


Abstract
The mammalian antibody repertoire is shaped by somatic hypermutation (SHM) and class switch recombination (CSR) of the immunoglobulin (Ig) loci of B lymphocytes. SHM and CSR are triggered by non-canonical, error-prone processing of G/U mismatches generated by activation-induced deaminase (AID). In birds, AID does not trigger SHM, but it triggers Ig gene conversion (GC), a ‘homeologous’ recombination process involving the Ig variable region and proximal pseudogenes. Because recombination fidelity is controlled by the mismatch repair (MMR) system, we investigated whether MMR affects GC in the chicken B cell line DT40. We show here that Msh6−/− and Pms2−/− DT40 cells display cell cycle defects, including genomic re-replication. However, although IgVλ GC tracts in MMR-deficient cells were slightly longer than in normal cells, Ig GC frequency, donor choice or the number of mutations per sequence remained unaltered. The finding that the avian MMR system, unlike that of mammals, does not seem to contribute towards the processing of G/U mismatches in vitro could explain why MMR is unable to initiate Ig GC in this species, despite initiating SHM and CSR in mammalian cells. Moreover, as MMR does not counteract or govern Ig GC, we report a rare example of ‘homeologous’ recombination insensitive to MMR.
INTRODUCTION
The antibody repertoire of higher organisms is firstly generated by VDJ recombination, followed by additional genetic modification through somatic hypermutation (SHM), immunoglobulin gene conversion (Ig GC) and class switch recombination (CSR). During SHM, nucleotide changes are introduced into the exons encoding the variable (IgV), N-terminal domain of the antibody heavy and light chains by a mechanism involving error-prone DNA polymerases [reviewed in (1,2)]. Although Ig GC serves the same purpose, mutations are not introduced directly but are copied from numerous pseudogene sequences located upstream on the same chromosome (3,4). In contrast, CSR involves the fusion of the IgV to a different constant (effector) region by double-strand break-induced region-specific recombination [reviewed in (5,6)].
SHM, Ig GC and CSR are all initiated by activation-induced deaminase (AID) (7–9), an enzyme expressed in antigen-stimulated B cells, which typically converts multiple cytosines in the Ig loci into uracils (2). Although uracil is generally highly efficiently repaired by base excision repair (BER), this process seems to be inefficient in antigen-stimulated B cells. Thus, some uracils persist until the next round of replication to give rise to C:G to T:A transition mutations (1,2). Others are removed by uracil–DNA glycosylase (UNG) (10–12), but the resulting abasic sites persist and are bypassed by translesion polymerases to yield all types of mutations at C:G base pairs (2,13). A third group of uracils is addressed by a poorly defined pathway, which involves MutSα (11,14), a heterodimer of mutS homologue 2 (MSH2) and MSH6 that normally initiates DNA mismatch repair (MMR) (15,16). It was proposed that MutSα detects G/U mismatches generated by AID and triggers an error-prone, long-patch repair process that introduces mutations at sites distal to those deaminated by AID (1,2). A related mechanism that involves MutSα and other factors was postulated to act at Ig switch regions to give rise to double-strand breaks that trigger CSR in the absence of UNG (11,17). The molecular mechanism of MMR-mediated diversification of Ig genes remains to be elucidated, but genetic experiments implicated exonuclease I (18), DNA polymerase η (19,20) and monoubiquitylated proliferating cell nuclear antigen (21,22) in this process. Interestingly, MutLα, a heterodimer of mutL homologue 1/postmeiotic segregation increased S cerevisiae 2 that acts immediately downstream of MutSα during MMR (15), plays no role in SHM [reviewed in (1)], although it can influence the chromosome rejoining pathway during CSR (23). The roles of UNG and MutSα in mammalian antibody diversification seem to be partially redundant, given that only their combined deficiency abrogates both CSR and SHM. Thus, in Ung−/− Msh2−/− or Ung−/− Msh6−/− mice, Ig lesions are limited to C:G to T:A transitions (11,24).
That antibody diversification can also involve Ig GC was first shown in chickens (3,4), and probably contributes to antibody diversification in most bird species (25) and rabbits (26), and perhaps in other species (25). The IgV exons, VJ in the light chain and VDJ in the heavy chain, are located downstream from an array of V, VJ and VDJ pseudogenes (referred to as ψV) that serve as donors in the gene conversion reactions. Ig GC replaces a contiguous stretch of 8 to >200 nucleotides and can thus introduce multiple base changes into the recipient V(D)J sequence (27), which may translate into amino acid replacements affecting the specificity and/or affinity of the antibody.
The chicken DT40 B cell lymphoma line undergoes constitutive AID-dependent Ig GC (28,29), and it is widely used to study antibody diversification (25,30,31) as well as DNA repair (31–33). Ig GC in DT40 cells is commonly used as a model for homologous recombination (HR) repair because the initiating event is well defined and requires HR factors, including the RAD51 paralogues XRCC2, XRCC3 and RAD51B (34), BRCA1 (35), BRCA2 (36) and RAD54 (37).
Unexpectedly, MMR does not seem to initiate Ig GC in DT40 cells, given that UNG inhibition or knockout largely eliminated Ig GC, accumulating instead C to T mutations (38,39). This implies that, unlike the mammalian enzyme (11,24), chicken MutSα does not recognize AID-generated G/U mismatches, that MMR-dependent processing of G/U mispairs does not take place in this system in the absence of UNG or that MMR-mediated processing of G/U mispairs in DT40 cells is mostly error-free, as seen in a proportion of AID-generated uracils in mouse B cells (40). However, even if MMR does not trigger Ig GC, it would be still predicted to affect its outcome; it should restrict it by preventing recombination between sequences that are too diverged (41–45), while, on the other hand, helping to repair mismatches arising through annealing of non-identical (homeologous) donor and recipient DNA sequences (31,33,46,47). Although MSH6-, MSH4- or MSH3-deficient cells were reported to undergo Ig GC [previously mentioned (7) as data not shown], the role of MMR in this process has not been studied in detail or compared with HR.
In this study, we describe the effect of MutSα and MutLα deficiency on the frequency, fidelity and overall quality of Ig GC in DT40 cells. Our data reveal that MMR is largely dispensable in this process, possibly because of its limited contribution towards processing of AID-generated uracils at the Ig locus. Moreover, we find that Ig GC represents a rare case of ‘homeologous’ recombination that is insensitive to the anti-recombination effects of MMR.
MATERIALS AND METHODS
Cell lines and transfections
DT40 IgM− CL18 (28) or its IgM+ derivative CL18c4 (34) was grown in 5% CO2 at 41°C in RPMI 1640 with glutamine (Wisent) supplemented with 10% FCS (Wisent), 1% chicken serum (Invitrogen), 50 µM β-MSH and antibiotics. DT40 Aid−/− was a kind gift of Dr H. Arakawa and Dr J.M. Buerstedde (7). Transfections for gene targeting and MSH6 complementation were as previously described (10,34). Retroviral particles for DT40 transduction were produced in HEK293T cells co-transfected with a plasmid for VSV-G, a plasmid for MLV gag/pol and the retroviral vector pMIG or pMIG-Ugi. HEK293T supernatant was harvested 48 h post-transfection, filtered and added to 106 DT40 cells in the presence of 8 µg/ml of polybrene in 24-well plates.
Gene targeting
The structure of the chicken Msh6 and Pms2 genes was determined by assembling the sequence of multiple polymerase chain reaction (PCR) products amplified from DT40 genomic DNA using oligonucleotides designed to span one or more introns based on the gene structure of the murine genes. The oligonucleotide sequences were based on the corresponding chicken cDNAs assembled from multiple expression sequence tags obtained from GeneBank™. The structures of the genes, targeting and screening strategies were confirmed by Southern blot. The targeting constructs were assembled in pBluescript II (Supplementary Figure S1). Antibiotic resistance cassettes were described earlier (48). The Msh6 targeting construct eliminates the last 20 amino acids encoded by exon 4 and most of the 3′-adjacent intron, including the splice donor site, and creates a frameshift. The Pms2 targeting construct eliminates exons 7 and 8 of the gene. Genomic DNA was purified using Puregene DNA isolation kit (Gentra) and digested with BamHI–EcoRI for screening Msh6 targeting or BamHI–XbaI for Pms2. Probes were labelled by PCR using 2 µM dATP/dAGT/dTTP and 50 µCi of [α-32P]dCTP and oligonucleotides OV1 (TGGGAGCAGGTAGTTTTGTG) and OV2 (CTCTGGGATGTCAGCAAGTC) for Msh6 and OJ43 (TGACAGGTTCTGCGTTCATA) and OJ44 (TAGGGCAGCATTCCATTC) for Pms2. Probes were purified using Illustra MicroSpin G-50 columns (GE Healthcare), and filters were hybridized in ExpressHyb (Clontech) and developed by PhosphorImager (GE Heathcare).
Growth and cell cycle analysis
Growth curves were generated by diluting exponentially growing cultures to 5–10 × 105 cells/ml that were kept in the log phase by appropriate dilutions. Trypan blue-excluded cells were periodically counted in a haemocytometer and corrected for dilution factor. Population doubling times were calculated from exponential curve fits of cell numbers versus time plots using Prism 5 (GraphPad software Inc). Cloning efficiency was estimated by fluorescence-activated cell sorting (FACS)-depositing single cells into 150 µl of normal culture medium in 96-well plates and counting the number of viable clones per plate after 2 weeks. DNA content profiles were determined by flow cytometry of 2 × 106 cells fixed in cold 70% ethanol, and stained in 0.5% Triton X-100, 100 mg/ml of RNase A and 60 mg/ml propidium iodide. S-phase profiles were determined by flow cytometry in cells pulsed for 30 min at 37°C in medium containing 10 µM BrdU (Sigma). Cells were fixed as described earlier in the text, and DNA was denatured in 2 N HCl 0.5% Triton X100, neutralized with 1 M Na2B4O7 pH 8.5 and labelled with anti-BrdU-fluorescein isothiocyanate (FITC) antibody (BD Biosciences) and propidium iodide. Apoptosis was determined by Annexin V-FITC staining (Sigma). Drug sensitivity was measured by culturing 50 000 cells for 24 h in 96-well plates in increasing concentrations of 6-thioguanine (Sigma). Viability was estimated by MTS reduction using Celltiter 96® Aqueous non-radioactive cell proliferation assay (Promega).
Antibody diversification assays
For IgM phenotype fluctuation analysis, homogeneous IgM+ or IgM− populations were obtained by FACS after blocking for 10 min in phosphate-buffered saline, 1% bovine serum albumin, 3% normal goat serum and staining for 30 min with anti-chicken IgM-FITC (Bethyl, 1:200). Dead cells were excluded by propidium iodide staining (10 µg/ml). Multiple 500 000 cell populations were cultured in 24-well plates for a given number of population doubling times, splitting 1:2 every 1 or 2 days, and their surface IgM phenotype determined by flow cytometry. Where indicated, the culture medium was supplemented with 1.25 nM Trichostatin A (TSA) (Sigma). Cells expressing Ugi or the control retrovirus were selected by GFP expression and stained using anti-chicken IgM-PE mAb (Southern biotech). Reduced UNG activity was confirmed by enzymatic assays as described previously (10).
Sequence analysis
IgM+ or IgM− cells (1–5 × 105) from the expanded DT40 populations were sorted by FACS, resuspended in DirectPCR (Viagen) and extracted with proteinase K. The variable region of the rearranged DT40 λ light chain locus was PCR amplified using KOD DNA polymerase (Novagen) [95°C 2 min + 8 × (95°C 20 s, touch down 68 >60°C 10 s, 70°C 1 min) + 23 × (95°C 15 s, 60°C 10 s, 70°C 1 min) + 72°C 5 min]. PCR products were A-tailed, cloned into pGEM-T-Easy (Promega) and sequenced by Macrogen Inc (Seoul, Korea) using the SP6 oligonucleotide. Sequences were aligned using Sequencher (Genecodes). Mutations were scored and assigned to all possible donor pseudogenes using Mutdet, a tailor-made software (D.F. and J.M.D.N., unpublished data) that assigned all possible gene conversion events following described criteria (10,34,38). Mutdet results were then manually verified. The pseudogene that could explain the most consecutive mutations and had the longest homology encompassing all mutations was scored as the donor. The maximum gene conversion tract was defined as the longest sequence fragment sharing perfect homology between each IgVλ sequence analysed and the pseudogene explaining the most consecutive mutations.
MMR and gel-shift assays
DT40 cell extracts were prepared by lysing the cells in isotonic lysis buffer [10 mM Tris–HCl, pH 7.5, 2 mM MgCl2, 3 mM CaCl2, 0.32 M sucrose, 1 mM dithiothreitol, 0.1 mM spermine, 0.5 mM spermidine, 0.3% IGEPAL CA-630 and complete inhibitor cocktail (Roche)]. For gel-shift assays, 10 µg of the respective DT40 cell extracts was incubated with 40 fmol of [32P]-labelled G/T, G/C or +1 duplexes (generated, respectively, by annealing the oligonucleotides CCA GAC GTC TGT TGA CGT TGG GAA GCT TGA G, CCA GAC GTC TGT CGA CGT TGG GAA GCT TGA G or CCA GAC GTC TGT CTG ACG TTG GGA AGC TTG A to CTC AAG CTT CCC AAC GTC GAC AGA CGT CTG G or CCA GAC GTC TGT CAA UGT TGG GAA GCT TGA G to CTC AAG CTT CCC AAC GTT GAC AGA CGT CTG G, all written 5′–3′) in 25 mM HEPES–KOH, pH 8.0, 0.5 mM ethylenediaminetetraacetic acid, 10% (v/v) glycerol, 0.5 mM dithiothreitol and 1 µg of Poly[d(I:C) − d(I:C)] for 20 min at room temperature in a final volume of 20 µl. Samples were separated on 5 or 6% TAE–polyacrylamide gels. The dried gels were exposed to a PhosphorImager screens.
A detailed procedure for the MMR assays has been described previously (49). Briefly, heteroduplex DNA substrates containing a G/T mismatch or a one nucleotide insertion (+1) within a SalI restriction site, or a G/U mispair within an AclI restriction site in the 46-bp polylinker of a pGEM13Zf(+) derivative, were constructed by primer extension, using the mismatch-containing 31mer oligonucleotides 5′-CCA GAC GTC TGT TGA CGT TGG GAA GCT TGA G-3′ for the G/T mismatch substrate, 5′-CCA GAC GTC TGT CTG ACG TTG GGA AGC TTG A-3′ for the +1 substrate or 5′CCA GAC GTC TGT CAA UGT TGG GAA GCT TGA G-3′ for the G/U substrate (the mispaired or inserted residue is highlighted in bold) and the single-stranded phagemid DNA as template. The strand discrimination signal was introduced by incubation with N.BstNBI, which introduced a specific nick into the complementary strand (3′ from the mismatch) at position 350 of the duplex. Isolation of the desired supercoiled heteroduplex substrates and the MMR assays were carried out as described, using 100 ng (47.5 fmol) heteroduplex DNA substrate and 80 μg of DT40 cell extracts in a volume of 25 μl. After 45 min of incubation at 37°C, the reactions were terminated by adding 10 µg of proteinase K and heat inactivation at 55°C for 2 h. After purification of the DNA on a Qiagen MinElute Reaction Cleanup Kit, samples were digested with SalI and DraI or AclI, respectively, treated with 10 µg of RNaseA overnight and then run on a 1% TAE–agarose gel containing 0.5 × Gel-Red (Biotium). To inhibit BER in assays using the G/U substrate, Thymine DNA glycosylase (TDG) was immunodepleted as previously described (49), and the extracts were pretreated for 10 min with 4U of Ugi (NEB) at 37°C to inhibit UNG.
Statistics
Statistical analysis was performed using Prism 5 (GraphPad software Inc).
RESULTS
Growth defects in MSH6- and PMS2-deficient DT40 cells
We targeted the MMR pathway in DT40 cells by disrupting either the Msh6 or Pms2 genes (Supplementary Figure S1). Loss of MSH6 selectively depletes MutSα, which plays an important role in both SHM and CSR, but not MutSβ, which is not involved in SHM or CSR (50–52), but which might affect other recombinogenic events. Similarly, loss of PMS2 depletes MutLα, which plays a role in CSR [reviewed in (1,2,5)], but it should not affect the other MLH1-containing heterodimer MutLγ (MLH1/MLH3), which participates in recombination (53). The chicken MSH6 and PMS2 proteins have not been characterized but share high similarity in primary sequence as well as in their structural and functional features with their mouse and human orthologues (Supplementary Figure S2).
To our surprise, Msh6−/− and Pms2−/− DT40 cells displayed major growth defects. Both cell lines had considerably longer lag phases (not shown) and a ∼2-fold increase in the population doubling times during exponential growth (Figure 1A and B). In addition, their cloning efficiencies were extremely low (Figure 1C). Because such drastic growth phenotypes have not been described in MMR-deficient cells from other organisms, we wanted to verify that they were indeed a consequence of the MMR deficiency. We, therefore, complemented the DT40 Msh6−/− line with human MSH6. This largely rescued growth and cloning efficiency (Figure 1A–C), which suggests that the phenotype is indeed caused by the MMR gene defect. The difference between wild type (WT) and hMSH6-complemented Msh6−/− cells could be caused by sequence divergence between chicken and human MSH6 (Supplementary Figure S2) and/or insufficient expression levels of the complemented cells. The heterozyogous Msh6+/− and Pms2+/− cells displayed only small increases in population doubling times, but both showed reduced cloning efficiency, albeit to a much lesser extent than the homozygous knockouts (Figure 1B and C). It seemed conceivable that the observed growth defects were related to DNA damage caused by AID. However, as doubly deficient Msh6−/− Aid−/− DT40 cells had similar growth defects to Msh6−/− cells (Figure 1D), this was not the case. Ablation of MSH6 or PMS2 in a second, IgM+, DT40 line CL18c4 led to similar phenotypes and showed an IgMdull subpopulation, which was constant over time and likely, another manifestation of the growth defects (Figure 1E). We, therefore, conclude that normal DT40 growth is dependent on the MMR pathway.
Figure 1.

Impaired proliferation in MMR-deficient DT40 cells. (A) Growth curves of log-phase cultures of DT40 CL18 cells, its Msh6−/− derivative 1015 and the latter clone expressing human Msh6 (hMsh6). Average cell numbers, corrected by dilution factor, of duplicate cultures ± standard error of the mean are plotted over time. (B) Population doubling times were calculated by exponential curve fit (R2 ≥ 0.95 in every case) of growth curves as in (A). Multiple measurements for both DT40 parental lines used in this work (CL18 and CL18c4) are plotted, as well as one or more measurements of at least two independent cell lines for each of the indicated genotypes. Mean population doubling times (DT) are indicated at the top. (C) Cloning efficiency determined as the proportion of clones arising per 96-well plate after single cell deposition. Mean + standard deviation (SD) of —six to nine plates from two to three experiments are plotted. (D) Growth curves as in (A) with DT indicated in brackets next to each cell line. (E) Flow cytometry profiles of the IgM+ DT40 cell line CL18c4 (Msh6+/+ and Pms2+/+ panels) along with representative Msh6+/− and Msh6−/− or Pms2+/− and Pms2−/− derivatives. In (B) an (C), *P < 0.05 ANOVA with Bonferroni’s post-test.
Cell cycle alterations in MSH6- and PMS2-deficient DT40
Unlike parental DT40, logarithmically growing Msh6−/− and Pms2−/− cells showed many Trypan blue-positive cells indicative of necrotic cell death (data not shown). Although we observed a trend towards higher spontaneous apoptosis in the Msh6−/− and Pms2−/− cells (Figure 2A), this was too small to account for the growth defects. Microscopic examination showed morphologically abnormalities in the Msh6−/− and Pms2−/− cultures, with numerous cells larger than normal. These cells had larger, often bi- or multilobular nuclei (Figure 2B), which suggested cell cycle checkpoint defects. Indeed, analysis of DNA content confirmed cell cycle alterations, with reduced proportion of cells in S-phase and a significant number of cells with >4n DNA content (Figure 2C). Complementation of Msh6−/− DT40 cells with hMSH6 corrected these defects to a large extent (Figure 2C). 2D cell cycle analysis of Msh6−/− cells using BrdU incorporation to distinguish cells in S-phase revealed a defect in DNA damage-induced G2/S checkpoint (Figure 2D), as previously described for MMR-deficient cells (54). More importantly, a second cycle of replication and a distinct cell population with 8n DNA content were visible in Msh6−/− cells, indicative of endore-duplication (Figure 2D). Thus, MMR-deficiency in DT40 causes cell cycle defects and re-replication that are most likely responsible for the impaired growth.
Figure 2.

Cell cycle defects in MMR-deficient DT40 cells. (A) Proportion of AnnexinV-positive cells in exponentially growing cultures of the indicated DT40 cell lines. Mean + SD of —five to seven measurements done on CL18 and two independently derived knockout cell lines are plotted. Statistical significance by Student’s unpaired two-tailed t-test. (B) Representative H&E staining images illustrating the presence of large cells with abnormal nuclear morphology in MSH6- and PMS2-deficient DT40. Bar, 20 µm. (C) Cell cycle profile of exponentially growing populations of DT40 cells. DNA was stained with propidium iodide and analysed by flow cytometry. 2n and 4n indicate the G1 and G2/M peaks, respectively (left panels). The population of cells with DNA content >4n was defined as >G2. The bar graphs summarize the mean + SD proportion of cells in each cell cycle stage obtained from seven replicates for CL18 and Msh6−/−, two replicates for Msh6−/− hMsh6 and three replicates for Pms2−/− (right panels). (D) 2D cell cycle analysis of DT40 cells pulsed with BrdU at 0, 5 or 24 h post-cisplatin treatment. BrdU incorporation into DNA was probed with FITC-labelled anti-BrdU antibody and DNA content by propidium iodide staining by flow cytometry. The proportion of cells in each gate is indicated. One representative of two experiments performed is shown.
MMR deficiency does not increase Ig gene conversion frequency
We analysed the frequency of Ig GC to test whether MMR counteracted this process, either through error-free repair of G/U mispairs or through its anti-recombination activity. The frequency of Ig GC can be estimated from fluctuation analysis of the surface IgM phenotype in DT40 (Figure 3A) (34). In brief, multiple subclones of the IgM− DT40 CL18 cell line, which has a frameshift in IgVλ preventing IgM expression (28), are expanded over time. Reversion of the frameshift by constitutive Ig GC allows re-expression of IgM with the median proportion of IgM+ cells in the clonal populations providing an estimation of Ig GC frequency. As the proportion of IgM+ cells increases with clonal expansion time, any difference in growth kinetics would affect these assays, which was a major concern in this case. Moreover, we could not obtain Msh6−/− or Pms2−/− single-cell clones (Figure 1C). We, therefore, modified the fluctuation assay by setting up multiple populations at exponential growth density (∼0.5 × 106 sIgM− cells/ml). These large starting populations bypassed the defect in cloning efficiency and minimized the distorting effects that the much longer lag phase in more diluted Msh6−/− and Pms2−/− cultures would have on Ig GC frequency. To account for the different growth rates, we expanded the cell populations for the same number of generations, which were calculated from the respective population doubling times. We confirmed that the median proportion of IgM+ cells increased linearly with the number of population doublings for DT40 WT as well as Msh6−/− cells (Figure 3B and C). When we used this method to compare the DT40 CL18 to multiple independent Msh6−/− or Pms2−/− derivatives, we observed interclonal variability. Nevertheless, although the median proportion of IgM+ cells arising from DT40 Msh6−/− was reduced for one line, it was not significantly different in three others (Figure 3D). Similarly, one DT40 Pms2−/− line generated only few IgM+ cells, but two others were significantly different neither from CL18 nor from their Pms2+/− parental lines. The variability in IgM+ proportion between lines could not be accounted for by variations in AID levels (Figure 3E; data not shown) nor did it correlate with their relative population doubling times. We suspect these variations to be because of different compensatory adaptations arising in the MMR-deficient lines during clonal expansion and growth. Importantly, Ig GC in Msh6−/− cells proceeded through its canonical mechanism. First, UNG deficiency caused by expressing the protein inhibitor Ugi in two independent DT40 Msh6−/− cell lines inhibited Ig GC as shown by a decrease in the frequency of IgM+ cells (Figure 3F) (38). Second, analysis of DT40 Msh6−/− Aid−/− lines showed that ablation of Msh6−/− did not result in spontaneous Ig GC in the absence of the initiating lesion even on treatment with Trichostatin A (Figure 3G), which exacerbates Ig GC in DT40 cells (55).
Figure 3.

No increase in Ig GC frequency in Msh6−/− or Pms2−/− DT40 cells. (A) Scheme of the frameshift reversion assay used to estimate IgVλ gene conversion frequency by measuring the proportion of DT40 cells gaining surface IgM expression over time. (B) Linear correlation between median proportion of IgM-gain cells and generation number (defined by population doubling time) for multiple DT40 CL18 subpopulations kept in exponential growth. The proportion of IgM+ cells arising from 24 subpopulations of IgM− cells was determined by flow cytometry at each indicated number of generations. Median values and data range for two independent experiments (circles and triangles) are plotted with linear fit shown. (C) Multiple IgM− subpopulations of DT40 CL18 (n = 24) and its Msh6−/− derivative line 1015 (n = 12) were sorted, independently expanded and analysed for IgM-gain as in (B) after 15, 30, 45 and 60 generations. The IgM+ proportion values are indicated for each subpopulation with medians plotted as horizontal bars. Linear fit of the medians is shown. (D) IgM-gain in multiple subpopulations of CL18 (n = 19), its Msh6−/− derivative lines 102 (n = 12), 1015 (n = 9), 1C1 (n = 19) and 1A5 (n = 9), Msh6−/− Aid−/− A5 (n = 12) and B1 (n = 12), Pms2−/− lines c20 (n = 20), c5 (n = 12) and c11 (n = 12) and the Pms2+/− c21 (n = 12) and c44 (n = 12) (parentals to Pms2−/− c5 and c11) after 70 generations was determined as in (C). The proportion of IgM+ cells in each subpopulation is plotted with medians indicated by horizontal lines and the value above. *P < 0.05 Kruskal–Walis non-parametric test with post-test. (E) Total lysates of DT40 cell lines were separated by sodium dodecyl sulphate–polyacrylamide gel electrophoresis, and AID and actin levels were analysed by western blotting. (F) Populations of 50 IgM− CL18 Msh6−/− cells expressing or not Ugi were sorted into 96-well plates and expanded for 45 generations. The proportion of IgM+ cells is plotted for each population (two shades of grey distinguish independent Msh6−/− lines) with median values indicated. (G) Three IgM− subpopulations of DT40 CL18, Msh6−/− line 1015 or Aid−/− Msh6−/− lines A5 and B1 were expanded for 45 generations in the presence of 1.25 nM TSA. The proportion of IgM+ cells in each subpopulation is plotted with median values indicated.
The frequency of Ig GC could be overestimated when normalized to population doubling times, which is influenced by cell death. Nevertheless, the BrdU pulse-chase experiments suggest that the cell cycle is longer in MMR-deficient than in WT DT40 cells given the persistence of a higher proportion of BrdU+ cells in Msh6−/− and Msh6+/− cells 24 h post-pulse (Figure 2D; data not shown). In any case, when logarithmically growing cells were expanded for the same length of time, Ig GC was still apparent, although its frequency in MMR-deficient DT40 cells was ∼2- to 4-fold lower than in the control population (e.g. compare Ig GC for CL18 at 60 generations to Msh6−/− 1015 at 30 generations in Figure 2C; data not shown).
We conclude that MSH6 and PMS2 are dispensable for Ig GC, and that their deficiency does not result in any obvious change in Ig GC mechanism (i.e dependence on AID and UNG). We cannot formally rule our that MSH6 and PMS2 might be required for optimal Ig GC (see later in the text), but our results show that their absence does not lead to an increase in Ig GC frequency.
MMR deficiency does not significantly affect the outcome Ig gene conversion
As the growth defects in MMR-deficient DT40 cells hinder accurate calculation of Ig GC frequency, we analysed the GC events by sequencing the IgVλ region amplified from IgM+ cells. This analysis provides information about multiple parameters that could be affected by MMR (Supplementary Figure S3), can be unequivocally scored and are less influenced by cell growth. Thus, we analysed the GC donor pseudogene usage, GC tract length, number of GC events per sequence and frequency of untemplated point mutations. We compared independent (not clonally related) Ig GC events obtained from multiple subpopulations of various DT40 lines grown for the same number of generations.
Ig GC in chickens prefers using the most similar pseudogene as donor (27). The pseudogene ψVL8 is most closely related in sequence to DT40 Vλ (∼90% overall, ∼97% around the frameshift insertion) (Supplementary Figure S4), and, as anticipated, it was most frequently used to correct the frameshift in DT40 CL18 (Figure 4A). This preference was retained in the Msh6−/− or Pms2−/− cells (Figure 4A). We observed slightly longer GC tracts in DT40 Msh6−/− cells than in CL18, but this difference was not significant, irrespective of whether the data from each independent Msh6−/− line were analysed separately or pooled (Figure 4B and C). A clearer trend towards longer Ig GC tract lengths in the Pms2−/− cells became statistically significant when the analysis was restricted to Ig GC events templated on ψVL8 (Figure 4C). This eliminates confounding effects, such as differences in the length, relative orientation or degree of homology of different pseudogenes with the IgVλ.
Figure 4.

Unaffected Ig gene conversion fidelity in Msh6−/− and Pms2−/−DT40. (A) Donor pseudogene usage. Proportion of IgVλ conversion events templated on each of the 25 IgV pseudogenes plotted for DT40 CL18, Msh6−/− and Pms2−/−. The data compile sequences from subpopulations coming from several fluctuation experiments (CL18 n = 3, Msh6−/− n = 6 and four independent lines, Pms−/− n = 3 and three independent lines). The distribution was not significantly different between the different genotypes (two-way ANOVA). (B) Maximum gene conversion tract length was determined for each event obtained from CL18 (three independent experiments, E1–E3) as well as from independent lines of Msh6−/− (two fluctuations compiled for lines 1015 and 1C1) or Pms2−/−. Tract length for each event is plotted with the mean and median (horizontal bars) values above. Differences are not significant by Kruskal–Walies non-parametric test. (C) All data from (B) were aggregated by genotype in box plots with 25–75% percentiles divided by the median and whiskers representing the minimum and maximum values. On the right, a similar plot considered only conversion tracts templated on pseudogene VL8. *P < 0.05 Kruskal–Walis non-parametric test with post-test. In (A–C), only independent events were considered (i.e. identical events found in sequences coming from the same subpopulation were excluded to prevent counting dynastically related events more than once). (D) The number of sequences containing one (white), two (grey) or three (black) conversion events were plotted as proportion of all sequences containing gene conversion events (n, indicated in the central circle). (E) The ratio of point mutations per mutated sequence was calculated for multiple data sets. The highest value in the Pms2−/− data set is biased by the presence of four sequences with multiple clonally related mutations. The empty triangle is the value for the same data set after removing these sequences. Clonally related sequences were not excluded in (D) and (E).
We then compared the number of mutagenic events per sequence. Should MMR-deficient cells be more permissive for recombination, one would expect more GC events per sequence. However, this was not the case (Figure 4D). Alternatively, if MutSα was recognizing AID-generated G/U mispairs to initiate faithful MMR, Msh6−/− cells would be expected to display more single base pair mutations at the IgV. This was also not the case. The data set of sequences obtained from IgM+ cells arising from IgM− populations had a similar ratio of point mutations per mutated sequence in CL18 and Msh6−/− or Pms2−/− lines (Figure 4E). A complementary experiment analysing mutations in the IgV of IgM-loss cells originating from the IgM+ Msh6−/− did not show any increase in mutations either (Table 1). Combining MSH6 deficiency with UNG inhibition by expressing Ugi in two independent CL18c4 Msh6−/− lines increased untemplated point mutation frequencies (Table 1), but not beyond what others and us have observed in UNG-deficient DT40 cells (38,39). Thus, MSH6 or PMS2 deficiency seems to lead to a small increase in the length of the GC tracts, but does not affect the donor preference or the number of mutagenic events per sequence at the IgVλ locus of DT40 cells during Ig GC.
Table 1.
Mutations in CL18c4-derived Msh6−/− DT40 cells sorted for IgM-loss
| Msh6+/+ |
Msh6−/− |
|||||
|---|---|---|---|---|---|---|
| Clone | CL18c4 | c414 |
c429 |
|||
| Population analysed | IgM− | IgMlow | IgMlow | IgMlow | IgMlow | |
| Ugia | Ugi | |||||
| Sequences | n | 83 | 69 | 24 | 72 | 24 |
| Mutated n (%) | 39 (47) | 10 (14) | 12 (50) | 3 (4) | 14 (58) | |
| Gene conversions | Independentb | 15 | 6 | 2 | 0 | 1 |
| Total | 15 | 6 | 3 | 0 | 1 | |
| Per mut seqc | 0.38 | 0.6 | 0.25 | - | 0.1 | |
| Point mutations | Independentc | 15 | 3 | 10 | 1 | 14 |
| Total | 16 | 4 | 12 | 1 | 15 | |
| Per mut seqc | 0.4 | 0.4 | 1 | 0.3 | 1.1 | |
| Ambiguousd | 8 | 4 | 2 | 2 | 2 | |
| Insertion/deletions | 8 | 0 | 0 | 0 | 0 | |
aThe endogenous UNG activity was inhibited in vivo during expansion of the populations by expressing the Ugi protein.
bIdentical events within a population were excluded to avoid recounting dynastically related events.
cAll, rather than independent, events were considered to calculate mutation load per mutated sequence.
dSingle base pair substitutions that could have been templated or untemplated.
MMR in DT40 cells is functional, and homologous recombination is heterology sensitive
The lack of effect of MMR-deficiency on Ig GC or mutation frequency was striking. We, therefore, asked whether MMR was active in DT40 cells. We measured their sensitivity to 6-thioguanine (6-TG), which, once incorporated into DNA and methylated, generates mismatches with C that cause futile MMR cycles, DNA fragmentation and cytotoxicity (56). As anticipated, WT DT40 cells were sensitive to 6-TG, whereas Msh6−/− cells were resistant (Figure 5A). Pms2−/− cells were also more resistant to 6-TG than WT cells, albeit to a lesser extent than Msh6−/− cells (Figure 5A). Together with the biochemical evidence described later in the text, these results show that MMR is active in DT40 cells. Despite this, DT40 cells fail to reject Ig GC intermediates, which must have heterologies. We, therefore, set out to test whether HR in DT40 cells might generally tolerate heterology, especially given that one of the key hallmarks of DT40 cells is their high efficiency of targeted integration (57) mediated by HR (32). To this end, we used gene targeting as an assay. The Msh6 disruption construct inactivated the first Msh6 allele with 35% efficiency. We generated a modified version of this construct containing 15 mutations within the 1764-bp-long 5′-arm (Figure 5B). This represents only 0.8% of heterology, well below the ∼10% divergence between Ig VL and the donor pseudogenes (3); yet, it was sufficient to completely abolish targeting (Figure 5C and D). Because of their negligible cloning efficiency, we were unable to formally demonstrate that gene targeting sensitivity to heterology in DT40 Msh6−/− and Pms2−/− cells is MMR-dependent. However, this has been well demonstrated in bacteria (58), yeast (59) and mammalian cells (44,60,61). Thus, our findings show that MMR is functional in DT40 cells, and that these cells tightly control the fidelity of HR in vivo, most likely through MMR.
Figure 5.
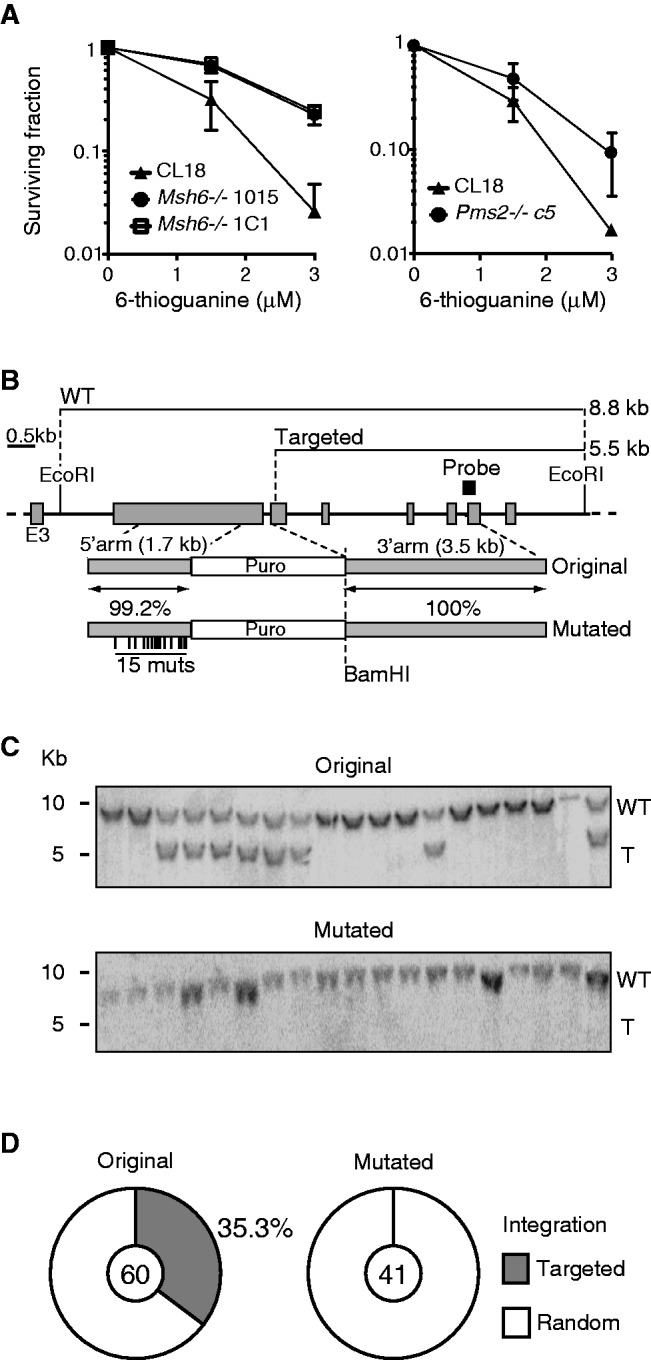
Mismatch sensing and heterology sensitive homologous recombination to in DT40. (A) Two independent Msh6−/− (left panel) and one Pms2−/− (right panel) lines and their respective parental DT40 cells were treated with increasing doses of 6-TG, and surviving fraction was determined 48 h later by MTS viability assays. Mean + SD of three experiments are plotted. (B) Partial scheme of chicken Msh6 and the fully homologous and mutated targeting constructs. The position of the mutations and homology between the constructs are indicated under each relevant segment. The restriction enzymes and probe used for Southern blot, as well as the expected fragment sizes, are shown. (C) Representative Southern blots of DNA from puromycin-resistant clones digested with EcoRI and BamHI. The position of DNA size markers (left), as well as the wild-type (WT) and targeted (T) fragments, are indicated. (D) Proportion of clones having targeted (grey) and random (white) integration of the homologous or mutated Msh6 constructs. The pie charts compile results from two independent experiments with the total number of clones analysed for each construct indicated in the centre.
Extracts of DT40 cells process G/U mispairs efficiently but predominantly by BER
To unequivocally demonstrate that MMR is biochemically active in DT40 cells and in an attempt to identify the possible cause(s) of the lack of effect of MutSα and MutLα loss on GC, we examined the substrate specificity of avian MMR. We first carried out gel-shift experiments with DT40 extracts. As shown in Figure 6A, mismatch-specific complexes were formed on incubation of the WT extracts with oligonucleotide heteroduplexes containing a G/T mispair or one extrahelical nucleotide (+1). The homoduplex G/C formed only unspecific complexes in these extracts (lane 1) (62). The fact that only the unspecific complexes were observed in extracts of the Msh6−/− cells (lanes 2, 3, 5, 6, 8, 9) confirmed that the cells lacked MutSα. Moreover, this result showed that MutSβ in DT40 cell extracts either does not bind the +1 substrate or is present in undetectable amounts. We failed to detect specific complexes with the G/U substrate even on supplementation of the extracts with Ugi. This implied either that, unlike the human protein (63), avian MutSα does not recognize this substrate, or that the uracil was removed from the oligonucleotide substrate by a glycosylase other than UNG. Pms2−/− extracts contained similar levels of MutSα to WT extracts (Supplementary Figure S5). To confirm that the binding affinity was reflected in repair efficiency, we carried out in vitro MMR assays using DT40 cell extracts and a phagemid heteroduplex containing a G/T mispair in a SalI restriction site, which makes it refractory to cleavage with this enzyme. The phagemid also contained a nick as a strand discrimination signal 3′ from the mispaired T. In this scenario, both MutSα and MutLα are absolutely required for successful MMR. Digestion of the unrepaired phagemid with DraI and SalI gives rise to fragments of 2484 and 694 bp. On correction of the G/T mismatch to G/C, the SalI site is restored, such that the 2484 fragment is cut into fragments of 1324 and1160 bp. Repair efficiency can thus be estimated from the ratio of the 2484 and 1324 (or 1160) fragments. As shown in Figure 6B, ∼30% of the G/T substrate was repaired in DT40 WT extracts (lane 2). In contrast, extracts of Pms2−/− (lane 3) and Msh6−/− (lane 5) cells were repair deficient. The lack of activity was not caused by poor quality of the extracts, given that MMR could be restored by the addition of recombinant human MutLα or MutSα, respectively (lanes 4 and 6). Importantly, although the WT extracts addressed the +1 substrate with similar efficiency to G/T (Figure 6C, compare lanes 2 and 4), we failed to detect repair of either substrate in extracts of Msh6−/− cells (lane 5), unless they were supplemented with recombinant MutSα (lane 6). This confirms the results of the gel-shift experiments and implies that MutSβ in DT40 extracts is either absent, or that it does not address an insertion/deletion loop of one extrahelical nucleotide, an intermediate arising during Ig GC, the processing of which is required to convert IgM− cells to IgM+ phenotype.
Figure 6.
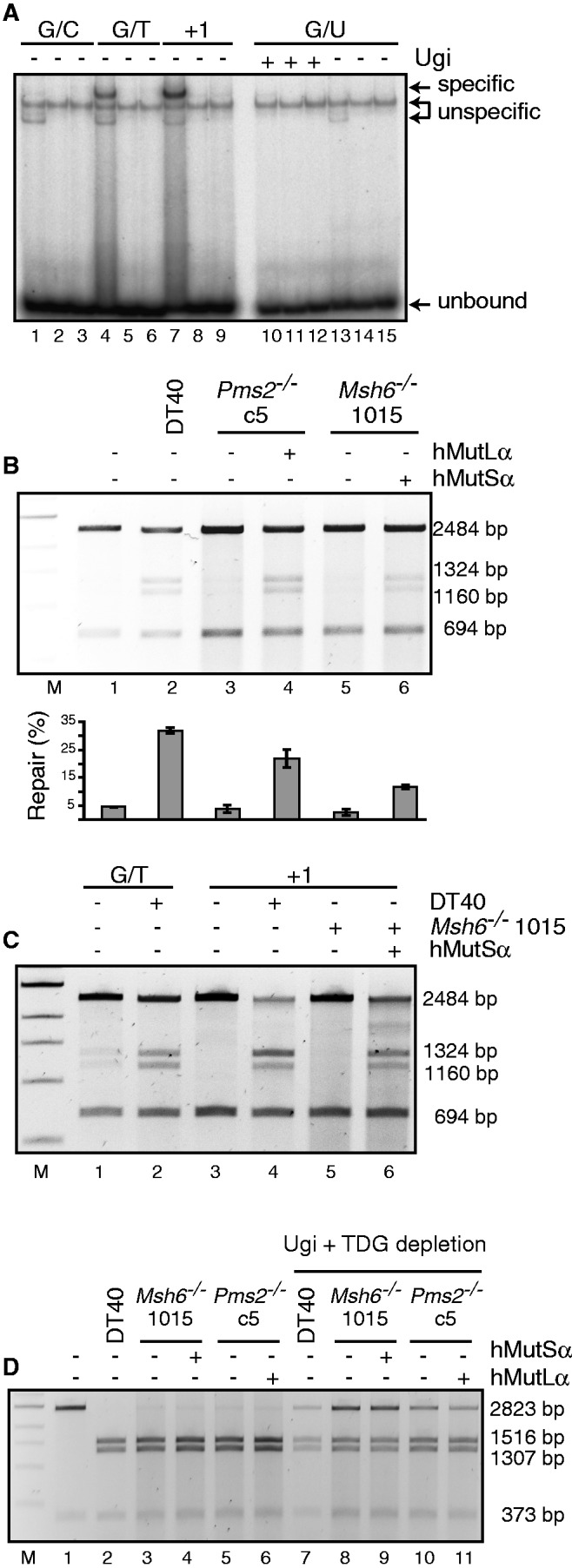
Mismatch binding and repair in DT40 cell extracts. (A) Gel-shift with DT40 WT cell extracts (lanes 1, 4, 7, 10 and 13), cell extracts of the Msh6−/− clones 1015 (lanes 2, 5, 8, 11 and 14) and 1c1 (lanes 3, 6, 9, 12 and 15). The extracts were incubated with the indicated substrates for 20 min at RT, run on a 5% TAE–polyacrylamide gel and exposed to a PhosphorImager screen. (B) MMR assays with extracts of DT40 WT, Pms2−/− and Msh6−/− cell lines. The extracts were incubated with a plasmid containing a G/T mismatch in a SalI restriction site and a nick in the T strand 350 nt 3′ from the mismatch. On repair of the mispaired T, the SalI restriction site is restored, and digestion with this enzyme gives rise to the 1324- and 1160-bp fragments in repair-proficient samples. The mean intensity + standard error proportion of these two product fragments over total signal from two experiments was quantified using ImageQuant and is plotted for each line below the gel. (C) Mismatch repair assay performed as in (B). Extracts of DT40 WT and Msh6−/− cells were incubated with a substrate containing a single-nucleotide insertion (+1). The G/T substrate served as a control of repair efficiency. (D) Mismatch repair assay carried out as described in (B) (lanes 2–6) or after immunodepletion of TDG and inhibition of UNG by the addition of Ugi (lanes 7–11). The substrate used in this assay contained a G/U mismatch in an AclI site and a nick in the U strand 350 nt 3′ from the mismatch. Repair of G/U to G/C restores the AclI site, and digestion with this enzyme gives rise to the 1516- and 1307-bp fragments in repair-proficient samples.
We also wanted to learn how the extracts processed a G/U mispair. As shown in Figure 6D (lanes 2–6), this substrate was quantitatively converted to G/C in all three extracts. The repair efficiency was diminished on TDG depletion and Ugi addition (lanes 7–11), but supplementation with recombinant MutSα or MutLα failed to increase it, which implied that MMR does not contribute to the processing of G/U mispairs in our assay, and that these mispairs are addressed almost exclusively—and efficiently—by BER. Intriguingly, the limited effect of Ugi addition and TDG depletion suggested that the extracts contain another efficient uracil DNA glycosylase. It could be TDG because we cannot estimate efficiency of its immunodepletion by the anti-human TDG antibody other than from the additive effect of UNG inhibition by Ugi and TDG immunodepletion on the in vitro MMR assay (Supplementary Figure S6), but we cannot exclude the presence of another enzyme, such as MBD4 (64), given that DT40 cells lack SMUG1 (65).
DISCUSSION
The near absence of Ig GC in UNG-deficient DT40 cells implied that MMR does not activate an alternative pathway of uracil processing for initiating Ig GC (10,39). This was unexpected, given that MMR compensates to a substantial extent for UNG-deficiency for SHM and CSR (11,24). Moreover, Ig GC is a process of homeologous recombination, which is believed to involve MMR in at least two stages (Supplementary Figure S2). To gain insights into the criteria that govern Ig GC, we disrupted the Msh6 or Pms2 genes in DT40 cells. In this way, we selectively inactivated two key MMR complexes, MutSα and MutLα, respectively, which have been clearly implicated in mammalian antibody diversification (14,23,52,66,67). At the same time, the knockouts do not affect the other MSH and MLH heterodimers (MutSβ and MutLγ), which have no role in these processes (50–52).
MMR and DT40 cell viability
MMR has not been studied in avian cells to date. The sequence and structure of MSH6 and PMS2 are conserved between chicken and their mammalian counterparts (Supplementary Figure S2), and our results show that MMR in DT40 cells is functionally similar to MMR in other species (it confers sensitivity to 6-TG, can repair mismatches in vitro and triggers a DNA damage-induced G2 checkpoint). However, unlike other eukaryotic MMR-deficient cells, the growth of the Msh6−/− and Pms2−/− DT40 B cells is greatly impaired. Moreover, they display abnormal morphology, most likely linked to spontaneous cell cycle alterations that include one round of endore-duplication. Based on the absence of a substantial increase in apoptosis and on the abnormal nuclear morphology, we posit that the 8n MMR-deficient DT40 cells undergo mitotic catastrophe (68). This unexpected phenotype contrasts with the viability of Msh6−/− and Pms2−/− mice (43,69) and with the fact that MSH2-, MSH3- or MSH6-deficient mouse B cells proliferate normally when stimulated in vitro (50,70,71). Nonetheless, germinal centre B lymphocytes in Msh2−/− mice show unusually high levels of genomic instability and compromised germinal centre reaction (66,71,72). Germinal centre B cells express low levels of p53 (73), which may distinguish them from in vitro activated B cells. That p53 may be implicated in this process is underscored by the finding that p53−/− Msh2−/− mouse embryo fibroblasts accumulate cells with 8n DNA content after genotoxic damage, which implies that MMR status and G1 checkpoint abrogation affect ploidy control (74). Given that DT40 cells do not express p53, and thus that their G1 checkpoint is compromised (75), they may be pre-disposed to re-replication (76), and this phenotype might be aggravated by the relaxation of the S (77) and G2/M (15,54) checkpoints in MMR-deficient cells. Our results thus support a role for MMR in the prevention of re-replication in checkpoint-deficient DT40 cells.
MMR and Ig gene conversion
Although Ig GC is mediated by HR factors (33,35–37), MMR in vertebrate cells impairs this process when sequence heterology exceeds 1% (42–44,78). Accordingly, MSH6- or PMS2-deficient cells have a hyper-recombinogenic phenotype both in yeast and mammals (43,78). We demonstrate here that MMR is functional in DT40 cells (Figures 5 and 6). We, therefore, expected to find an effect of MMR deficiency on Ig GC. Surprisingly, we found MutSα and MutLα to be largely dispensable for Ig GC in DT40 cells. This does not mean they do not normally play a role during Ig GC resolution. Indeed, we detected a significant trend towards longer GC tracts, most notably in PMS2-deficient cells, which confirms that MMR affects resolution of the heteroduplex intermediates arising during Ig GC (46,47,79). But as Ig GC can still proceed at near normal frequency in the absence of MMR, there must be alternative ways to resolve the GC. On the other hand, as Ig GC involves synapses between donor ψV and acceptor IgVλ sequences that are 3 to >10% divergent (3), we expected to see its frequency increased in Msh6−/− and/or Pms2−/− cells. Yet, this was not apparent in the fluctuation analyses, despite the caveat imposed by the cell growth defects of the MMR-deficient cells. Moreover, evidence of Ig GC should have been seen by sequencing the IgV as additional conversion events other than the one correcting the single nucleotide frameshift, but this was also not the case. In addition, Ig GC generally prefers to use as donor the ψV most similar to the IgV (3,27), which implies that the HR pathway for Ig GC senses homology. As in WT cells, the most frequently used donor pseudogene in Msh6−/− and Pms2−/− DT40 cells was ψV8, which is the closest sequence homologue to DT40 IgVλ. This suggests that the MMR defect in these cells did not bring about a relaxation in Ig GC stringency. MutSβ could compensate for the lack of MutSα, given that it has been shown to contribute to repressing recombination between divergent sequences in yeast (80). However, MutSα has a much more prominent role than MutSβ in preventing recombination in mammalian cells (43), which, combined with our findings that MSH6-deficient DT40 extracts lack the ability to correct +1 loops, make this scenario unlikely. Similarly, although MutLα prevents homeologous recombination in yeast (78), its absence is unlikely to be compensated for by MutLγ (81), despite the fact that the latter factor is involved in recombination control in mammalian cells (53). Another scenario whereby MSH6- or MMR-deficiency would not affect Ig GC frequency would be if HR in DT40 cells were less sensitive to MMR and more permissive to heterology. However, this was also not the case, as <1% heterology dramatically reduced gene targeting efficiency in these cells. That this was because MMR could not be formally demonstrated owing to the technical limitations caused by the deleterious effect of MMR deficiency on clonal expansion of DT40 cells. However, as anti-recombination is an evolutionary conserved function of MMR (44,58–61) and we show that MMR is active in DT40 cells, it is likely that it controls HR in birds. HR, but not Ig GC, being sensitive to heterologies in DT40, our results indicate a key difference between the pathways leading to gene targeting and Ig GC.
Why is the MMR system not involved in Ig GC in DT40 cells, when it controls HR and when it triggers SHM and CSR in mammalian cells? Our data suggest that part of the explanation might lie in the processing of AID-generated G/U mispairs. Purified human MutSα binds to G/U mispairs, albeit weaker than to G/T (63,82,83). We failed to detect a specific gel-shift with the G/U substrates in DT40 cell extracts, even though G/T and +1 substrates were bound efficiently (Figure 6A). This could mean either that chicken MutSα does not recognize G/U, or that the uracil was removed by glycosylases other than UNG present in the extracts, given that addition of Ugi had no effect. In an in vitro MMR assay, the G/U substrate was repaired extremely efficiently, and Ugi addition combined with TDG depletion brought about only a small reduction in efficiency, but this decrease was similar in MMR-proficient and MMR-deficient extracts. We, therefore, posit that the MMR system does not address G/U mispairs in DT40 extracts, possibly because of being outcompeted by BER factors. Should a similar situation prevail also in vivo, then it would explain why AID-induced G/U mispairs fail to trigger Ig GC in UNG-deficient, MMR-proficient cells (38,39). Still, this finding does not explain why MMR status does not affect Ig GC outcome in DT40 cells. It is possible that this is related to the recombinogenic phenotype of DT40 cells; as MMR is required to help maintain their genomic stability, MutSα and MutLα enzymes might be sequestered by HR in these cells in vivo and, therefore, unavailable to deal with GC events. Alternatively, the difference in sensitivity to heterology during gene targeting and Ig GC may indicate that the two processes proceed through different HR branches in DT40 cells. Our findings suggest that the Ig GC process in DT40 cells may represent a rare example of MMR-insensitive homeologous recombination.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–6.
FUNDING
Canadian Institutes of Health Research [MOP84543 to J.M.D.N.]; Canada Research Chair tier 2 (to J.M.D.N.). Michel Saucier fellowship through the Department of Microbiology and Immunology, Université de Montréal (to V.A.C. in part); Swiss National Science Foundation [310030A-118158 to J.J.]. Funding for open access charge: Institutional funds (to J.M.D.N.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are indebted to Michael Neuberger for discussions and support when the project was initiated at the MRC Laboratory of Molecular Biology, Cambridge, UK. They thank Jean-Marie Buerstedde, Hiroshi Arakawa, Katja Kratz, Stephanie Bergeron and Mariela Artola for reagents; Julian Sale and Anja Bielinsky for critical reading of the manuscript; and Tiago Chiavegatti for advice on statistics and Astrid Zahn for technical help.
REFERENCES
- 1.Di Noia JM, Neuberger MS. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 2007;76:1–22. doi: 10.1146/annurev.biochem.76.061705.090740. [DOI] [PubMed] [Google Scholar]
- 2.Peled JU, Kuang FL, Iglesias-Ussel MD, Roa S, Kalis SL, Goodman MF, Scharff MD. The biochemistry of somatic hypermutation. Annu. Rev. Immunol. 2008;26:481–511. doi: 10.1146/annurev.immunol.26.021607.090236. [DOI] [PubMed] [Google Scholar]
- 3.Reynaud CA, Anquez V, Grimal H, Weill JC. A hyperconversion mechanism generates the chicken light chain preimmune repertoire. Cell. 1987;48:379–388. doi: 10.1016/0092-8674(87)90189-9. [DOI] [PubMed] [Google Scholar]
- 4.Reynaud CA, Dahan A, Anquez V, Weill JC. Somatic hyperconversion diversifies the single Vh gene of the chicken with a high incidence in the D region. Cell. 1989;59:171–183. doi: 10.1016/0092-8674(89)90879-9. [DOI] [PubMed] [Google Scholar]
- 5.Chaudhuri J, Alt FW. Class-switch recombination: interplay of transcription, DNA deamination and DNA repair. Nat. Rev. Immunol. 2004;4:541–552. doi: 10.1038/nri1395. [DOI] [PubMed] [Google Scholar]
- 6.Stavnezer J, Guikema JEJ, Schrader CE. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 2008;26:261–292. doi: 10.1146/annurev.immunol.26.021607.090248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arakawa H, Hauschild J, Buerstedde JM. Requirement of the activation-induced deaminase (AID) gene for immunoglobulin gene conversion. Science. 2002;295:1301–1306. doi: 10.1126/science.1067308. [DOI] [PubMed] [Google Scholar]
- 8.Harris RS, Sale JE, Petersen-Mahrt SK, Neuberger MS. AID is essential for immunoglobulin V gene conversion in a cultured B cell line. Curr. Biol. 2002;12:435–438. doi: 10.1016/s0960-9822(02)00717-0. [DOI] [PubMed] [Google Scholar]
- 9.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102:553–563. doi: 10.1016/s0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 10.Di Noia J, Neuberger MS. Altering the pathway of immunoglobulin hypermutation by inhibiting uracil-DNA glycosylase. Nature. 2002;419:43–48. doi: 10.1038/nature00981. [DOI] [PubMed] [Google Scholar]
- 11.Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol. Cell. 2004;16:163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 12.Rada C, Williams GT, Nilsen H, Barnes DE, Lindahl T, Neuberger MS. Immunoglobulin isotype switching is inhibited and somatic hypermutation perturbed in UNG-deficient mice. Curr. Biol. 2002;12:1748–1755. doi: 10.1016/s0960-9822(02)01215-0. [DOI] [PubMed] [Google Scholar]
- 13.Di Noia JM, Williams GT, Chan DTY, Buerstedde J-M, Baldwin GS, Neuberger MS. Dependence of antibody gene diversification on uracil excision. J. Exp. Med. 2007;204:3209–3219. doi: 10.1084/jem.20071768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rada C, Ehrenstein MR, Neuberger MS, Milstein C. Hot spot focusing of somatic hypermutation in MSH2-deficient mice suggests two stages of mutational targeting. Immunity. 1998;9:135–141. doi: 10.1016/s1074-7613(00)80595-6. [DOI] [PubMed] [Google Scholar]
- 15.Jiricny J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell. Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 16.Kunkel TA, Erie DA. DNA mismatch repair. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 17.Stavnezer J, Schrader CE. Mismatch repair converts AID-instigated nicks to double-strand breaks for antibody class-switch recombination. Trends Genet. 2006;22:23–28. doi: 10.1016/j.tig.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 18.Bardwell PD, Woo CJ, Wei K, Li Z, Martin A, Sack SZ, Parris T, Edelmann W, Scharff MD. Altered somatic hypermutation and reduced class-switch recombination in exonuclease 1-mutant mice. Nat. Immunol. 2004;5:224–229. doi: 10.1038/ni1031. [DOI] [PubMed] [Google Scholar]
- 19.Delbos F, Aoufouchi S, Faili A, Weill JC, Reynaud CA. DNA polymerase eta is the sole contributor of A/T modifications during immunoglobulin gene hypermutation in the mouse. J. Exp. Med. 2007;204:17–23. doi: 10.1084/jem.20062131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeng X, Winter DB, Kasmer C, Kraemer KH, Lehmann AR, Gearhart PJ. DNA polymerase eta is an A-T mutator in somatic hypermutation of immunoglobulin variable genes. Nat. Immunol. 2001;2:537–541. doi: 10.1038/88740. [DOI] [PubMed] [Google Scholar]
- 21.Langerak P, Nygren AO, Krijger PH, van den Berk PC, Jacobs H. A/T mutagenesis in hypermutated immunoglobulin genes strongly depends on PCNAK164 modification. J. Exp. Med. 2007;204:1989–1998. doi: 10.1084/jem.20070902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roa S, Avdievich E, Peled JU, Maccarthy T, Werling U, Kuang FL, Kan R, Zhao C, Bergman A, Cohen PE, et al. Ubiquitylated PCNA plays a role in somatic hypermutation and class-switch recombination and is required for meiotic progression. Proc. Natl. Acad. Sci. USA. 2008;105:16248–16253. doi: 10.1073/pnas.0808182105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ehrenstein MR, Rada C, Jones AM, Milstein C, Neuberger MS. Switch junction sequences in PMS2-deficient mice reveal a microhomology-mediated mechanism of Ig class switch recombination. Proc. Natl. Acad. Sci. USA. 2001;98:14553–14558. doi: 10.1073/pnas.241525998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen HM, Tanaka A, Bozek G, Nicolae D, Storb U. Somatic hypermutation and class switch recombination in Msh6(−/−)Ung(−/−) double-knockout mice. J. Immunol. 2006;177:5386–5392. doi: 10.4049/jimmunol.177.8.5386. [DOI] [PubMed] [Google Scholar]
- 25.Arakawa H, Buerstedde J-M. Immunoglobulin gene conversion: insights from bursal B cells and the DT40 cell line. Dev. Dyn. 2004;229:458–464. doi: 10.1002/dvdy.10495. [DOI] [PubMed] [Google Scholar]
- 26.Weinstein PD, Anderson AO, Mage RG. Rabbit IgH sequences in appendix germinal centers: VH diversification by gene conversion-like and hypermutation mechanisms. Immunity. 1994;1:647–659. doi: 10.1016/1074-7613(94)90036-1. [DOI] [PubMed] [Google Scholar]
- 27.McCormack WT, Thompson CB. Chicken IgL variable region gene conversions display pseudogene donor preference and 5' to 3' polarity. Genes Dev. 1990;4:548–558. doi: 10.1101/gad.4.4.548. [DOI] [PubMed] [Google Scholar]
- 28.Buerstedde JM, Reynaud CA, Humphries EH, Olson W, Ewert DL, Weill JC. Light chain gene conversion continues at high rate in an ALV-induced cell line. EMBO J. 1990;9:921–927. doi: 10.1002/j.1460-2075.1990.tb08190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim S, Humphries EH, Tjoelker L, Carlson L, Thompson CB. Ongoing diversification of the rearranged immunoglobulin light-chain gene in a bursal lymphoma cell line. Mol. Cell. Biol. 1990;10:3224–3231. doi: 10.1128/mcb.10.6.3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arakawa H, Buerstedde JM. Activation-induced cytidine deaminase-mediated hypermutation in the DT40 cell line. Philos. Trans. R Soc. Lond. B Biol. Sci. 2009;364:639–644. doi: 10.1098/rstb.2008.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang ES, Martin A. Immunoglobulin gene conversion: Synthesizing antibody diversification and DNA repair. DNA Repair (Amst) 2007;6:1557–1571. doi: 10.1016/j.dnarep.2007.05.002. [DOI] [PubMed] [Google Scholar]
- 32.Sonoda E, Morrison C, Yamashita YM, Takata M, Takeda S. Reverse genetic studies of homologous DNA recombination using the chicken B-lymphocyte line, DT40. Philos. Trans. R Soc. Lond. B Biol. Sci. 2001;356:111–117. doi: 10.1098/rstb.2000.0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sale JE. Immunoglobulin diversification in DT40: a model for vertebrate DNA damage tolerance. DNA Repair (Amst) 2004;3:693–702. doi: 10.1016/j.dnarep.2004.03.042. [DOI] [PubMed] [Google Scholar]
- 34.Sale JE, Calandrini DM, Takata M, Takeda S, Neuberger MS. Ablation of XRCC2/3 transforms immunoglobulin V gene conversion into somatic hypermutation. Nature. 2001;412:921–926. doi: 10.1038/35091100. [DOI] [PubMed] [Google Scholar]
- 35.Longerich S, Orelli BJ, Martin RW, Bishop DK, Storb U. Brca1 in immunoglobulin gene conversion and somatic hypermutation. DNA Repair (Amst) 2008;7:253–266. doi: 10.1016/j.dnarep.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hatanaka A, Yamazoe M, Sale JE, Takata M, Yamamoto K, Kitao H, Sonoda E, Kikuchi K, Yonetani Y, Takeda S. Similar effects of Brca2 truncation and Rad51 paralog deficiency on immunoglobulin V gene diversification in DT40 cells support an early role for Rad51 paralogs in homologous recombination. Mol. Cell. Biol. 2005;25:1124–1134. doi: 10.1128/MCB.25.3.1124-1134.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bezzubova O, Silbergleit A, Yamaguchi-Iwai Y, Takeda S, Buerstedde J-M. Reduced X-ray resistance and homologous recombination frequencies in a RAD54−/− mutant of the chicken DT40 cell line. Cell. 1997;89:185–193. doi: 10.1016/s0092-8674(00)80198-1. [DOI] [PubMed] [Google Scholar]
- 38.Di Noia JM, Neuberger MS. Immunoglobulin gene conversion in chicken DT40 cells largely proceeds through an abasic site intermediate generated by excision of the uracil produced by AID-mediated deoxycytidine deamination. Eur. J. Immunol. 2004;34:504–508. doi: 10.1002/eji.200324631. [DOI] [PubMed] [Google Scholar]
- 39.Saribasak H, Saribasak NN, Ipek FM, Ellwart JW, Arakawa H, Buerstedde JM. Uracil DNA glycosylase disruption blocks Ig gene conversion and induces transition mutations. J. Immunol. 2006;176:365–371. doi: 10.4049/jimmunol.176.1.365. [DOI] [PubMed] [Google Scholar]
- 40.Roa S, Li Z, Peled JU, Zhao C, Edelmann W, Scharff MD. MSH2/MSH6 complex promotes error-free repair of AID-induced dU:G mispairs as well as error-prone hypermutation of A:T sites. PLoS One. 2010;5:e11182. doi: 10.1371/journal.pone.0011182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abuin A, Zhang H, Bradley A. Genetic analysis of mouse embryonic stem cells bearing Msh3 and Msh2 single and compound mutations. Mol. Cell. Biol. 2000;20:149–157. doi: 10.1128/mcb.20.1.149-157.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Wind N, Dekker M, Berns A, Radman M, te Riele H. Inactivation of the mouse Msh2 gene results in mismatch repair deficiency, methylation tolerance, hyperrecombination, and predisposition to cancer. Cell. 1995;82:321–330. doi: 10.1016/0092-8674(95)90319-4. [DOI] [PubMed] [Google Scholar]
- 43.de Wind N, Dekker M, Claij N, Jansen L, van Klink Y, Radman M, Riggins G, van der Valk M, van't Wout K, te Riele H. HNPCC-like cancer predisposition in mice through simultaneous loss of Msh3 and Msh6 mismatch-repair protein functions. Nat. Genet. 1999;23:359–362. doi: 10.1038/15544. [DOI] [PubMed] [Google Scholar]
- 44.Elliott B, Jasin M. Repair of double-strand breaks by homologous recombination in mismatch repair-defective mammalian cells. Mol. Cell. Biol. 2001;21:2671–2682. doi: 10.1128/MCB.21.8.2671-2682.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Evans E, Alani E. Roles for mismatch repair factors in regulating genetic recombination. Mol. Cell. Biol. 2000;20:7839–7844. doi: 10.1128/mcb.20.21.7839-7844.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen J-M, Cooper DN, Chuzhanova N, Férec C, Patrinos GP. Gene conversion: mechanisms, evolution and human disease. Nat. Rev. Genet. 2007;8:762–775. doi: 10.1038/nrg2193. [DOI] [PubMed] [Google Scholar]
- 47.Haber JE, Ira G, Malkova A, Sugawara N. Repairing a double-strand chromosome break by homologous recombination: revisiting Robin Holliday's model. Philos. Trans. R Soc. Lond. B Biol. Sci. 2004;359:79–86. doi: 10.1098/rstb.2003.1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Arakawa H, Lodygin D, Buerstedde JM. Mutant loxP vectors for selectable marker recycle and conditional knock-outs. BMC Biotechnol. 2001;1:7. doi: 10.1186/1472-6750-1-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fischer F, Baerenfaller K, Jiricny J. 5-Fluorouracil is efficiently removed from DNA by the base excision and mismatch repair systems. Gastroenterology. 2007;133:1858–1868. doi: 10.1053/j.gastro.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 50.Li Z, Scherer SJ, Ronai D, Iglesias-Ussel MD, Peled JU, Bardwell PD, Zhuang M, Lee K, Martin A, Edelmann W, et al. Examination of Msh6- and Msh3-deficient mice in class switching reveals overlapping and distinct roles of MutS homologues in antibody diversification. J. Exp. Med. 2004;200:47–59. doi: 10.1084/jem.20040355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martomo SA, Yang WW, Gearhart PJ. A role for Msh6 but not Msh3 in somatic hypermutation and class switch recombination. J. Exp. Med. 2004;200:61–68. doi: 10.1084/jem.20040691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wiesendanger M, Kneitz B, Edelmann W, Scharff MD. Somatic hypermutation in MutS homologue (MSH)3-, MSH6-, and MSH3/MSH6-deficient mice reveals a role for the MSH2-MSH6 heterodimer in modulating the base substitution pattern. J. Exp. Med. 2000;191:579–584. doi: 10.1084/jem.191.3.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lipkin SM, Wang V, Jacoby R, Banerjee-Basu S, Baxevanis AD, Lynch HT, Elliott RM, Collins FS. MLH3: a DNA mismatch repair gene associated with mammalian microsatellite instability. Nat. Genet. 2000;24:27–35. doi: 10.1038/71643. [DOI] [PubMed] [Google Scholar]
- 54.Hawn MT, Umar A, Carethers JM, Marra G, Kunkel TA, Boland CR, Koi M. Evidence for a connection between the mismatch repair system and the G2 cell cycle checkpoint. Cancer Res. 1995;55:3721–3725. [PubMed] [Google Scholar]
- 55.Seo H, Masuoka M, Murofushi H, Takeda S, Shibata T, Ohta K. Rapid generation of specific antibodies by enhanced homologous recombination. Nat. Biotechnol. 2005;23:731–735. doi: 10.1038/nbt1092. [DOI] [PubMed] [Google Scholar]
- 56.Nelson JA, Carpenter JW, Rose LM, Adamson DJ. Mechanisms of action of 6-thioguanine, 6-mercaptopurine, and 8-azaguanine. Cancer Res. 1975;35:2872–2878. [PubMed] [Google Scholar]
- 57.Buerstedde JM, Takeda S. Increased ratio of targeted to random integration after transfection of chicken B cell lines. Cell. 1991;67:179–188. doi: 10.1016/0092-8674(91)90581-i. [DOI] [PubMed] [Google Scholar]
- 58.Worth L, Clark S, Radman M, Modrich P. Mismatch repair proteins MutS and MutL inhibit RecA-catalyzed strand transfer between diverged DNAs. Proc. Natl. Acad. Sci. USA. 1994;91:3238–3241. doi: 10.1073/pnas.91.8.3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Selva EM, New L, Crouse GF, Lahue RS. Mismatch correction acts as a barrier to homeologous recombination in Saccharomyces cerevisiae. Genetics. 1995;139:1175–1188. doi: 10.1093/genetics/139.3.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Elliott B, Richardson C, Winderbaum J, Nickoloff JA, Jasin M. Gene conversion tracts from double-strand break repair in mammalian cells. Mol. Cell. Biol. 1998;18:93–101. doi: 10.1128/mcb.18.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.te Riele H, Maandag ER, Berns A. Highly efficient gene targeting in embryonic stem cells through homologous recombination with isogenic DNA constructs. Proc. Natl. Acad. Sci. USA. 1992;89:5128–5132. doi: 10.1073/pnas.89.11.5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiricny J, Hughes M, Corman N, Rudkin BB. A human 200-kDa protein binds selectively to DNA fragments containing G.T mismatches. Proc. Natl. Acad. Sci. USA. 1988;85:8860–8864. doi: 10.1073/pnas.85.23.8860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hughes MJ, Jiricny J. The purification of a human mismatch-binding protein and identification of its associated ATPase and helicase activities. J. Biol. Chem. 1992;267:23876–23882. [PubMed] [Google Scholar]
- 64.Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999;401:301–304. doi: 10.1038/45843. [DOI] [PubMed] [Google Scholar]
- 65.Di Noia JM, Rada C, Neuberger MS. SMUG1 is able to excise uracil from immunoglobulin genes: insight into mutation versus repair. EMBO J. 2006;25:585–595. doi: 10.1038/sj.emboj.7600939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frey S, Bertocci B, Delbos F, Quint L, Weill JC, Reynaud CA. Mismatch repair deficiency interferes with the accumulation of mutations in chronically stimulated B cells and not with the hypermutation process. Immunity. 1998;9:127–134. doi: 10.1016/s1074-7613(00)80594-4. [DOI] [PubMed] [Google Scholar]
- 67.Schrader CE, Edelmann W, Kucherlapati R, Stavnezer J. Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 1999;190:323–330. doi: 10.1084/jem.190.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vitale I, Galluzzi L, Castedo M, Kroemer G. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell. Biol. 2011;12:385–392. doi: 10.1038/nrm3115. [DOI] [PubMed] [Google Scholar]
- 69.Baker SM, Bronner CE, Zhang L, Plug AW, Robatzek M, Warren G, Elliott EA, Yu J, Ashley T, Arnheim N, et al. Male mice defective in the DNA mismatch repair gene PMS2 exhibit abnormal chromosome synapsis in meiosis. Cell. 1995;82:309–319. doi: 10.1016/0092-8674(95)90318-6. [DOI] [PubMed] [Google Scholar]
- 70.Kim N, Storb U. The role of DNA repair in somatic hypermutation of immunoglobulin genes. J. Exp. Med. 1998;187:1729–1733. doi: 10.1084/jem.187.11.1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vora KA, Tumas-Brundage KM, Lentz VM, Cranston A, Fishel R, Manser T. Severe attenuation of the B cell immune response in Msh2-deficient mice. J. Exp. Med. 1999;189:471–482. doi: 10.1084/jem.189.3.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Alabyev B, Manser T. Bcl-2 rescues the germinal center response but does not alter the V gene somatic hypermutation spectrum in MSH2-deficient mice. J. Immunol. 2002;169:3819–3824. doi: 10.4049/jimmunol.169.7.3819. [DOI] [PubMed] [Google Scholar]
- 73.Phan RT, Dalla-Favera R. The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature. 2004;432:635–639. doi: 10.1038/nature03147. [DOI] [PubMed] [Google Scholar]
- 74.Strathdee G, Sansom OJ, Sim A, Clarke AR, Brown R. A role for mismatch repair in control of DNA ploidy following DNA damage. Oncogene. 2001;20:1923–1927. doi: 10.1038/sj.onc.1204276. [DOI] [PubMed] [Google Scholar]
- 75.Takao N, Kato H, Mori R, Morrison C, Sonoda E, Sun X, Shimizu H, Yoshioka K-i, Takeda S, Yamamoto K. Disruption of ATM in p53-null cells causes multiple functional abnormalities in cellular response to ionizing radiation. Oncogene. 1999;18:7002–7009. doi: 10.1038/sj.onc.1203172. [DOI] [PubMed] [Google Scholar]
- 76.Vaziri C, Saxena S, Jeon Y, Lee C, Murata K, Machida Y, Wagle N, Hwang DS, Dutta A. A p53-dependent checkpoint pathway prevents rereplication. Mol. Cell. 2003;11:997–1008. doi: 10.1016/s1097-2765(03)00099-6. [DOI] [PubMed] [Google Scholar]
- 77.Brown KD, Rathi A, Kamath R, Beardsley DI, Zhan Q, Mannino JL, Baskaran R. The mismatch repair system is required for S-phase checkpoint activation. Nat. Genet. 2003;33:80–84. doi: 10.1038/ng1052. [DOI] [PubMed] [Google Scholar]
- 78.Erdeniz N, Nguyen M, Deschênes SM, Liskay RM. Mutations affecting a putative MutLalpha endonuclease motif impact multiple mismatch repair functions. DNA Repair. 2007;6:1463–1470. doi: 10.1016/j.dnarep.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bollag RJ, Elwood DR, Tobin ED, Godwin AR, Liskay RM. Formation of heteroduplex DNA during mammalian intrachromosomal gene conversion. Mol. Cell. Biol. 1992;12:1546–1552. doi: 10.1128/mcb.12.4.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nicholson A, Hendrix M, Jinks-Robertson S, Crouse GF. Regulation of mitotic homeologous recombination in yeast. Functions of mismatch repair and nucleotide excision repair genes. Genetics. 2000;154:133–146. doi: 10.1093/genetics/154.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cannavo E, Marra G, Sabates-Bellver J, Menigatti M, Lipkin SM, Fischer F, Cejka P, Jiricny J. Expression of the MutL homologue hMLH3 in human cells and its role in DNA mismatch repair. Cancer Res. 2005;65:10759–10766. doi: 10.1158/0008-5472.CAN-05-2528. [DOI] [PubMed] [Google Scholar]
- 82.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese LS. Structure of the human MutSalpha DNA lesion recognition complex. Mol. Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 83.Wilson TM, Vaisman A, Martomo SA, Sullivan P, Lan L, Hanaoka F, Yasui A, Woodgate R, Gearhart PJ. MSH2-MSH6 stimulates DNA polymerase eta, suggesting a role for A:T mutations in antibody genes. J. Exp. Med. 2005;201:637–645. doi: 10.1084/jem.20042066. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.