HumanHT-12 v3 Expression BeadChip (Illumina, San Diego, CA, USA).
Identification of periodically expressed genes—statistical analysis
Normalization across arrays was performed by robust multi-array average (RMA) from the Bioconductor package affy (10). As there are no probes specific for splice variants in the arrays used, for probes that hybridized with mRNAs corresponding to the same gene, only the probe having the highest variation was used for further analyses. The normalized data were then used to identify genes that showed cyclic behaviour. Genes that matched a cell cycle profile were identified using partial least squares (PLS) regression. Specifically, the gene expression matrix was regressed on to a sine and cosine function with periods equal to the estimated duration of the cell cycle. False discovery rates were computed using a modified Hotelling’s T-square statistic and resampling. Resampling (n = 1000) was done by assigning phase angles randomly to the time-steps, thus preserving the correlation within replicates of the same time-steps.
Assigning cell cycle phases
Assignment of cell cycle phases used the following protocol. First, each gene in the PLS model was assigned a phase angle by computing the arctangent of the gene’s first and second principle component. Second, we used a list of genes previously published to be predominantly expressed in G1/S, S, G2, G2/M and M/G1 phase [(2): Table 2 and the four M/G1 associated genes from Figure 2] and identified which of these genes were significant (q < 0.05) in our PLS model (Supplementary Figure S3A). Third, for each set of significant G1/S, S, G2, G2/M and M/G1 phase genes, we computed the set’s average phase angle and the corresponding standard deviation and used these values as parameters in a normal distribution to construct a phase angle probability density function for each of the five cell cycle phases (Supplementary Figure S3A). Fourth, we used these probability density functions to assign the most probable phase IDs to each of the significant (q < 0.05) genes in the PLS model. Fifth, if necessary, the standard deviation parameters in the phase angle probability density functions were increased to ensure that the phase assignments had the same order as the average phase angles for the five groups. Specifically, if any out of order phase assignments were detected, the lowest of the five standard deviations was increased by 25%, and the phase assignments were redone. This process was repeated until all phase assignments were in the order defined by the averages.
Figure 2.

HaCaT cell cycle genes include both generally active housekeeping and keratinocyte-specific genes. (A) Distributions of CpG content in promoters for all genes included in the PLS model (all, dashed line) and cell cycle expressed genes (CC, solid line). The lines are the kernel density estimates of the two CpG content distributions. (B) Fraction of LCG (black), ICG (grey) and HCG (white) promoters for the two groups in (A). Numbers above bars are the total number of non-redundant promoter regions. (C and D) Fraction of promoters that in HaCaT (C) or in seven different cell lines (D) contain exclusive H3K4me3 or H3K27me3 marks (K4, white; K27, dark grey), bivalent K4 and K27 marks (K4/K27, light grey), or no or inconsistent marks (None, diamonds). P-values are the results of binomial tests comparing the fraction of H3K4me3-marked genes in the two sets. (E and F) Fractions from (C: E) and (D: F) subdivided into LCG, ICG and HCG (black, dark grey and light grey lines) promoters. (G) GO biological process (BP) terms or KEGG pathways significantly enriched (P < 0.05) among HCG, ICG or LCG cell cycle genes subdivided into promoters with consistent H3K4me3 (white) or H3K27me3 (no significant terms) marks, or no or inconsistent marks (diamonds). Horizontal axes show Benjamini–Hochberg corrected P-values (–log10); numbers on the right show the expected (E) and observed (O) number of genes and the corresponding odds ratios (OR).
Promoter CpG analyses
Gene annotations were downloaded from the refGene table in the University of California, Santa Cruz (UCSC) Genome Browser database (human genome assembly version hg18) (11). Promoter regions were defined as the 1000 nt upstream of annotated transcription start sites. Overlapping regions corresponding to genes with multiple transcripts having identical or alternative start sites were merged into one region to create a non-redundant set of promoter regions. Based on this non-redundant set, CpG ratios and CpG promoter classes were calculated and assigned as previously described (12).
Promoter histone analyses
Processed peak data from chromatin immunoprecipitation sequencing (ChIP-seq) experiments of H3K4me3 and H3K27me3 in seven cell lines (GM12878, HUVEC, K562, NHEK, H1-hESC, HMEC and NHLF) were downloaded from UCSC (hg18) (13,14). Non-redundant promoter regions that contained overlapping H3K4me3 and H3K27me3 peaks in all seven cell lines were classified as H3K4me3 and H3K27me3 enriched, respectively; promoter regions that contained both marks in all seven cell lines were classified as bivalent; and promoter regions that did not contain any of the marks or only contained the marks in some of the cell lines were classified as having no or inconsistent marks.
Processed peak data from ChIP-seq experiments of H3K4me3 and H3K27me3 in HeLa and normal adult dermal fibroblasts (NHDF-Ad) were downloaded from UCSC (hg19; Broad Histone track) and remapped to hg18 by the UCSC liftOver tool.
HaCaT ChIP-seq
ChIP, library preparation and sequencing were done as described in the Supplementary Methods. The resulting input chromatin and H3K4me3 and H3K27me3 IP libraries contained ∼64 million (M), ∼42 M and ∼52 M sequence reads, respectively. We used cutadapt (15) to trim adapter sequences, bowtie (16) to align sequence reads from the human genome (version hg18) and discarded all reads that aligned to more than one genomic location. The ∼50 M, ∼32 M and ∼39 M aligned reads for the input, H3K4me3 and H3K27me3 libraries, respectively, were processed by SICER (17) to identify genomic regions that were significantly enriched for H3K4me3 and H3K27me3 in HaCaT (SICER used the input chromatin library as control to identify significant regions; see Supplementary Dataset S3 and S4).
Gene ontology and Kyoto Encyclopedia of Genes and Genomes analyses
Gene ontology (GO) term and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment was assessed by GOstats (18). For GO terms, the test was conditioned on the structure of the GO graph.
Data access
The microarray data from this publication have been submitted to the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress/) and were assigned the identifiers E-MTAB-454 (HaCaT) and E-TABM-1152 (HeLa). HaCaT ChIP-seq data are available from http://tang.medisin.ntnu.no/∼palsat/HaCaT-ChIP.tgz; bed-files with significant H3K4me3 and H3K27me3 regions are available as Supplementary Dataset S3 and S4.
RESULTS
Partial least squares regression identifies cell cycle-regulated transcripts
To overcome the problem of low synchronization of primary cells and to investigate potential cell type-related differences in cycling genes, we turned to the human keratinocyte cell line HaCaT—a non-tumorigenic, spontaneously immortalized cell line that exhibits apparently normal differentiation (19). Using a double thymidine block to arrest and, subsequently, release HaCaT cells at the G1/S boundary resulted in ∼90% of the cells re-entering the cell cycle and continuing as a uniform cohort through the cell cycle (Supplementary Figure S1). Based on the DNA content profile, we estimated the cell cycle duration to be ∼20 h. Although the cells gradually lost synchrony, such that 70% instead of 90% of the cells were synchronously entering the second S phase, the results were reproducible across three independent experiments. We, therefore, considered synchronized HaCaT cells a good starting point for gene expression analyses.
We were primarily interested in genes that showed consistent cyclic expression with a period equal to the estimated cell cycle duration of 20 h. We, therefore, used PLS (20) to identify genes that followed a sine or cosine pattern with a period of 20 h. Conceptually, this PLS analysis projects the time profile for all genes onto a plane described by the sine and cosine of the cell cycle. As a result, genes that are periodically expressed with high amplitude will be far from the origin, whereas genes that vary little or randomly will be close to the origin. Moreover, the phase angle of the cyclic variation will determine the direction from the origin such that genes that have the same temporal expression patterns will lie in the same direction from the origin on the plane.
By profiling RNA isolated from the synchronized cells at 3 h intervals for 33 h, and combining PLS analyses with resampling to compute false discovery rates (21), we identified 1249 Entrez genes that had significant periodic expression patterns during the HaCaT cell cycle (Figure 1, Supplementary Dataset S1). The synchronization experiments’ trajectory through the PLS cell cycle plane confirmed that the three replicates gave similar and reproducible results (Figure 1B). The trajectory also indicated the cells’ gradual loss of synchrony, as the samples were gradually approaching the origin—especially at later time points.
Figure 1.
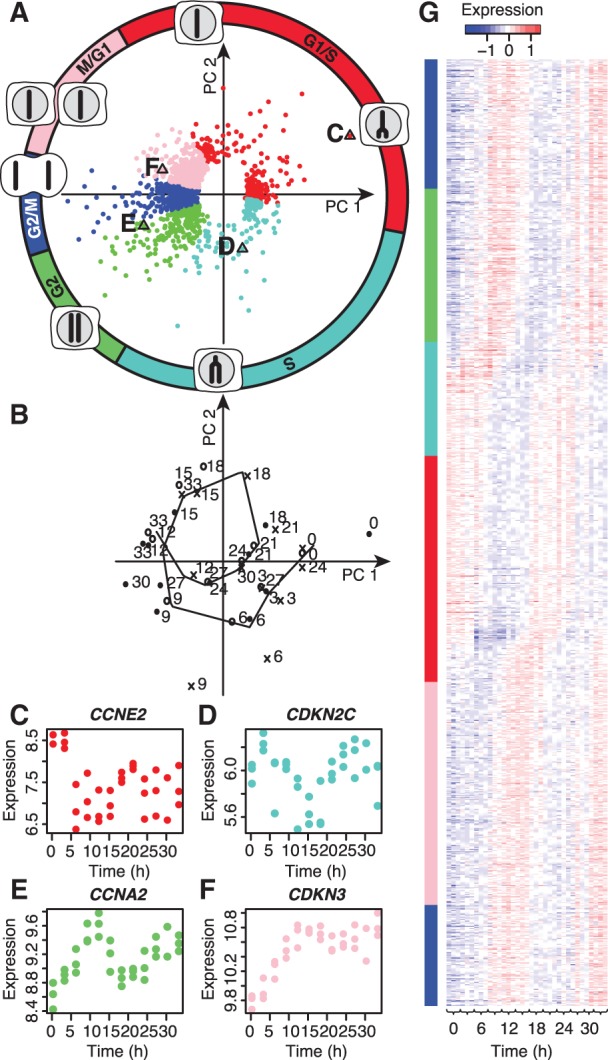
PLS regression identifies cyclic gene expression patterns. The loadings (A) and scores (B) plots show the contribution of each gene (loadings) to the location of each sample in the subspace of the PLS model (20) (scores; here, sine and cosine, period of 20 h). (A) Points show significant genes [q < 0.05; permutation test (21)] and their contribution to the PLS cell cycle model’s first and second components (PC1 and PC2). Colours show the cell cycle phase where the gene is predominantly expressed (see ‘Materials and Methods’ section). The circle and cell diagrams indicate early and late DNA replication, G2, mitosis and G1 as determined by the scores plot (B) and flow cytometry (Supplementary Figure 1). (B) Points show the location of the samples from the three replicates (point, circle and cross) within the PLS model’s first and second components; the line shows the replicates’ average trajectory; numbers show the sample time (h). (C–F) Expression profiles for selected genes from the loadings plot [labelled triangles in (A)]. (C) Cyclin E2 (CCNE2); (D) Cyclin-dependent kinase inhibitor 2C (CDKN2C); (E) Cyclin A2 (CCNA2); and (F) Cyclin-dependent kinase inhibitor 3 (CDKN3). (G) Heatmap showing the expression changes of the cell cycle genes relative to their median expression. The profiles are ordered according the genes’ phase angle in the PLS model in (A). Margin colours show the genes’ assigned cell cycle phase.
To further analyse the cell cycle genes, we used existing annotations (2) to subdivide the genes into five main groups that represented phases G1/S, S, G2, G2/M and M/G1 of the cell cycle (Figure 1; see ‘Materials and Methods’ section). The assigned phases showed good correspondence with previously published phases (Supplementary Table S1); the genes missing from our list had expression patterns that were either inconsistent between replicates, not cyclic, or consistently expressed in the first cell cycle only (Supplementary Figures S2–S4; Supplementary Discussion S1). We, therefore, concluded that our PLS model could robustly identify genes that showed consistent and reproducible cyclic expression patterns within the synchronized HaCaT cells.
HaCaT cell cycle genes include both generally active housekeeping and keratinocyte-specific genes
Having established a set of genes with cell cycle-dependent expression, we set out to characterize these genes further. As cell cycle control is a central housekeeping function, we expected our set of genes to be enriched for high-CpG (HCG) promoters—a hallmark of developmentally regulated genes and housekeeping genes (12,22). We also expected the cell cycle genes to be enriched for histone H3 lysine 4 tri-methylation (H3K4me3) within their promoters, as this epigenetic mark is associated with actively transcribed genes (23,24). Indeed, although the complete set of genes followed a bimodal distribution of HCG and low-CpG (LCG) promoters (12,22), the distribution for the cell cycle genes was shifted, such that a larger percentage had HCG promoters (Figure 2A and B; P = 2e-11, binomial test). Similarly, ChIP-seq of H3K4me3 and H3K27me3 marks in the HaCaT cells (see ‘Materials Methods’ section; Supplementary Dataset S2 and S3) showed that the cell cycle genes described here were enriched for H3K4me3 (Figure 2C). Moreover, the genes were also enriched for H3K4me3 marks that were present in and common for all of seven other cell lines (13,14) (Figure 2D), which is consistent with these genes having housekeeping functions common to many cell types.
The cell cycle genes that showed inconsistent or no histone modifications within their promoters were highly enriched among LCG genes (Figure 2E and F). Tissue-specific genes are commonly enriched among LCGs, which suggested that this subset of cell cycle expressed genes that have LCG promoters with no or inconsistent H3K4me3 modifications might be cell type-specific for HaCaT. To test this possibility, we first investigated whether all the human LCG genes without consistent H3K4me3 within the seven cell lines (LCG/NoH3K4me3) were enriched within GO categories or KEGG pathways (18). As expected, the set of all LCG/NoH3K4me3 genes was highly enriched for tissue specific functions, whereas the set of all HCG/H3K4me3 genes was enriched for housekeeping categories and pathways (Supplementary Figure S5, Supplementary Dataset S4). Second, by accounting for such background enrichment within the nine subgroups, we found both the HCG/H3K4me3 cell cycle genes and the intermediate CpG (ICG)/H3K4me3 cell cycle genes to be specifically enriched for cell cycle-related terms (Figure 2G). Moreover, the LCG/NoH3K4me3 cell cycle genes were specifically enriched for the KEGG cytokine–cytokine receptor interaction pathway, which is consistent with cytokines being important regulators of wound healing in general and keratinocyte proliferation in particular (25). Thus, at least some of the cell cycle genes seem to be cell type-specific for HaCaT cells.
Distinct cell types have exclusive cell cycle regulated genes related to the cells’ specific characteristics and functions
To further determine whether cell type-specific factors could explain differences in reported cycling genes, we analysed the overlap among the cell cycle-regulated genes identified in our and previous human studies (Figure 3). Although our study identified more cell cycle genes than did the foreskin fibroblast (FF) (3) and HeLa (2) studies (Figure 3A), the cell cycle genes we identified had clear cycling patterns in HaCaT. In contrast, the genes identified in the two previous studies but missed in our study had weak or inconsistent patterns in HaCaT; compare Figure 3B and 3C.
Figure 3.

Cell cycle genes that are common or exclusive for foreskin fibroblasts (FF) (3), HeLa (2) and HaCaT reveal characteristics that are specific for the three cell types. (A) Venn diagram showing the overlap in cell cycle genes between FF (BJ), HeLa (W) and HaCaT (33 h). (B) Heatmap showing the expression changes of the HaCaT cell cycle genes relative to their median expression. The genes are grouped according to their overlap with the cell cycle genes from the FF and HeLa studies and are ordered according the genes’ phase angle (from −π to π) in the HaCaT 33 h PLS model. Margin colours show the genes’ assigned cell cycle phase (red, G1/S; turquoise, S; green, G2; blue, G2/M; and pink, M/G1). (C) Heatmap showing the expression changes in HaCaT for genes identified as cell cycle regulated in FF or HeLa but not in HaCaT. The genes are grouped according to their overlap between the two studies and are ordered according to the genes’ phase angle in the HaCaT 33 h PLS model. (D) Median expression level in HaCaT of the genes common and exclusive for the three studies. The genes are grouped according to their overlap between the three studies. Boxes, horizontal black line, and circles show the first and third quartiles, the median and outliers; whiskers show the most extreme data points up to 1.5 times the interquartile range from the box. (E) KEGG pathways and GO molecular function (MF), biological process (BP) and cellular component (CC) terms that are overrepresented (P < 0.05) and unique for six of the seven subsets of cell cycle genes common or exclusive for FF, HeLa and HaCaT; see Supplementary Table S2 for pathways and terms overrepresented among cell cycle genes common for FF, HeLa and HaCaT.
Some of the differences between the studies were likely related to differences in microarray technologies and study designs. Microarray results are generally reproducible across platforms and laboratories for genes with medium and high expression (26), and the genes common for the three studies were generally highly expressed in HaCaT (Figure 3D). These genes were also strongly enriched for cell cycle functions (Supplementary Table S2), indicating that these common genes represent a core set of cell cycle genes. As for design, although previous studies have either not included or used poorly matched biological replicates, we included three direct biological replicates. This, combined with the reproducibly high level of synchronization for the HaCaT cells, gave our study better statistical power to identify cyclically expressed genes.
Although the cell cycle genes common for all three studies represented a core set of human cell cycle genes, the other subsets of cell cycle genes were at least partially related to cell type-specific functions and characteristics (Figure 3E). To illustrate, the GO term ‘vitamin D receptor binding’ was overrepresented among the cell cycle genes exclusive for HaCaT, which is consistent with vitamin D and the vitamin D receptor’s important functions in keratinocytes (27). In contrast, and consistent with HeLa’s cancer origin, ‘chronic myeloid leukaemia’ was the only term exclusive for the HeLa cell cycle genes. Further supporting this conclusion, the KEGG pathways ‘nucleotide excision repair’ and ‘base excision repair’ were exclusively overrepresented among the cell cycle genes common for FF and HaCaT, which suggests that these two DNA repair pathways are partially dysregulated in HeLa. Although we cannot exclude that technical differences underlie some of the differences in gene expression (see ‘Discussion’ section), distinct cell types do seem to have distinct cell cycle regulated genes. For some cells, such as HeLa and likely other cancers (3), these distinct cell cycle genes may reflect that the cells are in an abnormal dysregulated state. Nevertheless, for more normal non-tumorigenic cells, such as HaCaT, at least some of the distinct cell cycle genes are related to the cells’ specific functions.
Cell cycle genes marked with H3K27me3 are transcribed during DNA replication
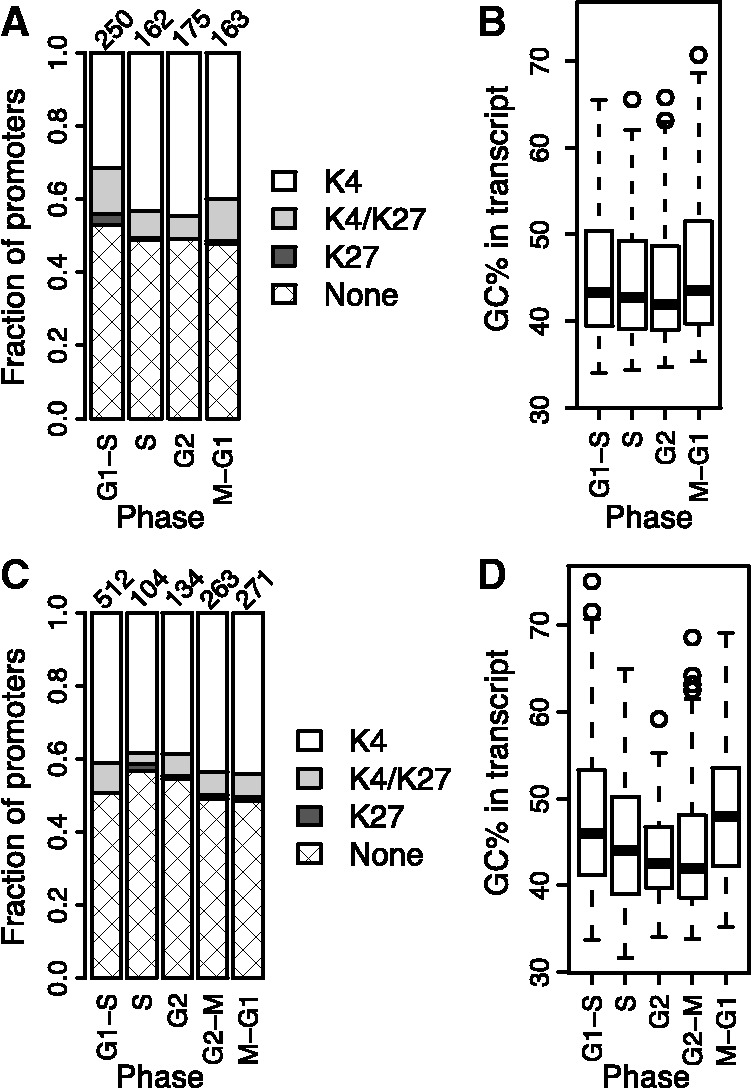
Although the cell cycle genes were generally enriched for H3K4me3, a fraction of the cell cycle genes were marked with H3K27me3 (Figure 2B)—the mark for genes silenced by Polycomb group proteins (28,29). Replication opens compacted chromatin, and although H3K27me3 marks are copied to daughter chromatin, the marks may initially be diluted, such that the normally silenced DNA is transiently accessible for transcription during this replication process. If this model was correct, we would expect that cell cycle genes marked with H3K27me3 were enriched among genes upregulated during DNA replication.
Indeed, the H3K27me3 marks in the HaCaT cells were differentially distributed among the cell cycle genes (Figure 4A). Of genes predominantly expressed in the G1/S and S phases, 37% had H3K4me3 marks and 12% had H3K27me3 marks. In contrast, of genes expressed in G2, G2/M and M/G1, 80% contained H3K4me3, whereas only 1% contained H3K27me3 marks. In total, 87% of the H3K27me3 marked cell cycle genes were expressed in G1/S or S (P = 5e-17; one-tailed binomial test). Similarly, a larger percentage of the G1/S and S phase genes was LCG (Figure 4B). The enrichment of H3K27me3 was not related to CpG content, as the G1/S and S phase genes showed similar enrichment for H3K27me3 independent of CpG content (Figure 4C). These patterns were also consistent within the previously published data from seven different cell lines (13,14) (Supplementary Figure S6).
Figure 4.

G1/S and S phase genes are enriched for H3K27me3 marks and show replication-related expression. (A) Distribution of HaCaT H3K4me3 and H3K27me3 marks in promoters of genes upregulated in G1/S, S, G2, G2/M and M/G1 phases; see Figure 2. (B) Fractions of LCG, ICG and HCG promoters for genes upregulated in the five cell cycle phases. (C) Distribution of histone marks from (A) subdivided by promoter CpG content. (D–F) GC% (D), replication timing (E) and expression level (F) of genes expressed in the five cell cycle phases. GC% is the GC% in the region defined by the gene’s annotated transcription start and end sites; replication timing is the median S/G1 ratio within embryonic stem cells (33) of regions overlapping each gene, such that values close to 100 and 0 represent early and late replication, respectively; expression is the gene’s average microarray expression. Boxes, horizontal black line, and circles show the first and third quartiles, the median and outliers; whiskers show the most extreme data points up to 1.5 times the interquartile range from the box. (G) Expression levels of specific subgroups of G1/S and S phase genes.
Genes upregulated in early S-phase tend to be replicated earlier than late S-phase genes
Replication occurs in distinct domains largely characterized by differences in GC content, and although these domains are partly reorganized during development, domains with high and low GC content are generally replicated early and late in S phase, respectively (30–35). If DNA replication facilitated transient transcription of silenced genes, we would, therefore, expect the genes upregulated during G1/S to generally have a higher GC content than the S phase and other cell cycle genes. Indeed, the G1/S genes had the highest overall GC content of the cell cycle genes (Figure 4D; P-value of 7e-3, two-sided un-paired Student’s t-test with unequal variance of difference in mean between G1/S and S phase genes) and replication-timing data from three different cell lines (30,33) indicated that the G1/S genes on average tended to be replicated earlier than the S phase genes (Figure 4E, Student’s t-test P-value of 1e-3; Supplementary Figure S7). Importantly, 63% of the cell cycle regulated genes reside in genomic regions previously shown to have stable replication timing across multiple cell types (31), and these genes had the same pattern in replication timing as the complete set of cell cycle genes (Supplementary Figure S8).
Consistent with some of the genes residing in silenced chromatin and being transiently expressed during replication, the G1/S and S genes had a general lower expression level than the genes expressed in the other phases (Figure 4F). Moreover, the genes marked with H3K27me3 in HaCaT had the lowest expression level of the G1/S and S phase genes (Figure 4G). The differences in expression between H3K27me3-marked genes and other subgroups were significant for all G1/S phase groups (P-values of 2e-16, 2e-9, 3e-5 and 0.01 for K4, HCG, ICG and LCG groups, respectively; two-sided un-paired Student’s t-test with unequal variance) and for the K4 and HCG S phase groups (P-values of 7e-4, 0.02, respectively; P-values of 0.1 and 0.3 for ICG and LCG groups, respectively).
Foreskin fibroblasts show similar replication-related expression of H3K27me3 marked genes
To investigate whether this replication-related cell cycle expression pattern could be found in other cell types, we first used our computational framework to reanalyse the previously published primary foreskin fibroblast data (3) (Supplementary Figure S9). As in the HaCaT cells, the majority (64%) of the genes that had significant cell cycle expression and were marked with H3K27me3 in fibroblasts were upregulated during DNA replication (Figure 5A); the mark was especially enriched for the G1/S genes (P = 0.009; one-tailed binomial test for enrichment). In contrast to HaCaT, the majority of the fibroblast cell cycle genes marked with H3K27me3 was also marked with H3K4me3. The fibroblast cell cycle genes also showed a similar trend in GC content as the HaCaT genes, although the difference between the genes expressed early (G1/S) and late (S) during DNA replication was not significant (Figure 5B; P = 0.1, Student’s t-test).
Figure 5.
H3K27me3 marks are enriched in genes upregulated during DNA replication in primary foreskin fibroblasts. (A) Distribution of H3K4me3 and H3K27me3 marks in promoters of genes upregulated in G1/S, S, G2 and M/G1 phases in primary foreskin fibroblasts synchronized by serum starvation [expression data from (3); ChIP-seq data from normal adult dermal fibroblasts (13,14)]. (B) GC% of the genes from (A). (C) Distribution of H3K4me3 and H3K27me3 marks in promoters of genes upregulated in G1/S, S, G2, G2/M and M/G1 phases in HeLa cells synchronized by nocodazole and mitotic shake-off [ChIP-seq data from HeLa (13,14)]. (D) GC% of the genes from (C). See Figures 2 and 4 for details on graphs.
Second, we measured the gene expression profiles in HeLa cells synchronized in the G2/M transition by nocodazole and mitotic shake-off and used PLS to identify genes with cell cycle-dependent expression (Supplementary Figures S10 and S11). Again, the majority (52%) of the cell cycle genes that were marked with H3K27me3 in HeLa was upregulated during DNA replication, but the differences between the phases were not significant (Figure 5C; P = 0.2). Moreover, the cell cycle genes again showed a similar trend in GC content as the HaCaT, such that the G1/S genes had a higher GC content than the S genes (Figure 5D; P = 0.01, Student’s t-test). Thus, replication-related expression of H3K27me3-marked genes does not seem to be a strong characteristic of all cell types. It is relevant to note, however, that the characteristic is absent in the most abnormal of the three cells, whereas HaCaT and the primary fibroblasts both have significant replication-related expression of H3K27me3-marked genes.
DISCUSSION
Previous studies of the gene expression patterns during the human cell division cycle have indicated marked differences between primary and cancer cells (3), but our results show that distinct cell types have distinct cell cycle expressed genes that are related to the cells’ functions. Specifically, by profiling the gene expression in synchronized human HaCaT keratinocytes, we have identified a set of cell cycle expressed genes containing known marks for cell type-specific expression, such as low CpG content in their promoters and few or inconsistent promoter histone marks. These genes were enriched for functions important for keratinocyte proliferation, such as cytokine–cytokine receptor interaction.
Cell cycle regulation is an important housekeeping function and accordingly, most of the cell cycle expressed genes identified in our study have high CpG promoters and the active chromatin mark H3K4me3 in multiple cell lines. Nevertheless, we have also shown that some genes with low CpG promoters or the silent chromatin mark H3K27me3 have cell cycle dependent expression. These genes are mostly expressed during S phase in a replication-related pattern, which suggests that DNA replication can open some epigenetically silenced loci for transcription.
Replication-dependent transcription is a mechanism that can achieve precise transcriptional regulation during the cell cycle, as normally silenced genes will only be transcribed when their loci are replicated in S-phase. Compared with the other cell cycle genes, the Polycomb-regulated cell cycle genes in HaCaT were significantly enriched for the GO terms ‘GO:0007267 cell–cell signalling’ and ‘GO:0046903 secretion’ (Benjamini–Hochberg-corrected P-values of 0.007 and 0.02, respectively). In HaCaT cells, replication-dependent transcription may, therefore, be a regulatory mechanism for coordinating cell–cell signalling with the cell cycle.
Lanzuolo et al. (36) have earlier reported that two Polycomb-repressed homeotic genes show replication-dependent expression in Drosophila S2 cells. Our results extend this observation to humans and show that many other genes with Polycomb-repressive marks have replication-dependent expression. We also note that cis-encoded RNAs do play a role in Polycomb-mediated targeting and regulation (37–39). Given that Polycomb regulates all transcripts in targeted loci, replication-dependent expression may be a mechanism for expressing such cis-encoded Polycomb-associated RNAs at existing Polycomb-regulated loci.
When comparing our list of HaCaT cell cycle genes with the cell cycle genes from the previous HeLa (2) and primary foreskin fibroblast studies (3), we found several differences, such that only 125 genes were common for all three studies (Figure 3A). Several technical factors, such as differences in microarray technology, analytical approach and experimental design, likely contributed to these differences, for example, the genes that were common for all three studies were highly expressed, whereas the genes that were exclusive for our study were lowly expressed in HaCaT. We could detect these HaCaT-specific genes because our three matched biological replicates gave increased statistical power to identify weak, but consistent, cyclic expression patterns.
In addition to these technical differences, our results also show that underlying biological differences between the cells can explain some of the differences in cell cycle expressed genes. Specifically, our results show that in addition to the core genes that have cell cycle-dependent expression in multiple cell types, some genes related to the cell type’s specific functions and characteristics also have cyclic expression. This is in contrast to a previous report, which indicated that such cell-type specific expression mostly reflects differences between primary and cancer cells (3). These cancer-related differences are evident in our analyses as well, but we also find that genes and pathways important to keratinocyte function, such as cytokine–cytokine receptor interaction (25) and vitamin D receptor binding, (27) have cell cycle-dependent expression. Both functions are important for normal keratinocyte proliferation, which makes it unlikely that these results are an artifact of HaCaT’s transformed state or technical differences between the studies. Instead, the cell cycle-dependent expression of these genes likely reflects that processing of proliferative signals from the extracellular environment should be coordinated with the cell cycle.
We should note that as we have measured expression changes in total cellular RNA, some of the expression changes we have detected could potentially be explained by cell cycle-dependent changes in RNA processing or RNA stability instead of RNA transcription. Moreover, our approach of chemically synchronizing cells can likely explain some of the changes in gene expression in our data, as the synchronization procedure likely triggers stress-related responses that could both give false-positive cyclic profiles and mask the cell-cycle-dependent expression of some true cell cycle genes. This masking effect could potentially explain why we did not detect the known cell-cycle genes CDKN1A and VEGFC to have cell cycle-dependent expression in HaCaT (Supplementary Figure S2C and D). Our analyses, including the good overlap between the genes we identified and known cell-cycle-expressed genes, strong enrichment for cell-cycle-related functions and overlap with previous studies, do suggest, however, that the majority of the genes we identified are true cell cycle genes.
In summary, by combining robust statistics with the power of matched biological replicates, we have identified a set of genes that show consistent cell cycle-dependent expression in HaCaT cells. These genes include both general cell cycle genes and genes specific for HaCaT function. Some of the cell cycle genes also have H3K27me3 marks—a histone modification commonly associated with Polycomb-silenced genes. We show that in both HaCaT and primary foreskin fibroblasts, these genes are upregulated during DNA replication, which is consistent with a mechanism where DNA replication can open some Polycomb-repressed loci for transcription.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2, Supplementary Figures 1–11, Supplementary Methods, Supplementary Datasets 1–4 and Supplementary References [2,3,18,30,31,33,40].
FUNDING
Norwegian Functional Genomics Program of the Norwegian Research Council (to H.E.K. and P.S.); Norwegian Cancer Association (to H.E.K.); Cancer Fund at St. Olav’s Hospital Trondheim (to H.E.K. and P.S.); Svanhild and Arne Must Fund for Medical Research (to H.E.K.); European Community program IHP-MCIF-99-1, contract HPMF-CT-2001-01290 (to J.P.-D.). Funding for open access charge: Norwegian Research Council.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank N.B. Liabakk for technical assistance and A.D. Riggs and O. Snøve Jr for critical reading. The sequencing service was provided by the Norwegian Sequencing Centre (www.sequencing.uio.no), a national technology platform hosted by the University of Oslo and supported by the ‘Functional Genomics’ and ‘Infrastructure’ programs of the Norwegian Research Council and the Southeastern Regional Health Authorities of Norway.
REFERENCES
- 1.Cho RJ, Huang M, Campbell MJ, Dong H, Steinmetz L, Sapinoso L, Hampton G, Elledge SJ, Davis RW, Lockhart DJ. Transcriptional regulation and function during the human cell cycle. Nat. Genet. 2001;27:48–54. doi: 10.1038/83751. [DOI] [PubMed] [Google Scholar]
- 2.Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol. Biol. Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bar-Joseph Z, Siegfried Z, Brandeis M, Brors B, Lu Y, Eils R, Dynlacht BD, Simon I. Genome-wide transcriptional analysis of the human cell cycle identifies genes differentially regulated in normal and cancer cells. Proc. Natl Acad. Sci. USA. 2008;105:955–960. doi: 10.1073/pnas.0704723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Iyer VR, Eisen MB, Ross DT, Schuler G, Moore T, Lee JC, Trent JM, Staudt LM, Hudson J, Jr, Boguski MS, et al. The transcriptional program in the response of human fibroblasts to serum. Science. 1999;283:83–87. doi: 10.1126/science.283.5398.83. [DOI] [PubMed] [Google Scholar]
- 5.Beyrouthy MJ, Alexander KE, Baldwin A, Whitfield ML, Bass HW, McGee D, Hurt MM. Identification of G1-regulated genes in normally cycling human cells. PLoS One. 2008;3:e3943. doi: 10.1371/journal.pone.0003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Mol. Biol. Cell. 1998;9:3273–3297. doi: 10.1091/mbc.9.12.3273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cho RJ, Campbell MJ, Winzeler EA, Steinmetz L, Conway A, Wodicka L, Wolfsberg TG, Gabrielian AE, Landsman D, Lockhart DJ, et al. A genome-wide transcriptional analysis of the mitotic cell cycle. Mol. Cell. 1998;2:65–73. doi: 10.1016/s1097-2765(00)80114-8. [DOI] [PubMed] [Google Scholar]
- 8.Laub MT, McAdams HH, Feldblyum T, Fraser CM, Shapiro L. Global analysis of the genetic network controlling a bacterial cell cycle. Science. 2000;290:2144–2148. doi: 10.1126/science.290.5499.2144. [DOI] [PubMed] [Google Scholar]
- 9.Rustici G, Mata J, Kivinen K, Lio P, Penkett CJ, Burns G, Hayles J, Brazma A, Nurse P, Bahler J. Periodic gene expression program of the fission yeast cell cycle. Nat. Genet. 2004;36:809–817. doi: 10.1038/ng1377. [DOI] [PubMed] [Google Scholar]
- 10.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 11.Rhead B, Karolchik D, Kuhn RM, Hinrichs AS, Zweig AS, Fujita PA, Diekhans M, Smith KE, Rosenbloom KR, Raney BJ, et al. The UCSC Genome Browser database: update 2010. Nucleic Acids Res. 2010;38:D613–D619. doi: 10.1093/nar/gkp939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, Schubeler D. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat. Genet. 2007;39:457–466. doi: 10.1038/ng1990. [DOI] [PubMed] [Google Scholar]
- 13.Rosenbloom KR, Dreszer TR, Pheasant M, Barber GP, Meyer LR, Pohl A, Raney BJ, Wang T, Hinrichs AS, Zweig AS, et al. ENCODE whole-genome data in the UCSC Genome Browser. Nucleic Acids Res. 2010;38:D620–D625. doi: 10.1093/nar/gkp961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 2011;17:10–12. [Google Scholar]
- 16.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics. 2009;25:1952–1958. doi: 10.1093/bioinformatics/btp340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Falcon S, Gentleman R. Using GOstats to test gene lists for GO term association. Bioinformatics. 2007;23:257–258. doi: 10.1093/bioinformatics/btl567. [DOI] [PubMed] [Google Scholar]
- 19.Boukamp P, Petrussevska RT, Breitkreutz D, Hornung J, Markham A, Fusenig NE. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988;106:761–771. doi: 10.1083/jcb.106.3.761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Johansson D, Lindgren P, Berglund A. A multivariate approach applied to microarray data for identification of genes with cell cycle-coupled transcription. Bioinformatics. 2003;19:467–473. doi: 10.1093/bioinformatics/btg017. [DOI] [PubMed] [Google Scholar]
- 21.Gidskehaug L, Anderssen E, Flatberg A, Alsberg BK. A framework for significance analysis of gene expression data using dimension reduction methods. BMC Bioinformatics. 2007;8:346. doi: 10.1186/1471-2105-8-346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc. Natl Acad. Sci. USA. 2006;103:1412–1417. doi: 10.1073/pnas.0510310103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 25.Werner S, Grose R. Regulation of wound healing by growth factors and cytokines. Physiol. Rev. 2003;83:835–870. doi: 10.1152/physrev.2003.83.3.835. [DOI] [PubMed] [Google Scholar]
- 26.Kuo WP, Liu F, Trimarchi J, Punzo C, Lombardi M, Sarang J, Whipple ME, Maysuria M, Serikawa K, Lee SY, et al. A sequence-oriented comparison of gene expression measurements across different hybridization-based technologies. Nat. Biotechnol. 2006;24:832–840. doi: 10.1038/nbt1217. [DOI] [PubMed] [Google Scholar]
- 27.Cianferotti L, Cox M, Skorija K, Demay MB. Vitamin D receptor is essential for normal keratinocyte stem cell function. Proc. Natl Acad. Sci. USA. 2007;104:9428–9433. doi: 10.1073/pnas.0702884104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Beisel C, Paro R. Silencing chromatin: comparing modes and mechanisms. Nat. Rev. Genet. 2011;12:123–135. doi: 10.1038/nrg2932. [DOI] [PubMed] [Google Scholar]
- 29.Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woodfine K, Fiegler H, Beare DM, Collins JE, McCann OT, Young BD, Debernardi S, Mott R, Dunham I, Carter NP. Replication timing of the human genome. Hum. Mol. Genet. 2004;13:191–202. doi: 10.1093/hmg/ddh016. [DOI] [PubMed] [Google Scholar]
- 31.Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl Acad. Sci. USA. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Farkash-Amar S, Lipson D, Polten A, Goren A, Helmstetter C, Yakhini Z, Simon I. Global organization of replication time zones of the mouse genome. Genome Res. 2008;18:1562–1570. doi: 10.1101/gr.079566.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Desprat R, Thierry-Mieg D, Lailler N, Lajugie J, Schildkraut C, Thierry-Mieg J, Bouhassira EE. Predictable dynamic program of timing of DNA replication in human cells. Genome Res. 2009;19:2288–2299. doi: 10.1101/gr.094060.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Costantini M, Bernardi G. Replication timing, chromosomal bands, and isochores. Proc. Natl Acad. Sci. USA. 2008;105:3433–3437. doi: 10.1073/pnas.0710587105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang CW, Lyou Y, Townes TM, Schubeler D, Gilbert DM. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lanzuolo C, Lo Sardo F, Diamantini A, Orlando V. PcG complexes set the stage for epigenetic inheritance of gene silencing in early S phase before replication. PLoS Genet. 2011;7:e1002370. doi: 10.1371/journal.pgen.1002370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Plath K, Fang J, Mlynarczyk-Evans SK, Cao R, Worringer KA, Wang H, de la Cruz CC, Otte AP, Panning B, Zhang Y. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- 38.Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pandey RR, Mondal T, Mohammad F, Enroth S, Redrup L, Komorowski J, Nagano T, Mancini-Dinardo D, Kanduri C. Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 2008;32:232–246. doi: 10.1016/j.molcel.2008.08.022. [DOI] [PubMed] [Google Scholar]
- 40.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.