


Abstract
The numerous discovered cases of domesticated transposable element (TE) proteins led to the recognition that TEs are a significant source of evolutionary innovation. However, much less is known about the reverse process, whether and to what degree the evolution of TEs is influenced by the genome of their hosts. We addressed this issue by searching for cases of incorporation of host genes into the sequence of TEs and examined the systems-level properties of these genes using the Saccharomyces cerevisiae and Drosophila melanogaster genomes. We identified 51 cases where the evolutionary scenario was the incorporation of a host gene fragment into a TE consensus sequence, and we show that both the yeast and fly homologues of the incorporated protein sequences have central positions in the cellular networks. An analysis of selective pressure (Ka/Ks ratio) detected significant selection in 37% of the cases. Recent research on retrovirus-host interactions shows that virus proteins preferentially target hubs of the host interaction networks enabling them to take over the host cell using only a few proteins. We propose that TEs face a similar evolutionary pressure to evolve proteins with high interacting capacities and take some of the necessary protein domains directly from their hosts.
INTRODUCTION
In recent years, the traditional view that transposable elements (TEs) are only a burden to their host organism has shifted. Although their parasitic nature is not questioned, the discoveries that many proteins originate from TEs and that TEs contributed to the invention of several key cellular machineries of multicellular organisms highlighted their significance in evolutionary innovations (1–3). The best known cases of TE domestication include the RAG protein of the immune system of vertebrates (4), CENP-B protein of centromeres (5–7), light sensing in plants (8), regulation of telomere length (9) or developmental regulation [PAX6 gene, (10)]. Although numerous cases of domestication have already been identified in model organisms, their real number remains unclear, as estimates range from thousands to dozens even in the well-characterized human genome [see (11) versus (12,13)]. Besides providing the raw material for novel genes, TEs also contributed to regulation and generation of allelic diversity: 25% of promoters in human genes contain TE sequences (14), whereas the activity of Helitrons, the eukaryotic rolling circle transposons [reviewed in (15)] and MuDR (MULE) transposons (16) resulted in the sometimes massive amplification of functional genes in their hosts (17–20). The ability of DNA transposons to mobilize fragments of DNA has even resulted in the development of highly efficient vectors (e.g. Sleeping Beauty transposon) for gene transfer (21).
Naturally occurring gene capturing has been well studied in the maize and rice genomes, where it occurs at a high rate, involves only particular repeat types like Helitrons or MuDR repeats and is a major force shaping the genome. During gene capturing, fragments or entire genes are incorporated into the transposon, and the subsequent amplification of the repeat results in a high copy number of the gene as well. However, Helitrons are unusual among other TEs in their ability to mobilize adjacent DNA [also the prokaryotic relatives of Helitrons are known to mobilize DNA fragments, including antibiotic resistance genes and virulence factors (22)], and even in Helitrons, the captured gene fragments only rarely contribute to the evolution of the transposon sequence itself (17).
As the global sequencing effort completed the genomes of most classic model organisms, attention has turned towards several other eukaryotic genomes, either owing to their phylogenetic importance [i.e. (23)], or owing to being important for a narrower research community (24). Besides providing key insights into genome organization and function, these studies have also revealed a large diversity of TEs that previously had not been appreciated: repeat classes that had been thought to be extinct in mammals were found [like DNA transposons in bats, (25)], entirely novel classes of repeats were discovered, e.g. Polintons (26,27), and the diversity of known families was greatly expanded (28,29).
Here, we investigate the incorporation of host genes into TE sequences, however, in a narrower sense than it was reported for Helitrons or MULEs; we focus only on those cases that ‘made it’ to the consensus sequence of the transposon and thus could influence its evolution. The incorporation of a protein domain into a TE has been described mostly in those cases, where it resulted in the emergence of a novel repeat type; in non-LTR repeats, the acquisition of an endonuclease, RNase H domain and an ORF1 protein in the early history of R2–R4-like repeats resulted in the emergence of their currently most common families [L1, I, Jockey, CR1; (30)]. Incorporation of genes has been documented in LTR retrotransposons, where the (multiple) transitions from the transposon state to a viral state were enabled by the acquisition—usually from other viruses—of envelope proteins (31). Also, the acquisition of a small number of proteins with unclear function by LTR elements has been described (32), and among DNA transposons, the acquisition of the helicase domain in Helitrons has been reported (15).
The main objective of this article is to search for cases of protein incorporation into TEs and test the systems-level properties of these proteins, i.e. whether the incorporated sequences originate from a random selection of host proteins, or TEs selectively incorporate genes with distinct properties within the cellular networks. Currently, cellular networks are well characterized only in a small number of model organisms; therefore, we use budding yeast (Saccharomyces cerevisiae) and the fruitfly (Drosophila melanogaster), as yeast is the eukaryote with the most-understood interactome, whereas among multicellular organisms, the Drosophila interactome is especially well characterized. Our results indicate that (i) the acquisition of host proteins by TEs is not a rare exceptional event but is relatively common; (ii) the incorporated genes participate in significantly more protein–protein interactions (PPIs) than expected by chance; (iii) are central within the interaction networks (i.e. have high betweenness and closeness centrality); and (iv) a considerable fraction of them are subject to selection, thus contribute to the evolution of the TEs.
MATERIALS AND METHODS
Data sources and homology search
We searched 4848 verified yeast ORF sequences and 13 909 Drosophila genes against RepBase (v.15.12), the main database of eukaryotic TEs. Only consensus sequences were used from RepBase to exclude dubious TEs from the analysis. In the case of Drosophila genes, we used only the core region of each gene, i.e. the regions that are present in all known alternative splicing products. This was necessary because TEs can occasionally be incorporated into transcripts by alternative splicing, and the functionality of such splice products is uncertain (33).The sequences of eukaryotic TEs were downloaded from RepBase [http://www.girinst.org, v. 15.12, (34)]. The sequences of yeast proteins were downloaded from the Saccharomyces Genome Database (http://www.yeastgenome.org); Drosophila genes were downloaded from FlyBase (http://flybase.org).
The DNA sequences of TEs were translated in all six frames, and the sequence comparisons between yeast and Drosophila proteins and the translated TE sequences were made with the jackhmmer tool of the hmmer package (35), with a bit score cutoff 27. In the homologous sequence fragments of TE, yeast and Drosophila proteins, we identified conserved domains using the Pfam database (36) v. 24, (http://pfam.sanger.ac.uk) with hmmscan. To decide whether the homology between a yeast protein and a TE sequence represents transposon domestication or the reverse process, the incorporation of a protein fragment into a TE, we implemented the following, automated protocol. First, we implemented the taxonomic tree of ∼180 000 taxa, using known phylogenetic relationships from the Pfam database (ncbi_taxonomy table). Next, we screened the Uniprot database (http://www.uniprot.org) with the sequence fragment from yeast or fly from a homologous pair of TE - yeast/Drosophila proteins, using jackhmmer (bit score threshold 27), excluding all matches of viral or TE origin. Using the species of the Swissprot hits, we identified a subtree on the global tree; in the cases where this resulted in no hits, we used Uniprot hits. We repeated the same procedure for the repeat fragment, using the 6-frame translated RepBase as the sequence database, and then compared the two trees (Figure 1B). The tree which the broader taxonomic span (i.e. contains the other) points to the source of the sequence (TE domestication versus protein incorporation into a TE). This method performs well in the case of protein incorporation events, where the phylogenetic spread of a protein domain is typically wide, but its homologue is present only in a small number of TEs; however, it is less reliable in the case of ancient TE domestications: in the case of common domains, e.g. Zinc fingers that are widespread both in TEs and host proteins, the sequence exchange events happened too long ago to reliably identify its source taxon, and also several independent domestication events may not be distinguished from each other, leading to a very broad phylogenetic distribution of such proteins. In the phylogenetic analysis of the activity-regulated cytoskeletal-associated protein (Arc) gene, multiple alignments were made with muscle (37), and the maximum likelihood phylogenetic tree (1000 bootstrap replications) was built with MEGA5 (38).
Figure 1.

(A) Examples of TEs with significant homology to yeast and Drosophila genes. Black bars indicate TE ORFs, gray bars indicate the location of the homologous yeast/Drosophila genes. Wherever present, we used the ORF coordinates provided by RepBase; for the repeats where RepBase provides no gene prediction, we predicted the ORFs with Glimmer3. (B) The workflow used to determine whether the homology between a TE and a gene is a result of domestication or the incorporation of a host protein into a TE.
Statistical analyses
We used Monte Carlo simulations to determine whether the number of genetic and protein interactions (degree), betweennes centrality and closeness centrality of the gene fragments that were incorporated into TEs are significantly different from the random expectation. First, we took 100 000 random samples of genes without replacement from the yeast and Drosophila data sets and determined the parameter of interest, i.e. the median node degree of the proteins in the sample. Next, we determined the number of random samples with higher median value than what we observed in the proteins homologous with TEs. Significance (p) was determined as p = (n + 1)/(N + 1), where n is the number of random samples with medians equal or higher than in the observed sample, and N is the total number of random samples. All analyses were carried out with perl scripts developed in-house. Betweenness centrality and closeness centrality were calculated with Pajek, a program for the analysis of large networks (http://pajek.imfm.si); in the analyses of yeast interactions, only those interactions were used where at least one of the interacting partners is a verified ORF.
Analysis of evolutionary rates in the captured proteins
We tested for significant selection in the incorporated proteins with two methods. Wherever the incorporated protein fragment was present in more than one TE family, we compared the two closest homologous families to decide whether their Ka/Ks ratio is significantly different than 1. We identified Ka and Ks values with PAML (39) and tested whether their ratio significantly differs from one with a likelihood ratio test: we fixed the Ka/Ks ratio at 1, and fitted a similar maximum-likelihood model to the alignment of the two sequences (40). The difference between the log-likelihoods of the two models was tested with Chi-square tests, to test whether the null model assuming neutral evolution (Ka/Ks = 1) in the TE protein performs significantly worse than the one where Ka and Ks could vary independently. In those cases where additional TE homologs could not be found, we applied a different (and less powerful) procedure; we searched the NCBI ref_mrna database with the captured fragment of the TE protein with tblastn. Using the best match to the TE fragment, we searched NCBI ref_mrna again, and using the results of the two searches identified an outgroup sequence, which is at least as distant (in terms of bit score) both to the TE fragment and its best homologue as they are to each other. These three sequences were used to construct an unrooted phylogenetic tree, and we identified the separate evolutionary rates for all of its branches with PAML. We tested whether the Ka/Ks ratio of the TE branch is significantly different from 1, similarly as for two homologous TEs, with a likelihood ratio test: we fixed the Ka/Ks ratio of the TE branch of the tree at 1 and fitted a similar maximum-likelihood model to the alignment of the three sequences, and the difference between two models was used to test whether the null model with Ka/Ks = 1 in the TE branch of the tree performs significantly worse than the one where the evolutionary rate was allowed to vary.
RESULTS
Identification of yeast and fly protein homologues in TEs and determination of their evolutionary origin
We identified 38 yeast proteins and 145 Drosophila proteins that show significant similarity to mobile elements (Figure 1, Supplementary Table S1, Supplementary UCSC Genome Browser track). By comparing the taxonomic distribution of the homologues of each protein with the taxonomic distribution of its corresponding TE sequence (see Methods and Figure 1B), we determined whether the matches are likely to be the result of domestication or capturing of a protein fragment by a TE. We identified only six cases that represent a domestication of a TE sequence, whereas the phylogenies of 108 genes support the protein incorporation scenario (Supplementary Table S1, see also Supplementary Figure S1A–C for examples). In 67 cases, the homologous sequences are so widespread both in the hosts and among TEs that our method could not infer whether the sequence was originating from a host or a TE, and in two cases, the phylogenies within TEs and hosts show no relationship (thus, horizontal transfer may be involved).
A particularly interesting case of domestication is the Arc gene of Drosophila (FBgn0033926). Arc genes in mammals received considerable attention in recent years because they are key regulators of synaptic plasticity required for normal brain functioning and long-term memory formation (41,42). In deuterostomes, they are present only in tetrapods and contain a domesticated fragment of a gag protein from a Gypsy retrotransposon (43). In Drosophila, the Arc genes are also expressed in neurons and regulate behavioral responses for stress (44), although unlike in mammals, they do not influence synaptic plasticity. Drosophila Arc genes also contain a domesticated fragment of a gag protein from an (insect) Gypsy retrotransposon and show homology to mammalian Arc genes (Figure 2). However, the absence of Arc-like genes in other protostomes than insects (and in other deuterostomes than tetrapods) together with their phylogeny (Figure 2) suggests that the gag proteins of Gypsy retrotransposons were recruited twice independently, and in both cases, the resulting ‘Arc’ genes gained functions in the neural system and can be seen as an example of ‘convergent domestication’.
Figure 2.

Evolution of Arc proteins. (A) Examples of Arc genes in vertebrates (human, mouse, chicken) and invertebrates (Drosophila melanogaster, Stomoxys calictrans, Drosophila silvestris). Black bars indicate the location of the regions that are homologous both to the domesticated Gypsy transposon and other Arc genes and contain a Retrotrans_gag conserved domain (pfam accession PF03732). (B) A maximum likelihood tree of the homologous regions of the Arc proteins and several Gypsy retrotransposons. Although the bootstrap support (1000 replications) is low for many branches, the presence of two Gypsy families between the Arc genes and the absence of Arc proteins in deuterostomes other than tetrapods and protostomes other than insects indicate that Gypsy gag proteins were domesticated twice independently.
Proteins incorporated into repeats occupy central positions in cellular networks
Most proteins do not operate in isolation but interact with other proteins and form multi-protein complexes, which perform a particular cellular function. Using PPIs from the BioGRID (v. 3.1.83) database for yeast (45) and from the FlyBase database (46) for Drosophila, we tested with Monte Carlo simulations whether the incorporated genes have distinct positions in the protein interaction network, i.e. whether the median number of protein interactions (degree) of the captured genes, their betweenness centrality (the fraction of shortest paths of the network that pass through a particular node) and closeness centrality (the inverse of the mean distance between node v and all other nodes reachable from it) are significantly higher than expected by chance. We found that the incorporated genes that are present in the PPI databases have significantly higher degree and centrality measures than expected by chance (Figures 3C and 4) both in yeast and Drosophila. To rule out any detection biases caused by the phylogenetic distance between Drosophila/yeast and the host species of TEs, we determined the divergence time between Drosophila/yeast and the TE hosts with the TimeTree application (47) and tested whether the network characteristics of incorporated proteins are positively correlated with divergence. None of the network parameters showed a significant correlation (Supplementary Figure S2).
Figure 3.

Characteristics of the genes that were incorporated into TEs. We performed Monte Carlo simulations to test whether fitness and network characteristics like degree (the number of interactions with other nodes), betweenness centrality (the fraction of shortest paths that pass through a node) and closeness centrality (the inverse of the mean distance between node v and all other nodes reachable from it) are significantly different in the incorporated genes than the random expectation. (A) Distribution of the median degree (PPIs) in 100 000 random samples, the arrow represents the median of the 35 Drosophila genes for which PPI information was available. (B) Distribution of the median betweenness centrality in 100 000 random samples, and the observed level in Drosophila. (C) Statistical summary of Monte Carlo simulations. We did not perform tests for the fitness effect for Drosophila gene knockouts, as we are not aware of studies that provide such data at a genomic scale.
Figure 4.

(A) The network of all PPIs of Drosophila genes with homology to a TE, for which PPI data were available in FlyBase (35 genes, highlighted in black). The median number of PPIs is 20. (B) An example of a PPI network for a randomly selected set of Drosophila genes (also 35 genes, highlighted in black). The median number of PPIs is 9, corresponding to the average of the random samples (see Figure 3).
An important question is whether the incorporated protein fragments/domains are themselves highly interacting or merely come from highly interacting proteins. Currently, this can be tested only indirectly because data on direct domain–domain interactions are sill limited and have been compiled only for proteins present in the PDB database (48), and thus represent only a small subset of all protein interactions. We used the 3did database (49), which contains 6260 interactions between 4302 Pfam domains of PDB entries, to test whether the Pfam domains found in the incorporated sequences are domains that interact with more domains than it would be expected from randomly selected ones. First, we searched for the presence of conserved protein domains in the protein fragments homologous to a TE using the Pfam database. Altogether, we identified 149 conserved domains in the yeast and fly genes homologous to a TE (Supplementary Tables S2 and S3), of which 74 are found in the protein incorporation cases. These represent only 37 different domains though, as frequently similar domains are incorporated into different TEs. From the 37 different Pfam domains of the incorporated proteins, 27 are present in the 3did database. Using Monte Carlo simulations, we found that the mean number of interactions of these domains with other Pfam domains is significantly higher (3.44, P = 0.023) than expected by chance (2.14 + /− 0.56), supporting the hypothesis that TEs pick up domains with higher number of interacting partners than the average.
Besides physical interactions, genetic interactions provide an alternative means to depict functional connections between genes. A genetic interaction is defined as the difference of the fitness effect of a double gene deletion mutant in comparison with the expected multiplicative effect of the two individual deletions. For example, an extreme case is a synthetic lethal interaction, a lethal double mutant phenotype where the individual deletion products of the two genes are both viable phenotypes. Recently, large-scale genetic interaction maps have become available for yeast [i.e. (50)], that enabled the characterization of the entire functional landscape of the yeast cell. Genetic interaction data for Drosophila are much less abundant and are available only for a small fraction (12%) of genes. Similarly to PPIs, we used genetic interactions deposited in the BioGRID and FlyBase databases and tested whether the incorporated genes have higher node degree, betweenness and closeness centrality in the genetic interaction network as well. We found that node degree and closeness centrality of the incorporated genes is significantly higher that the random expectation only in Drosophila, whereas betweenness centrality is not significantly different from the random expectation neither in yeast nor Drosophila (Figure 3B and C).
Finally, using Monte Carlo simulations, we tested whether the incorporated proteins interact more frequently with each other in the host cellular network than random proteins and whether they have related functions. Although the number of direct PPIs is low in both species, we found that their number is still much higher than expected between randomly chosen proteins (Figure 3), indicating that TEs are under selection to incorporate genes with particular functions. To test this, we estimated the enrichment of GO terms in them with GOrilla (51); however, the incorporated genes cannot be assigned to a single GO term either in yeast or Drosophila, the most enriched (P < 10−5) molecular function terms are metal ion binding in Drosophila and cofactor binding and glyceraldehyde-3-phosphate dehydrogenase activity in yeast (Supplementary Table S4).
Testing the functionality of proteins with captured gene fragments
Randomly inserting or deleting a sequence into a protein can lead to the loss of its function, and thus an important question concerning the TE proteins that captured a DNA fragment of their host is whether such proteins remained functional. Although domain rearrangements are relatively common in the evolution of genes (52), and recent studies show that mid-domain breaks can also result in functional proteins (53), it is not possible to prove in silico that a particular chimeric protein is or was functional (i.e. enzymatically active). Nevertheless, several lines of evidence indicate that many of these TE proteins contribute to the fitness of the TEs.
A functional protein is likely to be subject to purifying or adaptive selection. We tested for signatures of selection in the captured gene fragments of the TE proteins that have acquired such a gene fragment, and the length of the captured sequence was at least 50 aa residues (Supplementary Table S5). We used two methods (see Materials and Methods for details); whenever it was possible we compared the two closest homologous TE families sharing the same incorporated protein fragment, and in the remaining cases, we built an unrooted phylogenetic tree with the TE sequence, the closest RefSeq protein and an outgroup, and tested for selection on the TE branch of the tree. There is very little or no difference between the incorporated gene fragment and its closest RefSeq homolog (Ks < 0.01) in nine cases; thus, these sequences could not be tested reliably; additionally in 19 cases, the TE sequence and the closest RefSeq or RepBase homologs are so highly diverged (Ks > 10) that the saturation of synonymous substitutions also makes any tests of selection unreliable. From the 24 cases of protein incorporation where 0.01 < Ks < 10, we could detect significant selection in nine cases [P < 0.05, likelihood ratio test, (40)], with one case indicating adaptive evolution (hAT-N22_DR) and the remaining eight indicating purifying selection (Supplementary Table S5).
The incorporation of a new domain may result in a protein that contains conflicting molecular features, for example, the presence of both extracellular and nuclear domains within the same protein. To exclude those cases where the domain composition/structure already indicates a dysfunctional protein, we tested all TE proteins with a captured gene fragment with MisPred, a pipeline designed to detect mispredicted and abnormal proteins based on such conflicts (54). In the majority of the proteins with incorporated gene fragments, MisPred found no signs of abnormality, except five cases: BEL1-I_SM and Helitron-N3_ZM contain a truncated Pfam domain owing to mid-domain breaks, whereas the respective proteins in Gypsy-1-I_MI, Gypsy-40_Mad-I and SRV_MM-int contain transmembrane helices (55), which is unexpected in the case of TEs, as they are not part of membranes. In the case of Gypsy-1-I_MI, the transmembrane helix is not in the incorporated fragment; thus, it may be a misannotation or may have an unknown function. (We found additional four cases of proteins with transmembrane helices in TEs where the origin of the domain was unclear or owing to domestication).
Finally to investigate how the captured gene fragments influence the function of the TE proteins, we predicted the 3D structure of each chimeric protein that carries a fragment of a non-TE gene and is shorter than 500 amino acids (13 cases) with I-TASSER (56,57). Owing to the lack of sufficiently good templates in the Protein Data Bank (www.pdb.org), only three of the proteins have sufficiently high quality models (estimated TM-score ≥ 0.5) that also the positioning of the individual domains is likely to be correct, and therefore their function can be predicted with high confidence (Figure 5). These models were analysed with COFACTOR, a part of the I-TASSER pipeline that predicts the function and catalytic centers of proteins using their tertiary structure. Although in the three structures, the incorporated protein fragment provides a different functionality (oxidoreductase activity, methyltransferase activity - RNA binding, DNA binding; see Figure 5), the sequence of the incorporated fragment overlaps with the predicted binding sites of the proteins (Figure 5), further supporting the hypothesis that capturing host genes contributed to the emergence of new functions.
Figure 5.
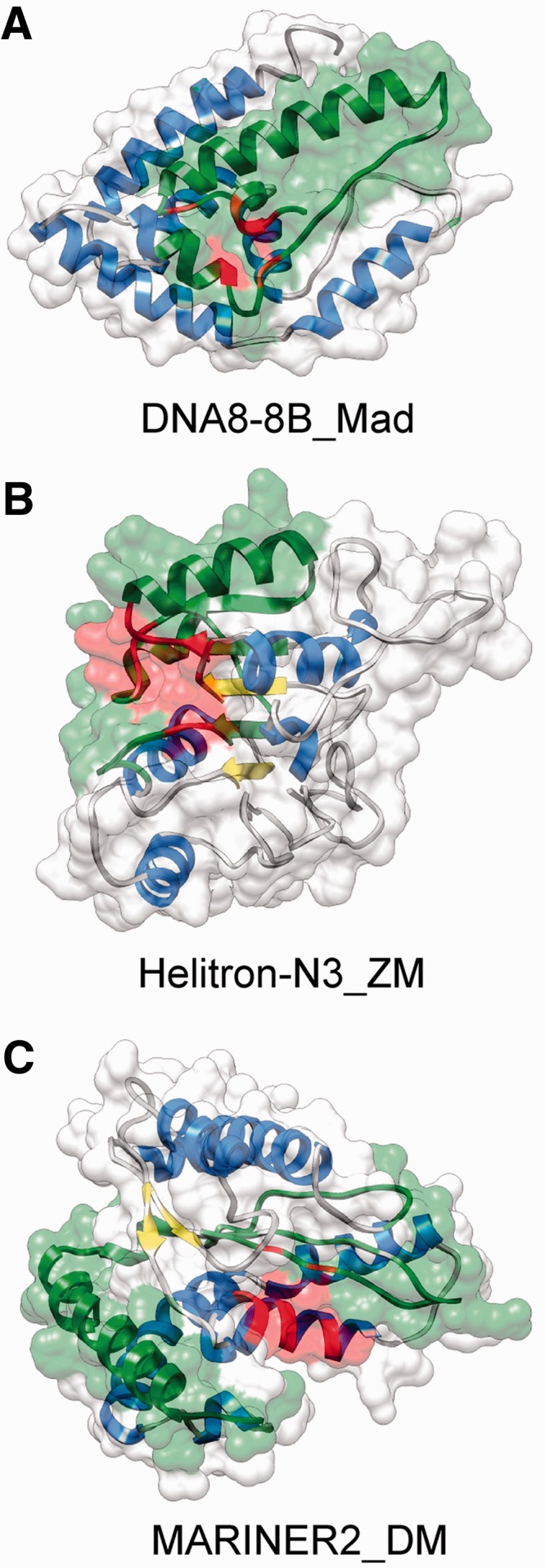
Examples of chimeric TE protein structures with an incorporated fragment of a host gene. The structures were predicted with I-TASSER, their function and catalytic centers were predicted with COFACTOR. In all cases, the estimated TM-score with the true topology is >0.5; thus, the models have an essentially correct topology. Alpha helices are highlighted with blue, beta sheets with yellow, green regions indicate the fraction of the sequence that is homologous to a non-TE protein and red highlights the catalytic core predicted by COFACTOR. (A) DNA8-8B_Mad from the apple genome, estimated TM score with the correct fold is 0.53. The incorporated fragment shows 80% sequence similarity to a short chain dehydrogenase (B9RTW7) with oxidoreductase activity (GO:0016491). (B) Helitron_N3_ZM from maize, estimated TM score is 0.64. The incorporated fragment shows 90% amino acid sequence similarity to maize fibrillarin (B6T4G7). The predicted highest scoring gene ontology terms for the molecular function of the protein are methyltransferase activity (GO:0008168) and RNA binding (GO:0003723). (C) MARINER2_DM transposon from Drosophila, estimated TM score is 0.74. The incorporated fragment is only 30% similar to the Drosophila gene CG18367-PA. The highest scoring molecular function GO term is DNA binding (GO:0003677).
DISCUSSION
Overall, our findings suggest that not only TE proteins contribute to the evolution of their hosts but the reverse process of protein ‘junkification’ might be also significant in explaining the origin and the diversity of the TE sequences. The central position of the homologues of the incorporated genes both in the yeast and Drosophila PPI networks suggests that TEs acquire genes with particular characteristics and function, and that these sequences are either not picked up randomly by the repeats or are not retained randomly in the repeats. Different TEs show variability in their insertion preferences; non-LTR retrotransposons, which do not move horizontally or are active in hosts with compact genomes, typically target gene-poor, AT-rich regions [i.e. L1s or Alus in the human genome, (12)] or heterochromatin [i.e. Ty5 retrotransposons in the yeast genome, (58)], most likely to minimize their deleterious effect on host fitness. In contrast, genomic parasites capable of horizontal transfer like DNA transposons, LTR retrotransposons and retroviruses either show little target selectivity, or preferentially insert near actively transcribed genes (59,60), which, in consequence, might be more likely incorporated into a TE. A test of this hypothesis would be if genes with homologues to different repeat classes would show different patterns, i.e. genes with homology to an LTR retrotransposon would be hubs of PPI networks, whereas genes with homology to non-LTR retrotransposons would not; however, the number of genes with homology to non-LTR retroelements is too low to allow a meaningful comparison.
An alternative hypothesis is that TEs incorporate genes fragments randomly, but only a small fraction of sequences—with high number of interactions and centrality—remain in the TE consensus owing to selection. A number of observations support this hypothesis. First, in the majority of the TEs, the captured protein fragment resides within the predicted genes of the TE and not in the intergenic regions of the repeat, have a similar strand orientation to the nearest TE protein (Supplementary UCSC Browser track) and in a significant fraction of the cases where there has been enough time to accumulate nucleotide differences between the host gene and the incorporated sequence (Ks > 0.01), significant selection could be detected (Supplementary Table S5). Second, all repeats in our analysis are consensus sequences that are present in multiple copies in their host genome; thus, the incorporation of foreign sequences clearly did not make these repeats dysfunctional. Third, research on virus-host interactions indicates that incorporation of proteins with high degree of interactions and centrality may be beneficial for the TEs. Recently, Calderwood et al. (61) demonstrated that the proteins of Epstein–Barr virus preferentially interact with hubs of human protein interaction networks, and this pattern was subsequently confirmed for many other viruses and even parasitic bacteria (62). The most likely cause of the preferential interaction with highly connected proteins is that targeting hubs of the hosts’ cellular network is the most efficient way of using its resources, i.e. diverting its pathways to the use of the parasite, especially if the parasite has only a few proteins to achieve this task. As autonomous TEs encode only few proteins (frequently only one), the efficient use of the host resources may favor the evolution of multifunctional proteins with abilities to interact with several pathways of the host interactome, and the simplest way to achieve this is to acquire such protein domains directly from the host. In addition, retroviruses and retrotransposons were shown to interact with overlapping sets of proteins (63), further corroborating this hypothesis.
As the identified cases of gene capturing did not happen in yeast or Drosophila, it raises the question to what extent the yeast and Drosophila homologues of the captured proteins have similar properties to the captured ones. We used these species to investigate the systems-level characteristics of the incorporated proteins because they have much better characterized interactomes than most other model organisms, and the fact that we observe a similar pattern in two very distantly related species, and also in domain–domain interactions, provides strong support for the generality of our results. Recent findings indicate that PPIs are highly conserved and evolve three orders of magnitude slower than protein sequences themselves (64), which explains the qualitatively similar results in the two species. However, it is unclear how far genetic interactions are conserved across species. Such comparisons are challenging owing to technological differences between model organisms (e.g. RNAi is used for gene knockdown in multicellular organisms while in in-frame deletion is used in yeast), and also the number of genes for which information is available is very different [see (65) for review]. The lack of a significant effect in yeast, and the low number of genes with known genetic interactions in fly indicates that any conclusions on genetic interactions cannot be readily generalized at this point.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–5, Supplementary Figures 1 and 2, Supplementary UCSC Genome Browser track.
FUNDING
Funding for open access charge: Hungarian Scientific Research Fund [PD83571 to G.A., NK77978 to A.Sz. and PD75261 to P.B.]; International Human Frontier Science Program Organization (to B.P.); ‘Lendület Program’ of the Hungarian Academy of Sciences to (to B.P.); NSF Career Award [DBI 0746198 to Y.Z.]; National Institute of General Medical Sciences [GM083107 and GM084222 to Y.Z.].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Csaba Pál for useful comments and suggestions.
REFERENCES
- 1.Volff J. Turning junk into gold: domestication of transposable elements and the creation of new genes in eukaryotes. Bioessays. 2006;28:913–922. doi: 10.1002/bies.20452. [DOI] [PubMed] [Google Scholar]
- 2.Jurka J, Kapitonov VV, Kohany O, Jurka MV. Repetitive sequences in complex genomes: structure and evolution. Annu. Rev. Genomics. Hum. Genet. 2007;8:241–259. doi: 10.1146/annurev.genom.8.080706.092416. 10.1146/annurev.genom.8.080706.092416. [DOI] [PubMed] [Google Scholar]
- 3.Feschotte C, Pritham EJ. DNA transposons and the evolution of eukaryotic genomes. Annu. Rev. Genet. 2007;41:331–368. doi: 10.1146/annurev.genet.40.110405.090448. 10.1146/annurev.genet.40.110405.090448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kapitonov VV, Jurka J. RAG1 core and V(D)J recombination signal sequences were derived from Transib transposons. PLoS Biol. 2005;3:e181. doi: 10.1371/journal.pbio.0030181. 10.1371/journal.pbio.0030181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smit AF, Riggs AD. Tiggers and DNA transposon fossils in the human genome. Proc. Natl Acad. Sci. USA. 1996;93:1443–1448. doi: 10.1073/pnas.93.4.1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kipling D, Warburton PE. Centromeres, CENP-B and Tigger too. Trends Genet. 1997;13:141–145. doi: 10.1016/s0168-9525(97)01098-6. [DOI] [PubMed] [Google Scholar]
- 7.Casola C, Hucks D, Feschotte C. Convergent domestication of pogo-like transposases into centromere-binding proteins in fission yeast and mammals. Mol. Biol. Evol. 2008;25:29–41. doi: 10.1093/molbev/msm221. 10.1093/molbev/msm221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin R, Ding L, Casola C, Ripoll DR, Feschotte C, Wang H. Transposase-derived transcription factors regulate light signaling in Arabidopsis. Science. 2007;318:1302–1305. doi: 10.1126/science.1146281. 10.1126/science.1146281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eickbush TH. Telomerase and retrotransposons: which came first? Science. 1997;277:911–912. doi: 10.1126/science.277.5328.911. [DOI] [PubMed] [Google Scholar]
- 10.Ivics Z, Izsvak Z, Minter A, Hackett PB. Identification of functional domains and evolution of Tc1-like transposable elements. Proc. Natl Acad. Sci. USA. 1996;93:5008–5013. doi: 10.1073/pnas.93.10.5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Britten R. Transposable elements have contributed to thousands of human proteins. Proc. Natl Acad. Sci. USA. 2006;103:1798–1803. doi: 10.1073/pnas.0510007103. 10.1073/pnas.0510007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 13.Zdobnov EM, Campillos M, Harrington ED, Torrents D, Bork P. Protein coding potential of retroviruses and other transposable elements in vertebrate genomes. Nucleic Acids Res. 2005;33:946–954. doi: 10.1093/nar/gki236. 10.1093/nar/gki236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jordan IK, Rogozin IB, Glazko GV, Koonin EV. Origin of a substantial fraction of human regulatory sequences from transposable elements. Trends Genet. 2003;19:68–72. doi: 10.1016/s0168-9525(02)00006-9. [DOI] [PubMed] [Google Scholar]
- 15.Kapitonov VV, Jurka J. Rolling-circle transposons in eukaryotes. Proc. Natl Acad. Sci. USA. 2001;98:8714–8719. doi: 10.1073/pnas.151269298. 10.1073/pnas.151269298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jiang N, Bao Z, Zhang X, Eddy SR, Wessler SR. Pack-MULE transposable elements mediate gene evolution in plants. Nature. 2004;431:569–573. doi: 10.1038/nature02953. 10.1038/nature02953. [DOI] [PubMed] [Google Scholar]
- 17.Yang L, Bennetzen JL. Distribution, diversity, evolution, and survival of Helitrons in the maize genome. Proc. Natl Acad. Sci. USA. 2009;106:19922–19927. doi: 10.1073/pnas.0908008106. 10.1073/pnas.0908008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pritham EJ, Feschotte C. Massive amplification of rolling-circle transposons in the lineage of the bat Myotis lucifugus. Proc. Natl Acad. Sci. USA. 2007;104:1895–1900. doi: 10.1073/pnas.0609601104. 10.1073/pnas.0609601104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Du C, Fefelova N, Caronna J, He L, Dooner HK. The polychromatic Helitron landscape of the maize genome. Proc. Natl Acad. Sci. USA. 2009;106:19916–19921. doi: 10.1073/pnas.0904742106. 10.1073/pnas.0904742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hanada K, Vallejo V, Nobuta K, Slotkin RK, Lisch D, Meyers BC, Shiu S, Jiang N. The functional role of pack-MULEs in rice inferred from purifying selection and expression profile. Plant Cell. 2009;21:25–38. doi: 10.1105/tpc.108.063206. 10.1105/tpc.108.063206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ivics Z, Izsvák Z. The expanding universe of transposon technologies for gene and cell engineering. Mob. DNA. 2010;1:25. doi: 10.1186/1759-8753-1-25. 10.1186/1759-8753-1-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Toleman MA, Bennett PM, Walsh TR. ISCR elements: novel gene-capturing systems of the 21st century? Microbiol. Mol. Biol. Rev. 2006;70:296–316. doi: 10.1128/MMBR.00048-05. 10.1128/MMBR.00048-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warren WC, Hillier LW, Marshall Graves JA, Birney E, Ponting CP, Grützner F, Belov K, Miller W, Clarke L, Chinwalla AT, et al. Genome analysis of the platypus reveals unique signatures of evolution. Nature. 2008;453:175–183. doi: 10.1038/nature06936. 10.1038/nature06936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Colbourne JK, Pfrender ME, Gilbert D, Thomas WK, Tucker A, Oakley TH, Tokishita S, Aerts A, Arnold GJ, Basu MK, et al. The ecoresponsive genome of Daphnia pulex. Science. 2011;331:555–561. doi: 10.1126/science.1197761. 10.1126/science.1197761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ray DA, Feschotte C, Pagan HJT, Smith JD, Pritham EJ, Arensburger P, Atkinson PW, Craig NL. Multiple waves of recent DNA transposon activity in the bat, Myotis lucifugus. Genome Res. 2008;18:717–728. doi: 10.1101/gr.071886.107. 10.1101/gr.071886.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pritham EJ, Putliwala T, Feschotte C. Mavericks, a novel class of giant transposable elements widespread in eukaryotes and related to DNA viruses. Gene. 2007;390:3–17. doi: 10.1016/j.gene.2006.08.008. 10.1016/j.gene.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 27.Kapitonov VV, Jurka J. Self-synthesizing DNA transposons in eukaryotes. Proc. Natl Acad. Sci. USA. 2006;103:4540–4545. doi: 10.1073/pnas.0600833103. 10.1073/pnas.0600833103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de la Chaux N, Wagner A. BEL/Pao retrotransposons in metazoan genomes. BMC Evol. Biol. 2011;11:154. doi: 10.1186/1471-2148-11-154. 10.1186/1471-2148-11-154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kojima KK, Kapitonov VV, Jurka J. Recent expansion of a new Ingi-related clade of Vingi non-LTR retrotransposons in hedgehogs. Mol. Biol. Evol. 2011;28:17–20. doi: 10.1093/molbev/msq220. 10.1093/molbev/msq220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malik HS, Burke WD, Eickbush TH. The age and evolution of non-LTR retrotransposable elements. Mol. Biol. Evol. 1999;16:793–805. doi: 10.1093/oxfordjournals.molbev.a026164. [DOI] [PubMed] [Google Scholar]
- 31.Malik HS, Henikoff S, Eickbush TH. Poised for contagion: evolutionary origins of the infectious abilities of invertebrate retroviruses. Genome Res. 2000;10:1307–1318. doi: 10.1101/gr.145000. [DOI] [PubMed] [Google Scholar]
- 32.Havecker ER, Gao X, Voytas DF. The diversity of LTR retrotransposons. Genome Biol. 2004;5:225. doi: 10.1186/gb-2004-5-6-225. 10.1186/gb-2004-5-6-225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gotea V, Makałowski W. Do transposable elements really contribute to proteomes? Trends Genet. 2006;22:260–267. doi: 10.1016/j.tig.2006.03.006. 10.1016/j.tig.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 34.Jurka J, Kapitonov VV, Pavlicek A, Klonowski P, Kohany O, Walichiewicz J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005;110:462–467. doi: 10.1159/000084979. 10.1159/000084979. [DOI] [PubMed] [Google Scholar]
- 35.Eddy SR. A new generation of homology search tools based on probabilistic inference. Genome Inform. 2009;23:205–211. [PubMed] [Google Scholar]
- 36.Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–D222. doi: 10.1093/nar/gkp985. 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Edgar RC. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics. 2004;5:113. doi: 10.1186/1471-2105-5-113. 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011;28:2731–2739. doi: 10.1093/molbev/msr121. 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007;24:1586–1591. doi: 10.1093/molbev/msm088. [DOI] [PubMed] [Google Scholar]
- 40.Yang Z, Bielawski JP. Statistical methods for detecting molecular adaptation. Trends Ecol. Evol. 2000;15:496–503. doi: 10.1016/S0169-5347(00)01994-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shepherd JD, Bear MF. New views of Arc, a master regulator of synaptic plasticity. Nat. Neurosci. 2011;14:279–284. doi: 10.1038/nn.2708. 10.1038/nn.2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korb E, Finkbeiner S. Arc in synaptic plasticity: from gene to behavior. Trends Neurosci. 2011;34:591–598. doi: 10.1016/j.tins.2011.08.007. 10.1016/j.tins.2011.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Campillos M, Doerks T, Shah PK, Bork P. Computational characterization of multiple Gag-like human proteins. Trends Genet. 2006;22:585–589. doi: 10.1016/j.tig.2006.09.006. 10.1016/j.tig.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 44.Mattaliano MD, Montana ES, Parisky KM, Littleton JT, Griffith LC. The Drosophila ARC homolog regulates behavioral responses to starvation. Mol. Cell. Neurosci. 2007;36:211–221. doi: 10.1016/j.mcn.2007.06.008. 10.1016/j.mcn.2007.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stark C, Breitkreutz B, Chatr-Aryamontri A, Boucher L, Oughtred R, Livstone MS, Nixon J, Van Auken K, Wang X, Shi X, et al. The BioGRID Interaction Database: 2011 update. Nucleic Acids Res. 2011;39:D698–D704. doi: 10.1093/nar/gkq1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McQuilton P, St Pierre SE, Thurmond J. FlyBase 101–the basics of navigating FlyBase. Nucleic Acids Res. 2012;40:D706–D714. doi: 10.1093/nar/gkr1030. 10.1093/nar/gkr1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hedges SB, Dudley J, Kumar S. TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics. 2006;22:2971–2972. doi: 10.1093/bioinformatics/btl505. 10.1093/bioinformatics/btl505. [DOI] [PubMed] [Google Scholar]
- 48.Berman H, Henrick K, Nakamura H, Markley JL. The worldwide Protein Data Bank (wwPDB): ensuring a single, uniform archive of PDB data. Nucleic Acids Res. 2007;35:D301–D303. doi: 10.1093/nar/gkl971. 10.1093/nar/gkl971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stein A, Céol A, Aloy P. 3did: identification and classification of domain-based interactions of known three-dimensional structure. Nucleic Acids Res. 2011;39:D718–D723. doi: 10.1093/nar/gkq962. 10.1093/nar/gkq962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Costanzo M, Baryshnikova A, Bellay J, Kim Y, Spear ED, Sevier CS, Ding H, Koh JLY, Toufighi K, Mostafavi S, et al. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. Gorilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu Y, Rasmussen MD, Kellis M. Evolution at the subgene level: domain rearrangements in the drosophila phylogeny. Mol. Biol. Evol. 2012;29:689–705. doi: 10.1093/molbev/msr222. 10.1093/molbev/msr222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rogers RL, Hartl DL. Chimeric genes as a source of rapid evolution in Drosophila melanogaster. Mol. Biol. Evol. 2012;29:517–529. doi: 10.1093/molbev/msr184. 10.1093/molbev/msr184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nagy A, Hegyi H, Farkas K, Tordai H, Kozma E, Bányai L, Patthy L. Identification and correction of abnormal, incomplete and mispredicted proteins in public databases. BMC Bioinformatics. 2008;9:353. doi: 10.1186/1471-2105-9-353. 10.1186/1471-2105-9-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Krogh A, Larsson B, von Heijne G, Sonnhammer EL. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 2001;305:567–580. doi: 10.1006/jmbi.2000.4315. 10.1006/jmbi.2000.4315. [DOI] [PubMed] [Google Scholar]
- 56.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc. 2010;5:725–738. doi: 10.1038/nprot.2010.5. 10.1038/nprot.2010.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinformatics. 2008;9:40. doi: 10.1186/1471-2105-9-40. 10.1186/1471-2105-9-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gao X, Hou Y, Ebina H, Levin HL, Voytas DF. Chromodomains direct integration of retrotransposons to heterochromatin. Genome Res. 2008;18:359–369. doi: 10.1101/gr.7146408. 10.1101/gr.7146408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bushman FD. Targeting survival: integration site selection by retroviruses and LTR-retrotransposons. Cell. 2003;115:135–138. doi: 10.1016/s0092-8674(03)00760-8. [DOI] [PubMed] [Google Scholar]
- 60.Liang Q, Kong J, Stalker J, Bradley A. Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis. 2009;47:404–408. doi: 10.1002/dvg.20508. 10.1002/dvg.20508. [DOI] [PubMed] [Google Scholar]
- 61.Calderwood MA, Venkatesan K, Xing L, Chase MR, Vazquez A, Holthaus AM, Ewence AE, Li N, Hirozane-Kishikawa T, Hill DE, et al. Epstein-Barr virus and virus human protein interaction maps. Proc. Natl Acad. Sci. USA. 2007;104:7606–7611. doi: 10.1073/pnas.0702332104. 10.1073/pnas.0702332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dyer MD, Murali TM, Sobral BW. The landscape of human proteins interacting with viruses and other pathogens. PLoS Pathog. 2008;4:e32. doi: 10.1371/journal.ppat.0040032. 10.1371/journal.ppat.0040032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Irwin B, Aye M, Baldi P, Beliakova-Bethell N, Cheng H, Dou Y, Liou W, Sandmeyer S. Retroviruses and yeast retrotransposons use overlapping sets of host genes. Genome Res. 2005;15:641–654. doi: 10.1101/gr.3739005. 10.1101/gr.3739005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Qian W, He X, Chan E, Xu H, Zhang J. Measuring the evolutionary rate of protein-protein interaction. Proc. Natl Acad. Sci. USA. 2011;108:8725–8730. doi: 10.1073/pnas.1104695108. 10.1073/pnas.1104695108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dixon SJ, Costanzo M, Baryshnikova A, Andrews B, Boone C. Systematic mapping of genetic interaction networks. Annu. Rev. Genet. 2009;43:601–625. doi: 10.1146/annurev.genet.39.073003.114751. 10.1146/annurev.genet.39.073003.114751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.