


Abstract
Maintenance of genomic integrity is essential to ensure normal organismal development and to prevent diseases such as cancer. Nuclear DNA is packaged into chromatin, and thus genome maintenance can be influenced by distinct chromatin environments. In particular, post-translational modifications of histones have emerged as key regulators of genomic integrity. Intense research during the past few years has revealed histone H4 lysine 20 methylation (H4K20me) as critically important for the biological processes that ensure genome integrity, such as DNA damage repair, DNA replication and chromatin compaction. The distinct H4K20 methylation states are mediated by SET8/PR-Set7 that catalyses monomethylation of H4K20, whereas SUV4-20H1 and SUV4-20H2 enzymes mediate further H4K20 methylation to H4K20me2 and H4K20me3. Disruption of these H4K20-specific histone methyltransferases leads to genomic instability, demonstrating the important functions of H4K20 methylation in genome maintenance. In this review, we explain molecular mechanisms underlying these defects and discuss novel ideas for furthering our understanding of genome maintenance in higher eukaryotes.
INTRODUCTION
Cells are equipped with genome maintenance pathways to avoid deleterious aberrations in their genetic information. This is of great importance, as cells are constantly exposed to conditions that can permanently alter the DNA sequence. Common causes of DNA alterations are exposure to ultraviolet radiation from the sun and endogenous metabolic processes, as well as errors during DNA replication. An essential player in the maintenance of genomic integrity is the DNA damage response (DDR), which is a network of interlinked signalling cascades that respond to cellular DNA damage (1). DNA is embedded in chromatin, and it has become increasingly clear that the chromatin environment plays an important role in the DDR. Histone proteins form the core of chromatin and their post-translational modifications (PTMs) are crucial for the regulation of diverse DNA-templated processes, such as DNA repair, DNA replication and mitosis (2–4). In this review, we focus on the regulation and functions of histone H4 lysine 20 methylation, which is emerging as a crucial modification to ensure genomic integrity both in the absence and presence of genotoxic stress.
CELL CYCLE-DEPENDENT METHYLATION OF HISTONE H4
Methylation of histone H4 was one of the first histone post-translational modifications to be discovered, nearly half a century ago, but it was not until recently that the catalysing enzymes were identified (5–7). In mammalian cells, the majority of histone H4 methylation is detected in the N-terminal tail on lysine 20 (H4K20). This methylation mark is evolutionarily conserved from yeast to human (7,8) and exists in three distinct states as mono-, di- and trimethylation. Each of these states results in distinct biological outputs: Mono- (H4K20me1) and dimethylated H4K20 (H4K20me2) are involved in DNA replication and DNA damage repair, whereas trimethylated H4K20 (H4K20me3) is a hallmark of silenced heterochromatic regions. In mammals, the different H4K20 methylation states are established through specific enzymes. There is only one known monomethyltransferase, SET8 (also known as PR-SET7) (5,6) and several di- and tri-methyltransferases of which SUV4-20H1 and SUV4-20H2 enzymes mediate the vast majority of these two modifications (7,9).
Mass spectrometry studies in proliferating cells have identified H4K20me2 as the most abundant methylation state on histone H4, found on ∼80% of all histone H4 molecules (9,10). In these proliferating cells, only very low levels of H4K20me1 and me3 are present together with a small fraction of unmodified histone H4. These measurements reflect the merge of a highly dynamic cell cycle regulation of the various H4K20 methylation states. Most strikingly are the fluctuations found for H4K20me1. This modification is declining during G1 phase, resulting in a very low level of H4K20me1 in the beginning of S phase. It accumulates during S and G2 phases resulting in a peak in M phase (10,11). This cell cycle-dependent regulation is mirrored in the abundance of the SET8 enzyme. In G1 and S phase, proteolytic degradation keeps SET8 at a low level, whereas the enzyme is stabilized in G2 and M phase, resulting in elevated levels of H4K20me1 (11–16).
Both H4K20me2 and me3 show less dramatic cell cycle differences and the modifications are present throughout the cell cycle (10). In mouse embryonic fibroblast cells (MEFs), SUV4-20H1 seems to have a preference to induce H4K20me2, whereas SUV4-20H2 mainly is responsible for H4K20me3. In vitro histone methyltransferase (HMT) assays indicate that both enzymes prefer H4K20me1 as substrate (G. S. and S. Dambacher, unpublished data) and SUV4-20H enzymes can, in vivo, indeed convert SET8-mediated H4K20me1 to H4K20me2 and me3. Consequently, loss of both SUV4-20H enzymes leads to strongly elevated levels of H4K20me1 (9).
The distinct localization of H4K20me2 and me3 suggest that these two methylation states have different functions. H4K20me3 is highly enriched at pericentric heterochromatin, telomeres, imprinted regions and repetitive elements, suggesting that this modification is involved in transcriptional silencing (7,17,18). H4K20me2, in contrast, is broadly distributed across the genome (7). Chromatin immunoprecipitation with massively parallel DNA sequencing (ChIP-seq) profiles are not yet available; however, the high abundance of this modification in bulk chromatin suggests that H4K20me2 is not specifically involved in transcriptional regulation and rather important for general chromatin-mediated processes.
The establishment of the H4K20 methylation states is not fully elucidated. We postulate that in proliferating cells, establishment of the different H4K20 methylation states follows the model shown in Figure 1: Mitotic chromosomes are not only highly enriched in H4K20me1 along chromosome arms but also contain H4K20me2. H4K20me3 nucleosomes are specifically enriched in pericentric heterochromatin of mitotic chromosomes. After mitosis, H4K20me1 levels decrease, presumably owing to conversion to H4K20me2 and me3 by SUV4-20H enzymes. In heterochromatin, HP1 is a targeting factor for SUV4-20H2 to mediate H4K20me3. It is currently unclear if targeting mechanisms for SUV4-20H1 exist, which would bring this enzyme to euchromatic regions. Early in G1 phase, the majority of H4K20me1 is lost owing to processive methylation to the H4K20me2 and me3 states; however, a minor fraction of this modification might be protected from conversion. In S phase, SET8 is kept at low levels, and newly incorporated histones therefore carry very low levels of H4K20 methylation. In late S/G2 phase, when SET8 levels increase, unmodified H4K20 becomes mono-methylated by SET8. Notably, this newly established modification is protected from further conversion by SUV4-20H enzymes until cells have passed through mitosis. As SUV4-20H enzymes are not degraded during G2/M phase, we hypothesize that there must be mechanisms that shield H4K20me1 from the SUV4-20H enzymes. An important protein in this context appears to be host cell factor 1 (HCF1) as knockdown of HCF1 leads to elevated levels of H4K20me2 on mitotic chromosomes (19). How HCF1 protects H4K20me1 from conversion by SUV4-20H enzymes is still unresolved. In addition, SUV4-20H enzymatic activity could be suppressed in G2/M phase, for example by post-translational modifications; however, such negative regulation has currently not been established and remains to be determined.
Figure 1.

Cell cycle regulated establishment of the different H4K20 methylation states. Resting cells in G1 or G0 phase have high levels of H4K20me3 at heterochromatic regions and carry H4K20me2 throughout the genome. H4K20me1 is restricted to specific genes. When cells enter S phase, new histone H4 molecules are incorporated, which lack H4K20 methylation (marked in white). As SET8 is kept at low levels, very little H4K20me1 is added during S phase. Towards the end of S phase and in G2, SET8 is stabilized and establishes H4K20me1 at nearly all new histone H4 molecules. This high level of H4K20me1 is preserved during mitosis and is probably protected (shielded) from conversion into H4K20me2 or me3 via currently unknown mechanisms. Directly after mitosis, in early G1, most of the H4K20me1 is then converted to H4K20me2 and me3 by SUV4-20H enzymes.
Histone lysine methylation is a relatively stable modification with a generally low turnover when compared with acetylation or phosphorylation. At present, it is unclear whether H4K20me2 and H4K20me3 can be actively removed. H4K20me1, in contrast, can be eliminated by the histone demethylase PHF8 (20). A functional consequence of this was suggested to be the removal of inhibitory H4K20me1 on a subset of E2F1-regulated gene promoters, which can support cell cycle progression past the G1-S transition (21). However, it is currently unclear whether the demethylation strategy is used under a variety of physiological settings. Alternatively, H4K20me1 can be removed by conversion into higher methylation states, as occurs during the cell cycle (see Figure 1).
Proper regulation of these dynamic and diverse fluctuations in H4K20 methylation throughout the cell cycle is of great importance to preserve cellular homeostasis. The responsible HMTs are therefore also emerging as key players in genomic maintenance. In the following sections, we will discuss the importance of the different H4K20 methylation states and the role of the H4K20 methyltransferases in maintenance of genomic integrity.
HISTONE H4 METHYLTRANSFERASES PLAY IMPORTANT ROLES TO ENSURE GENOMIC INTEGRITY
The link between histone H4 HMTs and genomic maintenance became evident when these enzymes were depleted by siRNA in human cell lines and removed genetically in experimental animal models. Genetic ablation of SET8 results in lethality in both fly (22) and mouse (11,23). RNA interference of SET8 also has severe consequences for the cells resulting in DNA double-strand breaks (DSBs), activation of DNA damage checkpoints, defective cell cycle progression and reduced cell proliferation (11,14,15,22,24). The pivotal role of SET8 in cellular homeostasis is underlined by the fact that massive DNA DSBs and checkpoint activation is observed very rapidly after depletion of SET8 (14).
The SUV4-20H methyltransferases have also been genetically ablated in the mouse (9). The phenotypic consequences are milder than after SET8 ablation; however, Suv4-20h double knock-out (DKO) mice show developmental delay and die perinatally (9). The exact cause of death is unclear; underdevelopment of the lung might be a contributing factor (G. S., unpublished data). MEFs lacking Suv4-20h enzymes are characterized by slow growth rates and early onset of senescence (9). This phenotype can be partially rescued by culturing the cells under low oxygen conditions, suggesting that oxidative stress is not well tolerated in Suv4-20h DKO cells. Altogether, these observations support the importance of histone H4K20 HMTs in maintenance of genomic stability.
EVIDENCE FOR CHROMATIN STRUCTURE REGULATION BY HISTONE H4 METHYLATION
The functional role of the H4 HMTs in genomic maintenance appears to be multifaceted, as the phenotypic outcome of the genetically modified animals differs. One of the key processes in genomic maintenance is modulation of chromatin compaction both in mitosis and during interphase. SET8 has been linked with regulation of chromatin structure, as improper mitotic chromosome condensation is observed in both SET8 knock-out (KO) flies and mice (11,22). The influence on chromatin structure is not restricted to mitosis, as MEFs from SET8 KO mice generally have a less compact chromatin structure when compared with WT (11). In addition, cellular studies demonstrate that ectopic expression of catalytically active SET8 results in a more compact chromatin structure in interphase cells (13,25). A recent study reported decreased chromatin compaction in interphase cells on ectopic expression of SET8 (12). This was suggested to be a consequence of increased H4K20me3 on specific histone gene promoters followed by transcriptional repression, loss of histone proteins and thereby chromatin decompaction (12). Although currently unresolved, these differences could be attributed to the duration of the experiments and/or the degree of over-expression of SET8. Future genetic knock-in studies are likely to shed light on this issue.
Chromatin undergoes a high degree of compaction during G2 and M phase to prepare for division. Based on the aforementioned observations and the cell cycle regulated nature of both the mark and the protein, we hypothesize that SET8 plays an important role in chromatin structure regulation. Such a role most likely involves monomethylated H4K20, as the compaction is dependent on the SET activity (13,25). The role of H4K20me1 in chromatin structure regulation can be multifaceted: the modification has previously been shown to function as a binding platform for different chromatin compaction factors such as Lethal 3 malignant brain tumour 1 (L3MBTL1) in interphase (26), as well as CAP-D3 and CAP-G2, two members of the mitotically operating condensin II complex (13,21).
Mammalian L3MBTL1 is a candidate tumour suppressor and the phenotypic alterations on depletion of the protein are rather similar to SET8 depletion, including DNA breaks, activation of the DDR, compromised replication fork progression and genomic instability (27), suggesting that these two proteins may be functionally linked. However, the L3MBTL1 KO mouse did not present with the aforementioned phenotypic alterations (28). This could be owing to compensatory mechanisms from within the L3MBTL family or from other chromatin compaction proteins, which is an issue that requires further clarification. In addition, the tumour cell line used by Gurvich et al. could be compromised in other ways and therefore show increased sensitivity to L3MBTL1 depletion.
Methylation of the H4K20 residue may also by itself provide structural support for the chromatin framework. Lu et al. (29) recently found that H4K20me2 and especially H4K20me3 enhance chromatin folding in in vitro nucleosomal array studies. The progressive methylation of K20 on newly synthesized H4 by SET8 (10) supports increased levels of H4K20me2 and me3, thereby directing compaction of the chromatin framework. It is currently difficult to validate the role of H4K20 in vivo in these processes, as it is technically difficult to create mammalian H4K20 KO cells for the required analysis. However, given that SET8-mediated H4K20me1 is required for H4K20me3, we consider it very likely that SET8 regulation of chromatin compaction can be mediated by H4K20me3.
The central role for histone H4 methylation in chromatin modulation and the importance of its tight regulation is further supported by recent studies perturbing the cell cycle oscillating SET8 levels. A number of studies have established that the CRL4(Cdt2) complex ubiquitylates and degrades chromatin-bound SET8 via a Proliferating cell nuclear antigen (PCNA)-interacting motif (PIP-box) in S phase. Inactivation of the PIP-box by mutagenesis results in a non-degradable form of SET8 (SET8ΔPIP), and ectopic expression of this has severe consequences for the cells. These include premature chromatin condensation, cell cycle progression failure and accumulation of cells in G2; re-replication; activation of the DDR; and cell death (12,13,16,25,30). The consequences of elevated SET8 levels are likely linked, and so far, most phenotypes are observed when elevating both wild-type and the stable PIP-box mutated version. Expression of both versions increases the level of H4K20me1 and induces a more compact chromatin structure. Cells expressing the stable PIP-box mutated version can proliferate through S phase but arrest in G2 (12,25), suggesting that cells are excluded from mitotic entry if their chromatin structure is too compact. The untimely compaction may be partly mediated via early recruitment of chromatin compaction factors during S phase (13). This highlights the importance of keeping SET8 levels tightly regulated to avoid untimely chromatin compaction, which may trigger the DDR and lead to cell death.
HISTONE H4 METHYLTRANSFERASES IN DNA REPLICATION
The H4K20 methylating enzymes are also emerging as important regulators of DNA replication aspects. DNA replication is initiated from origins of replication (ORIs) that are loaded with replication factors during mitosis and early G1. Direct involvement of H4 methyltransferases in the loading of replication origins is supported by three recent reports. These studies implicate both SET8 and Suv4-20h enzymes in DNA replication, as H4K20me2 and me3 seem to play important roles for recruitment of the Orc complex to replication origins (31–33). In addition, absence of H4K20me2 in Suv4-20h morpholino-treated zebrafish embryos leads to impaired development, a phenotype that is also found in Orc1 knockdown embryos (34). In the same line of argumentation, cells derived from Suv4-20h KO mice have a delayed S-phase entry, and the mice show developmental delay (9), which might be connected with a partial defect in DNA replication initiation pathways. SET8 has also been suggested to play a role in DNA replication. Expression of stable versions of SET8 promotes re-replication of the DNA, which suggests a potential role for SET8 as a positive regulator of origin licensing (12,30).
Disruption of SET8 also affects DNA replication; however, the phenotypes are different and more severe when compared with the S phase delay observed in Suv4-20h KO mice, suggesting that the distinct H4K20 methylation states regulate different aspects of DNA replication. Removal of SET8 methyltransferase activity leads to massive DNA damage during replication, and the observed genomic instability is largely dependent on active replication (14). Suppression of DNA replication, either with inhibitors blocking fork progression or RNA interference to replication initiation factors, can suppress the majority of DNA breakage occurring after SET8 depletion. Intriguingly, SET8 depletion has not been shown to affect loading of replication factors such as ORC complex members. In addition, depletion of replication factors such as components of the ORC complex does not result in rapid and marked loss of genomic integrity as observed following SET8 ablation. We therefore speculate that SET8 is involved in a more general regulation of chromatin structure around the ORIs that will support replication fork progression, whereas SUV4-20H-mediated me2/me3 appears to be more directly involved in Orc recruitment (Figure 2A).
Figure 2.

Role of histone H4 HMTs during replication. (A) H4K20me1 is added during G2 and M phase and shielded from conversion until G1 phase. In preparation for the following S phase, members of the Orc complex are recruited to ORI. Part of this involves binding between Orc1 and H4K20me2. We speculate that H4K20me1 contributes to a favourable chromatin structure around the ORI that support fork progression. (B) We hypothesise that SET8 directly supports the replication fork structure, either by itself or by methylation of one or more proteins that provide structural or functional support at the fork. Loss of SET8 results in dysfunctional replication and potentially exposure of DNA structures that are targeted and cut by endonucleases, thereby generating DSBs.
The defects in initiation of DNA replication in absence of SET8 or SUV4-20H enzymes are still not fully understood. In particular, the chromatin status of replication origins needs further investigation, as H4K20me1 and me2 were both detected on the same origin in U2OS cells in two independent studies (30,34). A possible explanation for these contrasting findings is that H4K20 methylation states are dynamically regulated at replication origins, and both H4K20me1 and H4K20me2 intermediate states can be detected. However, owing to the complexity of H4K20 methylation, more coherent studies, which would investigate all three H4K20 methylation states with validated antibodies, should help resolve this.
It is not understood how DNA replication issues on SET8 depletion result in DNA DSBs. We hypothesize that dysfunctional replication leads to DNA structures that cannot be corrected by the cellular DNA repair pathways in a timely manner. The aberrant and unresolved structures can be targeted by DNA endonucleases to cleave the otherwise strong phosphodiester bonds in the DNA strands. This is reminiscent of the marked endogenous DNA DSB formation following dysfunctional replication, which occurs when the checkpoint kinases CHK1 and WEE1 are inhibited or depleted (35–38). The nature and substrate of these nucleases remains to be determined, which are important future tasks.
Another intriguing aspect associated with DNA replication is the link between SET8 and PCNA. SET8 interacts directly with PCNA through a functional PCNA-interaction motif (14), and endogenous SET8 can be detected at sites of active DNA synthesis when cells are treated briefly with proteasome inhibitors (15). However, SET8 accumulation could be owing to the role of PCNA as a scaffold in SET8 degradation. Even if present at very low levels, SET8 can potentially perform important functions in the vicinity of the replication fork (Figure 2B). In support of this, it was very recently suggested that SET8 methylates PCNA directly to support FEN1-mediated processing of Okazaki fragments during DNA replication (39). Reduced Okazaki fragment processing could lead to loss of genomic integrity in the absence of SET8.
The DNA lesions around the replication fork created as a consequence of SET8 loss is sensed and targeted for repair by the Homologous recombination (HR) machinery. Proteins involved in HR repair, such as RPA and RAD51, are recruited to the fork area (14) in an attempt to repair defective structures. Remarkably, a significant fraction of the lesions generated on loss of SET8 can be suppressed by downregulation of key HR factors such as RAD51 and BRCA2 (14). Individual suppression of RAD51 or BRCA2 is distinct from the loss of SET8, as the loss of genomic integrity associated with RAD51 and BRCA2 depletion is marginal within the first 2 days after depletion. In contrast, SET8 depletion leads to rapid and widespread DNA damage appearing within 24 h. It is therefore possible that the DNA DSBs appearing after SET8 depletion are augmented by HR attempts that fail. HR failure can be owing to a complicated nature of lesions, which will prevent successful repair, or owing to the absence of a repair template in the form of a sister chromatid in early S phase. Depletion of HR proteins such as RAD51 inhibits the repair pathway and prevents the use of a dysfunctional pathway, which will prevent fork collapse.
Altogether, these data suggest a role for SET8 and H4K20 methylation during replication in vicinity of the replication fork or as regulators of the chromatin structure surrounding the ORIs/replication forks.
H4K20 HMTS AND EXOGENOUS DNA DAMAGE
Exogenous DNA damage initiates the DDR that mediates cell cycle arrest, repair and apoptosis depending on the type and amount of lesions. Methylated H4K20 is linked with the DDR in both yeast (8) and mammalian cells (40) as a binding platform for the repair factor 53BP1. The function of 53BP1 is not clear, but it is involved in programmed DNA damage in the immune system, and in response to exogenous DNA damage, it appears to promote end-joining repair and suppress HR (41,42). Depletion or KO of 53BP1 results in genomic instability in the absence of exogenous damage, suggesting a role for 53BP1 in genomic maintenance (43,44). 53BP1 recruitment to sites of DNA damage is complex and involves a large number of proteins as well as histone PTMs (Figure 3) (4,45). Peptide affinity studies suggest that 53BP1 has a high affinity for di- and to some degree mono-methylated H4K20 peptides (40). However, it has been difficult to provide in vivo evidence for the specificity of this binding. This is in part owing to the great abundance of dimethylated H4K20 in cells, which makes it difficult to prove changes in the H4K20me2 level at sites of DNA damage.
Figure 3.
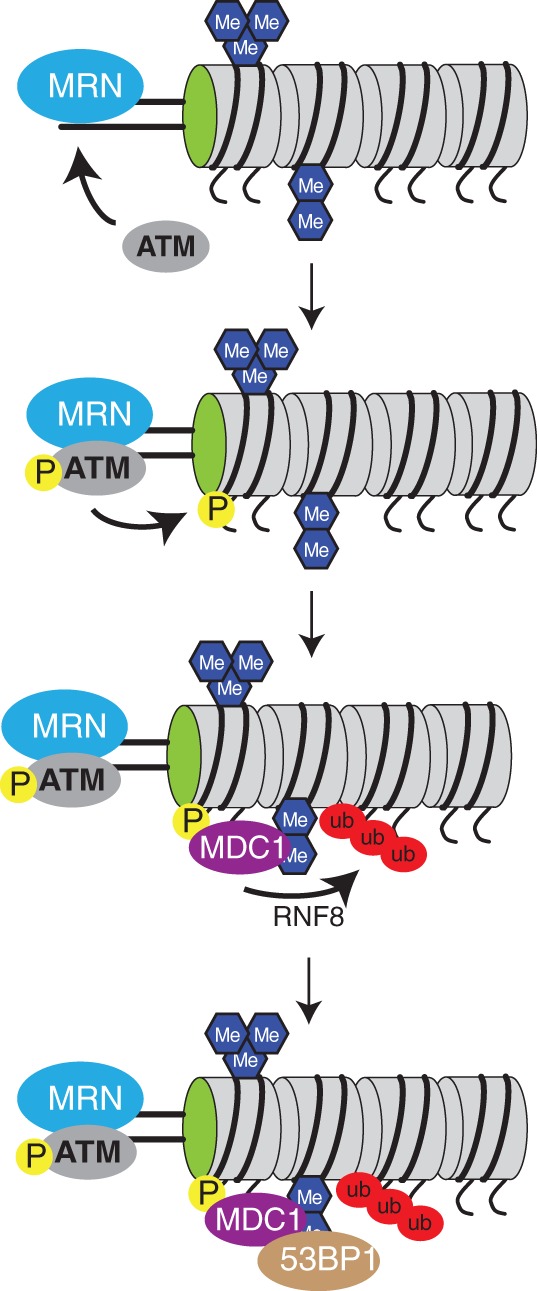
Recruitment of 53BP1 to DNA damage sites through chromatin. DSBs are recognized by the MRE11/RAD50/NBS1 (MRN) complex, which binds open DNA ends. Stable binding of the MRN complex leads to autophosphorylation of the ATM kinase, which then induces phosphorylation of the histone variant H2A.X in the vicinity of the DNA break. Phospho-H2A.X (γH2A.X) allows for accumulation of MDC1 and its partner protein RNF8, which in turn, establishes poly-ubiquitinylation of histones at the break site. 53BP1 is then stably recruited through multiple interactions, including binding to MDC1 and H4K20me2.
SUV4-20H1/2 ARE INVOLVED IN THE RESPONSE TO EXOGENOUS STRESS
Suv4-20h DKO mice represent a good system to investigate the function of H4K20me2 in 53BP1 recruitment and DNA damage repair. After DNA damage, 53BP1 and other DNA damage repair proteins accumulate at DSB sites within a very short time frame. Although 53BP1 can accumulate at DBS’s in Suv4-20h DKO cells, the formation of 53BP1 foci is significantly delayed (9). This suggests that H4K20me2 is not the only signal for 53BP1 recruitment to DSB’s. In this context, it is important to mention that 53BP1 is a large protein, which features interactions with other components of the DNA damage repair pathways, such as γH2A.X, MDC1 and BRCA1 (46–48). These proteins may be recruited independently of H4K20me2 and could facilitate 53BP1 recruitment to damage sites in absence of this modification. Based on the available data, we conclude that H4K20me2 is not primarily responsible for targeting of 53BP1 to damage sites. It rather represents an additional binding interface for 53BP1, which may be necessary for its stable chromatin association. 53BP1 has lower affinity for H4K20me1 and, therefore, in Suv4-20h DKO cells, which have a largely mono-methylated genome, 53BP1 binding to damage sites is less stable, which might explain the delayed repair foci formation. The delayed kinetics of 53BP1 recruitment seems to affect the efficient repair of DNA DSBs, which is demonstrated by elevated numbers of ionizing radiation (IR)-induced chromatid gaps in Suv4-20h DKO MEFs (9). Notably, Suv4-20h DKO cells are sensitive to different DNA damaging agents to a similar extent as 53BP1 mutant cells, suggesting that SUV4-20H enzymes are part of 53BP1-mediated DNA damage repair pathways.
The kinetics of 53BP1 foci formation might be important when cells have to choose a specific repair pathway. Recent reports have shown that 53BP1 may partly suppress resection of DNA ends at sites of DNA DSBs and thereby limit HR repair (41,42). This would reduce the likelihood of choosing HR repair in the presence of 53BP1. In Suv4-20 h DKO cells, the delayed or unstable 53BP1 binding to damage sites may result in higher levels of HR repair (Figure 4). In agreement with this assumption, sister chromatid exchange is elevated in Suv4-20h DKO ES cells (49).
Figure 4.
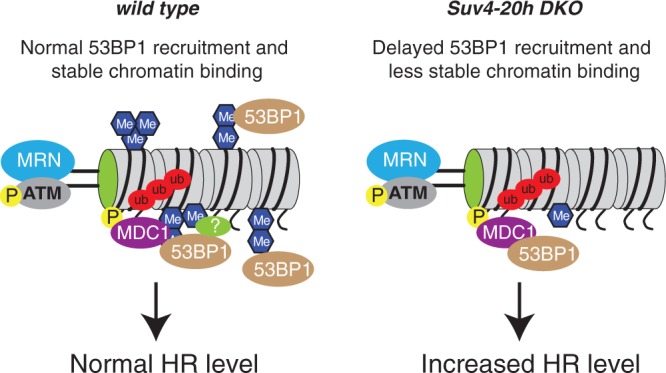
Role of SUV4-20H1/2 in recruitment of 53BP1. In wild-type cells, 53BP1 is stably recruited to DNA break sites through multiple interactions with e.g. MDC1 and H4K20me2. SET8-mediated H4K20me1 at DNA break sites may also contribute to 53BP1 recruitment; however, this link is not yet understood at the molecular level. Tight binding of 53BP1 suppresses long range resection of the DNA ends and therefore inhibits HR repair. High levels of H4K20me1, which is characteristic of Suv4-20h DKO cells, leads to less stable 53BP1 recruitment at break sites. 53BP1 can therefore not efficiently prevent end resection or inhibit HR repair, leading to elevated HR in SUV4-20H mutant cells.
H4K20me2 is a very abundant modification in fibroblast cells and, on DNA damage induction, no global upregulation of this modification can be detected (50). We therefore hypothesize that even at stochastic DNA damage sites, basal levels of H4K20me2 will be present. Why is 53BP1 not strongly binding to this very abundant modification in absence of DNA damage? One explanation is that H4K20me2 is normally not accessible owing to chromatin compaction and, only at DNA break sites, chromatin would be loose enough to expose this modification. 53BP1 can weakly bind to H4K20me2, but this interaction is not strong enough for stable recruitment and proper support of NHEJ repair. On damage, however, additional signals are accumulating at DNA double strand break sites, such as, e.g. phosphorylation of H2A.X and recruitment of MDC1, which generates additional affinity sites for 53BP1. We hypothesize that only synergistic interactions with these multiple signals will provide enough affinity for stable recruitment of 53BP1 to damage sites. Interestingly, very recent data suggest that 53BP1 recruitment to DNA damage sites may be negatively regulated by JMJD2A, which binds tightly to H4K20me2, thereby potentially shielding the mark (51). In Suv4-20 h DKO cells, the absence of H4K20me2 may therefore abrogate this shield allowing 53BP1 recruitment to accumulating γH2A.X, MDC1 and BRCA1 after DNA damage.
Recently, the notion of steady levels of H4K20 methylation was challenged by the finding that another putative H4K20 HMTase, MMSET, is recruited to sites of DNA damage (52). The authors of this study even detected an increase in all H4K20 methylation states at Sce-I-induced DSBs. This finding is very surprising, as thorough biochemical studies have demonstrated that the primary target for MMSET enzymatic activity is H3K36 (53). In addition, a very recent study investigated the function of MMSET in 53BP1 recruitment to laser-induced DNA damage (50). The authors could not detect any defects in 53BP1 recruitment in MMSET-deficient cells, and these data indicate that MMSET does not play a general role in DSB repair. Further experiments are therefore necessary to define its functions in particular damage repair pathways.
ROLE OF SET8 IN 53BP1 RECRUITMENT
SET8 may also play a role in 53BP1 function. A recent study suggested DNA damage-mediated binding of 53BP1 to monomethylated H4K20 (54). These data were based on recruitment of ectopically expressed SET8 to sites of laser-induced damage where de novo H4K20 was methylated as a prerequisite for 53BP1 recruitment. This was followed by a temporally delayed PCNA-mediated degradation of SET8 by CRL4(Cdt2) (54). Several other studies support the observation that SET8 is degraded in response to DNA damage, but none have been able to show an increase in H4K20 methylation or recruitment of 53BP1 before the degradation of SET8 (12,13,25,55). On the contrary, 53BP1 was previously shown recruited to sites of IR-induced damage even in the absence of SET8 (14,15). The observed differences could rely on multiple factors; one is the choice of cellular system; Oda and co-workers were using ectopically expressed SET8 compared with the DNA damage-mediated degradation and recruitment of 53BP1 that was performed at endogenous protein level. Depletion of SET8 markedly affects the experimental setup, as this leads to endogenous DNA damage and γH2A.X signalling, which perturbs analysis of the cellular responses to exogenous DNA damage. Second, Oda and co-workers applied laser-induced damage that generates strong, focal lesions, whereas the other studies were performed using IR, ultraviolet radiation or chemical drugs such as hydroxyurea (HU) or Methyl methanesulfonate (MMS), which creates different types of lesions. Finally, Oda and co-workers were using real-time microscopy and assaying recruitment minutes after the lesions had been inflicted, whereas the other studies were assaying effects observed at later time points. In conclusion, it is possible that there is a role for H4K20me1 in 53BP1 recruitment after DNA damage. We hypothesize that H4K20me1 is one of several modified histones/proteins that together constitute a binding platform used for 53BP1 recruitment to chromatin at sites of DNA damage.
IS THERE A FUNCTION FOR SET8 BEYOND 53BP1 RECRUITMENT?
It has recently become evident that SET8 may be more directly involved in the response to exogenous DNA damage (55,56). SET8 is rapidly degraded in response to DNA damage, also mediated by CRL4(Cdt2) ubiquitylation and proteasomal degradation via PCNA tethering of SET8 to chromatin (12,13,25,54). Expression of non-degradable SET8 however showed a sustained chromatin compaction in response to exogenous DNA damage (13,25). Modification and loosening of the chromatin structure in areas with DNA lesions is crucial for a proper repair (45), and it is possible that SET8 is degraded in response to DNA damage to allow decompaction and repair.
A major biological process regulated in response to exogenous DNA damage is transcription. Proper transcription requires opening of the chromatin structure and binding of transcription factors in promoter regions. The tumour suppressor p53 is a key DNA damage-induced transcription factor, and it was recently identified both as a SET8 substrate and a binding partner for L3MBTL1 (55,56). The chromatin compactor L3MBTL1 contains three MBT domains and uses the middle domain to bind H4K20me1 to compact chromatin (26). L3MBTL1 also uses this domain to bind methylated p53 (p53K382me1), a modification, which is catalysed by SET8 (55,56). The methylation of p53 on lysine 382 by SET8 is suggested to inhibit transcription of a subset of target genes, such as the CDK inhibitors p21 and PUMA. Under non-stressed conditions, this methylation can contribute to inhibit untimely transcription of p21 and prevent unscheduled cell cycle arrest (56). Recent data suggest that L3MBTL1 interacts with chromatin-bound p53K382me1, compacts the promoter regions of target genes and inhibits their transcription (55). DNA damage-induced degradation of SET8 would reduce the level of p53K382me1 and thereby also the level of chromatin-bound L3MBTL1, resulting in de-compaction of target gene promoters. Untimely transcription of p21 and unscheduled cell cycle arrest under non-stressed conditions challenges the proliferative capacity of the cell. This could also potentially cause unnecessary checkpoint activation and a DDR to eliminate the apparent, but non-existing obstruction.
SUMMARY AND CONCLUSIONS
During the past decade, there has been considerable progress in our understanding of the roles of histone H4 methylation and the catalysing enzymes. Cellular synchronization studies and mass spectrometry analysis have provided evidence for the highly cell cycle-regulated nature of H4K20, where especially mono- and trimethylation are fluctuating. We put forward a model suggesting collaboration between SET8 and the SUV4-20H methyltransferases in regulation of the methylation status of H4K20 on chromatin. This is crucial, as disruption of the dynamic fluctuations in H4K20 methylation during the cell cycle has severe consequences for the cells. Altogether, we propose a model where tight regulation of H4K20 HMTs and their marks are essential to maintain proper chromatin structure regulation, secure DNA replication, support the DDR and thereby maintain genomic stability.
Future in-depth analyses are necessary to better understand the molecular mechanisms behind these defects. This will require the development of novel tools that should allow investigation of SET8 and SUV4-20H enzymes at endogenous expression levels and in a variety of assays. Both enzymes are expressed at very low levels, and the development of specific antibodies, which would recognize these proteins in ChIP or immunofluorescence applications, has so far failed. Another important point to consider is that the three different H4K20 methylation states appear to have very different roles, and that these functions might also be modulated by additional modifications or proteins that occur in close vicinity. One example is binding of 53BP1 to a combinatorial signal of H4K20me2 in the context of γH2A.X and MDC1. It will be very interesting to identify combinatorial signals, which occur in the context of the other H4K20 methylation states and which could provide interaction interfaces for specific binding proteins that mediate the different downstream functions of H4K20 methylation.
FUNDING
The Lundbeckfoundation, The Danish Cancer Society, and The Danish Medical Research Council (to S.J. and C.S.S.); Research in the laboratory of G.S. is financed by the BMBF, SFB-TR5 Chromatin, SFB 684 and SPP1356. Funding for open access charge: Core funding.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors apologize to all authors whose work we could not cite owing to space limitations
REFERENCES
- 1.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461:1071–1078. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Beck DB, Oda H, Shen SS, Reinberg D. PR-Set7 and H4K20me1: at the crossroads of genome integrity, cell cycle, chromosome condensation, and transcription. Genes Dev. 2012;26:325–337. doi: 10.1101/gad.177444.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Lukas J, Lukas C, Bartek J. More than just a focus: the chromatin response to DNA damage and its role in genome integrity maintenance. Nat. Cell Biol. 2011;13:1161–1169. doi: 10.1038/ncb2344. [DOI] [PubMed] [Google Scholar]
- 5.Fang J, Feng Q, Ketel CS, Wang H, Cao R, Xia L, Erdjument-Bromage H, Tempst P, Simon JA, Zhang Y. Purification and functional characterization of SET8, a nucleosomal histone H4-lysine 20-specific methyltransferase. Curr. Biol. 2002;12:1086–1099. doi: 10.1016/s0960-9822(02)00924-7. [DOI] [PubMed] [Google Scholar]
- 6.Nishioka K, Rice JC, Sarma K, Erdjument-Bromage H, Werner J, Wang Y, Chuikov S, Valenzuela P, Tempst P, Steward R, et al. PR-Set7 is a nucleosome-specific methyltransferase that modifies lysine 20 of histone H4 and is associated with silent chromatin. Mol. Cell. 2002;9:1201–1213. doi: 10.1016/s1097-2765(02)00548-8. [DOI] [PubMed] [Google Scholar]
- 7.Schotta G, Lachner M, Sarma K, Ebert A, Sengupta R, Reuter G, Reinberg D, Jenuwein T. A silencing pathway to induce H3-K9 and H4-K20 trimethylation at constitutive heterochromatin. Genes Dev. 2004;18:1251–1262. doi: 10.1101/gad.300704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanders SL, Portoso M, Mata J, Bahler J, Allshire RC, Kouzarides T. Methylation of histone H4 lysine 20 controls recruitment of Crb2 to sites of DNA damage. Cell. 2004;119:603–614. doi: 10.1016/j.cell.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 9.Schotta G, Sengupta R, Kubicek S, Malin S, Kauer M, Callen E, Celeste A, Pagani M, Opravil S, De La Rosa-Velazquez IA, et al. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 2008;22:2048–2061. doi: 10.1101/gad.476008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pesavento JJ, Yang H, Kelleher NL, Mizzen CA. Certain and progressive methylation of histone H4 at lysine 20 during the cell cycle. Mol. Cell. Biol. 2008;28:468–486. doi: 10.1128/MCB.01517-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oda H, Okamoto I, Murphy N, Chu J, Price SM, Shen MM, Torres-Padilla ME, Heard E, Reinberg D. Monomethylation of histone H4-lysine 20 is involved in chromosome structure and stability and is essential for mouse development. Mol. Cell. Biol. 2009;29:2278–2295. doi: 10.1128/MCB.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Abbas T, Shibata E, Park J, Jha S, Karnani N, Dutta A. CRL4(Cdt2) regulates cell proliferation and histone gene expression by targeting PR-Set7/Set8 for degradation. Mol. Cell. 2010;40:9–21. doi: 10.1016/j.molcel.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Centore RC, Havens CG, Manning AL, Li JM, Flynn RL, Tse A, Jin J, Dyson NJ, Walter JC, Zou L. CRL4(Cdt2)-mediated destruction of the histone methyltransferase Set8 prevents premature chromatin compaction in S phase. Mol. Cell. 2010;40:22–33. doi: 10.1016/j.molcel.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jørgensen S, Elvers I, Trelle MB, Menzel T, Eskildsen M, Jensen ON, Helleday T, Helin K, Sørensen CS. The histone methyltransferase SET8 is required for S-phase progression. J. Cell Biol. 2007;179:1337–1345. doi: 10.1083/jcb.200706150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tardat M, Murr R, Herceg Z, Sardet C, Julien E. PR-Set7-dependent lysine methylation ensures genome replication and stability through S phase. J. Cell Biol. 2007;179:1413–1426. doi: 10.1083/jcb.200706179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu S, Wang W, Kong X, Congdon LM, Yokomori K, Kirschner MW, Rice JC. Dynamic regulation of the PR-Set7 histone methyltransferase is required for normal cell cycle progression. Genes Dev. 2010;24:2531–2542. doi: 10.1101/gad.1984210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalo S, Garcia-Cao M, Fraga MF, Schotta G, Peters AH, Cotter SE, Eguia R, Dean DC, Esteller M, Jenuwein T, et al. Role of the RB1 family in stabilizing histone methylation at constitutive heterochromatin. Nat. Cell Biol. 2005;7:420–428. doi: 10.1038/ncb1235. [DOI] [PubMed] [Google Scholar]
- 18.Regha K, Sloane MA, Huang R, Pauler FM, Warczok KE, Melikant B, Radolf M, Martens JH, Schotta G, Jenuwein T, et al. Active and repressive chromatin are interspersed without spreading in an imprinted gene cluster in the mammalian genome. Mol. Cell. 2007;27:353–366. doi: 10.1016/j.molcel.2007.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Julien E, Herr W. A switch in mitotic histone H4 lysine 20 methylation status is linked to M phase defects upon loss of HCF-1. Mol. Cell. 2004;14:713–725. doi: 10.1016/j.molcel.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Qi HH, Sarkissian M, Hu GQ, Wang Z, Bhattacharjee A, Gordon DB, Gonzales M, Lan F, Ongusaha PP, Huarte M, et al. Histone H4K20/H3K9 demethylase PHF8 regulates zebrafish brain and craniofacial development. Nature. 2010;466:503–507. doi: 10.1038/nature09261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu W, Tanasa B, Tyurina OV, Zhou TY, Gassmann R, Liu WT, Ohgi KA, Benner C, Garcia-Bassets I, Aggarwal AK, et al. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508–512. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakaguchi A, Steward R. Aberrant monomethylation of histone H4 lysine 20 activates the DNA damage checkpoint in Drosophila melanogaster. J. Cell Biol. 2007;176:155–162. doi: 10.1083/jcb.200607178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huen MS, Sy SM, van Deursen JM, Chen J. Direct interaction between SET8 and proliferating cell nuclear antigen couples H4-K20 methylation with DNA replication. J. Biol. Chem. 2008;283:11073–11077. doi: 10.1074/jbc.C700242200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Houston SI, McManus KJ, Adams MM, Sims JK, Carpenter PB, Hendzel MJ, Rice JC. Catalytic function of the PR-Set7 histone H4 lysine 20 monomethyltransferase is essential for mitotic entry and genomic stability. J. Biol. Chem. 2008;283:19478–19488. doi: 10.1074/jbc.M710579200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jørgensen S, Eskildsen M, Fugger K, Hansen L, Larsen MS, Kousholt AN, Syljuåsen RG, Trelle MB, Jensen ON, Helin K, et al. SET8 is degraded via PCNA-coupled CRL4(CDT2) ubiquitylation in S phase and after UV irradiation. J. Cell Biol. 2011;192:43–54. doi: 10.1083/jcb.201009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trojer P, Li G, Sims RJ, 3rd, Vaquero A, Kalakonda N, Boccuni P, Lee D, Erdjument-Bromage H, Tempst P, Nimer SD, et al. L3MBTL1, a histone-methylation-dependent chromatin lock. Cell. 2007;129:915–928. doi: 10.1016/j.cell.2007.03.048. [DOI] [PubMed] [Google Scholar]
- 27.Gurvich N, Perna F, Farina A, Voza F, Menendez S, Hurwitz J, Nimer SD. L3MBTL1 polycomb protein, a candidate tumor suppressor in del(20q12) myeloid disorders, is essential for genome stability. Proc. Natl Acad. Sci. USA. 2010;107:22552–22557. doi: 10.1073/pnas.1017092108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Qin J, Van Buren D, Huang HS, Zhong L, Mostoslavsky R, Akbarian S, Hock H. Chromatin protein L3MBTL1 is dispensable for development and tumor suppression in mice. J. Biol. Chem. 2010;285:27767–27775. doi: 10.1074/jbc.M110.115410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu X, Simon MD, Chodaparambil JV, Hansen JC, Shokat KM, Luger K. The effect of H3K79 dimethylation and H4K20 trimethylation on nucleosome and chromatin structure. Nat. Struct. Mol. Biol. 2008;15:1122–1124. doi: 10.1038/nsmb.1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tardat M, Brustel J, Kirsh O, Lefevbre C, Callanan M, Sardet C, Julien E. The histone H4 Lys 20 methyltransferase PR-Set7 regulates replication origins in mammalian cells. Nat. Cell Biol. 2010;12:1086–1093. doi: 10.1038/ncb2113. [DOI] [PubMed] [Google Scholar]
- 31.Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, Kouzarides T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell. 2010;143:470–484. doi: 10.1016/j.cell.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Beck DB, Burton A, Oda H, Ziegler-Birling C, Torres-Padilla ME, Reinberg D. The role of PR-Set7 in replication licensing depends on Suv4-20h. Genes Dev. 2012;26:2580–2589. doi: 10.1101/gad.195636.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vermeulen M, Eberl HC, Matarese F, Marks H, Denissov S, Butter F, Lee KK, Olsen JV, Hyman AA, Stunnenberg HG, et al. Quantitative interaction proteomics and genome-wide profiling of epigenetic histone marks and their readers. Cell. 2010;142:967–980. doi: 10.1016/j.cell.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 34.Kuo AJ, Song J, Cheung P, Ishibe-Murakami S, Yamazoe S, Chen JK, Patel DJ, Gozani O. The BAH domain of ORC1 links H4K20me2 to DNA replication licensing and Meier-Gorlin syndrome. Nature. 2012;484:115–119. doi: 10.1038/nature10956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Beck H, Nahse-Kumpf V, Larsen MS, O'Hanlon KA, Patzke S, Holmberg C, Mejlvang J, Groth A, Nielsen O, Syljuåsen RG, et al. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Mol. Cell. Biol. 2012;32:4226–4236. doi: 10.1128/MCB.00412-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dominguez-Kelly R, Martin Y, Koundrioukoff S, Tanenbaum ME, Smits VA, Medema RH, Debatisse M, Freire R. Wee1 controls genomic stability during replication by regulating the Mus81-Eme1 endonuclease. J. Cell Biol. 2011;194:567–579. doi: 10.1083/jcb.201101047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forment JV, Blasius M, Guerini I, Jackson SP. Structure-specific DNA endonuclease Mus81/Eme1 generates DNA damage caused by Chk1 inactivation. PLoS One. 2011;6:e23517. doi: 10.1371/journal.pone.0023517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sørensen CS, Syljuåsen RG. Safeguarding genome integrity: the checkpoint kinases ATR, CHK1 and WEE1 restrain CDK activity during normal DNA replication. Nucleic Acids Res. 2012;40:477–486. doi: 10.1093/nar/gkr697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Takawa M, Cho HS, Hayami S, Toyokawa G, Kogure M, Yamane Y, Iwai Y, Maejima K, Ueda K, Masuda A, et al. Histone lysine methyltransferase SETD8 promotes carcinogenesis by deregulating PCNA expression. Cancer Res. 2012;72:3217–3227. doi: 10.1158/0008-5472.CAN-11-3701. [DOI] [PubMed] [Google Scholar]
- 40.Botuyan MV, Lee J, Ward IM, Kim JE, Thompson JR, Chen J, Mer G. Structural basis for the methylation state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in DNA repair. Cell. 2006;127:1361–1373. doi: 10.1016/j.cell.2006.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat. Struct. Mol. Biol. 2010;17:688–695. doi: 10.1038/nsmb.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141:243–254. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fernandez-Capetillo O, Chen HT, Celeste A, Ward I, Romanienko PJ, Morales JC, Naka K, Xia Z, Camerini-Otero RD, Motoyama N, et al. DNA damage-induced G2-M checkpoint activation by histone H2AX and 53BP1. Nat. Cell Biol. 2002;4:993–997. doi: 10.1038/ncb884. [DOI] [PubMed] [Google Scholar]
- 44.Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol. Cell. Biol. 2003;23:2556–2563. doi: 10.1128/MCB.23.7.2556-2563.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Deem AK, Li X, Tyler JK. Epigenetic regulation of genomic integrity. Chromosoma. 2012;121:131–151. doi: 10.1007/s00412-011-0358-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eliezer Y, Argaman L, Rhie A, Doherty AJ, Goldberg M. The direct interaction between 53BP1 and MDC1 is required for the recruitment of 53BP1 to sites of damage. J. Biol. Chem. 2009;284:426–435. doi: 10.1074/jbc.M807375200. [DOI] [PubMed] [Google Scholar]
- 47.Wang B, Matsuoka S, Carpenter PB, Elledge SJ. 53BP1, a mediator of the DNA damage checkpoint. Science. 2002;298:1435–1438. doi: 10.1126/science.1076182. [DOI] [PubMed] [Google Scholar]
- 48.Ward IM, Minn K, Jorda KG, Chen J. Accumulation of checkpoint protein 53BP1 at DNA breaks involves its binding to phosphorylated histone H2AX. J. Biol. Chem. 2003;278:19579–19582. doi: 10.1074/jbc.C300117200. [DOI] [PubMed] [Google Scholar]
- 49.Benetti R, Gonzalo S, Jaco I, Schotta G, Klatt P, Jenuwein T, Blasco MA. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J. Cell Biol. 2007;178:925–936. doi: 10.1083/jcb.200703081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hartlerode AJ, Guan Y, Rajendran A, Ura K, Schotta G, Xie A, Shah JV, Scully R. Impact of Histone H4 Lysine 20 Methylation on 53BP1 Responses to Chromosomal Double Strand Breaks. PloS One. 2012;7:e49211. doi: 10.1371/journal.pone.0049211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mallette FA, Mattiroli F, Cui G, Young LC, Hendzel MJ, Mer G, Sixma TK, Richard S. RNF8- and RNF168-dependent degradation of KDM4A/JMJD2A triggers 53BP1 recruitment to DNA damage sites. EMBO J. 2012;31:1865–1878. doi: 10.1038/emboj.2012.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, Bergsagel PL, Wang L, You Z, Lou Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature. 2011;470:124–128. doi: 10.1038/nature09658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kuo AJ, Cheung P, Chen K, Zee BM, Kioi M, Lauring J, Xi Y, Park BH, Shi X, Garcia BA, et al. NSD2 links dimethylation of histone H3 at lysine 36 to oncogenic programming. Mol. Cell. 2011;44:609–620. doi: 10.1016/j.molcel.2011.08.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Oda H, Hubner MR, Beck DB, Vermeulen M, Hurwitz J, Spector DL, Reinberg D. Regulation of the histone H4 monomethylase PR-Set7 by CRL4(Cdt2)-mediated PCNA-dependent degradation during DNA damage. Mol. Cell. 2010;40:364–376. doi: 10.1016/j.molcel.2010.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.West LE, Roy S, Lachmi-Weiner K, Hayashi R, Shi X, Appella E, Kutateladze TG, Gozani O. The MBT repeats of L3MBTL1 link SET8-mediated p53 methylation at lysine 382 to target gene repression. J. Biol. Chem. 2010;285:37725–37732. doi: 10.1074/jbc.M110.139527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, Dutta S, Appella E, Gozani O. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol. Cell. 2007;27:636–646. doi: 10.1016/j.molcel.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]