


Abstract
Spontaneous lytic reactivation of Kaposi’s sarcoma–associated herpesvirus (KSHV) occurs at a low rate in latently infected cells in disease and culture. This suggests imperfect epigenetic maintenance of viral transcription programs, perhaps due to variability in chromatin structure at specific loci across the population of KSHV episomal genomes. To characterize this locus-specific chromatin structural diversity, we used MAPit single-molecule footprinting, which simultaneously maps endogenous CG methylation and accessibility to M.CviPI at GC sites. Diverse chromatin structures were detected at the LANA, RTA and vIL6 promoters. At each locus, chromatin ranged from fully closed to fully open across the population. This diversity has not previously been reported in a virus. Phorbol ester and RTA transgene induction were used to identify chromatin conformations associated with reactivation of lytic transcription, which only a fraction of episomes had. Moreover, certain chromatin conformations correlated with CG methylation patterns at the RTA and vIL6 promoters. This indicated that some of the diverse chromatin conformations at these loci were epigenetically distinct. Finally, by comparing chromatin structures from a cell line infected with constitutively latent virus, we identified products of lytic replication. Our findings show that epigenetic drift can restrict viral propagation by chromatin compaction at latent and lytic promoters.
INTRODUCTION
Kaposi’s sarcoma–associated herpesvirus (KSHV) infection causes sarcoma and lymphoproliferative disorders in immunocompromised individuals [reviewed in (1)]. In the latent phase, KSHV genomes reside within the nucleus as multicopy circular extrachromosomal elements or episomes. Both micrococcal nuclease cleavage patterns and immunoprecipitation of histone proteins demonstrate that episomal DNA is incorporated into nucleosomes (2–4). Lytic reactivation, thought to be triggered by multiple cellular stress pathways in vivo, is associated with changes in chromatin structure (3,4). Furthermore, histone deacetylase and DNA methyltransferase (DNMT) inhibitors can reactivate the lytic cycle in latently infected cell culture, as can phorbol esters, e.g. 12-O-tetradecanoylphorbol-13-acetate (TPA), which are protein kinase C agonists [reviewed in (5)]. Thus, episomal chromatin contributes to maintenance of the latent state and the repression of lytic transcription.
Chromatin intrinsically regulates gene expression, as many transcription factors exhibit reduced access to binding sites incorporated into nucleosomes [reviewed in (6)]. Furthermore, chromatin components often have post-synthesis chemical modifications (DNA methylation and acetylation, methylation, etc. of histones) that can activate or repress transcription locally, as well as recruit their own modifying enzymes. Chromatin modifications can thus constitute a self-perpetuating state. Such states are termed epigenetic if maintained throughout the cell cycle, as in the case of DNA methylation [reviewed in (7)]. Epigenetics underpin the differentiation of isogenic cells during development, as well as inactivation of one of two X chromosomes in female mammals. Imperfect maintenance of chromatin states, i.e. epigenetic drift, may contribute to aging and cancer [reviewed in (8)].
Epigenetic drift could be important for viral biology as well. DNA methylation gradually builds over much of the KSHV genome after de novo infection (3), indicating that epigenetic changes occur. However, the phenotypic consequences of this drift are unknown. Generally, DNA methylation is linked to transcriptional silencing [reviewed in (9)], suggesting that it could inactivate the virus. On the other hand, it might protect against the host innate immune response to unmethylated DNA (10).
Recently, it has become possible to characterize heterogeneity of chromatin structure at select loci within populations of mammalian nuclei by treatment with recombinant M.CviPI DNMT [reviewed in (11–13)]. This enzyme, which we cloned from Chlorella virus, methylates cytosine in GC dinucleotides to G-m5C (14). The methyl marks can be read by bisulfite genomic sequencing [BGS; (15)] and unequivocally distinguished from endogenous mammalian methylation at H-m5CG (by convention, H equals A, C, or T). As nucleosomes and DNA-bound factors block access of exogenous DNMT probes to DNA (16–19), the resultant GC methylation pattern is used to infer chromatin structure. The combination of high-resolution probing with M.CviPI and BGS is termed MAPit, for methylation accessibility probing for individual templates, developed by our laboratory (20). By sequencing clonally expanded individual molecules, MAPit allows detection of multiple chromatin states at selected loci within a cell population. This is in contrast to techniques such as chromatin immunoprecipitation, which report the population average [reviewed in (11)]. Although new, MAPit is rapidly being adapted to the study of diverse genomic loci (20–26). Related studies support the ability of MAPit to accurately map gaps between nucleosomes across the genome (27) and track heterogeneity of chromatin structure (28,29).
Hypothesizing that KSHV populations would exhibit epigenetic drift, we examined promoters and 5′ ends of genes of interest in latently infected cells by MAPit. The degree of diversity seen was surprising. Both fully closed and fully open conformations were present at three different KSHV promoters. Treatment with TPA and induced expression of the replication and transcription activator (RTA) were used to test which of the diverse chromatin structures found were linked to reactivation during lytic transcription. At the time point tested, only a subset of episomes appeared to have remodeled chromatin in response to lytic reactivation by both means. We also observed a subset of episomes associated with lytic replication, and identified a link between chromatin compaction, CG methylation and inhibition of reactivation. These findings suggest that latent virus is subject to progressive epigenetic inactivation.
MATERIALS AND METHODS
Cell culture
Body-cavity-based lymphoma cell line 1 (BCBL1) cells (30) were cultured in RPMI 1640 medium supplemented with 10% (v/v) fetal calf serum (FCS), 2 mM glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C under 5% CO2. TREx BCBL1-RTA cells for doxycycline-induced expression of RTA (31) were cultured in RPMI 1640 medium supplemented with 10% (v/v) FCS or tetracycline-free FCS, 2 mM glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C under 5% CO2. Telomerase-immortalized vein endothelial long-term–infected (TIVE-LTC) cells (32) were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% (v/v) FCS, 4.5 g/l glucose, 2 mM glutamine, 1 mM sodium pyruvate, 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C under 5% CO2.
For TPA-induced reactivation, BCBL1 cells were grown to a density of 2–3 × 105 cells/ml. TPA was added to a final concentration of 20 ng/ml and cells were incubated at standard conditions (37°C, 5% CO2) for 6 h. Cells were pelleted at 247g for 5 min and resuspended in the same volume of fresh RPMI medium and grown for another 6 h before nuclei isolation for MAPit. For reactivation by doxycycline-inducible RTA, TREx BCBL1-RTA cells were cultured for at least 2 days in tetracycline-free medium before performing the induction. Cells at a density of 4–5 × 105 per milliliter were then treated with 1 µg/ml doxycycline and 250 µM foscarnet for 6 h. Foscarnet was used to suppress viral lytic replication; quantification of viral load by real-time polymerase chain reaction (PCR) indicated that viral replication was largely blocked (not shown). Cells were pelleted at 247g for 5 min and resuspended in the same volume of fresh RPMI medium with 250 µM foscarnet and grown for another 6 h before nuclei isolation for MAPit. Uninduced BCBL1 or TREx BCBL1-RTA cells (treated with 250 µM foscarnet), respectively, served as the 0 h time point.
MAPit single-molecule footprinting
MAPit was performed as described previously (12,13). Briefly, BCBL1 or TREx BCBL1-RTA cells were harvested at a density of 3–6 × 105 cells/ml, washed twice with ice-cold phosphate-buffered saline (PBS), counted and resuspended at 106 cells/ml in PBS. Four to five million cells were stored as pellet at −80°C for later RNA extraction (see below). For each MAPit assay, 106 cells were pelleted and resuspended in 200 µl cold Buffer A [(10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES)-KOH, pH 7.5, 2 mM MgCl2, 10 mM KCl, 2 mM dithiothreitol (DTT), 0.2 mM phenylmethanesulfonyl fluoride (PMSF)]. Cells were incubated on ice for 10 min to release nuclei by hypotonic lysis, then vortexed 10 s at medium velocity and centrifuged for 10 s at 16 000g. To check for complete cellular lysis, an aliquot of nuclei in 0.2% (w/v) trypan blue were examined by light microscopy. Nuclei were only used for MAPit if the lysis was complete, and nuclei were evenly round and granular in appearance. Nuclear integrity was also confirmed in parallel by Hoechst staining, which dyes DNA, and fluorescence microscopy. Nuclei were washed twice with 500 µl cell resuspension buffer [20 mM HEPES-KOH, pH 7.5, 70 mM NaCl, 0.25 mM ethylenediaminetetraacetic acid (EDTA), 0.5 mM ethylene glycol tetraacetic acid (EGTA), 0.5% (v/v) glycerol, 10 mM DTT, 0.25 mM PMSF] and resuspended in 90 µl methylation buffer (cell resuspension buffer supplemented with 160 µM S-adenosyl-L-methionine).
TIVE-LTC cells were harvested at 100% confluency, washed twice with ice cold PBS, counted and resuspended at 106 cells/ml in PBS. Cells were pelleted (5 × 106 cells for each MAPit assay), washed once in 250 µl cell resuspension buffer and lysed in 100 µl cell lysis buffer [cell resuspension buffer plus 0.25% (v/v) Nonidet P-40] for 10 min on ice. Nuclei were pelleted at 1000g for 5 min at 4°C, then washed twice with 500 µl cell resuspension buffer and resuspended in 90 µl methylation buffer.
To methylate accessible GC sites, 30 U recombinant M.CviPI-maltose binding protein (MBP) fusion [New England Biolabs, hereafter M.CviPI; (14)] were added per 106 nuclei (based on cells counted after all washes completed), which were then incubated at 37°C for 15 min. Reactions were stopped by addition of an equal volume of 1% (w/v) sodium dodecyl sulfate (SDS), 100 mM NaCl, 10 mM EDTA and incubated overnight at 50°C with 100 µg/ml proteinase K. DNA was then phenol extracted (partitioned with an equal volume phenol:chloroform:isoamyl alcohol::25:24:1) and ethanol precipitated. A 0 U M.CviPI control reaction was also performed to gauge endogenous GC methylation (not shown; none was observed at any locus).
To ensure comparable results between experiments, M.CviPI activity was calibrated between aliquots of enzyme using methylation of a HaeIII site in the GRP78 promoter in BCBL1 cells as a standard. MAPit experiments were also conducted at 10 rather than 70 mM NaCl, with 3-fold higher and lower effective M.CviPI concentration, and over a 3-fold range in M.CviPI incubation time (Supplementary Figures S1 and S2). A non-linear relationship was seen between M.CviPI concentration and degree of methylation. This matches a simple model in which DNA molecules adopt multiple chromatin conformations that vary in accessibility to M.CviPI.
Restriction (R) endonuclease R.HaeIII probing of nuclei
Direct treatment of nuclei with R.HaeIII confirmed relative differences in accessibility between the LANA promoter and genomic loci (Supplementary Figure S3). Briefly, 4 × 105 nuclei were prepared as above and suspended in 100 µl 20 mM HEPES-KOH, pH 7.5, 70 mM NaCl, 10 mM MgCl2, 0.5% (v/v) glycerol, 10 mM DTT, 0.25 mM PMSF, plus 0-100 U R.HaeIII (New England Biolabs) or 50% (v/v) glycerol in 10 µl. After 30 min at 37°C, reactions were stopped by addition of an equal volume 1% (w/v) SDS, 100 mM NaCl, 10 mM EDTA and incubation for 20 min at 80°C. Genomic DNA was prepared as described above and percent digest quantified by real-time PCR.
Bisulfite genomic sequencing
To deaminate, 2–4 µg DNA was denatured for 5 min at 95°C in freshly made 0.3 N NaOH, then incubated for 6 h at 50°C in a saturated metabisulfite solution as previously described (33). Bisulfite-converted DNA was desulfonated and concentrated with a commercial kit (Zymo Research catalog no. D5026). Target loci were amplified from 20–100 ng bisulfite-treated DNA with 3 U HotStar Taq DNA polymerase (Qiagen) and 250 nM primers in 10 mM tris(hydroxymethyl)aminomethane (Tris)–HCl, pH 8.6, 50 mM KCl, 3 mM MgCl2 and 200 µM of each 2'-deoxynucleoside 5'-triphosphates (dNTP). Primers sequences are given in Supplementary Table S1. Amplification was performed in 2–3 separate reactions to minimize stochastic variation in PCR amplification, then pooled for cloning. Cycling conditions were 5 min at 95°C, followed by 40 cycles of 1 min at 95°C, 1 min at 56°C–60°C, (optimized for each primer pair), 3 min at 72°C, and a final extension of 5 min at 72°C.
Amplified DNA was separated from unincorporated primers by electrophoresis in 1% (w/v) agarose buffered with Tris-acetate-EDTA, excised and purified from the gel and ligated into a cloning vector using topoisomerase (TOPO TA; Invitrogen). Calcium-competent DH10b or TOP10 cells were transformed with 10–50 ng plasmid and plated on lysogeny broth (LB) medium with 100 µg/ml ampicillin and 40 µg/ml 5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside (X-gal) for blue/white screening. Colony PCR with M13 forward and reverse primers was also used to test cloning efficiency. Colonies containing the insert were suspended in 100 µl LB plus 100 µg/ml ampicillin and grown at 37°C overnight in 96-well plates without shaking. Glycerol was then added to 10% (v/v) and the plates were incubated at −80°C for at least 2 h, whereupon plasmid was subjected to TempliPhi™ rolling circle amplification and automated BigDye sequencing (Applied Biosystems) at the University of Florida Interdisciplinary Center for Biotechnology Research (http://www.biotech.ufl.edu).
Sequence analysis
The genomic sequences corresponding to BGS amplicons are listed in Supplementary Table S2. Sequence files in ABI format were aligned in Sequencher™ (Genecodes) and are available on request. MethylViewer [http://dna.leeds.ac.uk/methylviewer; (22)] was used to ascertain percent cytosine conversion and tabulate methylation status of each site in each sequence read (i.e. molecule). Molecules with <95% conversion of cytosines outside potential methylation sites were discarded. Molecules from the same initial DNA sample that had identical patterns of both methylation and unconverted cytosines were presumed to be duplicate clones, and all copies but one were then discarded. Molecules with either zero methylation or 100% conversion of cytosines were not screened in this way, as there were insufficient grounds to infer duplication. Duplicates made up 0–10% of molecules in each dataset.
The tables of methylation status were used to generate digital methylation maps for both CG and GC methylation for each molecule in Excel (Microsoft). Within the LANA, RTA and vIL6 promoters, CG methylation was rare; thus, GCG sites were counted as GC sites to increase resolution. For the amplicon overlapping the LANA open reading frame (ORF), GCG sites were excluded from the analysis. Digital methylation maps were clustered hierarchically using Cluster [http://rana.lbl.gov/EisenSoftware.htm; (34)], and printed as color maps with MapleTree (http://mapletree.sourceforge.net). Dendrograms were split by eye into clusters of interest. Molecules in each cluster originating from each experiment were counted. Expectation values for the observed distributions were computed as two-tailed Fisher’s exact tests. To generate the accessibility profile (Figure 2D), the fraction of sequence reads bearing accessible spans of 1–100 or 101–400 bp was computed at each base pair position. The difference in accessibility after TPA treatment was then subtracted from that before treatment. Likewise, to generate occupancy profiles (Figures 3D, 5D and 8D), the fraction of sequence reads bearing footprints of 4–39, 40–100 or 101–200 bp was computed at each base pair position. Footprints extending to the ends of the amplicons were not counted. To get the difference in occupancy, the fraction for one experimental condition was then subtracted from the fraction for another.
Figure 2.
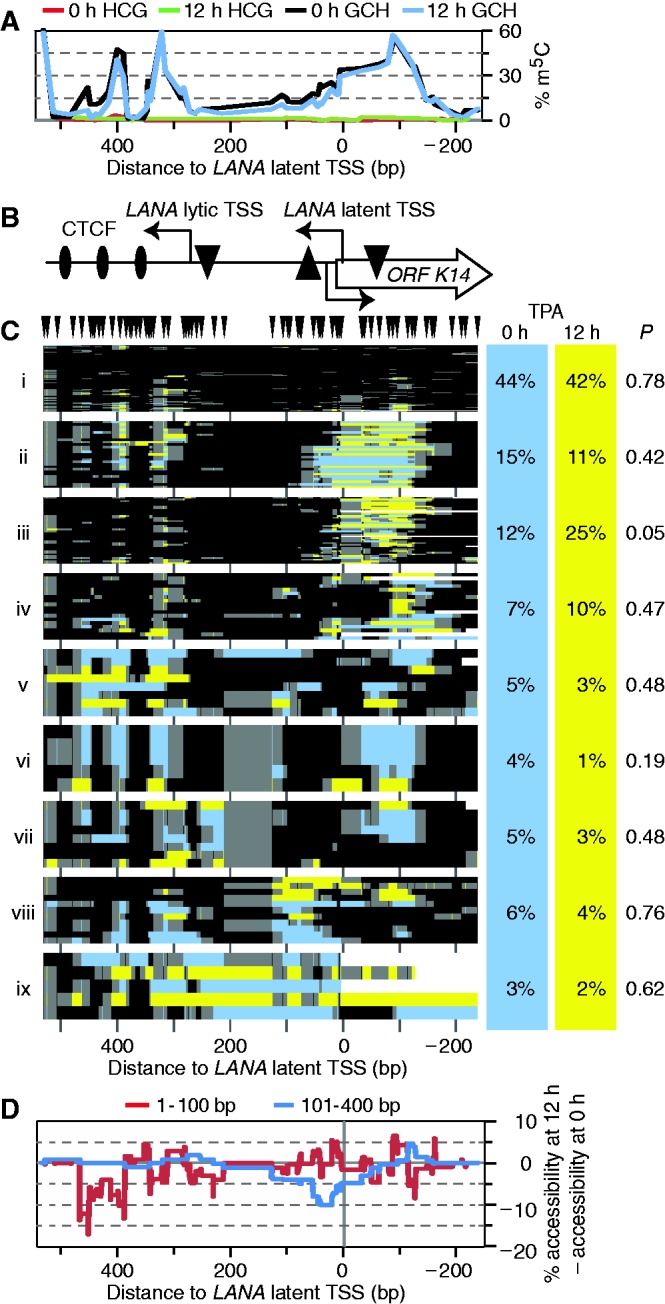
TPA treatment altered KLAR chromatin structure in ∼10% of BCBL1 episomes. Note that this KLAR amplicon, although overlapping the amplicon of Figure 1, was centered upstream to better observe accessible DNA at the latent TSS. (A) Profile of endogenous CG (0–h TPA treated, red; 12-h TPA treated, green) and exogenous GC methylation (0-h TPA treated, black; 12 h TPA treated, blue) across the locus. GCG sites were not counted. (B) Schematic of the analyzed amplicon. The LANA ORF begins left of the amplicon. The LANA latent and lytic TSSs are indicated. TATA boxes are indicated by triangles (upside down, latent and lytic LANA; righted, ORF K14). (C) All GC methylation plots for 0- and 12-h TPA treatment (blue and yellow, respectively), including GCG, as m5CG was rare in (A), were organized by unsupervised hierarchical clustering. GC sites are indicated above; plot is to scale with other panels. Dendrogram was split into nine fractions of interest. On right, proportion of sequence reads from each data set present in each cluster (columns do not sum to 100% owing to rounding). Expectation value P is the chance that as uneven a distribution of molecules from the two datasets would occur if molecules had been assigned to the cluster at random. (D) Difference in accessibility from 0- to 12-h of TPA treatment. Cumulative accessibility in two size ranges, 1–101 and 101–400 bp, was tallied at every position in each dataset, then the 0-h profile was subtracted from the 12-h profile.
Figure 3.
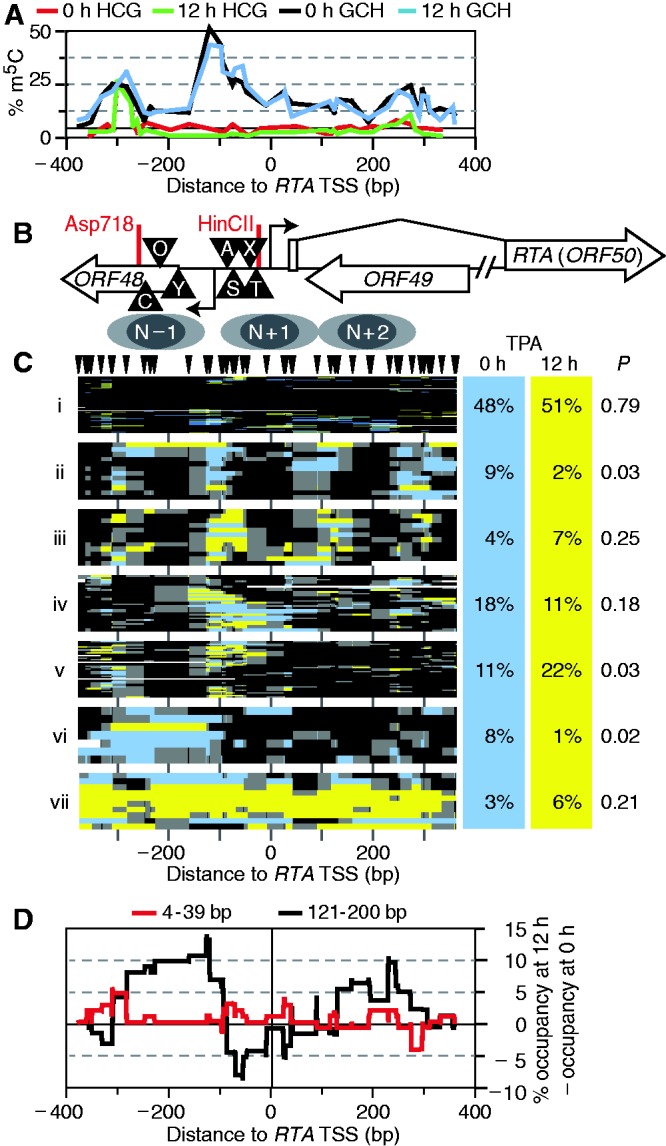
Induced lytic reactivation altered chromatin state frequency at RTA. (A) Profile of average endogenous CG (0-h TPA treatment, red; 12-h TPA treatment, green) and exogenous GC methylation (0-h TPA treatment, black; 12-h TPA treatment, blue) across the locus. GCG sites were not counted. Note locus of highest CG methylation coincided with a peak of GC methylation; elsewhere, CG methylation was minimal. (B) Diagram of RTA locus amplified. Triangles mark some of the known or suggested transcription factor–binding sites: C/EBP (C) (47), OCT-1 (O) (48), YY1 (Y) (49), AP-1 (A) (50), Sp1/3 (S) (2), XBP-1 (X) (51) and TATA (T). A second RTA TSS was recently reported roughly 500-bp upstream of this amplicon (52). Below, inferred nucleosome positions are indicated as two overlapping gray ovals, each 150-bp long. (C) Clustered GC methylation maps from 0-h (blue) and 12-h (yellow) treatment with TPA. Methylation of GC sites (including GCG, as m5CG was rare) was plotted as in Figure 1, to scale with panels (A, B and D). Position of GC sites is indicated by hashes at top. Maps from both data sets were sorted together by unsupervised, hierarchical clustering. Seven clusters of interest are presented. On right are given the proportion of sequence reads from each data set present in each cluster, and estimate of expectation value P assigned as in Figure 2. Columns do not sum to 100% due to rounding. (D) Difference in occupancy from 0 to 12 h of TPA treatment. Footprints in two size ranges, 4–39 and 121–200 bp, were counted at every position in each dataset, then the 0-h profile was subtracted from the 12-h profile. An intermediate size range, 40–120 bp, showed no difference between the data sets.
Figure 5.
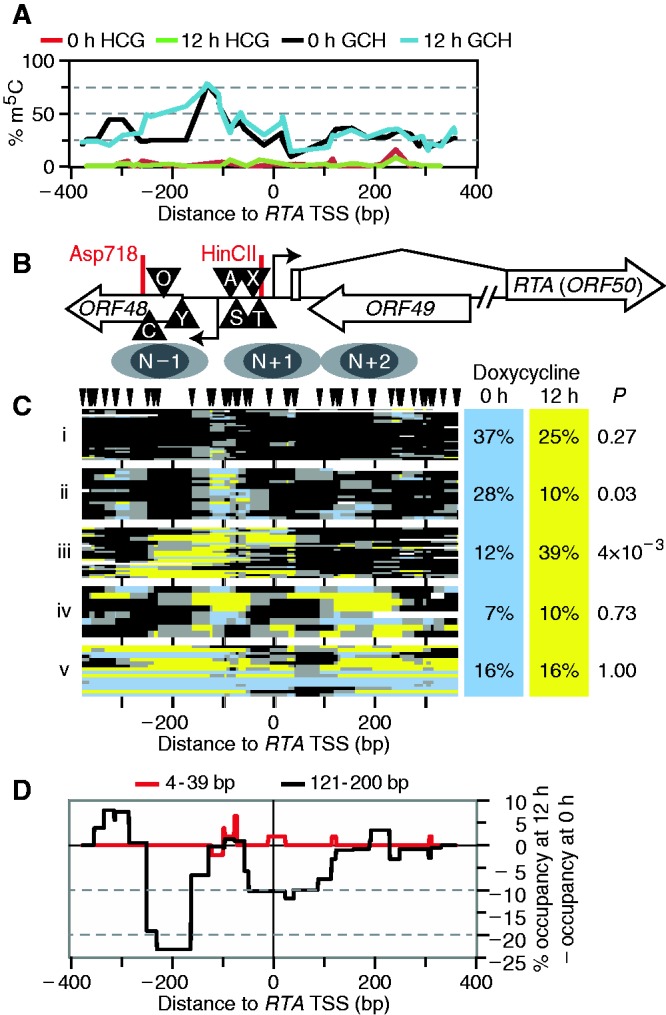
Expression of a doxycycline-inducible RTA transgene altered chromatin structure at the endogenous RTA promoter of 30–40% of episomes. (A) Profile of endogenous CG (uninduced, red; 12 h of doxycycline, green) and exogenous GC methylation (uninduced, black; 12 h of doxycycline, blue). GCG sites were not counted. (B) Schematic of the RTA locus, as in Figure 3B. (C) GC methylation plots for 0- and 12-h RTA induction (blue and yellow, respectively). GC sites are indicated above; plot is to scale with other panels. Dendrogram was split into five fractions of interest. On right, proportion of sequence reads from each data set present in each cluster, and expectation value P calculated, as in Figure 2. (D) Difference in footprint occupancy from 0 to 12 h of TPA treatment. Footprints in two size ranges, 4–39 and 121–200 bp, were counted at every position in each dataset, then the 0-h profile was subtracted from the 12-h profile.
Figure 8.
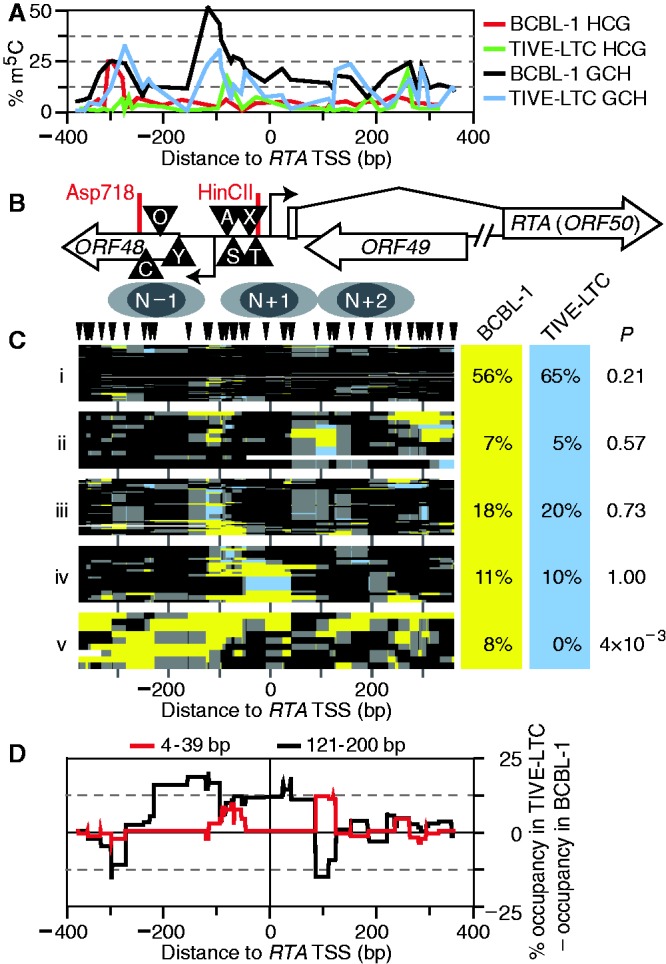
Silent chromatin at RTA promoter in TIVE-LTC cells. TPA-untreated BCBL1 MAPit reads from Figure 3 were compared with MAPit reads from TIVE-LTC cells. (A) Profile across RTA promoter of average endogenous CG (green) and exogenous GC (blue) methylation in TIVE-LTC cells, as well as CG (red) and exogenous GC (black) methylation in BCBL1 cells from Figure 2A for comparison. Note absence of ‘linker’ H-m5CG peak at −300 bp in TIVE-LTC cells, despite the presence of an overlapping G-m5CH peak, on left side of graph. (B) Diagram of RTA locus amplicon and inferred nucleosome positions, as in Figure 2, to scale with other panels. (C) Clustered GC (including GCG) methylation maps from BCBL1 (yellow) and TIVE-LTC (blue) presented as in Figure 2. Five clusters of interest are presented. On right, proportion of sequence reads from each data set present in each cluster, and estimate of expectation value P calculated as in Figure 2. (D) Difference in frequency of footprints of two size ranges from 0 to 12 h of TPA calculated as in Figure 2.
RNA preparation for real-time reverse transcription PCR (RT–PCR)
BCBL1 and TREx BCBL1-RTA cells were treated as described above in two independent experiments (including different stocks of cells). From 4 to 5 × 106 induced and uninduced cells, RNA was extracted using RNA Bee (TEL-TEST, Inc.) according to the manufacturer’s instructions. RNA was DNase I treated [10 µg RNA, 1 µl RNasin® (Promega), 1 × DNase I buffer, 2 µl (4 U) RNase-free DNase I (New England Biolabs) in 50 µl total volume] for 25 min at 37°C and recovered by ethanol precipitation after ∼16 h at −20°C. An aliquot of DNase I–digested RNA was tested for RNA quality by agarose gel electrophoresis and the rest stored at −80°C. RNA (1–3 µg) was reverse transcribed using oligo dT primer and Invitrogen Superscript III reverse transcriptase (RT) in a 20 µl reaction mixture as indicated by the manufacturer (20 µl reaction mixture per 1 µg RNA; for more RNA, the assay was scaled up accordingly). A reverse-transcription reaction without RT enzyme served as control (no RT).
Quantitative real-time PCR
According to the MIQE guidelines for quantitative real-time PCR [qPCR; (35)], we refer to reference genes instead of housekeeping genes and Cq (quantification cycle) instead of Ct (threshold cycle). All primers (Supplementary Table S1) were purchased from Integrated DNA Technologies, Inc. and tested for their efficiency (between 90 and 110%) and specificity (melting curve analysis).
Per 12 µl qPCR reaction, 0.5 µl complementary DNA was used. Primers were used at a final concentration of 200 nM. Two-step qPCR was performed with the Fast SYBR Green Master Mix and a StepOnePlus real-time PCR instrument (both Applied Biosystems). Cycle conditions were initially 95°C for 10 min, 40 cycles of 95°C for 15 s, 60°C for 1 min, followed by melting curve analysis.
For quantification of M.CviPI-mediated protection from R.HaeIII digest, 1 µg undeaminated DNA was digested with 10 U R.HaeIII, as per the enzyme manufacturer’s directions, or mock-treated with an equal volume of 50% (v/v) glycerol. For each sample, three wells with 10 ng DNA apiece were quantified by real-time PCR, as above.
RESULTS
Multiple chromatin conformations in the KSHV latency-associated region
LANA, vFLIP, vCyclin, Kaposin and 12 pre-microRNAs are highly expressed off polycistronic transcripts arising from a single KSHV latency-associated region (KLAR) in tumor cells. During latency, host RNA polymerase II initiates transcription of KLAR from the upstream latent transcription start site (TSS; Figure 1B). Additionally, a second downstream TSS is used during lytic reactivation (36). Three CCCTC-binding factor (CTCF) binding sites are located between these two TSSs and the LANA open reading frame (ORF73); their occupancy by the chromatin insulator CTCF (37) contributes to LANA expression, mediates chromosomal looping and plays a role in episomal maintenance (38,39). All KSHV-infected cells express LANA, which is required for replication and maintenance of viral episomes in latently infected cells [reviewed in (40)]. Yet, not all of the ∼80 episomes present in latently infected lymphoma cells need express LANA. We hypothesized that a subset of episomes might provide LANA function for the rest, which could be epigenetically inactivated.
Figure 1.

MAPit analysis of chromatin structural diversity. (A) MAPit workflow diagram, adapted from Darst et al. (13). Chromatin (e.g. nucleosomes) modulates accessibility of DNA sites to soluble proteins, such as (i) transcription factors (represented by shaded rectangle) in live cells, or (ii) exogenous M.CviPI DNMT in nuclei. Bisulfite treatment (iii) records M.CviPI accessibility pattern in the DNA sequence by conversion of unmethylated cytosine to uracil (cyan). Subsequent amplification and cloning (iv) allows recovery of this information from individual molecules. Note that endogenous and probe methylation sites are distinguished by sequence specificity of the respective DNMTs (respectively, cyan and magenta). The DNA sequences obtained are used to generate maps (v) of DNA methylation and accessibility. (B) MAPit at the KSHV gene LANA detected endogenous methylation and sporadic accessibility of the latent promoter. MAPit was performed in nuclei of BCBL1 cells. Top: map of genetic elements within the MAPit amplicon: LANA ORF (ORF73), three CTCF sites (ovals), lytic and latent TSSs (bent arrows) and TATA boxes (triangles). Middle: MethylViewer (22) display of MAPit results from control nuclei probed as indicated with 0 U or 30 U M.CviPI. Blue vertical ticks indicate positions of unconverted cytosines, excluding GC and CG sites (i.e. HCH); percent conversion of all HCH cytosines for each sequence clone is given on the right. Bottom (key): Circles and triangles, respectively, indicate CG and GC sites. Methylated sites are shaded. Methylated GCG sites are shaded gray to indicate their ambiguity; however, comparison of sequences from nuclei with and without M.CviPI treatment (middle, lower and upper panels) indicated that GCG sites within the LANA ORF were endogenously methylated. (C) Condensed view of the data in B, to scale with locus map at top. Each row of pixels represents one cloned sequence, i.e. molecule. Each plot tracks only GC or CG methylation; GCG sites are ignored. As indicated by key at bottom, spans of color mark ≥2 contiguous methylated sites; black marks ≥2 contiguous unmethylated sites; gray marks spans between methylated and unmethylated sites; white indicates missing or unaligned sequence. Positions of sites are indicated by circles and triangles (HCG and GCH, respectively). (D) Host promoters varied in degree of accessibility to M.CviPI. Maps of GC methylation at two representative promoters are shown, to scale with LANA amplicon. (E) MAPit at the three loci shown was semi-quantitative. At the HaeIII sites indicated in red at GAPDH, GRP78 and LANA, 10 of 20, 15 of 17 and 1 of 10 molecules had a methylated GC site, respectively (blue bars). R.HaeIII digestion followed by quantitative real-time PCR was used to measure bulk methylation of the same three sites in genomic DNA purified from M.CviPI-treated nuclei (red bars). Under these conditions, ∼95% of DNA from untreated nuclei (i.e. unmethylated) was digested (not shown). The concordance between BGS and R.HaeIII digest indicated that representation of methylated and unmethylated molecules in MAPit was unbiased.
As a single-molecule technique, MAPit provided a way to test this hypothesis directly. A key advantage of MAPit, in contrast to population average readouts, is the ability to discover distinct subpopulations without enrichment. Nuclei from the KSHV-infected pleural effusion lymphoma (PEL) cell line BCBL1 were incubated with 0 or 30 U M.CviPI, following the protocol outlined in Figure 1A. We cloned and sequenced 10 bisulfite-converted molecules encompassing the LANA promoter from each sample. The patterns of CG and GC methylation were drawn by MethylViewer (22), which represents methylated cytosines as ball-and-stick symbols (Figure 1B). Efficiency of conversion was estimated as percent HCH (H is any base but G) conversion in each sequence read (i.e. cytosine outside potential methylation sites; blue vertical ticks mark unconverted HCH). As expected, without M.CviPI addition, methylation of GCH sites was not above the background given by incomplete conversion. The M.CviPI-probed sample exhibited prominent accessibility above background between and adjacent to three known CTCF sites. Some molecules were also accessible near the latent TSS.
Although MethylViewer is excellent for comparing different methylation types on single molecules, such detailed graphs are unwieldy for large-scale analysis. We therefore devised a three-color map scheme (Figure 1C), used throughout the rest of this work. Patches of accessibility to DNMT (M.CviPI and endogenous DNMTs) received bright color (yellow and red, respectively), and patches of protection were colored black. Gray color denoted borders between methylated and unmethylated patches. We used unsupervised hierarchical clustering to order molecules, facilitating pattern recognition. Color maps for both GC and CG methylation status were clustered together. The 16 GCG sites were disregarded in this analysis, leaving 52 sites for GC and 36 for CG.
The data showed ‘banded’ CG methylation across almost all molecules in the LANA ORF. By contrast, the 10 molecules varied in degree and pattern of M.CviPI accessibility. This could reflect variation in chromatin structure between episomes. An alternate explanation would be a poor nuclear preparation, such that the M.CviPI probe did not enter all nuclei. However, this seems unlikely as nuclei appeared intact and homogenous by vital (trypan blue) and Hoechst staining (not shown). Moreover, all 10 clones of the LANA amplicon exhibited GC methylation above background unconverted HCH (Figure 1C, compare lower panels in 0 versus 30 U M.CviPI). In addition, we performed BGS at select host loci from the same sample. Two representative loci are shown (Figure 1D). At the more open locus, the promoter of GRP78, all 17 molecules sequenced had GC methylation. This locus was previously found to be highly accessible to a DNMT probe in a colorectal line (28,29).
Because MAPit requires amplification and cloning of bisulfite-converted DNA, it can be affected by sequence-specific biases in either step. We therefore measured M.CviPI methylation by R.HaeIII digestion of undeaminated DNA from the same samples used for MAPit (Figure 1E and Supplementary Figure S2). As R.HaeIII does not digest its site GGCC when the central cytosine is methylated, the concordance between R.HaeIII protection and BGS showed the latter to be unbiased. In turn, this meant that M.CviPI efficiently permeated nuclei and accessed the GRP78 promoter. We also probed nuclear DNA directly with R.HaeIII, without prior M.CviPI treatment (Supplementary Figure S3). The restriction enzyme, which binds DNA cooperatively as a dimer and thus is similar in size to the monomeric M.CviPI fusion protein, respectively, 74.2 and 83.7 kDa, was able to access and cut its sites on most molecules. Thus, variation at LANA was not due to uneven probing of nuclei.
As indicated by the second locus shown, the GAPDH promoter, heterogeneity of promoter chromatin structure was not unique to LANA. Variation in chromatin state between copies of the same gene can arise from several causes. Epigenetic variation occurs in X inactivation, imprinting and monoallelic expression. Dynamic variation occurs in periodic and sporadic cycles of chromatin remodeling and transcription [for instance, (41)]. However, none of these phenomena have been described for a viral gene such as LANA. In any case, the implication would be that the promoters in the closed conformation do not support transcription. Thus, as hypothesized, not all episomes continuously express LANA. Because all infected cells express LANA (32), this result implies heterogeneity of episomes within individual nuclei.
MAPit survey of LANA promoter architecture
In addition to the latent TSS, the LANA promoter uses a lytic TSS on reactivation (36). Furthermore, transcription starting at the latent TSS overlaps the lytic K14 promoter on the opposite strand. We asked whether MAPit could visualize changes in chromatin structure within this complex regulatory region during lytic reactivation induced by phorbol ester treatment. To test this, we cloned and sequenced 102 amplified deaminated LANA promoter molecules from M.CviPI-treated BCBL1 nuclei (0 h or uninduced), and 110 molecules from M.CviPI-treated nuclei harvested after 12 h of TPA treatment of cells (12 h or induced). Induced transcription of two early lytic genes, RTA (ORF50) and vIL6 (ORF K2), was demonstrated by real-time RT-PCR (Supplementary Figure S4). An early time point was chosen to obtain incomplete lytic reactivation. Otherwise, histone-depleted products of viral DNA replication would out-compete cloning of BGS products derived from chromatinized viral episomes (4,42). Hence, these induction conditions did not have an obvious effect on M.CviPI accessibility averaged across the population (Figure 2A and B).
We therefore asked whether differences might be detected in a single-molecule analysis. To determine which methylation patterns were enriched in one dataset relative to the other, we pooled all reads from the 0 - and 12-h TPA treatment time points and clustered them hierarchically as GC methylation plots (Figure 2C). We plotted accessibility to M.CviPI in each molecule as blue (0 h) or yellow (12 h) to mark its source and split the dendrogram into clusters of interest. To better visualize differences in chromatin structure between clusters, each was rescaled vertically. The fraction of molecules present in each cluster from each time point is given on the right. The KLAR amplicon in this experiment was positioned farther upstream from the LANA gene than that analyzed in Figure 1, allowing better characterization of accessibility at the LANA latent TSS. Strikingly, the three CTCF sites were seen to be occupied by footprints in almost all molecules. Even in cluster i, which had reduced M.CviPI methylation overall, the three CTCF sites were dimly outlined. Importantly, this demonstrates that M.CviPI permeated these nuclei, but was preferentially excluded from the latent TSS (especially compared with clusters iii and iv).
For each cluster, we found the probability P of having so skewed a distribution of molecules from the two samples by chance. Although clustering revealed several distinct chromatin conformations, TPA treatment increased representation of molecules only in cluster iii (by 13%) with borderline significance (P = 0.05). To further examine TPA-induced changes in LANA promoter chromatin structure, we calculated accessibility of lengths 1–100 and 101–400 bp occurring at each position, and subtracted accessibility before TPA treatment (0 h) from that after (12 h) (Figure 2D). TPA treatment decreased accessibility adjacent to the CTCF sites (1–100 bp, red curve) and just downstream of the LANA latent TSS (101–400 bp, blue curve) in ∼10–15% of the molecules, corresponding well with the number of molecules TPA increased in cluster iii. We conclude that TPA increased protection at the K14 TATA element, mainly in cluster iii.
Clustering also highlighted the large degree of diversity of chromatin structure at this locus. Both fully protected and almost fully accessible molecules were detected. Cluster ix was of particular interest as it appeared to be nearly nucleosome free. Complete chromatin unfolding and nucleosome displacement across a region of this size has not been observed at any human chromosomal locus studied by MAPit. We hypothesize that molecules of cluster ix originated from spontaneous lytic reactivation of KSHV, which occurs within 1–3% of PEL cells (30,43,44). As is the case for other herpesviruses, the KSHV genome is depleted for histones during lytic replication before being packaged into virions (4,42). These results indicated that MAPit could discover chromatin conformations comprising <∼5% of a complex population.
A subset of episomes undergoes changes in chromatin structure during reactivation
The master regulator of lytic transcription is the replication and transcription activator RTA (45). RTA directly activates several downstream viral genes, such as vIL6, as well as transcription of its own gene (46). We hypothesized that a subset of latent episomes would be poised to transcribe RTA (ORF50) and that these would have a distinct chromatin structure visible by MAPit. Induced reactivation would be predicted to shift these molecules to the actively transcribing state, which might involve nucleosome repositioning. We therefore cloned and sequenced 114 and 112 molecules from M.CviPI-treated nuclei harvested after 0- or 12-h TPA treatment, respectively (Figure 3).
Accessibility to CG and GC DNMTs was computed at each position. Within ORF48 (5′ of RTA), a peak of CG methylation was observed upstream (313–284 bp upstream of the RTA TSS) of a positioned nucleosome, hereafter N − 1 (Figure 3A and B). The same position had been previously shown to be endogenously methylated in BCBL1, although in a larger proportion of the population (53). A second notable feature of this plot was a broad peak in GC accessibility centered ∼100-bp upstream of the RTA TSS. An open chromatin structure was not expected to be so common in the latent state. Moreover, there was no obvious difference between GC accessibility profiles before and after TPA treatment (Figure 3A). Thus, the bulk of the population was unchanged. As expected, the GAPDH (Supplementary Figure S2C) and GRP78 control loci also showed no change in chromatin architecture in response to TPA (compare Supplementary Figure S5 with Figure 1D, upper).
As before, molecules from the two time points were clustered together to identify chromatin conformations enriched in either (Figure 3C). Cluster i, comprising ∼50% of all molecules, was, as at KLAR, largely (but not completely) inaccessible to M.CviPI and presumably transcriptionally inactive. Clusters ii–v all had an N − 1 footprint. Clusters ii and iii appeared similar to the eye: both featured three evenly spaced positioned nucleosome-sized footprints. However, cluster iii was more closely related to cluster v than ii in the original dendrogram (Supplementary Figure S6). The central N + 1 nucleosome in cluster ii covered several previously identified transcription factor binding sites upstream of the RTA TSS. The N + 1 and N + 2 nucleosomes seemed to shift slightly downstream in cluster iii versus ii. Cluster iv had a variably positioned N − 1 footprint, followed downstream by a large open gap, then 2–3 variably positioned nucleosome-sized footprints. Cluster v molecules shared a large M.CviPI-accessible linker upstream of N − 1, with chromatin that appeared to be denser downstream over the RTA transcribed region. Cluster vi lacked N − 1. Lastly, cluster vii was, just as cluster v found at KLAR, almost entirely accessible and again comprised ∼3% of all molecules.
Clusters ii, v and vi had P values below the usual threshold for significance (<0.05). Because clusters ii and vi decreased in response to TPA, they may represent transcriptionally poised states, primed to respond to activating stimuli. Cluster iv would then correspond to a transcriptionally active state or a thermodynamically stable stage in the cycle of RNA polymerase recruitment and release. Although it had too few molecules for statistical significance, cluster iii was also enriched for molecules from TPA-treated cells and might also represent a stage in the transcription cycle. Comparing the increase in cluster v to the decrease in clusters ii and vi, we conclude that at least 10% of episomes remodeled RTA chromatin in response to TPA. By the same token, a large fraction of molecules appeared not to have responded to TPA at this time point, including those of cluster i, which were highly compacted.
Several small footprints covering 1–2 GC sites occurred at 93, 86 and 74 bp upstream of the TSS (Supplementary Figure S6), which might represent some of the transcription factors reported to bind this area. To determine which footprints were more abundant in each dataset, we counted footprints of lengths 4–39 and 121–200 bp occurring at each position and subtracted footprint counts before TPA treatment from those after (Figure 3D). The result is difference in occupancy of factors within each footprint size range at each base pair position. This analysis revealed greater definition of the N − 1 footprint (broad peak in black spanning from −300 to −100 bp relative to the RTA TSS), repositioning of nucleosomes inside the transcribed region of RTA (sharp peaks on right), and changes in some sub-nucleosomal footprints flanking these areas (in red), but not changes in the putative transcription factor footprints. Nucleosome repositioning in the gene was consistent with the hypothesized shift from cluster ii to iii.
Together, these results suggested that only a subset of KSHV episomes had responded to TPA by reorganizing RTA promoter chromatin by 12 h of TPA induction. From this, one would predict a similar proportion of episomes to be expressing a downstream target of RTA, an immediate-early gene such as vIL6. We cloned 96 and 71 molecules amplified from bisulfite-converted DNA isolated from TPA-treated and untreated cells, respectively. Unlike at RTA, at the vIL6 promoter (within ORF2), there was a wide peak of accessibility to M.CviPI (Figure 4A and B). As with RTA, no change in bulk accessibility was seen on TPA treatment from the methylation plots alone. However, clustering of molecules once again showed otherwise. As at KLAR and RTA, there was both an abundant, highly inaccessible, likely silenced cluster (Figure 4C, cluster i) and a rare, highly accessible cluster (v), suggesting that the underlying chromatin behavior was episome wide. Again, neither of these two clusters changed significantly on TPA treatment. However, cluster iv, lacking two or more nucleosomes within the vIL6 promoter, did increase significantly from 8 to 24% of molecules after induction (P = 0.01). This suggests chromatin remodeling in response to activation by RTA. Moreover, these numbers were consistent with the estimate that ∼10% of episomes were transcribing RTA at this time point. The observation that 8% of molecules showed the same vIL6 chromatin accessibility during latency is in congruence with a recent report of low levels of vIL6 expression during latency (54). As at RTA, results were consistent with a model of heterogeneous reactivation of the lytic pathway, in which an open chromatin structure such as in Figure 3C, cluster vi is prerequisite. We conclude that chromatin structure reflects ability of episomes to respond to inducers of lytic transcription.
Figure 4.

TPA treatment altered chromatin structure at the vIL6 gene in 10–15% of BCBL1 episomes. (A) Profile of average endogenous CG (0-h TPA treatment, red; 12-h TPA treatment, green) and exogenous GC methylation (0-h TPA treatment, black; 12-h TPA treatment, blue) across the locus. (B) Diagram of vIL6 locus amplified. Little has been reported of the structure of the vIL6 promoter, except for the two shown TSSs (bent arrows) (74). (C) All GC methylation plots for 0- and 12-h TPA treatment (blue and yellow, respectively, including GCG) were organized by unsupervised hierarchical clustering. GC sites are indicated above; plot is to scale with other panels. Dendrogram was split into five fractions of interest. On right, proportion of sequence reads from each data set present in each cluster, and expectation value P calculated as in Figure 2.
As a protein kinase C agonist, TPA induces weak signaling that may resemble a pathway for spontaneous KSHV reactivation in vivo. To further examine the extent to which the fraction of KSHV episomes with relatively inaccessible chromatin (i.e. in cluster i) was resistant to lytic reactivation, we performed an experiment with TREx BCBL1-RTA cells. These cells, containing an integrated doxycycline-inducible RTA transgene, overexpress RTA after 12 h of treatment with doxycycline and hence undergo efficient reactivation (31,55). We collected cells after 12 h of induction and performed MAPit, using primers specific to the wild-type KSHV RTA promoter. We cloned 51 and 45 molecules amplified from bisulfite-converted DNA isolated from doxycycline-treated and untreated cells, respectively (Figure 5). Fold induction of lytic genes was 30- and 6-fold higher for RTA and vIL6 transcripts, respectively, in doxycycline-treated TREx BCBL1-RTA cells than had been observed in parental BCBL1 cells treated with TPA (Supplementary Figure S4).
Several differences in RTA promoter chromatin architecture were also observed between the cell lines. In the TREx BCBL1-RTA cell line, the upstream peak of CG methylation was absent (compare Figures 5A and 3B, red and green curves), and a footprint spanned the two GC sites within the Sp1 binding site in 10–20% of molecules [Figure 5B and C, under triangle labeled ‘S’; (2)]. Although this footprint was slightly more common on RTA induction, the increase did not pass the threshold for significance (P = 0.4).
As before, we noted a subset of episomes with highly accessible chromatin, cluster v. These were more abundant than we had observed in the parent cell line BCBL1, possibly due to higher basal expression of RTA in the untreated TREx BCBL1-RTA cells. Consistent with this, 5.6-fold more RTA transcript was observed in uninduced (tetracycline-free FCS) TREx BCBL1-RTA versus BCBL1 cells (not shown).
Three clusters of molecules with intermediate accessibility to M.CviPI were found, ii–iv (Figure 5C). As seen in the presence of phorbol ester, induction of RTA in trans caused a significant shift in abundance between two clusters (ii and iii). However, in this case, the fraction of molecules affected was larger (30%, i.e. increase in cluster iii versus decrease in clusters i and ii), and the doxycycline-induced state (cluster iii) was more accessible to M.CviPI than had been seen with TPA induction (Figure 3, cluster iv). Displacement of N − 1 occurred in 20% of molecules (Figure 5D), consistent with the shift of at least 20% of molecules to cluster iii. Increased chromatin remodeling in TREx BCBL1-RTA is consistent with the higher and more persistent levels of RTA expression than are observed with TPA-induced reactivation (31,56,57). Alternatively, dissimilarities between the two experimental systems may reflect differences in transcription factor and/or co-activator recruitment. TPA brings AP-1 to the promoter, but direct RTA induction acts through C/EBP (47,50).
As in the BCBL1 cells assayed in Figure 3, a large fraction of cloned sequences showed promoters with compact chromatin, inaccessible to M.CviPI (Figure 5C, cluster i). The fraction of KSHV episomes with closed chromatin did not significantly decrease in response to RTA induction in trans despite 30-fold increased RTA expression as compared with TPA induction. Thus, MAPit indicated heterogeneous reactivation of RTA in highly induced TREx BCBL1-RTA cells, just as in TPA-treated BCBL1 cells.
Epigenetic diversity in latent KSHV
Our results suggested that the promoters of LANA, RTA and vIL6 existed in at least two states across the population of episomes. The first had compact chromatin and was non-responsive for lytic reactivation, and the second exhibited various open conformations. However, it was not yet clear whether these states were epigenetic or reflected periodic or sporadic cycles of chromatin remodeling and transcription. Heritability of chromatin conformations identified by MAPit can be inferred by correlations between GC methylation and CG methylation, which is epigenetic. Indeed, a patch of endogenous CG methylation occurred upstream of the RTA promoter in 25% of BCBL1 molecules sequenced (Figures 3A and 6A and B). Note that clusters shown in Figure 6B were based solely on HCG methylation, i.e. excluding all GCG sites. Thus, they do not correspond to the GCH methylation clusters in Figure 3C. To test whether the latter were epigenetically distinct, we counted molecules with upstream m5CG patches in each M.CviPI methylation-defined cluster (Figure 6C). This feature was defined as methylation of 2–3 of the CG sites 284–313 bp upstream of the RTA TSS.
Figure 6.
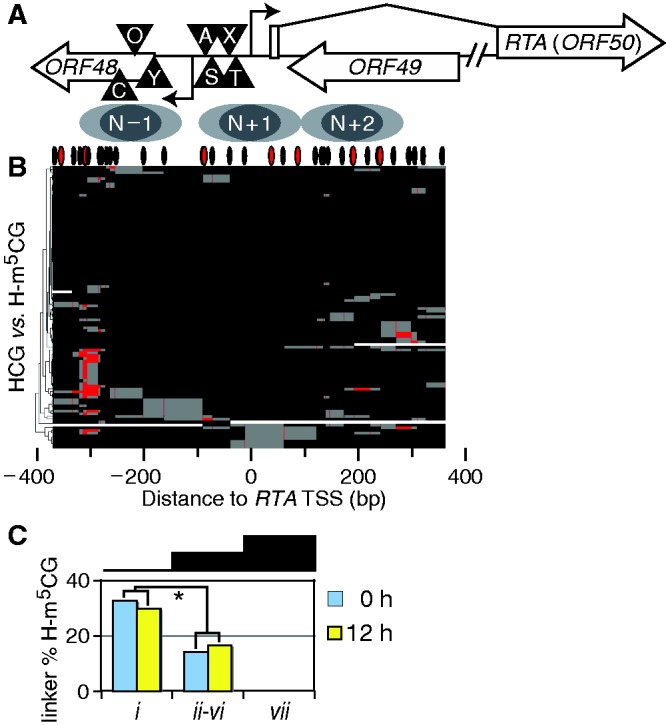
Correlations between CG methylation and chromatin structure observed by MAPit. (A) Schematic of the analyzed amplicon. Symbols are as defined in Figure 3B. (B) CG methylation plot indicating localized patch of CG methylation upstream of RTA TSS. These are CG methylation plots of the same molecules as Figure 3C, 0-h treatment (not counting GCG). Unsupervised hierarchical clustering revealed that ∼25% of molecules shared a patch of CG methylation (red) ∼300 bp upstream of the RTA TSS. Positions of CG sites are indicated by hashes at top. (C) Link between CG methylation and MAPit structure at RTA. All molecules with 2–3 methylated CG sites within the linker were counted for each MAPit cluster defined in Figure 2C, as a percent of the total molecules in the cluster for each time point. Clusters ii–vi were grouped because some had as few as three molecules from a time point, increasing variance observed. Asterisk indicates data used to calculate P < 0.01 that constant CG methylation between clusters could generate observed distribution.
From both TPA-treated and untreated cells, 30% of cluster i molecules in Figure 3C had upstream m5CG patches. However, only 15% of molecules from clusters ii–vi and 0% from cluster vii had upstream m5CG patches. Clearly, lytic induction did not affect abundance of upstream m5CG patches in either group of molecules. Pooling the two experimental conditions, we computed P < 0.01 that CG methylation would differ so much by chance between clusters i and ii–vi. Hence, the presence of m5CG patches upstream of RTA was anti-correlated with openness of nearby chromatin to GC methylation. Furthermore, epigenetic ‘fingerprinting’ showed that changes in cluster abundance on TPA treatment were primarily due to flux in molecules lacking the upstream CG methylation (Supplementary Table S3). This indicated that upstream-methylated RTA promoters were not significantly remodeled in response to TPA. Thus, CG methylation fingerprinting indicates that different MAPit patterns arose from epigenetically distinct subpopulations at RTA. These data indicate an epigenetic basis for the chromatin structural diversity observed at RTA, implying long-term stability.
Altered chromatin dynamics on constitutively latent episomes
Unlike BCBL1, TIVE-LTC do not reactivate either spontaneously or in response to TPA (32). We therefore decided to study chromatin conformation in TIVE-LTC as a model for purely latent episomes, which are transcriptionally silenced except for the LANA region. For comparison with 94 TPA-untreated LANA reads from latent BCBL1, we cloned and sequenced 107 from TIVE-LTC cells. To determine the extent to which TIVE-LTC and BCBL1 episomes had chromatin structures in common, we pooled all GC methylation plots from the two datasets and clustered them hierarchically (Figure 7).
Figure 7.
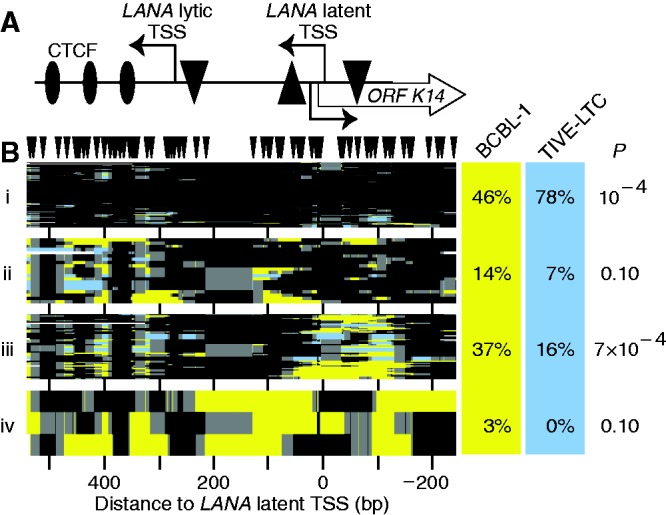
Chromatin structure at KLAR in TIVE-LTC compared with BCBL1 cells. (A) Schematic of the locus. The LANA ORF begins left of the amplicon. The latent and lytic TSSs are indicated. TATA boxes are indicated by triangles with orientations as in Figure 1A. (B) All GC methylation plots for BCBL1 and TIVE-LTC (yellow and blue, respectively, including GCG) were organized by unsupervised hierarchical clustering. GC sites are indicated above; plot is to scale with the other panel. Dendrogram was split into four fractions of interest. On right, proportion of sequence reads from each data set present in each cluster, and estimate of expectation value P calculated as in previous figures. Column for TIVE-LTC cells does not sum to 100% owing to rounding.
We divided the dendrogram into clusters of interest as before and calculated P values for under- or overrepresentation of molecules in each cluster from each cell type. There was a significant increase in locus-wide protection (46–78%) of LANA in TIVE-LTC compared with BCBL1 cells (cluster i, P = 10−4). This was not caused by reduced nuclear permeation by M.CviPI, as the control locus GAPDH was, if anything, more open in TIVE-LTC cells (Supplementary Figure S2C). TIVE-LTC cells also produced fewer molecules in which nucleosomes were shifted upstream to cover the latent TSS (Figure 7B, cluster ii). In addition, there was a significant reduction from 37 to 16% in the number of molecules with accessible chromatin over the latent TSS in BCBL1 cells (cluster iii, P = 7 × 10−4). However, CTCF occupancy was unaltered between the cell lines, as observed from molecules exhibiting accessible DNA around the CTCF sites (clusters ii and iii). Finally, we observed no highly accessible molecules in the TIVE-LTC cells (cluster iv). Combining data from both the LANA and RTA loci (below) yielded P = 0.01 for the presence of highly open presumably lytic molecules in BCBL1, but not in TIVE-LTC, cells.
We also compared the 114 TPA-untreated RTA reads from BCBL1 to 105 from M.CviPI-treated TIVE-LTC nuclei, and plotted the percent methylation at each site in the amplicon (Figure 8A). Surprisingly, KSHV episomes from TIVE-LTC cells, which except for the LANA region are transcriptionally silenced (32), had less overall endogenous CG methylation than did those from BCBL1 cells at the RTA locus, and lacked the upstream peak (Figure 8A, −313 to −284). The GC accessibility profile was different as well. TIVE-LTC episomes had a different nucleosome organization with less accessible chromatin at the RTA promoter (Figure 8A, −200 to +100 relative to the RTA TSS), but slightly more observed downstream (Figure 8A, between +100 and +200 bp from the RTA TSS). Subtracting BCBL1 footprints from TIVE-LTC footprints (Figure 8D), we found differences in frequency at which nucleosomes were footprinted in the RTA promoter (broad black peaks between −250 and +100 bp). Increased positioning of the N + 1 nucleosome centered upstream of the RTA TSS in TIVE-LTC cells corresponded with an increase in an accessible linker downstream, which was occupied by sub-nucleosomal footprints of variable size (red peak centered at +109, cf. cluster iii in panel C). Thus, the bulk accessibility data suggested that the RTA locus was more compact in TIVE-LTC than in BCBL1 cells. This compaction of the RTA promoter may account for the inability of TIVE-LTC cells to spontaneously reactivate.
DISCUSSION
We applied single-molecule MAPit footprinting to interrogate the chromatin structures of viral episomes in KSHV-infected cells of lymphoid and endothelial origin, which contain ∼80 and 5 copies per cell, respectively. Three loci that differed with respect to expression were analyzed: the highly transcriptionally active LANA promoter, the inactive but poised RTA promoter, and the moderately active but highly RTA-responsive vIL6 promoter (58). At all three loci, MAPit revealed that KSHV episomes within latently infected PEL cells exist in a complex mix of chromatin conformations. Our data suggest that, as evidenced by chromatin architecture, only a fraction of episomes participate in latent gene expression and in reactivation at any given time. Links between nucleosome positioning and CG methylation provided novel evidence that epigenetic drift restricts ability of virus to reactivate lytic transcription. We discuss how this drift may affect important steps within the γ-herpesvirus life cycle, such as long-term maintenance of latency and the rate and ability to reactivate lytic replication.
Heterogeneity in chromatin accessibility
MAPit was originally developed as a probe of heterogeneity in chromatin remodeling (28). We have now detected diverse chromatin states at two human and three viral loci by MAPit. These results were reproducible, verified by restriction endonuclease digestion and consistent with previous single-molecule studies of promoter function (12,28,29).
Heterogeneity in chromatin architecture can occur at even constitutively active promoters, as transcription factors dissociate, bind, recruit remodelers and so forth. However, molecules without accessibility to M.CviPI are presumably inaccessible to other soluble factors as well. Thus, it is difficult to envision how the transcriptional machinery could be recruited to the M.CviPI-inaccessible copies of the promoters GAPDH, LANA and so forth. On the other hand, the half-life of this compacted state in live cells is unknown. It may or may not in all cases be due to long-term silencing. However, as discussed below, at the RTA promoter, at least some episomes were in an epigenetically distinct state with reduced local accessibility to M.CviPI.
Broad peaks of accessibility to M.CviPI, i.e. nucleosome-free regions typically associated with transcriptionally active or poised promoters, were identified near each of the TSSs. In latently infected cells, the NFR at the LANA promoter co-localizes with active histone modifications (H3K9/K14ac and H3K4me3), but lacks detectable repressive marks (H3K27me3 and H3K9me3) (3,4), (R.R, unpublished data). In the absence of stable repressive marks, it is surprising that the NFR was essentially absent from 40 to 50% of episomes in BCBL1 (Figure 2C) and a larger fraction (∼80%) in TIVE-LTC cells (Figure 7B). However, at present, it is not clear if the inaccessible fraction either lacked positive marks or, alternatively, possessed positive marks and lacked an NFR needed to support assembly of the preinitiation complex.
Mechanism of lytic reactivation
Spontaneous viral reactivation occurs as a result of leaky RTA expression. We analyzed chromatin structures during lytic reactivation by both TPA and induction of a conditional RTA transgene. Both latent and lytic episomes were observed. The latter, comprising ∼3% (16% in TREx BCBL1-RTA cells with higher basal RTA expression) of molecules sequenced, were distinguished by their high chromatin accessibility and were absent in TIVE-LTC, in which KSHV cannot reactivate. We hypothesize that these highly GC-methylated reads derive from products of lytic replication and therefore largely lack nucleosomes. Although certain related viruses have been reported to incorporate nucleosomes during lytic replication (59,60), our results suggest that lytically replicated KSHV DNA is largely protein free.
Some of the other clusters might correspond to chromatin conformations present earlier in the lytic cycle. Indeed, MAPit revealed open chromatin at the RTA promoter in latently infected BCBL1 cells. Nucleosomes N − 1 and N + 1 were both occasionally displaced (Figure 3C, clusters iv and vi; Figure 5, clusters iii and iv). To link any of these observed conformations with transcription, RTA was induced either by TPA or by RTA overexpression and resulting changes in chromatin states were determined. Under the two conditions, different classes of intermediate-open promoter molecules were observed to change in abundance at RTA. Those that decreased might have been poised for reactivation, whereas those that increased may represent stages in cycles of active transcription.
In both induction experiments, a pool of molecules with a partly accessible RTA promoter was depleted in response to lytic reactivation (cluster ii in Figures 3 and 5). Promoters with this chromatin architecture were therefore probably primed or poised to respond to activators of RTA. Such molecules comprised 10–20% of the total population. As spontaneous reactivation is rare, it follows that many of the intermediates we observed required some additional event to progress to reactivation. Interestingly, the promoters for RTA and vIL6, both targets of RTA, have been shown to have trimethylation of both H3K4 and H3K27 (3,4) (R.R., unpublished data), the bivalent mark associated with developmentally poised genes (61). Trimethylation of H3K27, which is associated with transcriptional repression, is lost quickly on RTA (and vIL6) promoter activation (3,4). Loss of this mark may allow episomes with an open RTA promoter to progress to lytic reactivation. Alternatively, the mark may be ancillary. In either case, our results show that reactivation as monitored by increased chromatin accessibility is limited to a subset of episomes, even when induced by overexpression of RTA. Increased accessibility in a limited subset of KSHV copies is consistent with the finding that histone H3 binding was not depleted at the RTA and vIL6 promoters under identical conditions of reactivation (4). An uneven distribution of episomes potent for reactivation between cells might contribute to the low rate of spontaneous reactivation, as seen both in BCBL1 cells and in endothelial cells within KS tumors (62).
Cytosine methylation on linker DNA in KSHV
Bands of CG methylation separated by a nucleosome particle length of DNA occurred across nearly all episomes in the population downstream of the three CTCF sites in the LANA gene (Figure 1B and C). It was unlikely coincidence that GC methylation peaked in the same locations (our unpublished observations). CTCF is known to organize arrays of nucleosomes (63). We therefore propose that nucleosomes downstream of CTCF in the LANA ORF are highly positioned. Another potential example would be the methylation patch just upstream of the N − 1 nucleosome in the RTA promoter. A corollary conclusion is that DNMTs methylate linker, but not nucleosomal DNA, in KSHV. This conclusion contradicts a report of preferential methylation of nucleosomal DNA within Arabidopsis (64). Curiously, only linkers between nucleosomes were methylated. No patch of CG methylation appeared in an open TSS or between CTCF sites. Nor were all linkers sites of CG methylation: although the 5′ end of RTA ORF was occupied by nucleosomes at varying translational positions, little CG methylation occurred there. Finally, TIVE-LTC cells, which appeared to have the highest nucleosome density (i.e. least accessibility to M.CviPI) did not develop patches of CG methylation. Evidently, although CG methylation was restricted to linker DNA, it had other unknown determinants as well, post-translational modifications of histones perhaps. Indeed, several DNMTs interact with histone tails [reviewed in (65)].
Epigenetic diversity in KSHV
The discovery of partial CG methylation upstream of RTA provided a means to test whether the diverse chromatin states that we observed in BCBL1 episomes were epigenetic. Günther and Grundoff (3) showed that repression of RTA in latency was independent of DNA methylation in PEL and SLK cells. We have now observed the same in TIVE-LTC cells (Figure 8A). Thus, we do not conclude that methylation of 2–3 sites upstream of the RTA promoter affects transcription of the locus. However, we hypothesize that, as DNA methylation is epigenetic, the sites can be used to ‘fingerprint’ distinct epigenetic lineages within the BCBL1 viral population.
At the RTA promoter, the degree of upstream CG methylation correlated with the chromatin conformation identified by MAPit. Whereas 31% of episomes in the most compact chromatin state (Figure 3C, cluster i) had the upstream patch of CG methylation, only 15% of molecules in the intermediate-open conformations (clusters ii–vi) did (Figure 6B). This uneven distribution suggested that there were at least two epigenetic states within latently infected BCBL1 cells. Furthermore, these states likely differed in their propensity to reactivate lytic expression, as cluster i molecules were unchanged by addition of TPA.
Because the TREx BCBL1-RTA cells, cloned from BCBL1, lacked the upstream patch of CG methylation, we could not test whether resistance to direct RTA induction was epigenetic. However, it was interesting to note that ∼40% of TREx BCBL1-RTA endogenous RTA promoters were in a compact state, and the proportion in this state was not significantly affected by induction of RTA in trans.
Several latent states—L1, L2 and L3—have been defined in a close relative to KSHV, the Epstein–Barr virus (EBV). These states are distinguished by expression of different subsets of the latent genes. In EBV, CTCF restricts spread of DNA methylation (66). Rare events in which CTCF function is compromised could allow EBV to drift between the three latent states. Likewise, it has been proposed that KSHV might have multiple levels of latency (67). Our data suggest three distinct latent states (Figure 9). The deepest level of latency, L0, was seen in TIVE-LTC, which had completely lost their ability to reactivate. Interestingly, TIVE-LTC cells had little trimethylation of H3K4 at the RTA promoter (R.R., unpublished data), lacking the bivalent state previously identified there (3,4). The RTA promoter was occasionally accessible in TIVE-LTC, but never as much as the most open RTA promoters in BCBL1. Either lack of H3K4 trimethylation, presence of H3K27 trimethylation or some other local factor may have prevented the degree of promoter chromatin disassembly needed for reactivation of the gene.
Figure 9.
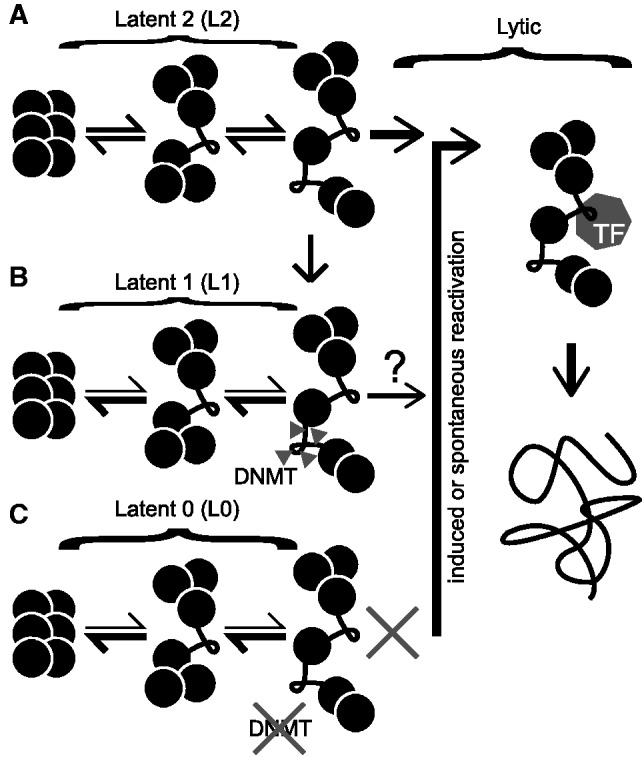
Model for three distinct epigenetic states of KSHV defined by MAPit at RTA promoter. (A) In the most open latent state, Latent 2 (L2), unfolding of higher-order chromatin structure transiently exposes the TSS and an upstream linker. With induced or spontaneous reactivation, transcription factors (polygon labeled TF) replace repressors (not shown) near the TSS. This leads to lytic replication, producing nucleosome-free DNA (far right). (B) The exposed linker in L2 can be CG methylated (triangles) by endogenous DNMTs. This methylation correlates with a shift to another epigenetic state, Latent 1 (L1), in which chromatin tends to be more closed. L1 virus responds to phorbol ester induction of lytic transcription weakly or not at all. (C) A third state, Latent 0 (L0), occurred in TIVE-LTC cells. Although in these cells, different stages of chromatin folding were observed, neither DNMTs nor transcription factors were able to access internucleosomal linker DNA, perhaps due to the presence (or absence) of specific histone modifications. L0 cannot be induced to reactivate by phorbol ester.
The next level of latency, L1, occurring in BCBL1 episomes, was associated with CG methylation upstream of the RTA locus and tended to have a closed chromatin structure. The third level of latency, L2, had the most open structure at RTA, with the promoter poised for reactivation. We have not yet determined the range of chromatin conformations accessed by each epigenetic state, only that the L1 state is more likely to have a closed chromatin conformation at RTA. It seems likely that the same episomes have compact chromatin at the LANA and vIL6 promoters, given that the proportions in the closed state were the same at all three loci.
We hypothesize that infrequent methylation of RTA promoter DNA accompanied chromatin compaction and the transition from L2 to L1. Our findings suggest that the gradual methylation of episomes following infection (3) is linked to phenotypic changes. DNA methylation, at least at the RTA promoter, was associated with loss of both chromatin plasticity (Supplementary Table S3) and accessibility to soluble factors (e.g. M.CviPI). Progressive silencing of KSHV episomes would serve as a natural defense for the host cell. It could eventually lead to repression of LANA and loss of infection, a phenomenon observed in vitro (68,69). These results support a model originally proposed by Grundhoff and Ganem (2004) (68) in which lytic reactivation, albeit infrequent, is required for life-long persistence of KSHV infection. Recently, Kobiler et al. (70), using the Brainbow cassette to trace the lineage of viral genomes during lytic replication, demonstrated that only 6–10 genomes within each cell (i.e. <1%) gave rise to progeny virus. Our observations suggest that a similar restriction may apply to KSHV reactivation from latency.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–3, Supplementary Figures 1–6 and Supplementary References [71–73].
FUNDING
National Institutes of Health [R01CA095525 and R01CA155390 to M.P.K., R01CA088763 to R.R.]; Department of Defense [BC097648 to M.P.K.]; Florida Department of Health [2BT01 to M.P.K.]. Funding for open access charge: NIH [R01CA155390 to M.P.K. and R01CA088763 to R.R.].
Conflict of interest statement. The author M.P.K. has indicated that he shares royalties for the M.CviPI enzyme used as chromatin probe in the article with co-inventors.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Amber L. Delmas and Marc F. Schetelig for performing some of the cloning, Nancy H. Nabilsi for primer design and suggestions with the manuscript, Mayank Talwar for aid with cell culture and Jae Jung (USC) for providing TREx BCBL1-RTA cells.
REFERENCES
- 1.Ganem D. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu. Rev. Pathol. 2006;1:273–296. doi: 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- 2.Lu F, Zhou J, Wiedmer A, Madden K, Yuan Y, Lieberman PM. Chromatin remodeling of the Kaposi's sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 2003;77:11425–11435. doi: 10.1128/JVI.77.21.11425-11435.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Günther T, Grundhoff A. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 2010;6:e1000935. doi: 10.1371/journal.ppat.1000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Toth Z, Maglinte DT, Lee SH, Lee HR, Wong LY, Brulois KF, Lee S, Buckley JD, Laird PW, Marquez VE, et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010;6:e1001013. doi: 10.1371/journal.ppat.1001013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv. Cancer Res. 2007;97:81–109. doi: 10.1016/S0065-230X(06)97004-3. [DOI] [PubMed] [Google Scholar]
- 6.Radman-Livaja M, Rando OJ. Nucleosome positioning: how is it established, and why does it matter? Dev. Biol. 2010;339:258–266. doi: 10.1016/j.ydbio.2009.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Probst AV, Dunleavy E, Almouzni G. Epigenetic inheritance during the cell cycle. Nat. Rev. Mol. Cell. Biol. 2009;10:192–206. doi: 10.1038/nrm2640. [DOI] [PubMed] [Google Scholar]
- 8.Holliday R. Epigenetics of aging. In: Tollefsbol TO, editor. Perspectives in Aging and Epigenetics. New York, NY: Springer; 2010. pp. 447–455. [Google Scholar]
- 9.Zemach A, Zilberman D. Evolution of eukaryotic DNA methylation and the pursuit of safer sex. Curr. Biol. 2010;20:R780–R785. doi: 10.1016/j.cub.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 10.Takeshita F, Leifer CA, Gursel I, Ishii KJ, Takeshita S, Gursel M, Klinman DM. Cutting edge: role of toll-like receptor 9 in CpG DNA-induced activation of human cells. J. Immunol. 2001;167:3555–3558. doi: 10.4049/jimmunol.167.7.3555. [DOI] [PubMed] [Google Scholar]
- 11.Pondugula S, Kladde MP. Single-molecule analysis of chromatin: changing the view of genomes one molecule at a time. J. Cell. Biochem. 2008;105:330–337. doi: 10.1002/jcb.21849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pardo CE, Darst RP, Nabilsi NH, Delmas AL, Kladde MP. Simultaneous single-molecule mapping of protein-DNA interactions and DNA methylation by MAPit. Curr. Protoc. Mol. Biol. 2011 doi: 10.1002/0471142727.mb2122s95. Chapter 21, Unit 21.22.21-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Darst RP, Pardo CE, Pondugula S, Gangaraju VK, Nabilsi NH, Bartholomew B, Kladde MP. Simultaneous single-molecule detection of endogenous C-5 DNA methylation and chromatin accessibility using MAPit. Methods Mol. Biol. 2012;833:125–141. doi: 10.1007/978-1-61779-477-3_9. [DOI] [PubMed] [Google Scholar]
- 14.Xu M, Kladde MP, Van Etten JL, Simpson RT. Cloning, characterization and expression of the gene coding for a cytosine-5-DNA methyltransferase recognizing GpC. Nucleic Acids Res. 1998;26:3961–3966. doi: 10.1093/nar/26.17.3961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frommer M, MacDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc. Natl Acad. Sci. USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kladde MP, Simpson RT. Positioned nucleosomes inhibit Dam methylation in vivo. Proc. Natl Acad. Sci. USA. 1994;91:1361–1365. doi: 10.1073/pnas.91.4.1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kladde MP, Xu M, Simpson RT. Direct study of DNA-protein interactions in repressed and active chromatin in living cells. EMBO J. 1996;15:6290–6300. [PMC free article] [PubMed] [Google Scholar]
- 18.Dechassa ML, Sabri A, Pondugula S, Kassabov SR, Chatterjee N, Kladde MP, Bartholomew B. SWI/SNF has intrinsic nucleosome disassembly activity that is dependent on adjacent nucleosomes. Mol. Cell. 2010;38:590–602. doi: 10.1016/j.molcel.2010.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kelly TK, Miranda TB, Liang G, Berman BP, Lin JC, Tanay A, Jones PA. H2A.Z maintenance during mitosis reveals nucleosome shifting on mitotically silenced genes. Mol. Cell. 2010;39:901–911. doi: 10.1016/j.molcel.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kilgore JA, Hoose SA, Gustafson TL, Porter W, Kladde MP. Single-molecule and population probing of chromatin structure using DNA methyltransferases. Methods. 2007;41:320–332. doi: 10.1016/j.ymeth.2006.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wolff EM, Byun HM, Han HF, Sharma S, Nichols PW, Siegmund KD, Yang AS, Jones PA, Liang G. Hypomethylation of a LINE-1 promoter activates an alternate transcript of the MET oncogene in bladders with cancer. PLoS Genet. 2010;6:e1000917. doi: 10.1371/journal.pgen.1000917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pardo CE, Carr IM, Hoffman CJ, Darst RP, Markham AF, Bonthron DT, Kladde MP. MethylViewer: computational analysis and editing for bisulfite sequencing and methyltransferase accessibility protocol for individual templates (MAPit) projects. Nucleic Acids Res. 2011;39:e5. doi: 10.1093/nar/gkq716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delmas AL, Riggs BM, Pardo CE, Dyer LM, Darst RP, Izumchenko E, Monroe M, Hakam A, Kladde MP, Siegel EM, et al. WIF1 is a frequent target for epigenetic silencing in squamous cell carcinoma of the cervix. Carcinogenesis. 2011;32:1625–1633. doi: 10.1093/carcin/bgr193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.You JS, Kelly TK, De Carvalho DD, Taberlay PC, Liang G, Jones PA. OCT4 establishes and maintains nucleosome-depleted regions that provide additional layers of epigenetic regulation of its target genes. Proc. Natl Acad. Sci. USA. 2011;108:14497–14502. doi: 10.1073/pnas.1111309108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Andreu-Vieyra C, Lai J, Berman BP, Frenkel B, Jia L, Jones PA, Coetzee GA. Dynamic nucleosome-depleted regions at androgen receptor enhancers in the absence of ligand in prostate cancer cells. Mol. Cell. Biol. 2011;31:4648–4662. doi: 10.1128/MCB.05934-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang X, Noushmehr H, Han H, Andreu-Vieyra C, Liang G, Jones PA. Gene reactivation by 5-aza-2′-deoxycytidine-induced demethylation requires SRCAP-mediated H2A.Z insertion to establish nucleosome depleted regions. PLoS Genet. 2012;8:e1002604. doi: 10.1371/journal.pgen.1002604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bell O, Schwaiger M, Oakeley EJ, Lienert F, Beisel C, Stadler MB, Schübeler D. Accessibility of the Drosophila genome discriminates PcG repression, H4K16 acetylation and replication timing. Nat. Struct. Mol. Biol. 2010;17:894–900. doi: 10.1038/nsmb.1825. [DOI] [PubMed] [Google Scholar]
- 28.Jessen WJ, Hoose SA, Kilgore JA, Kladde MP. Active PHO5 chromatin encompasses variable numbers of nucleosomes at individual promoters. Nat. Struct. Mol. Biol. 2006;13:256–263. doi: 10.1038/nsmb1062. [DOI] [PubMed] [Google Scholar]
- 29.Gal-Yam EN, Jeong S, Tanay A, Egger G, Lee AS, Jones PA. Constitutive nucleosome depletion and ordered factor assembly at the GRP78 promoter revealed by single molecule footprinting. PLoS Genet. 2006;2:e160. doi: 10.1371/journal.pgen.0020160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Renne R, Zhong W, Herndier B, McGrath M, Abbey N, Kedes D, Ganem D. Lytic growth of Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 1996;2:342–346. doi: 10.1038/nm0396-342. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura H, Lu M, Gwack Y, Souvlis J, Zeichner SL, Jung JU. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 2003;77:4205–4220. doi: 10.1128/JVI.77.7.4205-4220.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.An FQ, Folarin HM, Compitello N, Roth J, Gerson SL, McCrae KR, Fakhari FD, Dittmer DP, Renne R. Long-term-infected telomerase-immortalized endothelial cells: a model for Kaposi's sarcoma-associated herpesvirus latency in vitro and in vivo. J. Virol. 2006;80:4833–4846. doi: 10.1128/JVI.80.10.4833-4846.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darst RP, Pardo CE, Ai L, Brown KD, Kladde MP. Bisulfite sequencing of DNA. Curr. Protoc. Mol. Biol. 2010 doi: 10.1002/0471142727.mb0709s91. Chapter 7, Unit 7.9.1-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc. Natl Acad. Sci. USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, et al. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009;55:611–622. doi: 10.1373/clinchem.2008.112797. [DOI] [PubMed] [Google Scholar]
- 36.Pearce M, Matsumura S, Wilson AC. Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi's sarcoma-associated herpesvirus originate from a common promoter. J. Virol. 2005;79:14457–14464. doi: 10.1128/JVI.79.22.14457-14464.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, Neiman PE, Lobanenkov VV. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol. Cell. Biol. 1993;13:7612–7624. doi: 10.1128/mcb.13.12.7612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stedman W, Kang H, Lin S, Kissil JL, Bartolomei MS, Lieberman PM. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 2008;27:654–666. doi: 10.1038/emboj.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kang H, Wiedmer A, Yuan Y, Robertson E, Lieberman PM. Coordination of KSHV latent and lytic gene control by CTCF-cohesin mediated chromosome conformation. PLoS Pathog. 2011;7:e1002140. doi: 10.1371/journal.ppat.1002140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lieberman PM, Hu J, Renne R. In: Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K, editors. Cambridge: Cambridge University Press; 2007. [PubMed] [Google Scholar]
- 41.Harper CV, Finkenstadt B, Woodcock DJ, Friedrichsen S, Semprini S, Ashall L, Spiller DG, Mullins JJ, Rand DA, Davis JR, et al. Dynamic analysis of stochastic transcription cycles. PLoS Biol. 2011;9:e1000607. doi: 10.1371/journal.pbio.1000607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Oh J, Fraser NW. Temporal association of the herpes simplex virus genome with histone proteins during a lytic infection. J. Virol. 2008;82:3530–3537. doi: 10.1128/JVI.00586-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lukac DM, Renne R, Kirshner JR, Ganem D. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology. 1998;252:304–312. doi: 10.1006/viro.1998.9486. [DOI] [PubMed] [Google Scholar]
- 44.Chang J, Renne R, Dittmer D, Ganem D. Inflammatory cytokines and the reactivation of Kaposi's sarcoma-associated herpesvirus lytic replication. Virology. 2000;266:17–25. doi: 10.1006/viro.1999.0077. [DOI] [PubMed] [Google Scholar]
- 45.Lan K, Kuppers DA, Verma SC, Robertson ES. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting RTA: a potential mechanism for virus-mediated control of latency. J. Virol. 2004;78:6585–6594. doi: 10.1128/JVI.78.12.6585-6594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Deng H, Young A, Sun R. Auto-activation of the RTA gene of human herpesvirus-8/Kaposi's sarcoma-associated herpesvirus. J. Gen. Virol. 2000;81:3043–3048. doi: 10.1099/0022-1317-81-12-3043. [DOI] [PubMed] [Google Scholar]
- 47.Wang SE, Wu FY, Yu Y, Hayward GS. CCAAT/enhancer-binding protein-alpha is induced during the early stages of Kaposi's sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J. Virol. 2003;77:9590–9612. doi: 10.1128/JVI.77.17.9590-9612.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sakakibara S, Ueda K, Chen J, Okuno T, Yamanishi K. Octamer-binding sequence is a key element for the autoregulation of Kaposi's sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 2001;75:6894–6900. doi: 10.1128/JVI.75.15.6894-6900.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang PJ, Chen LW, Shih YC, Tsai PH, Liu AC, Hung CH, Liou JY, Wang SS. Role of the cellular transcription factor YY1 in the latent-lytic switch of Kaposi's sarcoma-associated herpesvirus. Virology. 2011;413:194–204. doi: 10.1016/j.virol.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 50.Wang SE, Wu FY, Chen H, Shamay M, Zheng Q, Hayward GS. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 2004;78:4248–4267. doi: 10.1128/JVI.78.8.4248-4267.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dalton-Griffin L, Wilson SJ, Kellam P. X-box binding protein 1 contributes to induction of the Kaposi's sarcoma-associated herpesvirus lytic cycle under hypoxic conditions. J. Virol. 2009;83:7202–7209. doi: 10.1128/JVI.00076-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gray KS, Allen RD, III, Farrell ML, Forrest JC, Speck SH. Alternatively initiated gene 50/RTA transcripts expressed during murine and human gammaherpesvirus reactivation from latency. J. Virol. 2009;83:314–328. doi: 10.1128/JVI.01444-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen J, Ueda K, Sakakibara S, Okuno T, Parravicini C, Corbellino M, Yamanishi K. Activation of latent Kaposi's sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc. Natl Acad. Sci. USA. 2001;98:4119–4124. doi: 10.1073/pnas.051004198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chandriani S, Ganem D. Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 2010;84:5565–5573. doi: 10.1128/JVI.02723-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kedes DH, Ganem D. Sensitivity of Kaposi's sarcoma-associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J. Clin. Invest. 1997;99:2082–2086. doi: 10.1172/JCI119380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Paulose-Murphy M, Ha NK, Xiang C, Chen Y, Gillim L, Yarchoan R, Meltzer P, Bittner M, Trent J, Zeichner S. Transcription program of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus) J. Virol. 2001;75:4843–4853. doi: 10.1128/JVI.75.10.4843-4853.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Arias C, Walsh D, Harbell J, Wilson AC, Mohr I. Activation of host translational control pathways by a viral developmental switch. PLoS Pathog. 2009;5:e1000334. doi: 10.1371/journal.ppat.1000334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bellare P, Ganem D. Regulation of KSHV lytic switch protein expression by a virus-encoded microRNA: an evolutionary adaptation that fine-tunes lytic reactivation. Cell Host Microbe. 2009;6:570–575. doi: 10.1016/j.chom.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nitzsche A, Paulus C, Nevels M. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 2008;82:11167–11180. doi: 10.1128/JVI.01218-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Placek BJ, Huang J, Kent JR, Dorsey J, Rice L, Fraser NW, Berger SL. The histone variant H3.3 regulates gene expression during lytic infection with herpes simplex virus type 1. J. Virol. 2009;83:1416–1421. doi: 10.1128/JVI.01276-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell. 2006;125:315–326. doi: 10.1016/j.cell.2006.02.041. [DOI] [PubMed] [Google Scholar]
- 62.Staskus KA, Zhong W, Gebhard K, Herndier B, Wang H, Renne R, Beneke J, Pudney J, Anderson DJ, Ganem D, et al. Kaposi's sarcoma-associated herpesvirus gene expression in endothelial (spindle) tumor cells. J. Virol. 1997;71:715–719. doi: 10.1128/jvi.71.1.715-719.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fu Y, Sinha M, Peterson CL, Weng Z. The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 2008;4:e1000138. doi: 10.1371/journal.pgen.1000138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiang CZ, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nat. Rev. Genet. 2009;10:161–172. doi: 10.1038/nrg2522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hashimoto H, Vertino PM, Cheng X. Molecular coupling of DNA methylation and histone methylation. Epigenomics. 2010;2:657–669. doi: 10.2217/epi.10.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tempera I, Wiedmer A, Dheekollu J, Lieberman PM. CTCF prevents the epigenetic drift of EBV latency promoter Qp. PLoS Pathog. 2010;6:e1001048. doi: 10.1371/journal.ppat.1001048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tempera I, Lieberman PM. Chromatin organization of gammaherpesvirus latent genomes. Biochim. Biophys. Acta. 2010;1799:236–245. doi: 10.1016/j.bbagrm.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gründhoff A, Ganem D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J. Clin. Invest. 2004;113:124–136. doi: 10.1172/JCI200417803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Skalsky RL, Hu J, Renne R. Analysis of viral cis elements conferring Kaposi's sarcoma-associated herpesvirus episome partitioning and maintenance. J. Virol. 2007;81:9825–9837. doi: 10.1128/JVI.00842-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kobiler O, Lipman Y, Therkelsen K, Daubechies I, Enquist LW. Herpesviruses carrying a Brainbow cassette reveal replication and expression of limited numbers of incoming genomes. Nat. Commun. 2010;1:146. doi: 10.1038/ncomms1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Melnikov AA, Gartenhaus RB, Levenson AS, Motchoulskaia NA, Levenson Chernokhvostov VV. MSRE-PCR for analysis of gene-specific DNA methylation. Nucleic Acids Res. 2005;33:e93. doi: 10.1093/nar/gni092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yuan CC, Miley W, Waters D. A quantification of human cells using an ERV-3 real time PCR assay. J. Virol. Methods. 2001;91:109–117. doi: 10.1016/s0166-0934(00)00244-5. [DOI] [PubMed] [Google Scholar]
- 73.Lei X, Bai Z, Ye F, Xie J, Kim CG, Huang Y, Gao SJ. Regulation of NF-κB inhibitor IκBα and viral replication by a KSHV microRNA. Nat. Cell. Biol. 2010;12:193–199. doi: 10.1038/ncb2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Deng H, Song MJ, Chu JT, Sun R. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi’s sarcoma-associated herpesvirus) J. Virol. 2002;76:8252–8264. doi: 10.1128/JVI.76.16.8252-8264.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.