


Abstract
Hybridization of nucleic acids on solid surfaces is a key process involved in high-throughput technologies such as microarrays and, in some cases, next-generation sequencing (NGS). A physical understanding of the hybridization process helps to determine the accuracy of these technologies. The goal of a widespread research program is to develop reliable transformations between the raw signals reported by the technologies and individual molecular concentrations from an ensemble of nucleic acids. This research has inputs from many areas, from bioinformatics and biostatistics, to theoretical and experimental biochemistry and biophysics, to computer simulations. A group of leading researchers met in Ploen Germany in 2011 to discuss present knowledge and limitations of our physico-chemical understanding of high-throughput nucleic acid technologies. This meeting inspired us to write this summary, which provides an overview of the state-of-the-art approaches based on physico-chemical foundation to modeling of the nucleic acids hybridization process on solid surfaces. In addition, practical application of current knowledge is emphasized.
INTRODUCTION
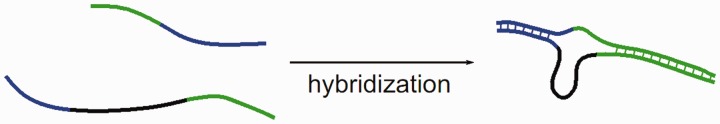
Hybridization of nucleic acids on a solid surface is a key process used in a broad range of technologies. Usually a DNA oligonucleotide (probe) is immobilized on a glass slide or a micrometer-sized bead, with the oligonucleotide acting as a trap for a complementary target sought in a sample. If a complementary target exists in the sample, a duplex is typically formed on binding of the two nucleic acid strands. The number of duplexes of a particular kind is supposed to reflect the concentration of the corresponding target. However, the targets within a typical biological experiment are within a complex mixture of RNA sequences. And as well as hybridization between a target and its complementary probe sequence, there are other reactions taking place within an experiment. For example, sequences can fold into a secondary structure or hybridize to partially complementary RNA in solution.
A number of microarray experiments have shown that the extent of hybridization of the complementary probe and target strongly varies from duplex to duplex (e.g. 1,2). Specifically, given equal concentration of several targets and a fixed concentration of probes, the number of duplexes will vary depending on the sequence of the probe–target duplex. This phenomenon is also likely responsible for the lack of correspondence between absolute target concentration and quantification produced by the 454 next-generation sequencing (NGS, 3), although other factors such as NGS probe-specific biases must also be considered.
It is highly desirable to be able to predict the number of probe-target duplexes formed at a given target concentration. Such a prediction would essentially mean a probe response function, which could be used to measure target concentration from the known number of probe-target duplexes. The knowledge of the response function is critical for quantitative applications of the microarray and NGS technologies. The advantage of a physics-based algorithm is that one could set a physical error bar based on the data and clearly distinguish useful datasets [i.e. those that best match to the expected isotherm (4)] from the problematic ones.
Held et al. (5) conducted the first systematic attempt to use physico-chemical principles to determine the microarray probe response function for the popular Affymetrix Genechip. Further attempts have been carried out since (e.g. 6–8), with many of the approaches taking advantage of ‘spiked-in’ Genechip data, in which the concentration of particular transcripts is already established (9). The models of surface hybridization are typically related to similar models of hybridization in solution, and use either modeling parameters (hybridization, folding energies, etc) taken from the studies conducted in a bulk solution, or obtained by fitting a model to a training dataset. To our knowledge no physico-chemical modeling of NGS technologies has been attempted at the same level of detail seen for microarray studies, although investigation of sources of noise (10) and bias in NGS data is an active field of research (11). Moreover, modeling work for microarrays has benefitted from stable protocols, whereas NGS technologies and protocols are still in a period of rapid evolution, with substantially revised versions released each year.
In spite of the heavy use of microarrays and NGS by the biological community, there are many questions pertinent to hybridization of nucleic acids on the surface that remain unanswered.
Do we clearly understand hybridization process on solid surfaces?
What are the stages of this process?
Which response functions are precise enough to be practically useable?
Are there any alternative approaches to quantify nucleic acid targets?
A group of 15 researchers from all over the world gathered for a 2-day workshop at the Max Planck Institute for Evolutionary Biology in Ploen, Germany (http://www.evolbio.mpg.de/ploenworkshop/) to exchange ideas about potential solutions to these questions. This area of research is interdisciplinary, and the meeting brought together researchers analysing topics ranging from the chemical bonding expected for individual sequences, the characteristics of populations of nucleic acids, all the way to attempts to understand the causes of outliers seen across many tens of thousands of microarray experiments.
This present report summarizes the state of the field, showing latest achievements of particular groups, their future directions and the relevance of their findings to biologists as well as other scientists. We attempted to make sections of this report as self contained as possible, therefore each section may be read independently from the others. Each section is prefixed with a short italicized text for non-specialists.
HIDDEN SECRETS OF A HUNDRED THOUSAND MICROARRAYS
Several artifacts were discovered by massive analysis of 100 000 microarrays from various research laboratories. These artifacts are associated with molecular biology protocols, runs of multiple G nucleotides in microarray probes and fluorescent light scattering.
The tremendous success of microarray technology has led to thousands of publications, describing the results from hundreds of thousands of microarray observations of messenger RNA. The vast majority of these data are now available for meta-analysis—researchers typically upload a description of their results and the associated data to repositories such as the Gene Expression Omnibus (GEO, 12) following publication of their own analysis. There are now >100 000 microarray hybridizations within GEO.
Affymetrix Genechips consists of a large number of 25-mer single-stranded DNA probes, and transcripts are measured through a set of probes. Each probe-set consists of typically 11 perfect-match (PM) and mismatch (MM) probes, with a MM probe having its central base complementary to that found in the partner PM probe. Every microarray experiment usually undergoes several pre-processing steps (13,14). The pre-processing of a microarray requires a correction for background signals as well as normalization of the data and the calculation of an expression measure through condensing the multiple PM and MM values into a single measure of the expression of the transcript (15,16).
A. Harrison and colleagues have performed a comparative study of the differences between various pre-processing protocols. This study has determined that the calculation of the expression measure is the dominant cause of variation in the lists of genes reported to be differentially expressed in experiments (17), consistent with the findings of (18). Expression measure estimates such as robust multiarray averaging (RMA) (15) and GC-content corrected RMA (16) use algorithms based on the observed data values to identify and eliminate outlier values; however, these algorithms make no use of prior knowledge regarding the reliability of probes. Adapting these algorithms to do so would result in more robust analyses. In reality, some probes are noticeably less responsive to target concentration than the others, being either unresponsive (no hybridization signal) or invariant (same hybridization signal) across many observations. The consistency of these results means that they do not appear as noise, which is a random process, but as erroneous signal, i.e. a systematic error. Mining large surveys of microarrays is now shedding light on the nature of outliers observed in microarray data (19).
Through examining correlations between the probes within an exon, A. Harrison’s research is already finding cases where many of the existing expression measure calculations may be missing interesting biophysical effects. The research has found a large family of correlated outlier probes, the sequences of which contain a common run of four or more contiguous guanines (20). Because of the widespread correlations with other such probes, they are not measuring the same signal as the rest of the probes in the probeset to which they have been assigned. Thus, one should place little reliance on any probe containing four consecutive guanines, regardless of whether its value appears to be in agreement with others in the probeset. A. Harrison and colleagues suggest that this ‘4G’ signature results from guanine molecules in four adjacent probes interacting to form a structure that resembles a G-quadruplex (21). The existence of these correlations in intensities seen across many experiments in GEO suggests there is something in these experiments that causes thousands of G-quadruplexes to change together, something that varies from experiment to experiment. Possibilities include changes in concentration of different cations, pH variations, even ethanol contamination—all of which may show subtle variations from experiment to experiment due to random and, likely, small differences in the use of the protocol when running the microarrays. Another feature that might affect the microarray-to-microarray variation in the extent of quadruplex formation is the life-history of the microarray before being run in the experiment: a microarray in a cold dark environment for a long time may well form many G-quadruplexes on its surface whereas a microarray that is heated strongly immediately before being used is expected to have fewer G-quadruplexes.
A family of sequence motifs, centered on GCCTCCC and related to the preparation of target, also confuses the interpretation of data from microarray experiments (22). The T7-binding domain is essential for the first interaction with the RNA polymerase and the start of transcription in the 3′ in vitro transcription Microarray protocol. A small spacer sequence is added between the T7 binding site and the oligo-d(T) stretch, and this spacer is transcribed as a leader sequence for all copies of the amplified RNA. This T7 spacer sequence causes hybridization artifacts on those probes containing sub-sequences complementary to part or all of the spacer sequence—this is why probe sequences containing motifs such as CCTCC do not work in some cases. Unfortunately, there are no rules presently for eliminating these motifs in the probe design of Affymetrix microarrays.
A rather different discovery is that all probes adjacent to the edge of an array are correlated with edge probes, even though these edge probes are present only as controls and the adjacent probes are meant to be measuring biologically meaningful expression. Furthermore, many probes across the entire array are correlated with these edge probes. At first sight, therefore, the presence of high correlations between biologically relevant probes and these control probes is surprising. The answer lies in the intensity of neighboring probes: all the lower-intensity edge probes are adjacent to bright probes, while all the affected non-control probes lie next to high-intensity probes. Presumably light from the brightest probes ‘spills over’ into neighboring probes (23). The cause of this effect is currently unclear: it may be due to focusing problems, lens aberrations or perhaps even the diffraction limit. The amount of blurring varies from CEL (i.e., raw intensity) file to CEL file, but usually dominates the signal of dim neighbors of bright probes, and means that the measurements from affected probes are correlated. A. Harrison and colleagues have shown that this blurring is causing misleading intensities being reported for some probes. The discovery of such technical defects is consistent with an earlier work (24).
MODELING EACH STEP IN A MICROARRAY EXPERIMENT
C.J.B.’s group aims to contribute to the ultimate development of practical algorithms for inferring absolute target concentrations based on Langmuir adsorption theory applied to microarrays. H.B. provides a comprehensive treatment of the physico-chemical processes involved in the hybridization step, including the effects of non-specific binding, bulk hybridization and probe and target molecule folding (25).
H.B. and colleagues aim at disentangling the complex nature of microarray hybridization process by addressing selected effects in separate studies to understand their nature, to judge the effect size and finally to develop models and algorithms that allow suitable calibration of the raw data. Particularly, they studied the global relation between the levels of non-specific background and specific hybridization (26), the effect of washing, described below, (6), the effect of target depletion due to surface hybridization (27), the effect of probe sequence and of special sequence motifs (28–30) and of RNA-quality (31) using physical models of surface hybridization. For example, it was found that incremental changes in non-specific background entail opposite sign incremental changes in the effective specific-binding constant (26). This effect, which they refer to as the ‘up-down’ effect, results from the subtle interplay of competing interactions between the probes and specific and non-specific targets at the chip surface and in bulk solution. Existing heuristic normalization techniques that do not exclude absent probes, level intensities instead of expression values and/or use low variance criteria for identifying invariant sets of probes lead to biased results in expression analysis. It was also found that the extent of the up-down effect is modified if RNA targets are replaced by DNA targets, in that microarray sensitivity and specificity are improved via a decrease in the non-specific background, which effectively amplifies specific binding.
Developing a practical algorithm to estimate specific target abundances based on Langmuir-like models and using only information available in a given biological experiment is a challenging problem. Two promising developments in this direction have been the ‘Hook Curve’ formalism (32,33) and the Inverse Langmuir Method (4). The ‘Hook Curve’ (Figure 1) combines hybridization data of pairs of probes that bind the same transcript with different affinities such as the PM/MM probe pairs on microarrays. The plot of the log–intensity difference versus their logged mean intensity provides curves of characteristic hook-like shape, the dimensions of which enable parameterization of the Langmuir isotherm in a chip- and probe-specific fashion (32,33). In addition to practical expression analysis, this approach allows distinguishing between different hybridization mechanisms such as local and global depletion of targets in supernatant solution (27) and to identify different effects causing chip-to-chip intensity variance such as scanner settings or non-specific background levels (see Figure 1a and b). Recently the hook approach was applied to judge RNA-quality using microarray data (31). Here, probe pairs with different distances to the 3′-end of the transcripts are used for mutual referencing. It was demonstrated that decomposition of the probe signals into contributions due to specific and non-specific hybridization and consideration of saturation behavior might be essential for proper quality control of the RNA used for hybridization (see Figure 1c).
Figure 1.

Schematic illustration of the hook plot that presents the log-intensity difference of probe pairs interrogating the same transcript (such as PM and MM probes) taken from one array hybridization as a function of their mean log-intensity (panel a). The probes in the increasing part, the maximum and the decaying part are dominated by non-specific (N), specific (S) hybridization and by asymptotic saturation (as), respectively. The horizontal position of the hook plot, its width and height vary in a characteristic fashion owing to different experimental effects (panel b, see arrows): optical scaling of the intensity owing to changes of the scanner settings or the labeling, shift the whole hook curve in horizontal direction; alterations of the non-specific background level owing to changes of the amount of RNA and/or of its composition, alter the width of the curve and shift only the increasing part in horizontal direction; modifications of the MM design and/or of the hybridization conditions (i.e. the ionic strength), change the vertical dimensions of the hook and, finally, alterations of the post-hybridization washing efficiency mainly affect the height and width of the hook curve. The plots in the right part of panel b compare pairs of hook curves taken from an experimental series referring to the effects schematically illustrated in the left part. The thick curves are experimental data and the thin curves are theoretical hook curves calculated according to the competitive Langmuir binding model. Panel c shows the so-called degradation ‘hooks’: The thick data-curves are calculated using probe intensity data of two different arrays hybridized with RNA of different quality as plots of the smoothed log-3′/5′-intensity ratio-versus-mean where the 3′ and 5′ values are mean log-intensity values using the three probes from each probe set nearest the 3′ and 5′ ends of the respective transcript, respectively. Good-quality RNA (orange curve, RIN is the RNA Integrity Number) shows a lower maximum than bad-quality RNA (blue curve), indicating a decreased 3′/5′-intensity gradient of probes along the transcript. The log-intensity difference vanishes for predominantly non-specifically hybridized probes that are insensitive for RNA quality. Also for saturated probes, the obtained signal difference decreases [see (31) for details].
The importance of the washing step in the hybridization protocol
Washing is among the factors that potentially distort expression measures (34,35). Experiments on microarrays were conducted using altered protocols for washing, scanning and staining (6). It turns out that the effect of washing scales inversely with the binding constant of targets and gradually removes especially weakly bound non-specific targets. The intensity decays obtained as a function of the number of washing cycles are compatible with a heterogeneous energy landscape of bound transcripts. Interestingly, the study also reports indications that fluorescent markers attached to the bound targets drastically increase their washing yield compared with non-labeled ones. This result possibly explains partly contradictory findings on the effect of washing of specific targets, which presumably are caused by different labeling techniques (6,34,36).
The importance of washing, in conjunction with Langmuir adsorption theory, has been combined in a physico-chemical model (37), which incorporates both a broad sweep of the chemical reactions occurring during the hybridization step and the dissociation effects of the post-hybridization washing step (see Table 1). This model aims to predict the observed coverage fraction 0 ≤ θ(x) ≤ 1 of fluorescently labeled target-bound molecules on each probe feature of a microarray as a function of the molar concentration x of specific target in the hybridizing solution. Assuming the depletion of target molecules from the supernatant solution due to hybridization to be negligible, the coverage fraction is found to take the form of the hyperbolic response function
| (1) |
where the three parameters α, β and K are functions of chemical reaction rates and the molar concentrations of the various non-specific target species present in the hybridization solution. The model has been extended to incorporate target depletion (27). The theoretical prediction for this local depletion effect was verified experimentally using binding isotherms of PM and MM probes. Thereby the weaker MM-probes reveal a downwards-curved ‘delayed’ binding isotherm compared with that of the competing PM probes. Such downwards-curved isotherms were observed also in independent studies that presumably reflect competitive depletion effects on the respective microarrays (38).
Table 1.
Reactions associated with hybridization of nucleic acids strands
| In bulk solution | Reaction |
|---|---|
| Non-specific hybridization | S + N ↔ S.N |
| Specific target folding | S ↔ S’ |
| At the microarray surface | |
| Specific hybridization | P + S ↔ P.S |
| Non-specific hybridization | P + N ↔ P.N |
| Probe folding | P ↔ P’ |
| During the washing phase | |
| Dissociation of specific duplexes | P.S ↔ P (+ S) |
| Dissociation of non-specific duplexes | P.N ↔ P (+ N) |
A typical behavior predicted by the model for a PM/MM pair of features on an Affymetrix microarray is illustrated in Figure 2, together with an example of a fit to data from the Affymetrix U95 Latin Square spike-in experiment (39). The parameter α corresponds to the signal due to non-specific hybridization at a specific target concentration of zero. Competitive hybridization entails that contribution of non-specific signal decreases asymptotically to zero as the specific target concentration x → ∞. The hybridization model predicts that the asymptotic coverage fraction of bound targets, α + β, is θ(∞) = 1 before washing. The asymptote is reduced considerably as a result of bound targets being removed during washing, even though the washing process is ostensibly intended to remove only unbound targets (6). As the MM probes have a weaker binding affinity, the asymptote of the MM response curve is always below that of the PM signal. The parameter K is an effective equilibrium constant, and is in general larger for a PM probe than for its partner MM probe. Although the three parameters in Eq. (1) are in principle determined from physical parameters including chemical equilibrium constants and the composition of the non-specific background, in practice this information is not known and must be inferred from the available information such as the probe sequences and the observed distribution of intensities over the entire microarray.
Figure 2.

Typical behavior of the coverage function θ(x) for a PM/MM pair of probes plotted on linear (upper) and log (lower) scales before (left) and after (middle) post-hybridization washing is accounted for. The coverage fraction θ is the fraction of probe molecules on a feature that have formed probe–target duplexes; x is the concentration of target RNA in the hybridization solution specific to the PM probe sequence. The right-hand plots are measured fluorescence intensities in arbitrary units for one of the probes of the Affymetrix U95 Latin Square spike-in experiment, together with fits of these data to the response function Eq. (1). The PM responses are shown in black and the MM in red.
POLYMER PHYSICS OF DNA ARRAYS
The oligonucleotides in DNA microarrays are short polyelectrolyte chains terminally grafted to a surface. Their hybridization isotherms and kinetics reflect two regimes: (i) sparsely grafted chains when chain–surface interactions dominate and (ii) densely grafted brushes when chain–chain interactions are dominant. A. Halperin, A.B. and co-workers used polymer science insights to develop modifications of the Langmuir hybridization isotherms to allow for electrostatic and excluded volume interactions as well as their dependence on grafting density, ionic strength and the length of the probes, targets and spacers.
DNA microarrays are polymeric systems, and polymer physics thus affords insights concerning modeling their hybridization behavior. Exploiting polymer physics direction is tractable for oligonucleotide chips where the probe chains are terminally anchored to the surface via covalent bonds. Exploiting polymer physics direction is tractable for oligonucleotide chips where the probe chains are terminally anchored to the surface via covalent bonds. The structure of microarrays of this format is well defined, independent of time and of the probe sequence, and thus amenable to theoretical modeling. The polymeric approach extends the theory of terminally anchored chains, as developed for inert flexible synthetic polymers, to allow for hybridization reactions and the associated changes in the charge and rigidity. It distinguishes between two regimes. Sparsely grafted chains in the ‘mushroom’ regime do not interact with each other. In this case, the hybridization behavior is affected by the impenetrability of the wall and its effect on the number of accessible chain configurations. In the ‘brush’ regime, densely grafted chains crowd each other and stretch out to lower the repulsive interactions between them. In turn, the steric and electrostatic interactions vary with the degree of hybridization. The main outcome of the theory, as developed by Halperin et al. (40), are hybridization isotherms relating the fraction of hybridized probes θ to the target concentration ct, temperature T and the bulk equilibrium constant Kpt(T) for various situations characterized by different surface interaction free energy densities γint(θ), Eq. (2)
| (2) |
The precise form of γint(θ) differs with the length of the spacer chains (41), the relative length of probes and targets (42), the grafting density of the probes density and the ionic strength (43). This Langmuir-type isotherm serves as a basis for the description of competitive hybridization at the surface and in the bulk (43,44). The hybridization isotherms predicted by the theory were confirmed for short synthetic DNA targets (40-mer) by Fiche et al. using Surface Plamson Resonance imaging apparatus with precise temperature control in the 15°C–85°C range (45). The experiments using dedicated microarrays in controlled conditions, yielded equilibrium DNA melting curve for several synthetic short DNA duplexes as a function of ionic strength (46, Figure 3). Furthermore, for the same type of duplexes, the precise determination of melting temperatures enabled exploration of denaturant effects (47) as well as the detection of single point mutations from homozygous and heterozygous samples (48) and for low abundance mutations (49).
Figure 3.

Collapsing master curves for all salt concentrations. The probes are grafted by self-assembling of thiols. Enthalpy ΔH0 and entropy ΔS0 are extracted from linear fits [adopted from (46)].
DNA ARRAYS FROM FIRST PRINCIPLES
Often empirical and statistical methods are used to make sense of microarray data, but up to now microarrays have resisted interpretation from first principles. Generally this is attributed to surface effects that substantially change the hybridization properties of DNA from its well-known bulk properties. To test this hypothesis, the A. Ott group uses microarrays made in the lab to investigate the detection of single nucleotide mismatches.
The arrays in the A.O.’s lab are synthesized from dendrimer surface linkers using optically directed deprotection (50). Experimental results from a single target-sequence hybridizing to the complete set of single nucleotide polymorphisms on the microarray can be accurately described using a molecular zipper model with next-neighbor parameters from bulk (51). The results can directly be mapped (52) to the well-known empirical ‘Position-Dependent Nearest-Neighbor Model’ (53). Observations of substantial deviations from the Langmuir isotherm likely result from limitations in synthesis fidelity that come with the optical deprotection chemistry. Erroneous sequences on the array also need to be taken into account for theoretical interpretation, as their contribution is not negligible.
In most real applications, the length-distribution of the oligonucleotide targets, produced from biological material, is poorly controlled. The length of the hybridizing strands in solution is crucial to the DNA-detection fidelity because the number of false conformations that may bind in competition grows about exponentially with the length of the hybridizing strands.
To get a better insight into the hybridization process of two strands of unequal length, Trapp et al. considered a DNA microarray with additional bases in the probe sequence motifs (54). The target solution contains only one target species of a specific length. This is to avoid target–target interactions and competitive effects. On hybridization, a bulged loop (referred to as loop in the following) occurs in the probe (see Figure 4). Figure 5 shows the experimental microarray fluorescence intensity as a function of loop length and position for a hybridization temperature of 317 K. Strongest and weakest fluorescence intensities are normalized to 1 and 0, respectively. The number of additional bases varies from 1 to 13 bases resulting in loops of the same length. The additional loop bases are inserted at 20 different positions along the probe motif. Thus, 260 different probes are considered.
Figure 4.
Bulging loop on a probe.
Figure 5.

Signal intensity as a function of loop length and position, hybridization temperature 317 K.
In Figure 6, the experimental data are averaged over loop length and loop position. This is to get a better overview of the fluorescence intensity dependence on loop length and loop position. The stability of the duplexes with a bulged loop decreases monotonically with increasing length of the inserted loop sequence. The fluorescence intensity of the sequence with the largest loop of 13 bases is reduced to 60% of the PM fluorescence intensity. Moreover, the stability of a duplex also depends on the position where the additional loop sequence is inserted into the probe motif, with the duplex stability highest for bulged loops in the middle or end positions. The fluorescence intensity variation as a function of loop position along the duplex is weak (only 10%) when compared with the fluorescence intensity as a function of loop length. To reproduce our experimental results, we use a molecular zipper model at thermal equilibrium. In the zipper model introduced before (51,52), duplex opening was only possible from both ends of the duplex because of the high stacking barrier in the middle of the strand. However, duplex opening at the loop position must be permitted so as to account for the loops being studied. Figure 7 shows the comparison between the experimental data and the model. The upper part of the figure shows the hybridization fluorescence intensity as a function of loop length, and the lower part shows the fluorescence intensity dependence on loop position. Taking into account unavoidable sequence defects, the model can reproduce our data well. Details can be found in (54).
Figure 6.

Averaged signal intensity over loop length (top) and loop position (bottom).
Figure 7.

Comparison between the experimental data and the model.
In conclusion, in simple situations, microarray data can be understood from first principles. It is sometimes not the presence of the microarray surface that changes the properties of DNA molecular recognition, rather it seems that it is the physics of DNA itself that can be the limiting factor to the detection process of DNA microarrays. A duplex formed out of two strands of unequal length is stable and contributes significantly to the fluorescence intensity of a DNA microarray. This makes the hybridization process of DNA a complex matter. In practical terms, bulged loops occurring in DNA microarray experiments need to be taken into account for a deeper understanding of microarray data.
PHYSICS OF MISMATCHES
What do we know about the hybridization parameters of mismatched probe–target duplexes? E.C. and J.H. investigate whether the parameters developed earlier for bulk solution hybridizations (i.e. the Nearest Neighbor Model) are applicable for mismatched duplexes on the microarray surface.
A powerful algorithm allowing a full computation of hybridized fraction of target sequence in solution at the full transcriptome scales was recently introduced (55). The algorithm shows that at typical concentrations and temperatures used in biological experiments, many RNA fragments are almost fully hybridized, therefore effectively depleted from the solution. This provides new criteria for probe selections in microarrays. E.C. and J.H. group have also been developing algorithms to interpret microarray signal intensities from physico-chemical principles (55,56). Recently they designed a series of microarray experiments to access nearest-neighbor hybridization parameters from microarray data (57). They show an excellent predictability of the mismatched duplex signal intensity-based thermodynamic models over four orders of magnitude in the measured fluorescence scale (i.e. the full dynamical range of the system), see Figure 8. This figure shows the experiment with a single target sequence at concentration c (=100 pM), hybridizing to a custom microarray containing a PM spot and spots with one or two MMs. Several thousands of mismatching probes were used in the experiments on different target sequences, so that the thermodynamic parameters could be obtained with high accuracy. The intensity of the mismatching probes decreases as predicted by the following equation:
| (3) |
Figure 8.

Comparison of experimental data (filled symbols) and expected isotherm (dashed line). In this plot, I is the measured fluorescence intensities from the microarray spots, while ΔΔG is the hybridization free energy as obtained from the nearest-neighbor model measured with respect to the PM free energy, for which ΔΔG = 0. The experiments are obtained from an Agilent custom array [for more details see (57)].
Having obtained good estimates of the hybridization parameters, one can think about applications to molecular diagnostics (58). That article considers mixtures of two DNA sequences t (the wild type) and t' (the mutant) differing by a single nucleotide with respect to t. It shows that by using appropriately designed arrays and DNA hybridization thermodynamic parameters, it is possible to accurately quantify the presence of t' even at low relative concentrations. This paves the way to applications in virology or cancer where somatic mutations (often occurring in low amount) are known to have a crucial influence on the evolution of a disease.
OPTIMIZING SUBOPTIMAL PROBES
Common rules for design of selective probes suggest avoiding complementary stretches exceeding 25% of the probe total length. C.G. put these rules to test and discovered them to be not conservative enough.
The group of C.G. attempts to improve the selectivity of microarray probes. A common guideline implemented in software for the construction of DNA microarrays has been that avoiding complementary stretches of more than 15 nucleotides in a 50- or 60-mer probe, or 75% of the probe in total, will eliminate sequence-specific cross-hybridization reactions (e.g. 59). However, solution simulation of the hybridization behavior of pairs of oligonucleotide probes and their targets suggests that even a complementary stretch of sequence 9 nucleotides in length has the potential to give rise to specific cross-hybridization. Competition between the intended binding partner and thermodynamically nearest suboptimal targets designed by a common software has been examined (60), but the original 15-mer design criterion (61) remains widely used either as an initial sequence screen or as the sole predictor of cross-hybridization (62) and is largely unexamined in isolation from other factors, despite improvements in our facility with printing and handling microarrays in the intervening decade. To explore the effect of minimal partial matches in a microarray context, C.G. and colleagues designed a set of binding partners to a 50-mer oligonucleotide probe. Each target was designed to be anticomplementary to the probe, with the exception of a complementary stretch from 6 to 21 nucleotides in length. Solution melting experiments with these oligonucleotides found partial duplexes stable in the prevailing range of hybridization temperatures used in microarray experiments. Such duplexes formed when only 12 bp of complementary sequence were present. Surface hybridization experiments have confirmed that a signal indistinguishable from a full-strength signal arising from a low copy number PM can be obtained from sequences that form only a partial duplex (63).
A representative PM oligonucleotide pair with balanced GC content to represent a microarray probe and target were selected. Several permutations of the target were created such that a single continuous stretch of sequence complementary to probe was preserved, while ensuring that the sequence flanking the complementary region did not permit extension of the partial duplex across a mismatch or mismatches. This resulted in a series of partially complementary pairs derived from the original PM pair. Pairs with partially complementary stretches of 6, 9, 12, 15, 18 and 21 nucleotides were investigated. For each, a version of the sequence was created with the complementary stretch near the center of the oligo pair (central), and a second with the complementary stretch near one of the ends of the pair (terminal). Figure 9 shows the signal due to formation of duplexes between partially complementary probe and target pairs on the microarray surface. Figure 9A shows that specific signal for the target in isolation exceeds background signal when a complementary 12 nucleotides stretch is present. Figure 9B measures signal owing to the same partially complementary target in the presence of an equimolar concentration of unlabeled PM target. Here, the signal does not exceed background until an 18 nucleotides complementary stretch is present. In a microarray experiment, it is not known at the start whether a PM for the target is present, and in the absence of a PM competitor, a specific partial match may give rise to signal that suggests the presence of a PM in low concentration. Microarray and other molecular capture strategies that rely on a 15 nucleotides lower bound to completely eliminate specific cross-hybridization may not be sufficiently conservative.
Figure 9.

Surface-attached 50-mer probe response to designed anticomplementary 50-mer targets, bearing a continuous complementary stretch of varying length (6, 9, 12, 15, 18, or 21 nucleotides). The placement of the complementary stretch is central (diamonds) or terminal (squares). Dotted line indicates the average background intensity across the experiment; solid line indicates the signal intensity due to hybridization of a perfectly matched 50-mer target. (A) target alone and (B) in presence of an equal concentration of unlabeled PM.
PSEUDO-LANGMUIR AND OTHER HYBRIDIZATION REGIMES
Hybridization on the microarray surface is more complicated than simple chemisorption, as one might expect. The R.L. group identified distinct hybridization behaviors that depend on the salt concentration and on how many probes are grafted per unit surface area.
R.L. and coworkers are focusing on understanding the molecular organization and interactions that take place on DNA-modifed surfaces, and how these influence hybridization performance in technologies such as DNA microarrays. Recently, this group has investigated a model oligonucleotide system in which 20-mer probes were hybridized with 18-mer targets as a function of probe coverage S0 and solution ionic strength (salt concentration) CB (64,65). These studies sought to quantify cooperative influence of these two ‘electrostatic’ variables on hybridization, with S0 determining surface charge concentration and thus repulsion between the surface layer and incoming targets, and CB modulating this repulsion through electrostatic screening. Electrochemical methods were used to precisely monitor extents of hybridization to electroactively labeled target molecules. Hybridization yields were quantified once a stable signal was reached, which was presumed reflective of equilibrium.
The principal conclusions of these studies can be summarized by a diagram of hybridization behaviors, Figure 10. At highest coverage and lowest ionic strength, hybridization signals were too weak for detection owing to dominance of electrostatic repulsions between the probe layer and the target oligonucleotides. Due to the weak signals, for these operationally ‘non-hybridizing’ (NH) conditions, the dependence of hybridization on S0 and CB could not be established. An increase in ionic strength, by addition of more salt, from the NH regime led to detectable hybridization in the ‘electrostatically suppressed’ (SH-E) regime. SH-E behavior was characterized by sensitivity of hybridization yields to both CB and S0, where this sensitivity could be modeled as primarily reflecting electrostatics (40,43,64–66). Further increases in ionic strength, provided probe coverage was sufficiently high, led to ‘packing constraint-suppressed’ (SH-P) conditions. In this regime, increase in probe coverage suppressed hybridization much more severely than expected on basis of pure electrostatics. This strong suppression was attributed to additional penalties derived from packing constraints. In comparison, increase in ionic strength from the SH-E regime but at lower probe coverage led to ‘pseudo-Langmuir’ (PL) behavior. In the unusual PL regime, changes in probe coverage S0 only minimally impacted hybridization yields; in other words, the hybridization probability for a given probe was nearly independent on proximity to neighboring probes. Such independence on binding site coverage would be expected for true Langmuir conditions (67); however, in these experiments, and indeed in microarray applications, probe molecular dimensions are too large compared with probe–probe separation for interactions to be absent. Rather, independence of hybridization signals with regard to S0 suggests that interactions between probes are, within the PL regime, insensitive to their spacing. A tentative explanation for this observation may derive from probe conformational flexibility that maintains energetics of probe–probe interactions, and hence their impact on hybridization, approximately constant over this coverage range. Lastly, at sufficiently sparse coverage, where average intermolecular separations far exceed the length of the probes so that interactions among them are largely prevented, true Langmuir behavior is expected, though was not explored in the studies.
Figure 10.

A map of surface hybridization regimes as a function of probe coverage S0 and salt concentration CB, and as reported in (65). See text for details.
SURFACE HETEROGENEITY
B.M.P.’s most recent work centers on the problem of how and where attachments to the underlying microarray surface can be made (68). For DNA mounted on surfaces for microarrays, microbeads and nanoparticles, the nature of the random attachment of oligonucleotide probes to an amorphous surface substrate gives rise to a locally inhomogeneous density of probes.
The fluctuations of the probe surface density are inherent to all common surface or bead platforms, regardless if they exploit either an attachment of pre-synthesized probes or probes synthesized in situ on the surface. The surface density affects the local electrostatics that have been long appreciated to effect the surface binding free energies and so hybridization efficiencies (69). B.M.P. and colleagues (68) recently demonstrated the crucial role of the probe surface density fluctuations in performance of DNA arrays.
To account for the fluctuations of the probe surface density, Vainrub and B.M.P. start with a DNA array hybridization isotherm that accounts for the electrostatic polarization field near the surface (66). The isotherm describes the electrostatic repulsion between the DNA target and probe array arising owing to the large negative charge of DNA phosphate backbone in terms of a homogeneous probe surface density, i.e. in the mean-field approximation. It assumes that the repulsion energy is according to the following equation:
| (4) |
Here NP is the surface density of probes, ZP and ZT are the lengths of probe and target expressed in a number of bases, θ is the extent of hybridization (0 ≤ θ ≤ 1, and w is the electrostatic interaction parameter that depends on the NaCl concentration in the hybridization solution. This leads to the hybridization isotherm:
| (5) |
Here C0 is the concentration of assayed DNA targets, ΔH0 and ΔS0 are the enthalpy and entropy of double helix formation in solution.
They then modeled the density fluctuations of a disordered 2D surface as a simple random Poisson distribution that corresponds to a very high temperature Boltzmann distribution, relevant to how the attachment sites were frozen out of the cooling glass substrate.
On a regular 2D lattice, each probe can have from m = 0 to 6 neighboring probes. For random probe attachment, the probability pm that the probe has m neighbors is as follows:
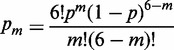 |
(6) |
Here p = sNp is the probability for a hexagonal cell to be occupied by the probe. Now the local surface density Nm fluctuates depending on the number m of the probes in six surrounding cells. Incorporating this distribution, they derived the corresponding array hybridization isotherm that includes a counter-ion screened electrostatic repulsion between the assayed DNA and a random probe array on the surface. They write their model in a form convenient to find the melting curve θm = θm(T):
| (7) |
This describes the hybridization yield θm to the probes that have m neighboring probes. Hybridization to the total array is then given by summing over the Poisson distribution of probe site possibilities:
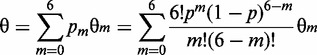 |
(8) |
The calculated melting curves for short synthetic 40- and 19-mer targets were found to be in excellent agreement with published experimental results for arrays with both pre-synthesized and in situ synthesized oligonucleotide probes (Figure 11). The approach developed allows one to accurately predict the melting curves of DNA arrays using only the known sequence dependent hybridization enthalpy and entropy from solution and the experimental macroscopic surface density of probes.
Figure 11.

Theoretical and experimental melting curves on glass. Experiment: PM (circles) and single MM (triangles) 19-mer oligonucleotides synthesized on glass surface. Theory: fluctuating surface coverage (solid lines) and homogeneous (dashed lines) density of probes.
SURFACE CHEMICAL IMAGING OF INDIVIDUAL MICROARRAY SPOTS
What is the true composition of the microarray surface and oligonucleotide spots? L.J.G. and D.W. Grainger bombard the microarray surface with positive bismuth ions and record mass spectra of the ions ricocheted from the microarray to determine actual species of molecules on the surface.
Fabrication of the nucleic acid microarray itself continues to present challenges for improving the reproducibility, sensitivity and quantification. Work by L.J.G. and D.W. Grainger has focused on direct analysis of individual printed DNA microarray spots on both model and commercial arraying surfaces using a variety of analytical techniques including imaging comparisons from time-of-flight secondary ion mass spectrometry (ToF-SIMS), providing chemical details, and epifluorescence microscopy providing spatial density details. The microarray printing process commonly involves spotting nanoliter amounts of DNA solution on glass substrates coated to bind nucleic acids. These microarray spots are the basis for the microarray assay ‘answer’ and importantly can have many significant variations arising from several factors affecting assay answer. These include DNA probe chain length substrate surface chemistry printing solutions and washing (all mentioned above), as well as printer type and evaporation processes during spot drying. Imaging ToF-SIMS yields a mass spectrum from molecules at the microarray spot surface with micrometer to sub-micrometer lateral resolution. This information can be correlated to other imaging modes (e.g. high resolution fluorescence) to provide spot distribution and density information directly related to how spots capture target.
Figure 12 shows examples of printed single spot heterogeneity as followed by the phosphate ion peak in ToF-SIMS for spots with different concentrations of DNA. Phosphate peaks have been shown to mirror DNA base presence in ToF-SIMS imaging of DNA microarrays (70), and exhibit strong ToF-SIMS signal intensity for better visual contrast in images. Brighter regions in the images in Figure 12 indicate higher DNA concentration, and exhibit a wide variability in the types of heterogeneity within the spots, reflecting differences in DNA density and distribution. Furthermore, no heterogeneity was similar across the three different printed DNA concentrations.
Figure 12.

ToF-SIMS images of the P ion in single printed DNA spots show large variability of DNA concentration in and across the microarray spots. Spots are printed from 20 and 40 μM DNA concentration drops (from left to right) with 100% of the DNA tagged with Cy3. Image size (400 × 400 μm). The Cy3 fluorescence images of the same spots are shown for comparison.
ion in single printed DNA spots show large variability of DNA concentration in and across the microarray spots. Spots are printed from 20 and 40 μM DNA concentration drops (from left to right) with 100% of the DNA tagged with Cy3. Image size (400 × 400 μm). The Cy3 fluorescence images of the same spots are shown for comparison.
Printed spots with higher DNA surface densities show lower target hybridization efficiencies (70). Hence, variability in DNA probe surface densities within the spots likely results in variable amounts of target capture at different areas in the spots, and total target capture within any given spot. More controlled experiments using a model self-assembled DNA monolayers on gold showed that target hybridization efficiency changes with surface probe density (71). Variation in printed DNA density within spots seen with imaging ToF-SIMS and high-resolution fluorescence analysis can be easily missed by lower resolution scanners often used to analyse commercial microarray results. As a result, assay data from spots that lack consistency in probe density reflect highly variable target capture efficiency and variable reliability.
DNA-METER: DIRECTLY CALIBRATED PROBES
Every probe on the microarray surface is a sensor for its target. ‘DNA-meter’ is an approach that renders every microarray probe as an individual analytical device, whose characteristics are experimentally determined during a calibration process. The multitude of probes is calibrated simultaneously in a series of several hybridizations.
Since 2000, the goal of the A.P.–P.A.N.–D.T. group is to bring microarray technology from a semi-quantitative technique into the realm of analytical chemistry and molecular diagnostics. The key issues to be addressed are probe response function and selectivity. As defined in the ‘Introduction’ section of this article, the probe response function refers to the dependence of probe signal intensity on concentration of target sequences. Probe selectivity refers to the extent of cross-reaction between targets and non-target sequences. To address the first issue, an ability to accurately predict the behaviors of microarray probes is required, which has been a problem since the beginning of the microarray technology (1990s) (72). As research progressed, it became evident that neither heuristic approaches (61,73–78) nor solution-based melting thermodynamics (35,53,79–84) could accurately predict probe behavior on the microarray surface (1,83,85,86). The selectivity issue is an open-ended problem because the number of potential targets is extremely large and theoretical/experimental evaluation of such target space seems intractable.
The unpredictable probe response function problem was circumvented by directly calibrating microarray probes using a dilution series of a sample of biologically relevant complexity and concentration [details of the method are in (Pozhitkov et al.; submitted for publication)]. The new approach has been already successfully applied (87). Briefly, a set of samples representing sequences obtained from gene expression or a genomic study is mixed together into one pooled sample; a dilution series is prepared from this sample and several microarrays are hybridized. To decrease probe variability, the microarray contains probes in 10-fold replicates because a single replicate is too noisy. On hybridization to the dilution series, the probes response is recorded as a calibration curve. For instance, 25-mer probes on the Agilent microarray respond with a power curve, i.e. Freundlich isotherm y = axb (Figure 13, panel A). The characteristics of the calibration curve, such as parameter b and goodness of fit R2, enable the researcher to discard probes that are non-responsive. For example, a low value for the parameter b would cause a high error in back-calculating target concentrations. Detailed analysis of the curves as well as curve selection guidelines based on expected error is presented in (87). After appropriate probes are selected, calculation of relative target concentrations is possible. Figure 13, panel B shows back-calculation of the relative concentrations. Apparently good resolution of concentrations is attainable.
Figure 13.

Calibration curve for a probe (panel A); grey lines represent individual probe replicates, black line—averages. Histogram of concentrations determined from signal intensities and calibrated probes (panel B); open circles represent true concentrations.
The direct calibration of probes makes microarray similar to a regular analytical instrument, which has a known (recorded) response function. Through the response function, one translates signal intensities into concentrations, estimates errors and accounts for the probe non-linearity, which is essential for further biological interpretation.
NON-HYBRIDIZATION EFFECTS ON MICROARRAY AND NGS SIGNALS
A calibration and an understanding of quantitative signals are prerequisites for the analysis of large-scale profiling data to find biologically relevant patterns. To this end, the research group of D.P.K. pursues the identification and removal of signal distortions in measurements of gene activities from multiple platforms, i.e. microarrays and NGS.
It is now increasingly clear that measurements of target abundances by NGS or microarrays are also affected by other sources of signal variation. These need to be understood both in attempts to improve and model the measurement process itself, and also for getting a more meaningful and reliable quantification readout. This is a prerequisite for sensitive de novo expression pattern discovery, and advanced profiling of alternative gene transcripts, which requires truly quantitative signals for individual exons and exon–exon junctions.
D.P.K. and colleagues determined that one of the major sources of signal variation is sample processing. Most protocols for quantitative profiling of targets by NGS or microarrays include one or several steps for target selection, target amplification, transcription, fragmentation and labeling. These are critical factors substantially contributing to inter-laboratory variation in both RNA-Seq and microarray results. The limited processivity of enzymes is known to create 3′- or 5′-biases for 3′-anchored and random primed transcription, respectively (88). Non-uniform fragmentation contributes considerably to signal variation along the transcript. For both microarrays and RNA-Seq, such variation has been observed to be about two orders of magnitude. Both qualitative and quantitative profiling methods are affected by protocol choices. The reverse-transcription step has, for example, recently been identified as being responsible for a multitude of false-positive negative-strand signals—this suggests that direct hybridization of chemically or end-labeled messenger RNA to microarrays, and future label-free NGS protocols should considerably improve detection and quantification accuracy (89).
The combined effects of these confounding factors can be studied with help of large-scale calibration experiments, with the aim of improving our understanding and modeling for the subsequent extraction of the most meaningful signal. For such calibration experiments, synthetic spike-ins are one valuable option (39) although it is not easy to ensure that synthetic spike-ins are representative of realistic biological samples (90).
Interestingly, there are biological situations that provide another form of calibration experiment. For instance, while aneuploidy is badly tolerated in humans and many other higher organisms, leading to death or serious disease like Down’s syndrome (chromosome 21 trisomy), plants are surprisingly tolerant of additional chromosome copies. It has recently been shown that genes on these additional chromosomes exhibit a direct dosage response with only a small percentage of genes forming an exception where dosage compensation or other regulatory mechanisms interfere (91). This can be seen in a clear average trend that emerges when plotting the differences between a genotype with three copies of chromosome 5 compared with the normal wild-type control as a function of the gene expression level (see Figure 14). The magenta lines represent this trend average and variation for genes on the triplicate chromosome 5. The orange lines show the zero average and variation of genes on the other chromosomes present only in duplicate. In an ideal system, the magenta line should start at zero (1.5 × 0 = 0) and rise to a constant level of log2(1.5), until perhaps saturation sets in. The strongly non-linear response observed, however, cannot be explained with our current models, and provides an opportunity for testing further modeling advances. It is worth pointing out that while RNA-Seq data do not exhibit a similar strongly varying trend at high expression levels, they show higher variation (10) and stronger distortions for lower expression levels (data not shown).
Figure 14.

M(A) plot of the average expression differences M between chromosome 5 trisomic Arabidopsis thaliana plants and disomics (y-axis) as a function of average expression A (x-axis).
In general, both the average differential signal response and also the average signal variation turn out to be a function of the expression level. While both vary, they vary differently, leading to an intensity-dependent bias in differential expression detection. This means that functional groups with genes of non-uniformly distributed expression levels will have different likelihoods of being identified as differentially expressed. Transcription factors, for example, are enriched in genes of low expression levels. A reduced sensitivity to differential expression at low expression levels will thus lead to an underestimation of differentially expressed transcription factors. This particularly affects studies where hundreds to thousands of genes change, and scientists seek under- or over-represented functional groups in these genes to suggest biological processes involved.
An ongoing development of our understanding of the whole measurement process will thus support the improvement and validation of advanced hybridization models, while also leading to quantitative readouts of reduced bias, which are critical for probing complex biological systems. Improved models for the interpretation of arbitrary probes (92) should also allow the application of advanced high-density high-resolution microarrays that can take advantage of probes targeting the most informative sequence regions rather than designing arrays with a limited number of probes selected for similar thermodynamic properties.
SUMMARY
Through a dynamic interplay between experimental and theoretical studies, we are developing an increasingly sophisticated understanding of the nuances of hybridization of nucleic acids measured using high-throughput technologies. Although models and experiments of hybridization in solution provide useful first-order descriptions, there are subtle differences between solution and solid surface hybridization. Also, a collection of probes on a solid surface may not resemble the ideal of a uniform ‘bed of nails’ on which there are no interactions between equally spaced neighbors.
The physical properties of an ensemble of same-sequence strands of nucleic acids may show a range of hybridization phases, dependent on the density of probes, the ionic concentration, electrostatic distributions, and other characteristics. The spatial distribution of probes is heterogeneous and chemical imaging now enables the local density of probes to be monitored. A densely packed ensemble of identical sequences may result in non-canonical interactions between nucleic acids, such as Hoogsteen hydrogen bonds, which then allow higher-order structures such as tetrads to form.
Chemical binding on solid surface can be described using classical models such as Langmuir adsorption isotherms. However, analysis of collections of nucleic acids has demonstrated the need for further considerations, and the development of alternative isotherms. This work has highlighted the need to include surface effects resulting from polymer physics, the multiple steps in experiments such as washing at different ionic considerations, bulk hybridization as well as folding of individual sequences. Moreover, measurements of probes with different sequence compositions enable a determination of the physics of mismatches, how the efficiency of duplex formation depends on the position of particular sequences within the probes, as well as the determination of cross-hybridization expected for sub-optimal probes.
FUTURE OUTLOOK
The reader may wonder what is coming up in the field and how one has to conduct microarray or NGS experiments properly in the light of the knowledge described here. The main contribution of this article is to report on the progress being made towards ‘what really happens on the arrays’ and to point towards rules, which in turn will be fed into the refinement of analysis algorithms. The transcriptomics field is a broad community, and one of its strengths is that different groups are providing solutions, or addressing problems, from different perspectives. Such research may lead to an elegant description of the hybridization process, similar to the Carnot’s ideal engine, thus reducing complicated models to simple equations. However, direct studies of the microarray surface may find ‘Tunguska’ distribution of probes, thus challenging many contemporary theoretical models.
‘Uncertainty principles’ of transcriptomics experiments may need to be established. In practice, experiments conducted by biologists using microarrays and NGS necessarily cannot control for all the physical effects when measuring a heterogeneous mixture of competing sequence. However, the work described here will be hopefully of benefit to the designers of future microarrays. There are sub-sequences, such as contiguous runs of guanine, which should be considered in probe selection. Similarly, considerations of grafting density versus probe and target length, probe accessibility and surface treatments, help to improve sensitivity and selectivity. We note, though, that it may prove impossible to explicitly take into account all of the effects at a probe-specific level.
In short, it is too early to completely replace the microarrays with the new fashion of NGS. Microarrays have their limitations but many of their measurements can be interpreted with rationales from physics and chemistry. This provides a solid base from which experiments of several conditions can be compared with each other, and reliable inferences made from the data. There is an active field of algorithm development leading to increasingly accurate inferences of transcript abundances—interested biologists are directed to repositories of such methods, such as http://bioconductor.org. Moreover, meta-analysis of large collations of experiments determines known outliers, and their causes, and many of the resulting problems from the outliers are being treated effectively via bioinformatics and biostatistical tools within the software repositories, for example through the usage of alternative annotation descriptors.
It is important to remember that there are a number of experimental issues that affect transcriptomics experiments of any flavor, irrespective of whether they are microarray or NGS. For example, the technical variance of both microarrays and NGS is several times smaller than the biological variance. It is therefore a moot point which technology is being used if the experiment of interest does not contain sufficient biological replicates to enable inferences of biological signals to be made. It may also be similarly a moot point to make simple comparisons between the technical variances of NGS and microarray technologies—microarrays have many but limited number of probes sampling a broad range of transcripts, whereas RNA-seq experiments may use up many of their counts on highly expressed transcripts, leaving only a few counts for low-expressed genes, and for these genes, the signals may get lost in the Poisson noise. Indeed, the use of some capture arrays within NGS experiments emphasizes the need for a greater understanding of the physical and statistical characteristics of all transcriptomics technologies.
Transcriptomics experiments of all flavors may also be affected by systematic biases introduced during sample preparation. These biases result from transforming populations of RNA from a cell into an amplified library of reverse-transcribed complementary DNA fragments of about the same length. Moreover, target–target hybridization within the supernatant solution may bedevil the inferences made from transcriptomics experiments. Discrepancies in the protocol before measurement may also need greater standardization to enable reliable inferences to be made from the data.
We are beginning to see the development of physico-chemical treatment of the data from NGS experiments. It is already clear that the protocols being used within experiments leave a significant mark in the data. The rapid development of sequencing technologies and protocols may thus make detailed modeling of experiments premature. We envisage that history may repeat itself—the development of market leaders will lead to the community identifying the need to use physico-chemical–based analysis to model ‘Spike-In’ measurements, such as we saw with Affymetrix microarrays and the microarray community. We expect that insights about high-density ensembles of nucleic acids gained from microarrays will be of use to scientists building models of the data produced from NGS technologies.
Given the availability of nucleic acid synthesis and purification technologies, we propose to generate benchmark target mixtures consisting of hundreds to thousands of species of known biologically relevant concentration. The new benchmark target mixture will expand possibilities for modeling of hybridization physical chemistry on different platforms (including NGS) and provide new insights complementing the decade-old Affymetrix Latin Square dataset. With available high-throughput computing techniques, the future modeling of the nucleic acids hybridization on the surface need to develop alternative approaches capable of considering fates of every target, hybridizing specifically or non-specifically to a corresponding microarray probe or an NGS bead.
The work described here highlights rapid progress being made in our understanding of the physico-chemical properties of high-throughput measurements of nucleic acids on a solid surface. Increasingly accurate determination of target concentrations is now being achieved, allowing us to consider microarrays and NGS experiments as analytical experiments. Together with the theoretical advances, a practical calibration of such devices is indispensable. An example from another field: although the physics of old-fashioned vacuum tubes or modern ubiquitous transistors is well known, a precise theoretical description of the actual device’s performance is not feasible. Hence, every device of this sort comes with its empiric averaged characteristic curves.
FUNDING
German Academy of Natural Sciences (Leopoldina), Max-Planck Society, Agilent and Molecular Devices for supporting the Workshop (to A.P. and D.T.); NIH [EB-001473 and NESAC/BIO, NIH Grant No. EB-002027 to L.J.G.]; National Science Foundation grant [DBI 10388671] and National Science Foundation grant CREST HRD 1241701. (to P.A.N.); National Science Foundation of the United States [DMR-1206754 to R.L.]; Vienna Science and Technology Fund (WWTF), Baxter AG, Austrian Research Centres Seibersdorf, and the Austrian Centre of Biopharmaceutical Technology (to D.P.K.). Funding for open access charge: Max Planck Society.
Conflict of interest statement. None declared.
REFERENCES
- 1.Pozhitkov A, Noble PA, Domazet-Loso T, Nolte AW, Sonnenberg R, Staehler P, Beier M, Tautz D. Tests of rRNA hybridization to microarrays suggest that hybridization characteristics of oligonucleotide probes for species discrimination cannot be predicted. Nucleic Acids Res. 2006;34:e66. doi: 10.1093/nar/gkl133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mei R, Hubbell E, Bekiranov S, Mittmann M, Christians FC, Shen MM, Lu G, Fang J, Liu WM, Ryder T, et al. Probe selection for high-density oligonucleotide arrays. Proc. Natl. Acad. Sci. USA. 2003;100:11237–11242. doi: 10.1073/pnas.1534744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amend AS, Seifert KA, Bruns TD. Quantifying microbial communities with 454 pyrosequencing, does read abundance count? Mol. Ecol. 2010;19:5555–5565. doi: 10.1111/j.1365-294X.2010.04898.x. [DOI] [PubMed] [Google Scholar]
- 4.Mulders GCWM, Barkema GT, Carlon E. Inverse Langmuir method for oligonucleotide microarray analysis. BMC Bioinformatics. 2009;10:64. doi: 10.1186/1471-2105-10-64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Held GA, Grinstein G, Tu Y. Modeling of DNA microarray data by using physical properties of hybridization. Proc. Natl. Acad. Sci. USA. 2003;100:7575–7580. doi: 10.1073/pnas.0832500100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Binder H, Krohn K, Burden CJ. Washing scaling of GeneChip microarray expression. BMC Bioinformatics. 2010;11:291. doi: 10.1186/1471-2105-11-291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li S, Pozhitkov A, Brouwer M. A competitive hybridization model predicts probe signal intensity on high density DNA microarrays. Nucleic Acids Res. 2008;36:6585–6591. doi: 10.1093/nar/gkn740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li S, Pozhitkov A, Brouwer M. Linking probe thermodynamics to microarray quantification. Phys. Biol. 2010;12:048001. doi: 10.1088/1478-3975/7/4/048001. [DOI] [PubMed] [Google Scholar]
- 9. Latin Square Data for Expression Algorithm Assessment. http://www.affymetrix.com/support/technical/sample_data/datasets.affx.
- 10.Łabaj PP, Leparc GG, Linggi BE, Markillie LM, Wiley HS, Kreil DP. Characterization and improvement of RNA-Seq precision in quantitative transcript expression profiling. Bioinformatics. 2011;27:i383–i391. doi: 10.1093/bioinformatics/btr247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bullard JH, Purdom E, Hansen KD, Dudoit S. Evaluation of statistical methods for normalization and differential expression in mRNA-Seq experiments. BMC Bioinformatics. 2010;11:94. doi: 10.1186/1471-2105-11-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barrett T, Troup DB, Wilhite SE, Ledoux P, Rudnev D, Evangelista C, Kim IF, Soboleva A, Tomashevsky M, Edgar R. NCBI GEO: mining tens of millions of expression profiles—database and tools update. Nucleic Acids Res. 2007;35:D760–D765. doi: 10.1093/nar/gkl887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 14.Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–193. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- 15.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu Z, Irizarry R, Gentleman R, Martinez Murillo F, Spencer F. A model-based background adjustment for oligonucleotide expression arrays. J. Am. Stat. Assoc. 2004;99:909–917. [Google Scholar]
- 17.Harrison AP, Johnston CE, Orengo CA. Establishing a major cause of discrepancy in the calibration of Affymetrix GeneChips. BMC Bioinformatics. 2007;8:195. doi: 10.1186/1471-2105-8-195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Harr B, Schlötterer C. Comparison of algorithms for the analysis of Affymetrix microarray data as evaluated by co-expression of genes in known operons. Nucleic Acids Res. 2006;34:e8. doi: 10.1093/nar/gnj010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Upton GJ, Sanchez-Graillet O, Rowsell J, Arteaga-Salas J, Graham N, Stalteri M, Memon F, May S, Harrison A. On the causes of outliers in Affymetrix GeneChip data. Brief. Funct. Genomic Proteomic. 2009;8:199–212. doi: 10.1093/bfgp/elp027. [DOI] [PubMed] [Google Scholar]
- 20.Upton GJ, Langdon WB, Harrison AP. G-spots cause incorrect expression measurement in Affymetrix microarrays. BMC Genomics. 2008;9:613. doi: 10.1186/1471-2164-9-613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Langdon WB, Upton GJ, Harrison AP. “Probes containing runs of guanines provide insights into the biophysics and bioinformatics of Affymetrix GeneChips”. Brief. Bioinformatics. 2009;10:259–277. doi: 10.1093/bib/bbp018. [DOI] [PubMed] [Google Scholar]
- 22.Kerkhoven RM, Sie D, Nieuwland M, Heimerikx M, Brugman W, Lieftink C, De Ronde J, Velds A. Platform specific T7-amplification biases revealed by analysis of thousands of microarray data sets with implications for cross-platform comparisons. Proc. Am. Assoc. Cancer Res. Annu. Meet. 2007;48:879. [Google Scholar]
- 23.Upton GJ, Harrison AP. The detection of blur in Affymetrix GeneChips. Stat. Appl. Genet. Mol. Biol. 2010;9 doi: 10.2202/1544-6115.1590. Article 37. [DOI] [PubMed] [Google Scholar]
- 24.Suárez-Fariñas M, Pellegrino M, Wittkowski KM, Magnasco MO. Magnasco: a “corrective make-up” program for microarray chips. BMC Bioinformatics. 2005;6:294. doi: 10.1186/1471-2105-6-294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Binder H. Thermodynamics of competitive surface adsorption on DNA microarrays. J. Phys. 2006;18:S491–S523. [Google Scholar]
- 26.Binder H, Bruecker J, Burden CJ. Non-specific hybridization scaling of microarray expression estimates - a physico–chemical approach for chip-to-chip normalization. J. Phys. Chem. B. 2009;113:2874–2895. doi: 10.1021/jp808118m. [DOI] [PubMed] [Google Scholar]
- 27.Burden CJ, Binder H. Physico-chemical modelling of target depletion during hybridization on oligonucleotide microarrays. Phys. Biol. 2010;7:016004. doi: 10.1088/1478-3975/7/1/016004. [DOI] [PubMed] [Google Scholar]
- 28.Binder H, Kirsten T, Hofacker I, Stadler PF, Loeffler M. Interactions in oligonucleotide duplexes upon hybridisation of microarrays. J. Phys. Chem. B. 2004;108:18015–18025. [Google Scholar]
- 29.Binder H, Fasold M, Glomb T. Mismatch- and G-stack modulated probe signals on SNP-microarrays. PLoS One. 2009;4:e7862. doi: 10.1371/journal.pone.0007862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fasold M, Stadler PF, Binder H. G-stack modulated probe intensities on expression arrays–sequence corrections and signal calibration. BMC Bioinformatics. 2010;11:207. doi: 10.1186/1471-2105-11-207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fasold M, Binder H. Estimating RNA-quality using GeneChip microarrays. BMC Genomics. 2012;13:186. doi: 10.1186/1471-2164-13-186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Binder H, Krohn K, Preibisch S. ‘Hook’-calibration of GeneChip-microarrays: Chip characteristics and expression measures. Algorithms Mol. Biol. 2008;3:11. doi: 10.1186/1748-7188-3-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Binder H, Preibisch S. ‘Hook’-calibration of GeneChip -microarrays: theory and algorithm”. Algorithm Mol. Biol. 2008;3:12. doi: 10.1186/1748-7188-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Burden CJ, Pittelkow YE, Wilson SR. Adsorption models of hybridisation and post-hybridisation behaviour on oligonucleotide microarrays. J. Phys. 2006;18:5545–5565. [Google Scholar]
- 35.Held GA, Grinstein G, Tu Y. Relationship between gene expression and observed intensities in DNA microarrays—a model study. Nucleic Acids Res. 2006;34:e70. doi: 10.1093/nar/gkl122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pozhitkov AE, Stedtfeld RD, Hashsham SA, Noble PA. Revision of the nonequilibrium thermal dissociation and stringent washing approaches for identification of mixed nucleic acid targets by microarrays. Nucleic Acids Res. 2007;35:e70. doi: 10.1093/nar/gkm154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burden CJ. Understanding the physics of oligonucleotide microarrays: the Affymetrix spike-in data reanalysed. Phys. Biol. 2008;5:016004. doi: 10.1088/1478-3975/5/1/016004. [DOI] [PubMed] [Google Scholar]
- 38.Pozhitkov AE, Boube I, Brouwer MH, Noble PA. Beyond Affymetrix arrays: expanding the set of known hybridization isotherms and observing pre-wash signal intensities. Nucleic Acids Res. 2010;38:e28. doi: 10.1093/nar/gkp1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Burden CJ, Pittelkow YE, Wilson SR. Statistical analysis of adsorption models for oligonucleotide microarrays. Stat. Appl. Genet. Mol. Biol. 2004;3 doi: 10.2202/1544-6115.1095. Article 35. [DOI] [PubMed] [Google Scholar]
- 40.Halperin A, Buhot A, Zhulina EB. On the hybirdization isotherms of DNA microarrays: the langmuir model and its extensions. J. Phys. 2006;18:S463–S490. [Google Scholar]
- 41.Halperin A, Buhot A, Zhulina EB. Hybridization at a surface: the role of spacers in DNA microarrays. Langmuir. 2006;22:11290–11304. doi: 10.1021/la0616606. [DOI] [PubMed] [Google Scholar]
- 42.Halperin A, Buhot A, Zhulina EB. Brush effects on DNA chips: thermodynamics, kinetics and design guidelines. Biophys. J. 2005;89:796–811. doi: 10.1529/biophysj.105.063479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Halperin A, Buhot A, Zhulina EB. Sensitivity, specificity and the hybridization isotherms of DNA chips. Biophys. J. 2004;86:718–730. doi: 10.1016/S0006-3495(04)74150-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Halperin A, Buhot A, Zhulina EB. Hybridization isotherms of DNA microarrays and the quantification of mutation studies. Clin. Chem. 2004;50:2254–2262. doi: 10.1373/clinchem.2004.037226. [DOI] [PubMed] [Google Scholar]
- 45.Fiche JB, Buhot A, Calemczuk R, Livache T. Temperature effects on DNA chip experiments from surface plasmon resonance imaging: isotherms and melting curves. Biophys. J. 2007;92:935–946. doi: 10.1529/biophysj.106.097790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fuchs J, Fiche JB, Buhot A, Calemczuk R, Livache T. Salt concentration effects on equilibrium melting curves from DNA microarrays. Biophys. J. 2010;99:1886–1895. doi: 10.1016/j.bpj.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fuchs J, Dell'Atti D, Buhot A, Calemczuk R, Mascini M, Livache T. Effects formamide of formamide on the thermal stability of DNA duplexes on biochips. Anal. Biochem. 2010;397:132. doi: 10.1016/j.ab.2009.09.044. [DOI] [PubMed] [Google Scholar]
- 48.Fiche JB, Fuchs J, Buhot A, Calemczuk R, Livache T. Point mutation detection by surface plasmon resonance imaging coupled with a temperature scan method in a model system. Anal. Chem. 2008;80:1049–1057. doi: 10.1021/ac7019877. [DOI] [PubMed] [Google Scholar]
- 49.Pingel J, Buhot A, Calemczuk R, Livache T. Temperature scans/cycles for the detection of low abundant DNA point mutations on microarrays. Biosens. Bioelectron. 2012;31:554–557. doi: 10.1016/j.bios.2011.10.012. [DOI] [PubMed] [Google Scholar]
- 50.Naiser T, Mai T, Michel W, Ott A. Versatile maskless microscope projection photolithography system and its application in light-directed fabrication of DNA microarrays. Rev. Sci. Instrum. 2006;77:063711. [Google Scholar]
- 51.Naiser T, Kayser J, Mai T, Michel W, Ott A. Stability of a surface-bound oligonucleotide duplex inferred from molecular dynamics: a study of single nucleotide defects using DNA microarrays. Phys. Rev. Lett. 2009;102:218301. doi: 10.1103/PhysRevLett.102.218301. [DOI] [PubMed] [Google Scholar]
- 52.Naiser T, Kayser J, Mai T, Michel W, Ott A. Position dependent mismatch discrimination on DNA microarrays - experiments and model. BMC Bioinformatics. 2008;9:509. doi: 10.1186/1471-2105-9-509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang L, Miles MF, Aldape KD. A model of molecular interactions on short oligonucleotide microarrays. Nat. Biotechnol. 2003;21:818–821. doi: 10.1038/nbt836. [DOI] [PubMed] [Google Scholar]
- 54.Trapp C, Schenkelberger M, Ott A. Stability of double-stranded oligonucleotide DNA with a bulged loop: a microarray study. BMC Biophys. 2011;4:20. doi: 10.1186/2046-1682-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.vanDorp MGA, Berger F, Carlon E. Computing equilibrium concentrations for large heterodimerization networks Phys. Rev. 2011 doi: 10.1103/PhysRevE.84.036114. E84, 036114. [DOI] [PubMed] [Google Scholar]
- 56.Berger F, Carlon E. From hybridization theory to microarray data analysis: performance evaluation. BMC Bioinformatics. 2011;12:464. doi: 10.1186/1471-2105-12-464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hadiwikarta W, Walter J, Hooyberghs J, Carlon E. Probing hybridization parameters from microarray experiments: nearest-neighbor model and beyond. Nucl. Acids Res. 2012;40:e138. doi: 10.1093/nar/gks475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hooyberghs J, Carlon E. Hybridisation thermodynamic parameters allow accurate detection of point mutations with DNA microarrays. Biosens. Bioelectr. 2010;26:1692–1695. doi: 10.1016/j.bios.2010.07.039. [DOI] [PubMed] [Google Scholar]
- 59.Chou HH, Hsia AP, Mooney DL, Schnable PS. Picky: oligo microarray design for large genomes. Bioinformatics. 2004;20:2893–28902. doi: 10.1093/bioinformatics/bth347. [DOI] [PubMed] [Google Scholar]
- 60.Chou HH, Trisiriroj A, Park S, Hsing YI, Ronald PC, Schnable PS. Direct calibration of PICKY-designed microarrays. BMC Bioinformatics. 2009;10:347. doi: 10.1186/1471-2105-10-347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kane MD, Jatkoe TA, Stumpf CR, Lu J, Thomas JD, Madore SJ. Assessment of the sensitivity and specificity of oligonucleotide (50mer) microarrays. Nucleic Acids Res. 2000;28:4552–4557. doi: 10.1093/nar/28.22.4552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lemoine S, Combes F, Le Crom S. An evaluation of custom microarray applications: the oligonucleotide design challenge. Nucleic Acids Res. 2009;37:1726–1739. doi: 10.1093/nar/gkp053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Garhyan J, Gharaibeh RZ, McGee S, Gibas CJ. The illusion of specific capture: surface and solution studies of suboptimal oligonucleotide hybridization. In review. 2012 doi: 10.1186/1756-0500-6-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gong P, Levicky R. DNA surface hybridization regimes. Proc. Natl. Acad. Sci. USA. 2008;105:5301–5306. doi: 10.1073/pnas.0709416105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Irving D, Gong P, Levicky R. DNA surface hybridization: comparison of theory and experiment. J. Phys. Chem. B. 2010;114:7631–7640. doi: 10.1021/jp100860z. [DOI] [PubMed] [Google Scholar]
- 66.Vainrub A, Pettitt BM. Coulomb blockage of hybridization in two-dimensional DNA arrays. Phys. Rev. E Stat, Nonlin Soft. Mat. Phys. 2002;66:41905. doi: 10.1103/PhysRevE.66.041905. [DOI] [PubMed] [Google Scholar]
- 67.Langmuir I. The constitution and fundamental properties of solids and liquids. J. Am. Chem. Soc. 1916;38:2221–2295. [Google Scholar]
- 68.Vainrub A, Pettitt BM. Accurate prediction of binding thermodynamics for DNA on surfaces. J. Phys. Chem. B. 2011;115:13300–13303. doi: 10.1021/jp208141g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vainrub A, Pettitt BM. Thermodynamics of association to a molecule immobilized in an electric double layer. Chem. Phys. Lett. 2000;323:160–166. [Google Scholar]
- 70.Lee CY, Harbers GM, Grainger DW, Gamble LJ, Castner DG. Fluorescence, XPS, and ToF-SIMS surface chemical state image analysis of DNA microarrays. J. Am. Chem. Soc. 2007;129:9429–9438. doi: 10.1021/ja071879m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lee CY, Nguyen PC, Grainger DW, Gamble LJ, Castner DG. Structure and DNA hybridization properties of mixed nucleic acid/maleimide-ethylene glycol monolayers. Anal. Chem. 2007;79:4390–4400. doi: 10.1021/ac0703395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Marshall E. Getting the noise out of gene arrays. Science. 2004;306:630–631. doi: 10.1126/science.306.5696.630. [DOI] [PubMed] [Google Scholar]
- 73.Letowski J, Brousseau R, Masson L. Designing better probes: effect of probe size, mismatch position and number on hybridization in DNA oligonucleotide microarrays. J. Microbiol. Methods. 2004;57:269–278. doi: 10.1016/j.mimet.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 74.Li X, He Z, Zhou J. Selection of optimal oligonucleotide probes for microarrays using multiple criteria, global alignment and parameter estimation. Nucleic Acids Res. 2005;33:6114–6123. doi: 10.1093/nar/gki914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.He Z, Wu L, Li X, Fields MW, Zhou J. Empirical establishment of oligonucleotide probe design criteria. Appl. Environ. Microbiol. 2005;71:3753–3760. doi: 10.1128/AEM.71.7.3753-3760.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liebich J, Schadt CW, Chong SC, He Z, Rhee SK, Zhou J. Improvement of oligonucleotide probe design criteria for functional gene microarrays in environmental applications. Appl. Environ. Microbiol. 2006;72:1688–1691. doi: 10.1128/AEM.72.2.1688-1691.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Relógio A, Schwager C, Richter A, Ansorge W, Valcárcel J. Optimization of oligonucleotide-based DNA microarrays. Nucleic Acids Res. 2002;30:e51. doi: 10.1093/nar/30.11.e51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chou CC, Chen CH, Lee TT, Peck K. Optimization of probe length and the number of probes per gene for optimal microarray analysis of gene expression. Nucleic Acids Res. 2004;32:e99. doi: 10.1093/nar/gnh099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Matveeva OV, Shabalina SA, Nemtsov VA, Tsodikov AD, Gesteland RF, Atkins JF. Thermodynamic calculations and statistical correlations for oligo-probes design. Nucleic Acids Res. 2003;31:4211–4217. doi: 10.1093/nar/gkg476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Heim T, Tranchevent LC, Carlon E, Barkema GT. Physical-chemistry-based analysis of Affymetrix microarray data. J. phys. chem. B. 2006;110:22786–22795. doi: 10.1021/jp062889x. [DOI] [PubMed] [Google Scholar]
- 81.Mei R, Hubbell E, Bekiranov S, Mittmann M, Christians FC, Shen MM, Lu G, Fang J, Liu WM, Ryder T, et al. Probe selection for high-density oligonucleotide arrays. Proc. Natl. Acad. Sci. USA. 2003;100:11237–11242. doi: 10.1073/pnas.1534744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rouillard JM, Zuker M, Gulari E. OligoArray 2.0: design of oligonucleotide probes for DNA microarrays using a thermodynamic approach. Nucleic Acids Res. 2003;31:3057–3062. doi: 10.1093/nar/gkg426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Luebke KJ, Balog RP, Garner HR. Prioritized selection of oligodeoxyribonucleotide probes for efficient hybridization to RNA transcripts. Nucleic Acids Res. 2003;31:750–758. doi: 10.1093/nar/gkg133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tanaka F, Kameda A, Yamamoto M, Ohuchi A. Design of nucleic acid sequences for DNA computing based on a thermodynamic approach. Nucleic Acids Res. 2005;33:903–911. doi: 10.1093/nar/gki235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lockhart DJ, Dong H, Byrne MC, Follettie MT, Gallo MV, Chee MS, Mittmann M, Wang C, Kobayashi M, Horton H, et al. Expression monitoring by hybridization to high-density oligonucleotide arrays. Nat. Biotechnol. 1996;14:1675–1680. doi: 10.1038/nbt1296-1675. [DOI] [PubMed] [Google Scholar]
- 86.Wodicka L, Dong H, Mittmann M, Ho MH, Lockhart DJ. Genome-wide expression monitoring in Saccharomyces cerevisiae. Nat. Biotechnol. 1997;15:1359–1367. doi: 10.1038/nbt1297-1359. [DOI] [PubMed] [Google Scholar]
- 87.Czypionka T, Cheng J, Pozhitkov A, Nolte AW. Transcriptome changes after genome-wide admixture in invasive sculpins (Cottus) Mol. Ecol. 2012;21:4797–4801. doi: 10.1111/j.1365-294X.2012.05645.x. [DOI] [PubMed] [Google Scholar]
- 88.Leparc GG, Tüchler T, Striedner G, Bayer K, Sykacek P, Hofacker IL, Kreil DP. Model-based probe set optimization for high-performance microarrays. Nucleic Acids Res. 2009;37:e18. doi: 10.1093/nar/gkn1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu WH, Høvik H, Olsen I, Chen T. Strand-specific transcriptome profiling with directly labeled RNA on genomic tiling microarrays. BMC Mol. Biol. 2011;12:3. doi: 10.1186/1471-2199-12-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Irizarry RA, Cope L, Wu Z. Feature-level exploration of the Choe et al. Affymetrix GeneChip control dataset. Genome Biology. 2006;7:404. doi: 10.1186/gb-2006-7-8-404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huettel B, Kreil DP, Matzke M, Matzke AJ. Effects of aneuploidy on genome structure, expression, and interphase organization in Arabidopsis thaliana. PLoS Genet. 2008;4:e1000226. doi: 10.1371/journal.pgen.1000226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mueckstein U, Leparc GG, Posekany A, Hofacker I, Kreil DP. Hybridization thermodynamics of NimbleGen microarrays. BMC Bioinformatics. 2010;11:35. doi: 10.1186/1471-2105-11-35. [DOI] [PMC free article] [PubMed] [Google Scholar]